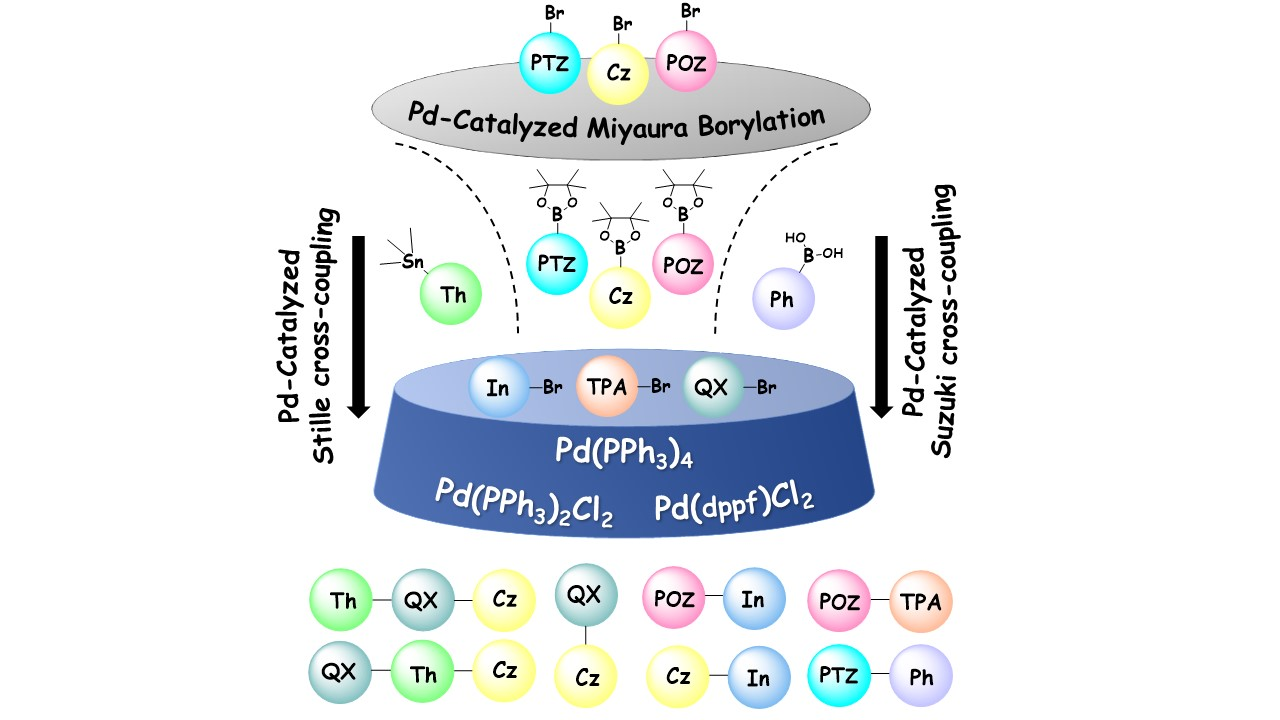
3.2. Synthetic Procedures
3.2.1. Synthesis of 10-octyl-10H-phenothiazine (1a)
Sodium hydroxide (2.0 g, 35.7 mmol) was added to a solution of 10H-phenothiazine (2.4 g, 11.9 mmol), 1-bromooctane (3.5 g, 17.9 mmol), and potassium iodide (catalytic) in 50 mL dimethyl formamide (DMF). The reaction mixture was stirred for 5 h at room temperature, and then 200 mL of water was added. The crude product was extracted with chloroform (3 × 50 mL), and the organic layer was washed with saturated ammonium chloride aqueous solution and then water. The organic layer was dried over anhydrous sodium sulfate. After removing the solvent, the residue was purified by column chromatography on silica gel by using n-Hexane as an eluent to obtain 3.4 g (90%) of compound 1a as a colorless oil. 1H-NMR (850 MHz, CDCl3) δ: 7.12–7.14 (m, 4H, ArH), 6.89 (td, 2H, J = 7.65, 0.85 Hz, ArH), 6.85 (d, 2H, J = 7.65 Hz, ArH), 3.83 (t, 2H, J = 7.65 Hz, N-CH2), 1.79 (qn, 2H, J = 7.65 Hz, CH2), 1.42 (qn, 2H, J = 7.65 Hz, CH2), 1.22–1.32 (m, 8H, 4CH2), 0.86 (t, 3H, J = 7.65 Hz, CH3). 13C-NMR (213 MHz, CDCl3) δ: 145.3, 127.4, 127.1, 124.9, 122.3, 115.4, 47.4, 31.7, 29.2, 29.2, 27.0, 26.9, 22.6, 14.1. IR (cm−1): C-H Olefinic 3066, aliphatic 2923, 2852, C=C stretch 1594, 1571. MS (ESI): m/z calcd for C20H26NS 312.2 [M+1]+, found 312.1.
3.2.2. Synthesis of 10-octyl-10H-phenothiazine-3-carbaldehyde (2a)
To an ice-cooled flask containing N,N-dimethylformamide (0.5 g, 0.5 mL, 7 mmol), phosphoryl chloride (0.3 g, 0.2 mL, 2.1 mmol) was added dropwise with stirring. After the addition, the solution was stirred at room temperature for 90 min. Then, the flask was cooled again in an ice bath, and compound 1a (0.3 g, 1 mmol) was added. The reaction mixture was warmed gradually to run at 75 °C for 12 h. Then, it was cooled to room temperature, poured into ice water, basified with a saturated aqueous solution of potassium carbonate, extracted with chloroform (4 × 30 mL, washed, dried with magnesium sulfate, evaporated, and purified by column chromatography using petroleum ether:ethyl acetate (98:2) as an eluent to obtain 0.25 g (75%) of compound 2a as a yellow oil. 1H-NMR (850 MHz, CDCl3) δ: 9.79 (s, 1H, CHO), 7.64 (dd, 1H, J = 8.5, 1.7 Hz, ArH), 7.58 (d, 1H, J = 1.7 Hz, ArH), 7.15–7.17 (m, 1H, ArH), 7.11 (dd, 1H, J = 7.65, 0.85 Hz, ArH), 6.96 (td, 1H, J = 7.65, 0.85 Hz, ArH), 6.87–6.90 (m, 2H, ArH), 3.88 (t, 2H J = 7.65 Hz, N-CH2), 1.81 (qn, 2H, J = 7.65 Hz, CH2), 1.43 (qn, 2H, J = 7.65 Hz, CH2), 1.21–1.33 (m, 8H, 4CH2), 0.86 (t, 3H, J = 7.65 Hz, CH3). 13C-NMR (213 MHz, CDCl3) δ: 190.1, 150.8, 143.5, 131.0, 130.1, 128.4, 127.6, 127.5, 125.0, 123.8, 123.6, 115.9, 114.8, 48.0, 31.7, 29.7, 29.2, 29.1, 26.8, 26.7, 22.6, 14.1. IR (cm−1): C-H aliphatic 2924, 2853, C-H aldehyde 2723, C=O aldehyde 1683, C=C stretch 1597, 1573. MS (ESI): m/z calcd for C21H26NOS 340.2 [M+1]+, found 340.2.
3.2.3. Synthesis of 7-bromo-10-octyl-10H-phenothiazine-3-carbaldehyde (3a)
In an ice bath, compound 2a (5.8 g, 17 mmol) was dissolved thoroughly in dry THF (85 mL), followed by the addition of N-bromosuccinimide (NBS) (3.2 g, 18. Mmol). The reaction system stirred for 1 h at 0 °C and then left at room temperature to stir until completion. The workup and purification process was handled as described above. The product was extracted with chloroform, dehydrated with Na2SO4, and purified by re-crystallization with EtOH to give a yellow-orange oil (7.0 g, 98%). 1H-NMR (850 MHz, CDCl3): δ in ppm 0.86 (t, 3H, J = 7.2 Hz, CH3), 1.23–1.31 (m, 8H, 4CH2), 1.42 (quint, 2H, J = 7.6 Hz, CH2), 1.78 (quint, 2H, J = 7.4 Hz, CH2), 3.85 (t, 2H, J = 7.2 Hz, CH2), 6.71 (d, 1H, J = 8.7 Hz, ArH), 6.9 (d, 1H, J = 8.5 Hz, ArH), 7.22 (d, 1H, J = 2.3 Hz, ArH), 7.25 (dd, 1H, J = 8.6, 2.3 Hz, ArH), 7.57 (d, 1H, J = 1.9 Hz, ArH), 7.65 (dd, 1H, J = 8.5, 1.9 Hz, ArH), 9.8 (s, 1H, CHO). 13C-NMR (214 MHz, CDCl3): δ in ppm 14.07 (CH3), 22.57 (CH2), 26.57 (CH2), 26.7 (CH2), 29.07 (CH2), 29.13 (CH2), 31.66 (CH2), 48.02 (NCH2), 114.97 (CH), 115.78 (C), 117.06 (CH), 124.34 (C), 126.09 (C), 128.43 (CH), 129.75 (CH), 130.2 (CH), 130.24 (CH), 131.21 (C), 142.58 (C), 150.3 (C), 189.03 (CHO). IR (cm−1): C-H olefinic 3051, C-H aliphatic 2943, 2852, C-H aldehyde 2725, C=O 1684, C=C 1593, 1563. MS (ESI): m/z calcd for C21H25BrNOS 418.1 [M+1]+ and 420.1 [M+3]+, found 418.0 and 420.0, respectively.
3.2.4. Synthesis of 3-bromo-10-octyl-7-(1,4,5-triphenyl-1H-imidazol-2-yl)-10H-phenothiazine (4)
To a solution of compound 3a (0.4 g, 1 mmol) and phenylamine (0.1 g, 1.5 mmol) in glacial acetic acid, benzil (0.2 g, 1 mmol), and ammonium acetate (0.4 g, 5 mmol) was added. The mixture was stirred at 110 °C for 12 h. After cooling, the solution was poured into a copious amount of water and then neutralized with sodium bicarbonate solution. The dark precipitate was filtered and washed with water. The product was then purified by column chromatography on silica gel by using petroleum ether:ethyl acetate (97:3) as an eluent, affording compound 4 as an off-white powder (0.5 g (82%), m.p. 145 °C). 1H-NMR (850 MHz, DMSO-d6) δ: 7.47–7.48 (m, 2H, ArH), 7.32–7.37 (m, 5H, ArH), 7.28–7.30 (m, 5H, ArH), 7.23–7.26 (m, 4H, ArH), 7.18 (t, 1H, J = 7.65 Hz, ArH), 7.14 (dd, 1H, J = 8.5, 1.7 Hz, ArH), 7.12 (d, 1H, J = 1.7 Hz, ArH), 6.91–6.94 (m, 2H, ArH), 3.80 (t, 2H, J = 6.8 Hz, N-CH2), 1.61 (qn, 2H, J = 7.65 Hz, CH2), 1.33 (qn, 2H, J = 7.65 Hz, CH2), 1.18–1.23 (m, 8H, 4CH2), 0.83 (t, 3H, J = 7.65 Hz, CH3). 13C-NMR (213 MHz, DMSO-d6) δ: 145.4, 144.8, 143.8, 136.1, 131.7, 131.6, 130.7, 130.1, 130.0, 129.7, 129.5, 129.4, 129.2, 128.9, 128.6, 128.1, 126.9, 126.8, 125.9, 122.8, 118.1, 115.9, 114.4, 47.0, 31.5, 29.0, 28.9, 26.4, 22.5, 14.4. IR (cm−1): C-H olefinic 3061, C-H aliphatic 2922, 2854, C=C stretch 1600. MS (ESI): m/z calcd for C41H39BrN3S 684.2 [M+1]+ and 686.2 [M+3]+, found 684.2 and 686.2, respectively.
3.2.5. Synthesis of 10-octyl-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-7-(1,4,5-triphenyl-1H-imidazol-2-yl)-10H-phenothiazine (5)
A solution of 4 (0.7 g, 1 mmol), bis(pinacolato)diboron (0.3 g, 1.1 mmol), potassium acetate (0.3 g, 2.5 mmol), and Bis(triphenylphosphine)palladium(II) dichloride (0.02 g, 0.02 mmol) in dry toluene (40 mL) was refluxed at 110 °C under an N2 atmosphere for 18 h. After being cooled to room temperature, the mixture was poured into water (40 mL), and the organic layer was separated. The aqueous layer was extracted with dichloromethane (3 × 40 mL), and the combined organic layers were dried over anhydrous sodium sulphate. The organic solvent was evaporated to dryness, and the crude product was purified by silica gel column chromatography, eluting with petroleum ether:ethyl acetate (94:6) to obtain 0.4 g (60%) of a yellow solid (m.p.: 140 °C). 1H-NMR (850 MHz, CDCl3) δ: 7.61 (d, 2H, J = 7.65 Hz, ArH), 7.58 (dd, 1H, J = 8.5, 1.7 Hz, ArH), 7.51 (d, 1H, J = 1.7 Hz, ArH), 7.21–7.34 (m, 10H, ArH), 7.13–7.14 (m, 3H, ArH), 7.07 (m, 2H, ArH), 6.80 (d, 1H, J = 8.5 Hz, ArH), 6.67 (d, 1H, J = 8.5 Hz, ArH), 3.80 (t, 2H, J = 7.65 Hz, NCH2), 1.76 (qn, 2H, J = 7.65 Hz, CH2), 1.39 (qn, 2H, J = 7.65 Hz, ArH), 1.34 (s, 12H, 4CH3), 1.25–1.30 (m, 8H, 4CH2), 0.89 (t, 3H, J = 7.65 Hz, CH3). 13C-NMR (213 MHz, CDCl3) δ: 147.24, 146.09, 144.66, 136.95, 134.11, 133.76, 131.12, 130.67, 130.51, 129.20, 128.43, 128.35, 128.20, 127.97, 127.76, 127.64, 127.47, 126.67, 124.57, 123.50, 114.69, 114.62, 83.73, 47.49, 31.76, 29.21, 29.18, 26.81, 26.64, 24.85, 22.65, 14.15. IR (cm−1): C-H aliphatic 2924, 2854, C=C stretch 1580. MS (ESI): m/z calcd for C47H51BN3O2S 732.4 [M+1]+, found 732.6.
3.2.6. Synthesis of 10-octyl-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-10H-phenothiazine-3-carbaldehyde (6a)
Potassium acetate (4.2 g, 0.03 mol) and Pd(dppf)Cl2 (0.4 g, 5 × 10−4 mol) were added to the mixture of the brominated derivatives 3a (4.2 g, 0.01 mol) and bis(pinacolato)diboron (3.1 g, 0.012 mol) in dry 1,4-dioxane (100 mL). The reactions took place under a nitrogen atmosphere with reflux at 90 °C for 34 h. The mixtures were left to cool down to room temperature before performing extraction with CHCl3 (20 mL × 5–6 times). After extraction, the organic layers were dried with anhydrous Na2SO4 and worked up as usual. Purification with column chromatography, using the system PE/EA (9.5:0.5) as an eluent, afforded the desired pure compound as a yellow oil (3.5 g, 74%). 1H-NMR (850 MHz, CDCl3): δ in ppm 9.78 (s, 1H, CHO), 7.61 (dd, 1H, J = 8.4, 1.7 Hz, ArH), 7.58 (dd, 1H, J = 8.1, 1.1 Hz, ArH), 7.55 (d, 1H, J = 1.7 Hz, ArH), 7.52 (d, 1H, J = 1.1 Hz, ArH), 6.88 (d, 1H, J = 8.4 Hz, ArH), 6.84 (d, 1H, J = 8.2 Hz, ArH), 3.87 (t, 2H, J = 7.3 Hz, CH2), 1.79 (quint, 2H, J = 7.4 Hz, CH2), 1.41 (quint, 2H, J = 7.4 Hz, CH2), 1.32 (s, 12H, 4CH3), 1.21–1.3 (m, 8H, 4CH2), 0.85 (t, 3H, J = 7.3 Hz, CH3). 13C-NMR (214 MHz, CDCl3): δ in ppm 190.12 (CHO), 150.25 (C), 145.92 (C), 134.39 (CH), 133.9 (CH), 131.18 (C), 129.96 (CH), 128.42 (CH), 125.14 (C), 122.96 (C), 115.26 (CH), 114.92 (CH), 83.88 (2C), 48.01 (NCH2), 31.72 (CH2), 29.18 (CH2), 29.13 (CH2), 26.75 (CH2), 26.64 (CH2), 24.85 (4CH3), 22.63 (CH2), 14.13 (CH3). IR cm−1: C-H olefinic 3043, C-H aliphatic 2954, 2853, C-H aldehyde 2724, C=O 1686, C=C 1602, 1578.
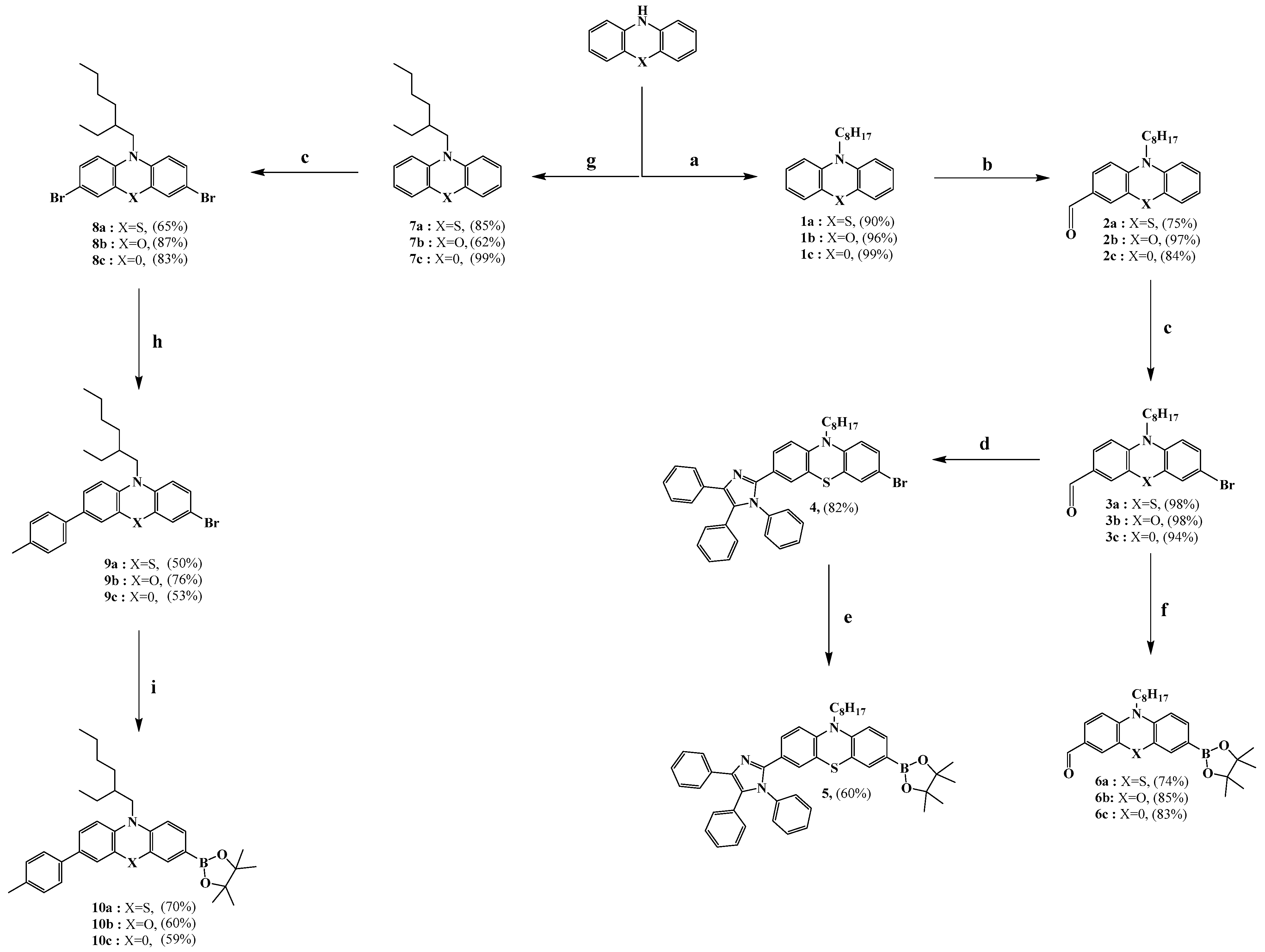
3.2.7. Synthesis of 5-bromo-1-ethyl-2,3,3-trimethyl-3H-indolium iodide (11)
N-alkylation of the indolenine derivative was carried out through the dissolution of a mixture of 5-bromo-2,3,3-trimethylindolenine (14.3 g, 60 mmol) and iodoethane (28.0 g, 180 mmol) in 150 mL acetonitrile at room temperature, which was then refluxed at 85 °C for 48 h. Then, the CH3CN was evaporated using a rotary evaporator. Pure brown crystals 11 (21.2 g, 89.6%) with an m.p. of 220.5 °C were obtained by concentrating the residue in acetone and re-precipitating with diethyl ether (five times). 1H-NMR (600 MHz, DMSO-d6): δ in ppm 8.18 (d, 1H, J = 1.7 Hz, ArH), 7.94 (d, 1H, J = 8.5 Hz, ArH), 7.85 (dd, 1H, J = 8.6, 1.8 Hz, ArH), 4.47 (q, 2H, J = 7.3 Hz, CH2), 2.81 (s, 3H, CH3), 1.54 (s, 6H, 2CH3), 1.42 (t, 3H, J = 7.4 Hz, CH3). 13C-NMR (214 MHz, DMSO-d6): δ in ppm 197.08, 144.68, 140.48, 132.37, 127.34, 123.25, 117.71, 54.86, 43.8, 22.11, 14.37, 13.01.IR, cm−1: C-H olefinic 3049,C-H aliphatic 2985, 2969, C=N 1605, C=C 1451.
3.2.8. Synthesis of (E)-5-bromo-2-(4-(dimethylamino)styryl)-1-ethyl-3,3-dimethyl-3H-indolium iodide (12)
5-bromo-1-ethyl-2,3,3-trimethyl-3H-indolium iodide (10 mmol, 3.9 g) and 4-dimethylaminobenzaldehyde (12 mmol, 1.6 g) were added to 50 mL ethanol. A catalytic amount of piperidine (1 mmol, 0.1 g, 0.1 mL) was added and refluxed at 60 °C overnight. It was then cooled to room temperature, evaporated, and finally purified by reprecipitation with diethyl ether (3–5 times) to yield 5.2 g (99%) of dark green crystals (m.p.: 219 °C). 1H-NMR (850 MHz, DMSO-d6): δ (ppm) 8.35 (d, 1H, J = 15.5 Hz, =CH), 8.12–8.11 (m, 3H, 3ArH), 7.74 (dd, 1H, J = 8.5, 1.9 Hz, ArH), 7.67 (d, 1H, J = 8.5 Hz, ArH), 7.22 (d, 1H, J = 15.5 Hz, =CH), 6.89 (d, 2H, J = 9.1 Hz, 2ArH), 4.5 (q, 2H, J = 7.3 Hz, CH2), 3.18 (s, 6H, 3CH2), 1.75 (s, 6H, 3CH2), 1.35 (t, 3H, J = 7.3 Hz, CH3). 13C-NMR (213.77 MHz, DMSO-d6): δ (ppm) 178.55 (C=N), 155.17 (CH), 154.81 (C), 145.01 (C), 140.18 (C), 131.64 (2CH), 126.21 (CH), 122.44 (C), 120.15 (C), 115.19 (2CH), 112.35 (CH), 104.11 (2CH), 51.01 (C), 40.57 (CH2), 40.01 (2CH3), 26.29 (2CH3), 13.09 (CH3). IR (cm−1): C-H olefinic 3073, 3016, C-H aliphatic 2972, 2911, C=N 1612, 1591, C=C 1566, 1523. HRMS (ESI): m/z calc. mass for C22H26BrN2 is 397.12739 [M]+ and 399.12534 [M+2]+, found 397.12741 and 399.12519.
3.2.9. Synthesis of 5-bromo-1-ethyl-3,3-dimethyl-2-((E)-3-((E)-1,3,3-trimethylindolin-2-ylidene)prop-1-en-1-yl)-3H-indol-1-ium iodide (13)
5-Bromo-1-ethyl-2,3,3-trimethyl-3H-indolium iodide (11.8 g, 30 mmol) and (E)-2-(1,3,3-trimethylindolin-2-ylidene)acetaldehyde (7.2 g, 36 mmol) were dissolved in a dry flask containing 100 mL of acetic anhydride and left under reflux with stirring at 90 °C for 24 h. After cooling to room temperature, the mixture was dried by vacuum. Purification of the product was carried out by reprecipitation with diethyl ether (three times). The product was obtained in a quantitative yield (17.3 g, 99%) as a dark blue powder (293 °C). 1H-NMR (850 MHz, DMSO-d6): δ (ppm) 8.33 (dd, 1H, J = 13.5 Hz, =CH), 7.93 (d, 1H, J = 2 Hz, ArH), 7.66 (d, 1H, J = 7.3 Hz, ArH), 7.63 (dd, 1H, J = 8.3, 2 Hz, ArH), 7.51 (d, 1H, J = 7.9 Hz, ArH), 7.47 (ddd, 1H, J = 8, 8, 1.1 Hz, ArH), 7.41 (d, 1H, J = 8.4 Hz, ArH), 7.33 (ddd, 1H, J = 7.3, 7.3, 0.9 Hz, ArH), 6.53 (d, 1H, J = 13.6 Hz, =CH), 6.47 (d, 1H, J = 13.3 Hz, =CH), 4.13 (q, 2H, J = 7.2 Hz, CH2), 3.68 (s, 3H, CH3), 1.70 (s, 6H, 2CH3), 1.69 (s, 6H, 2CH3), 1.30 (t, 3H, J = 7.3 Hz, CH3). 13C-NMR (213.77 MHz, DMSO-d6): δ (ppm) 175.05 (C=N), 172.35 (C=N), 149.71 (CH), 142.87 (C), 142.55 (C), 140.93 (C), 140.72 (C), 131.35 (CH), 128.59 (CH), 125.78 (2CH), 125.54 (CH), 122.43 (CH), 117.13 (C), 112.88 (CH), 111.81 (CH), 103.74 (CH), 101.99 (CH), 49.05 (C), 48.82 (C), 38.92 (CH2), 31.64 (CH3), 27.24 (2CH3), 27.16 (2CH3), 21.05 (C), 12.06 (CH3). IR (cm−1): C-H olefinic 3015, C-H aliphatic 2991, 2968, 2941, C=N 1607, C=C 1549, 1403. HRMS (ESI): m/z calc. mass for C26H30BrN2 is 449.15869 [M]+ and 451.15664 [M+2]+, found 449.15840 and 451.15628, respectively.
3.2.10. General Procedure of the Suzuki Coupling Reaction for the Synthesis of (14, 15a–c), (15a), (15b), (15c)
Before adding any of the reactants or reagents, a mixture of toluene/methanol solvents (1:1) was brought to an inert condition by degassing and purging Ar gas for several minutes. The boronic ester, brominated compound, catalyst (Pd(dppf)Cl2), and base (K2CO3) were added to the inert mixture of solvents in the following proportions (1.5:1:0.1:5). The mixture was purged again with Ar and then left to reflux at 80 °C until completion.
(E)-2-(4-(dimethylamino)styryl)-1-ethyl-5-(7-formyl-10-octyl-10H-phenoxazin-3-yl)-3,3-dimethyl-3H-indol-1-ium iodide (14)
A mixture of 12 (1.3 g, 2.5 mmol), 6b (1.8 g, 3.8 mmol), Pd(dppf)Cl2 (0.2 g, 0.3 mmol), and K2CO3 (1.7 g, 12.5 mmol) was added to the inert solvent mix (Toluene/MeOH, 10:10 mL). The reaction vessel was left to reflux under argon for 24 h. The reaction mixture was then evaporated and filtered by DCM, and the filtrate was finally purified by column chromatography, with a gradual increase in polarity from CHCl3 to CHCl3:MeOH (7:3). The compound was obtained in a low yield (0.5 g, 26%) as a dark blue solid (m.p.: 248 °C). 1H-NMR (850 MHz, DMSO-d6): δ (ppm) 9.69 (s, 1H, CHO), 8.35 (d, 1H, J = 15.5 Hz, =CH), 8.12 (d, 1H, J = 1.6 Hz, ArH), 8.11 (broad d, 2H, J = 6.7 Hz, 2ArH), 7.82 (dd, 1H, J = 8.5, 1.7 Hz, ArH), 7.74 (d, 1H, J = 8.5 Hz, ArH), 7.47 (dd, 1H, J = 8.3, 1.8 Hz, ArH), 7.36 (dd, 1H, J = 8.4, 2.1 Hz, ArH), 7.27 (d, 1H, J = 15.5 Hz, =CH), 7.24 (d, 1H, J = 2.1 Hz, ArH), 7.04 (d, 1H, J = 1.8 Hz, ArH), 6.92 (dd, 2H, J = 8.5, 5.4 Hz, 2ArH), 6.90 (d, 2H, J = 9.1 Hz, 2ArH), 4.56 (q, 2H, J = 7.2 Hz, CH2), 7.71 (t, 2H, J = 8.0 Hz, CH2), 3.18 (s, 6H, 2CH3), 1.81 (s, 6H, 2CH3), 1.16 (quint, 3H, J = 7.4 Hz, CH3), 1.44 (quint, 2H, J = 7.5 Hz, CH2), 1.40 (t, 3H, J = 7.3 Hz, CH3), 1.36 (quint, 2H, J = 7.9 Hz, CH2), 1.31–1.26 (m, 4H, 2CH2), 0.87 (t, 3H, J = 7.0 Hz, CH3). 13C-NMR (213.77 MHz, DMSO-d6): δ (ppm) 189.95 (CHO), 178.43 (C=N)), 154.30 (C), 154.50 (C), 144.22 (C), 144.04 (C), 143.72 (C), 139.86 (C), 138.22 (C), 138.03 (C), 133.09 (C), 130.83 (C), 129.65 (C), 129.63 (C), 129.20 (CH), 126.18 (CH), 122.62 (CH), 122.42 (C), 120.33 (2CH), 113.69 (CH), 113.45 (CH), 113.34 (CH), 113.02 (CH), 112.21 (CH), 111.86 (CH), 104.53 (CH), 50.98 (C), 43.18 (CH2), 40.56 (CH2), 39.91 (2CH3), 31.22 (CH2), 28.80 (CH2), 28.71 (CH2), 26.42 (2CH3), 25.97 (CH2), 24.60 (CH2), 22.07 (CH2), 13.97 (CH3), 13.26 (CH3). IR (cm−1): C-H olefinic 3052, C-H aliphatic 2922, 2851, C=O 1671, C=N 1608, C=C 1567, 1502. HRMS (ESI): m/z calc. mass for C44H50O2N3 is 640.38975 [M]+, found 640.38899.
1-ethyl-5-(7-formyl-10-octyl-10H-phenothiazin-3-yl)-3,3-dimethyl-2-((E)-3-((E)-1,3,3-trimethylindolin-2-ylidene)prop-1-en-1-yl)-3H-indol-1-ium iodide (15a)
The mixture of 13 (1.6 g, 2.7 mmol), 6a (1.9 g, 4.1 mmol), Pd(dppf)Cl2 (0.2 g, 0.3 mmol), and K2CO3 (1.9 g, 13.5 mmol) was refluxed in inert toluene/MeOH (10:10 mL) for half an hour. After completion, the mixture was evaporated, extracted with CHCl3, and then reprecipitated from DCM by PE. A pure dark violet solid of compound 15a was obtained with a yield of 2.1 g (93%) and an m.p. of 172 °C. 1H-NMR (850 MHz, DMSO-d6): δ (ppm) 9.81 (s, 1H, CHO), 8.35 (t, 1H, J = 13.4 Hz, =CH), 7.98 (d, 1H, J = 1.6 Hz, ArH), 7.74 (dt, 2H, J = 8.4, 1.7 Hz, 2ArH), 7.64 (d, 1H, J = 7.3 Hz, ArH), 7.63 (d, 1H, J = 1.9 Hz, ArH), 7.61 (dd, 1H, J = 8.5, 2.1 Hz, ArH), 7.59 (d, 1H, J = 2.2 Hz, ArH), 7.51 (d, 1H, J = 8.4 Hz, ArH), 7.48–7.45 (m, 2H, 2ArH), 7.31 (dt, 1H, J = 7.4, 1.8 Hz, ArH), 7.21 (d, 1H, J = 8.5 Hz, ArH), 7.17 (d, 1H, J = 8.6, ArH), 6.5 (d, 1H, J = 13.4 Hz, =CH), 6.50 (d, 1H, J = 13.5 Hz, =CH), 4.19 (q, 2H, J = 7.1 Hz, CH2), 4.00 (t, 3H, J = 7 Hz, CH2), 3.66 (s, 3H, CH3), 1.75 (s, 6H, 2CH3), 1.71 (s, 6H, 2CH3), 1.41 (quint, 2H, CH2), 1.34 (t, 3H, J = 7.3 Hz, CH3), 1.28 (quint, 2H, J = 7.8, CH2), 1.24–1.19 (m, 8H, 4CH2), 0.82 (t, 3H, J = 6.9 Hz, CH3). 13C-NMR (213.77 MHz, DMSO-d6): δ (ppm) 190.65, 174.45, 173.11, 149.79, 149.49, 142.70, 142.30, 141.63, 140.88, 140.64, 135.87, 134.84, 130.96, 130.42, 128.65, 127.79, 126.55, 126.20, 125.32, 125.11, 123.32, 123.22, 122.46, 120.55, 116.92, 115.69, 111.63, 111.58, 103.11, 102.47, 49.03, 48.93, 47.03, 39.07, 31.12, 28.63, 28.48, 27.42, 27.34, 26.06, 25.94, 22.6, 13.99, 12.32. IR (cm−1): C-H olefinic 3030, C-H aliphatic 2926, 2854, C=N 1682, C=O 1557, C=C 1446, 1413. HRMS (ESI): m/z calc. mass for C47H54N3OS+ is 708.3982 [M]+, found 708.3790.
1-ethyl-5-(7-formyl-10-octyl-10H-phenoxazin-3-yl)-3,3-dimethyl-2-((E)-3-((E)-1,3,3-trimethylindolin-2-ylidene)prop-1-en-1-yl)-3H-indol-1-ium iodide (15b)
The starting materials 13 (1.4 g, 2.5 mmol) and 6b (1.8 g, 3.8 mmol) were mixed with the Pd(dppf)Cl2 catalyst (0.2 g, 0.3 mmol) and K2CO3 base (1.7 g, 12.5 mmol) in toluene/MeOH (10:10 mL) and refluxed at 80 °C under Ar for 24 h. The reaction mixture was then evaporated, extracted with chloroform, and reprecipitated from DCM by PE:EA (1:1). Purified 15b was obtained with a yield of 1.9 g (93%) as a dark violet-blue powder (m.p: 223 °C). 1H-NMR (850 MHz, CDCl3): δ (ppm) 9.67 (s, 1H, CHO), 8.41 (t, 1H, J = 13.3 Hz, =CH), 7.52 (dd, 1H, J = 8.2, 1.7 Hz, ArH), 7.46 (d, 1H, J = 1.6 Hz, ArH), 7.39 (td, 1H, J = 7.9, 1.0 Hz, ArH), 7.36 (d, 1H, J = 7.4 Hz, ArH), 7.33 (dd, 1H, J = 8.2, 1.8 Hz, ArH), 7.25–7.22 (m, 3H, ArH & 2 = CH), 7.16 (d, 1H, J = 8.2 Hz, ArH), 7.13 (d, 1H, J = 7.9 Hz, ArH), 7.10 (d, 1H, J = 1.8 Hz, ArH), 7.07 (dd, 1H, J = 8.2, 2.1 Hz, ArH), 6.90 (d, 1H, J = 2.1 Hz, ArH), 6.61 (d, 1H, J = 8.3 Hz, ArH), 6.55 (d, 1H, J = 8.3 Hz, ArH), 4.32 (q, 2H, J = 7.2 Hz, CH2), 3.79 (s, 3H, CH3), 3.56 (t, 2H, J = 8.1 Hz, CH2), 1.74 (s, 6H, 2CH3), 1.72 (s, 6H, 2CH3), 1.50 (t, 3H, J = 7.2 Hz, CH3), 1.45 (quint, 2H, J = 7.5 Hz, CH2), 1.39 (quint, 2H, J = 7.3 Hz, CH2), 1.34–1.28 (m, 8H, 4CH2), 0.89 (t, 3H, J = 7.0 Hz, CH3). 13C-NMR (213.77 MHz, CDCl3): δ (ppm) 189.75 (CHO), 174.16 (C=N), 173.00 (C), 150.49 (C), 145.15 (C), 145.01 (C), 142.84 (C), 141.65 (C), 141.06 (C), 140.59 (C), 138.92 (C), 137.33 (C), 134.49 (C), 131.13 (C), 130.14 (C), 129.00 (2CH), 127.01 (CH), 125.42 (CH), 122.37 (CH), 122.13 (CH), 120.05 (CH), 114.20 (CH), 114.05 (CH), 112.64 (CH), 111.21 (CH), 110.94 (CH), 110.81 (CH), 105.16 (CH), 104.88 (CH), 49.10 (C), 48.95 (C), 44.51 (CH2), 40.43 (CH2), 32.79 (CH3), 31.88 (CH2), 29.45 (CH2), 29.36 (CH2), 28.31 (2CH3), 28.27 (2CH3), 26.97 (CH2), 25.20 (CH2), 22.74 (CH2), 14.22 (CH3), 13.11 (CH3). IR (cm−1): C-H olefinic 3033, C-H aliphatic 2923, 2852, C=N 1672, C=O 1553, C=C 1504, 1416. HRMS (ESI): m/z calc. mass for C47H54O2N3 is 694.42105 [M]+, found 694.42011.
1-ethyl-5-(6-formyl-9-octyl-9H-carbazol-3-yl)-3,3-dimethyl-2-((E)-3-((E)-1,3,3-trimethylindolin-2-ylidene)prop-1-en-1-yl)-3H-indol-1-ium iodide (15c)
The following amounts of the reactant, catalyst, and base were used: 13 (1.6 g, 2.8 mmol), 6c (1.8 g, 4.2 mmol), Pd(dppf)Cl2 (0.2 g, 0.3 mmol), and K2CO3 (1.9 g, 14.0 mmol) were mixed with toluene/MeOH (10:10 mL) in an inert environment and refluxed at 80 °C for half an hour. The purification process was conducted after evaporation and extraction with CHCl3 through reprecipitation from DCM by PE. Compound 15c was obtained as a dark violet solid with a yield of 2.25 g (100%) and an m.p. of 181 °C. 1H-NMR (850 MHz, DMSO-d6): δ (ppm) 10.10 (s, 1H, CHO), 8.89 (d, 1H, J = 1.3 Hz, ArH), 8.75 (d, 1H, J = 1.6 Hz, ArH), 8.38 (t, 1H, J = 13.4 Hz, =CH), 8.14 (d, 1H, J = 1.6 Hz, ArH), 8.04 (dd, 1H, J = 8.5, 1.4 Hz, ArH), 7.95 (dd, 1H, J = 8.5, 1.7 Hz, ArH), 7.91 (dd, 1H, J = 8.2, 1.7 Hz, ArH), 7.83 (d, 1H, J = 8.5 Hz, ArH), 7.81 (d, 1H, J = 8.5 Hz, ArH), 7.65 (d, 1H, J = 7.3 Hz, ArH), 7.59 (d, 1H, J = 8.3 Hz, ArH), 7.48–7.45 (m, 2H, 2ArH), 7.31 (dt, 1, J = 6.8, 1.9 Hz, ArH), 6.55 (d, 1H, J = 13.4 Hz, =CH), 6.50 (d, 1H, J = 13.4 Hz, =CH), 4.52 (t, 2H, J = 7.1 Hz, CH2), 4.24 (quint, 2H, J = 7.2 Hz, CH2), 3.67 (s, 3H, CH3), 1.81 (s, 6H, 2CH3), 1.72 (s, 6H, 2CH3), 1.38 (t, 3H, J = 7.3 Hz, CH3), 1.31–1.26 (m, 4H, 2CH2), 1.23–1.16 (m, 8H, 4CH2), 0.81 (t, 3H, J = 7.2 Hz, CH3). 13C-NMR (213.77 MHz, DMSO-d6): δ (ppm) 191.92, 174.27, 173.18, 149.41, 144.13, 142.72, 141.69, 140.58, 140.48, 138.17, 131.88, 128.63, 128.42, 127.37, 127.15, 125.86, 125.23, 123.88, 123.06, 122.52, 122.45, 121.09, 119.09, 111.72, 111.50, 110.64, 110.10, 102.93, 102.53, 49.12, 48.86, 42.77, 31.17, 28.73, 28.61, 28.54, 27.48, 27.37, 26.41, 22.03, 13.95, 12.38. IR (cm−1): C-H olefinic 3014, C-H aliphatic 2927, 2854, C=N 1682, C=O 1594, C=C 1557, 1447. HRMS (ESI): m/z calc. mass for C47H54N3O+ is 676.4261 [M]+, found 676.4304.
3.2.11. Synthesis of 7-(4-(diphenylamino)phenyl)-10-octyl-10H-phenothiazine-3-carbaldehyde (16)
4-Bromo-N,N-diphenylaniline (0.8 g, 2.5 mmol), 6a (1.2 g, 2.5 mmol), Pd(PPh3)4 (0.1 g, 0.1 mmol), and 2M of K2CO3 (2.5 mL) were mixed with degassed and inert toluene (10 mL). The mixture was refluxed at 90 °C under an Ar atmosphere for 20 h. The mixture underwent evaporation, extraction with DCM, dehydration with Na2SO4, and, finally, purification with column chromatography, using PE:EA (9.5:0.5) as an eluent to obtain a yellow semisolid compound (0.7 g, 49%). 1H-NMR (850 MHz, CDCl3): δ (ppm) 9.79 (s, 1H, CHO), 7.64 (dd, 1H, J = 8.4, 1.9 Hz, ArH), 7.59 (d, 1H, J = 1.9 Hz, ArH), 7.39 (d, 2H, J = 8.6 Hz, 2ArH), 7.35 (d, 1H, J = 7.7 Hz, ArH), 7.31 (d, 1H, J = 2.1 Hz, ArH), 7.27 (dd overlapped with solvent, 4H, J = 8.3, 7.4 Hz, 4ArH), 7.12 (d, 4H, J = 7.6 Hz, 4ArH), 7.11 (d, 2H, J = 8.6 Hz, 2ArH), 7.04 (t, 2H, J = 7.4 Hz, 2ArH), 6.90 (dd, 2H, J = 8.4, 5.5 Hz, 2ArH), 3.90 (t, 2H, J = 7.0 Hz, CH2), 1.84 (quint, 2H, J = 7.5 Hz, CH2), 1.46 (quint, 2H, J = 7.5 Hz, CH2), 1.34 (quint, 2H, J = 7.5 Hz, CH2), 1.30–1.25 (m, 6H, 3CH2), 0.87 (t, 3H, J = 7.1 Hz, CH3). 13C-NMR (213.77 MHz, CDCl3): δ (ppm) 190.13, 150.62, 147.73, 147.31, 142.19, 136.36, 133.55, 131.11, 130.28, 129.42, 128.55, 127.27, 125.76, 125.53, 124.67, 124.57, 124.16, 123.98, 123.12, 116.20, 114.80, 48.21, 31.85, 29.32, 29.28, 26.96, 26.85, 22.74, 14.23. IR (cm−1): C-H olefinic 3033, C-H aliphatic 2952, 2925, C=O 1686, C=C 1581.
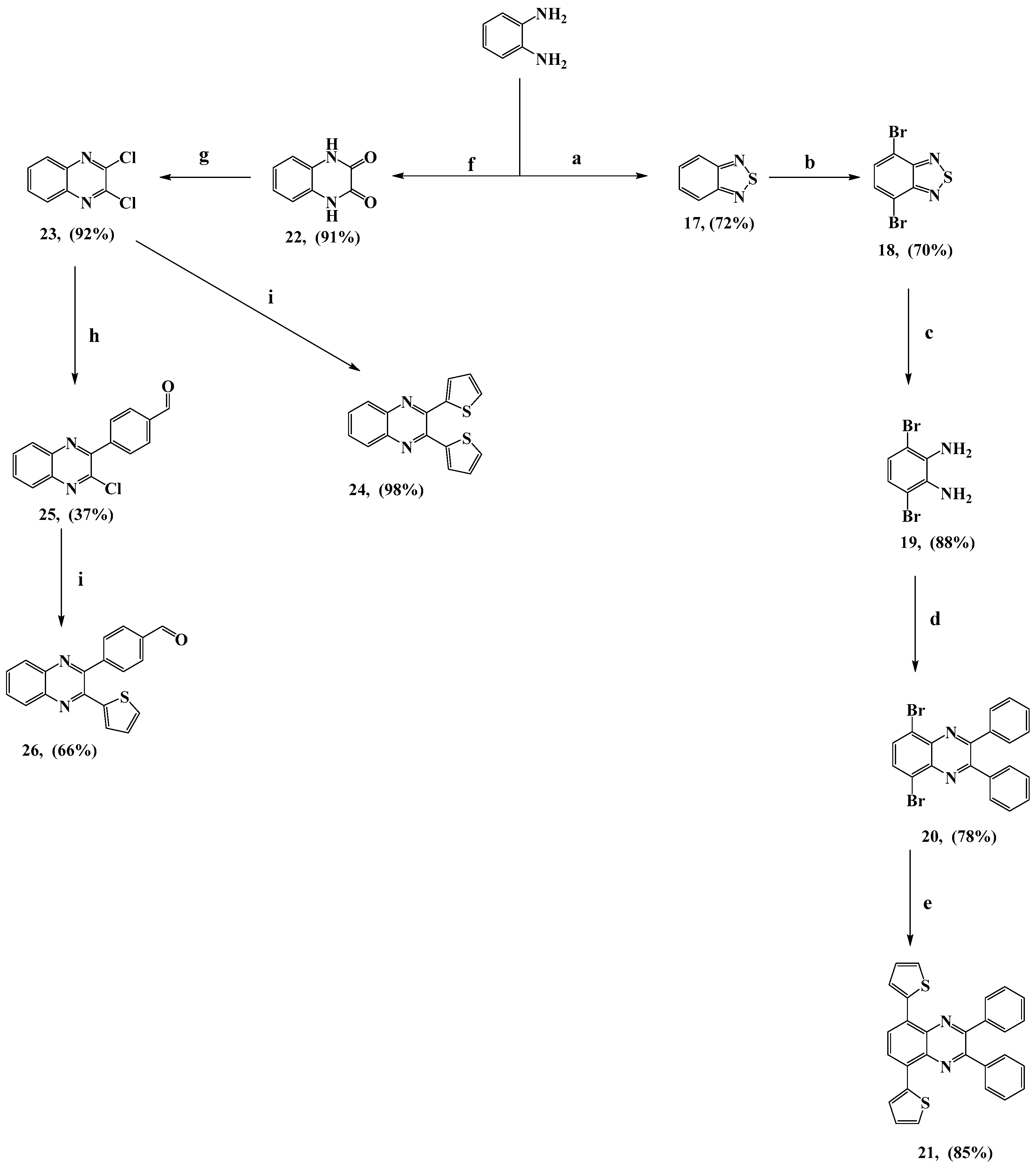
3.2.12. Benzo[c][1,2,5]thiadiazole (17)
To a suspension of the compound o-phenylenediamine (5 g, 46.7 mmol, 1.0 eq) in dry toluene (100 mL), thionyl chloride (SOCl2) (24.6 g, 15 mL, 206.9 mmol, 4.4 eq) was added. It was then refluxed for 3 h, followed by the addition of SOCl2 (5 mL, 1.5 eq) and dry pyridine (1 mL), dropwise. The reaction continued for an additional 19 h while monitoring the reaction by TLC. Toluene and an excess amount of SOCl2 were removed under vacuum, and the residue was collected, poured into water, extracted using EA (50 mL × 4–5 times), washed with brine, and dried over Na2SO4. The solvent was removed under reduced pressure to afford the crude product. Therefore, the purification of the crude product by silica gel column chromatography, using PE/EA (50:1) as an eluent, gave 17 as white needles (4.6 g, 72%) (m.p.: 43 °C). 1H-NMR (600 MHz, CDCl3) δ ppm: 7.99 (dd, 2H, J = 6.6, 3.0 Hz, ArH), 7.57 (dd, 2H, J = 6.6, 3.0 Hz, ArH). 13C-NMR (150 MHz, CDCl3) δ ppm: 121.66, 129.40, 154.90. IR (cm−1): C-H olefinic 3061, C=C 1519, 1478.
3.2.13. 4,7-Dibromobenzo[c][1,2,5]thiadiazole (18)
Compound 17 (5 g, 36.7 mmol, 1.0 eq) was suspended in hydrobromic acid (HBr, 47%) (100 mL). It was then refluxed with stirring, and bromine (Br2) (5.7 mL, 110.3 mmol, ~3.0 eq) was added in a dropwise manner for about 3 h. Then, it was refluxed for another 2 h while monitoring the reaction by TLC. The precipitate was filtered off and recrystallized from a mixture of acetic acid (AcOH) and EtOH to yield 18 as white needles (7.5 g, 70%) (m.p.: 189 °C). 1H-NMR (850 MHz, CDCl3) δ ppm: 7.73 (s, 2H, ArH). 13C-NMR (213 MHz, CDCl3) δ ppm: 114.04, 132.52, 153.09. IR (cm−1): C-H olefinic 3079, 3046, C=C 1587, 1498, 1475.
3.2.14. 3,6-Dibromobenzene-1,2-diamine (19)
Compound 18 (2 g, 6.8 mmol, 1.0 eq) was dissolved in THF (108 mL) and EtOH (40 mL). It was then cooled to 0 °C, and sodium borohydride (NaBH4) (4.4 g, 116 mmol, 17.0 eq) was added in a portion wise manner. Then, the mixture was stirred at RT for 20 h while monitoring the reaction by TLC. The solvent under vacuum was removed after the reaction was complete; the mixture was extracted with diethyl ether (Et2O) (40 mL × 4–5 times), washed with brine, and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure to afford the crude product. Then, purification by silica gel column chromatography, using PE/EA (5:1) as an eluent, gave 19 as a pale-yellow solid (1.6 g, 88%) (m.p. 89 °C). 1H-NMR (600 MHz, CDCl3) δ ppm: 6.84 (s, 2H, ArH), 3.79 (br, s, 4H, 2NH2). 13C-NMR (213 MHz, CDCl3) δ ppm: 109.83, 123.40, 133.86. IR (cm−1): NH2 3396, 3366, N-H bend 1647, C=C 1444.
3.2.15. 5,8-Dibromo-2,3-diphenylquinoxaline (20)
Compound 19 (13.1 g, 49.4 mmol, 1.0 eq) and benzil (10.4 g, 49.7 mmol, 1.0 eq) were mixed in toluene (180 mL) and glacial AcOH (120 mL). It was then refluxed with stirring for 3 h while monitoring the reaction by TLC. It was then cooled to room temperature, followed by solvent removal under vacuum. The residue obtained was extracted from an aqueous solution using CHCl3 (100 mL × 4–5 times), followed by the usual work-up above to afford the crude product. Recrystallization from a mixture of CHCl3/ MeOH (1:4) afforded 20 as white needles (16.9 g, 78%) (m.p. 216 °C). 1H-NMR (850 MHz, CDCl3) δ ppm: 7.92 (s, 2H, ArH), 7.66 (d, 4H, J = 7.65 Hz, ArH), 7.41 (t, 2H, J = 7.65 Hz, ArH), 7.36 (t, 4H, J = 7.65 Hz, ArH). 13C-NMR (213 MHz, CDCl3) δ ppm: 123.85, 128.52, 129.72, 130.38, 133.24, 138.04, 139.48, 154.28. IR (cm−1): C-H olefinic 3064, 3061, C=C 1584, 1454.
3.2.16. 2,3-Diphenyl-5,8-di(thiophen-2-yl)quinoxaline (21)
To a mixture of compound 20 (0.7 g, 1.5 mmol, 1.0 eq) and tributyl(thiophen-2-yl)stannane (1.7 g, 4.5 mmol, 3.0 eq) in dry toluene (20 mL), Pd(PPh3)2Cl2 (0.1 g, 0.1 mmol, 0.05 eq) was added under a nitrogen atmosphere. It was then refluxed for 2 h at 110 °C while monitoring the reaction by TLC. After reaction was complete, the mixture was cooled to room temperature, followed by solvent removal under vacuum. The residue obtained was extracted from an aqueous solution using CHCl3 (20 mL × 4–5 times), followed by the usual work-up to afford the crude product. Therefore, the crude product was purified by silica gel column chromatography, using PE/DCM (3:1) as an eluent, to obtain 21 as an orange solid (0.6 g, 85%) (m.p. 208 °C). Rf = 0.395 (eluent: PE/DCM, 3:1). 1H-NMR (850 MHz, CDCl3) δ ppm: 8.14 (s, 2H, ArH), 7.88 (dd, 2H, J = 3.4, 0.85 Hz, ArH), 7.75 (dd, 4H, J = 7.65, 0.85 Hz, ArH), 7.52 (dd, 2H, J = 5.1, 0.85 Hz, ArH), 7.38–7.41 (m, 6H, ArH), 7.19 (dd, 2H, J = 5.1, 3.4 Hz, ArH). 13C-NMR (213 MHz, CDCl3) δ ppm: 126.58, 126.77, 127.22, 128.37, 129.03, 129.15, 130.62, 131.44, 137.37, 138.82, 138.89, 151.87. IR (cm−1): C-H olefinic 3064, C=C 1633, 1467, 1421.
3.2.17. Quinoxaline-2,3(1H,4H)-dione (22)
The compound o-phenylenediamine (10. 5 g, 96.6 mmol, 1.0 eq) and oxalic acid dihydrate (13.6 g, 107.6 mmol, 1.1 eq) were refluxed in aqueous HCl (4 N, 100 mL) for 2 h at 100 °C while monitoring the reaction by TLC. It was then cooled to room temperature; then, the precipitate was filtered, washed several times with water, and dried to obtain 22 as a gray microcrystalline solid (14.2 g, 91%) (m.p. > 400 °C). 1H-NMR (600 MHz, DMSO-d6) δ ppm: 11.90 (s, 2H, 2NH), 7.12 (dd, 2H, J = 6.0, 3.6 Hz, ArH), 7.07 (dd, 2H, J = 6.0, 3.6 Hz, ArH). 13C-NMR (213 MHz, DMSO-d6) δ ppm: 115.15, 123.02, 125.62, 155.20. IR (cm−1): C-H olefinic 3034, C=O 1671, C=C 1500, 1473.
3.2.18. 2,3-Dichloroquinoxaline (23)
To a solution of compound 22 (11.8 g, 72.5 mmol, 1.0 eq) in POCl3 (77.9 g, 508.1 mmol, 47.5 mL, ~7.0 eq), DMF (1.2 g, 16.1 mmol, 1.3 mL, 0.22 eq) was added. It was then refluxed for 3 h at 110 °C while monitoring the reaction by TLC. Then, it was cooled to room temperature and slowly poured into ice water. Then, the precipitate was filtered, washed with water, and dried to obtain pure 23 as a gray solid (13.3 g, 92%) (m.p. 154 °C). 1H-NMR (600 MHz, CDCl3) δ ppm: 8.03 (dd, 2H, J = 6.6, 3.6 Hz, ArH), 7.81 (dd, 2H, J = 6.6, 3.6 Hz, ArH). 13C-NMR (213 MHz, CDCl3) δ ppm: 128.36, 131.38, 140.71, 145.50. IR (cm−1): C-H olefinic 3043, C=C 1557, 1530, 1485.
3.2.19. 2,3-Di(thiophen-2-yl)quinoxaline (24)
To a mixture of 23 (5.9 g, 30 mmol, 1.0 eq) and tributyl(thiophen-2-yl)stannane (33.6 g, 90 mmol, 3.0 eq) in dry DMF (250 mL), Pd(PPh3)2Cl2 (1.0 g, 1.5 mmol, 0.05 eq) was added under nitrogen atmosphere. It was then refluxed for 1 h at 110 °C while monitoring the reaction by TLC. After the reaction was complete, the temperature was lowered to get the mixture to room temperature, followed by the addition of water to stop the reaction. Following CHCl3 extraction of the product (50 mL × 4–5 times), it was washed with brine and dried over anhydrous Na2SO4. Then, the solvent was removed under reduced pressure to obtain the crude product, and it was purified by column chromatography on silica gel, using PE/DCM (5:2) as an eluent, to obtain compound 24 as a yellow solid (8.7 g, 98%) (m.p. 145 °C). 1H-NMR (600 MHz, CDCl3) δ ppm: 8.08 (dd, 2H, J = 6.6, 3.6 Hz, ArH), 7.72 (dd, 2H, J = 6.6, 3.6 Hz, ArH), 7.50 (dd, 2H, J = 4.8, 1.2 Hz, ArH), 7.25 (dd, 2H, J = 3.6, 1.2 Hz, ArH), 7.04 (dd, 2H, J = 4.8, 3.6 Hz, ArH). 13C-NMR (213 MHz, CDCl3) δ ppm: 127.72, 128.98, 129.04, 129.49, 130.29, 140.75, 141.56, 146.79. IR (cm−1): C-H olefinic 3094, C=C 1550, 1521, 1474.
3.2.20. 4-(3-Chloroquinoxalin-2-yl)benzaldehyde (25)
Compound 23 (0.5 g, 2.5 mmol, 1.0 eq), 4-formylphenylboronic acid (0.6 g, 3.8 mmol, 1.5 eq), K2CO3 (0.3 g, 2.5 mmol, 1.0 eq), and Pd(PPh3)4 (0.1 g, 0.1 mmol, 0.03 eq) were mixed in mixture solvent of dry toluene/MeOH 5:1 (60 mL) under an argon atmosphere and a drying system of CaCl2 with stirring at ambient temperature. Then, the reaction mixture was heated for 20 h at 80 °C and monitored by TLC. After the reaction was complete, the solvent was removed under vacuum and extracted by distilled water and EA (30 mL × 4–5 times). The combined organic extract was washed with brine and dried over anhydrous Na2SO4. Then, the solvent was removed at reduced pressure to afford the crude product and was purified by silica gel column chromatography, using PE/EA (9:1) as an eluent, to remove the residue of the start. This was followed by increasing the polarity of the eluent gradually until 8:2 to obtain the desired compound 25 as a white powder (0.3 g, 37%) (m.p. 167 °C). 1H-NMR (850 MHz, CDCl3) δ ppm: 10.14 (s, 1H, CHO), 8.17 (dd, 1H, J = 7.65, 0.85 Hz, ArH), 8.09 (dd, 1H, J = 7.65, 1.7 Hz, ArH), 8.04–8.07 (m, 4H, ArH), 7.83–7.86 (m, 2H, ArH). 13C-NMR (213 MHz, CDCl3) δ ppm: 128.38, 129.48, 129.67, 130.60, 130.97, 131.67, 136.97, 141.04, 141.46, 142.49, 145.77, 151.73, 191.92. IR (cm−1): C-H olefinic 3042, C-H aldehyde 2819, 2733, C=O 1698, C=C 1604, 1558, 1479. HRMS (ESI): m/z cacld for C15H10ClN2O 269.0482 [M+1]+ and 271.0638 [M+3]+, found 269.0485 and 271.0458, respectively.
3.2.21. 4-(3-(Thiophen-2-yl)quinoxalin-2-yl)benzaldehyde (26)
To a mixture of compound 25 (0.2 g, 0.7 mmol, 1.0 eq) and tributyl(thiophen-2-yl)stannane (0.6 g, 1.5 mmol, 2.0 eq) in dry DMF (30 mL), Pd(PPh3)2Cl2 (0.03 g, 0.04 mmol, 0.05 eq) was added under an argon atmosphere. It was then refluxed for 2 h at 110 °C while monitoring the reaction by TLC. After the reaction was complete, the temperature was lowered to get the mixture to room temperature, followed by the addition of water to stop the reaction. Following CHCl3 extraction of the product (30 mL × 4–5 times), it was washed with brine and dried over anhydrous Na2SO4. Then, the solvent was removed under reduced pressure to obtain the crude product and was purified by column chromatography on silica gel, using PE/EA (9:1) as an eluent, to obtain 26 as a yellow powder (0.2 g, 66%) (m.p. 157 °C). 1H-NMR (850 MHz, CDCl3) δ ppm: 10.13 (s, 1H, CHO), 8.15 (dd, 1H, J = 8.5, 0.85 Hz, ArH), 8.12 (dd, 1H, J = 8.5, 0.85 Hz, ArH), 8.01 (d, 2H, J = 8.5 Hz, ArH), 7.82 (d, 2H, J = 8.5 Hz, ArH), 7.79–7.80 (m, 1H, ArH), 7.75–7.77 (m, 1H, ArH), 7.44 (dd, 1H, J = 4.25, 0.85 Hz, ArH), 6.90 (dd, 1H, J = 5.1, 4.25 Hz, ArH), 6.79 (dd, 1H, J = 3.4, 0.85 Hz, ArH). 13C-NMR (213 MHz, CDCl3) δ ppm: 128.04, 129.00, 129.12, 129.95, 130.13, 130.28, 130.30, 130.40, 131.06, 136.77, 140.31, 141.38, 141.95, 145.09, 146.67, 151.13, 191.97. IR (cm−1): C-H olefinic 3082, C-H aldehyde 2851, 2754, C=O 1694, C=C 1603, 1530. HRMS (ESI): m/z cacld for C19H13N2OS 317.0749 [M+1]+, found 317.0740.
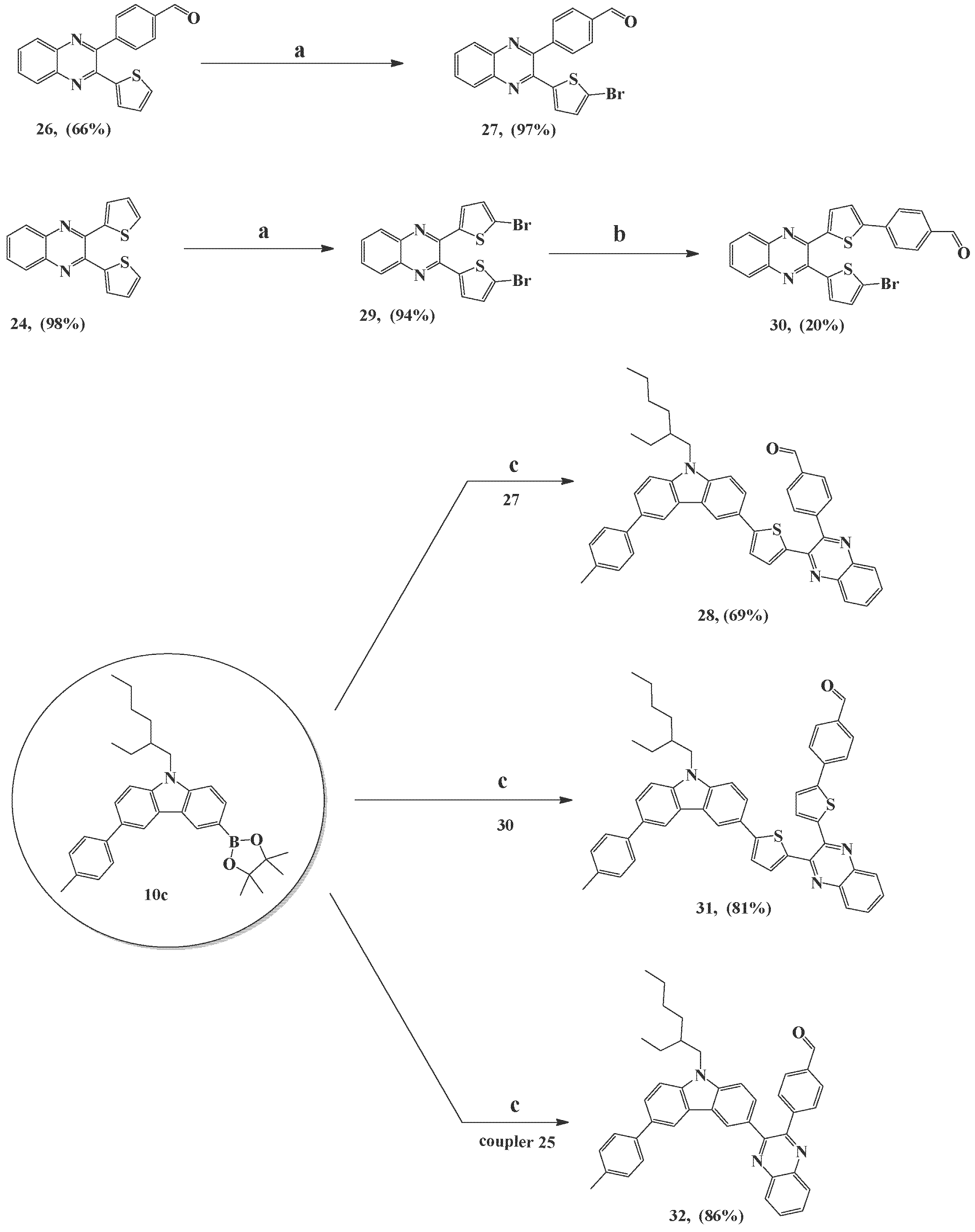
3.2.22. 4-(3-(5-Bromothiophen-2-yl)quinoxalin-2-yl)benzaldehyde (27)
To a solution of compound 26 (0.3 g, 0.9 mmol, 1.0 eq) in DMF (12 mL), a solution of NBS (0.2 g, 1.3 mmol, 1.5 eq) in DMF (3 mL) was added in a dropwise manner. It was then stirred for 24 h in the dark at room temperature while monitoring the reaction by TLC. After completion, the reaction was stopped by adding ice. The precipitate was filtered off and washed several times with distilled water to obtain compound 27 as a yellow powder (0.3 g, 97%) (m.p. 177 °C). 1H-NMR (850 MHz, CDCl3) δ ppm: 10.14 (s, 1H, CHO), 8.10 (t, 2H, J = 7.65 Hz, ArH), 8.03 (d, 2H, J = 7.65 Hz, ArH), 7.82 (d, 2H, J = 7.65 Hz, ArH), 7.80 (t, 1H, J = 7.65 Hz, ArH), 7.76 (t, 1H, J = 7.65 Hz, ArH), 6.83 (d, 1H, J = 4.25 Hz, ArH), 6.45 (d, 1H, J = 4.25 Hz, ArH). 13C-NMR (213 MHz, CDCl3) δ ppm: 117.90, 128.98, 129.25, 130.14, 130.21, 130.26, 130.50, 130.97, 131.13, 136.87, 140.47, 141.33, 144.00, 144.96, 145.54, 150.67, 191.91. IR (cm−1): C-H olefinic 3057, C-H aldehyde 2832, 2738, C=O 1698, C=C 1602, 1530. HRMS (ESI): m/z cacld for C19H12BrN2OS 394.9854 [M+1]+ and 397.0010 [M+3]+ found 394.9854 and 396.9837, respectively.
3.2.23. 4-(3-(5-(9-(2-Ethylhexyl)-6-p-tolyl-9H-carbazol-3-yl)thiophen-2-yl)quinoxalin-2-yl)benzaldehyde (28)
Compound 27 (0.3 g, 0.8 mmol, 1.0 eq), 9-(2-ethylhexyl)-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-6-p-tolyl-9H-carbazole 10c (0.6 g, 1.1 mmol, 1.5 eq), K2CO3 (1.5 g, 10.6 mmol, 14.0 eq), and Pd(PPh3)4 (0.04 g, 0.04 mmol, 0.05 eq) were mixed in a degassed aqueous solvent of toluene/EtOH/H2O 2:1:1 (80 mL) under an argon atmosphere with stirring at ambient temperature. Then, the reaction mixture was heated for 2 h at 80 °C and monitored by TLC. After the reaction was complete, the solvent was removed under vacuum and extracted by distilled water and DCM (30 mL × 4–5 times). The combined organic extract was washed with brine and dried over anhydrous Na2SO4. Then, the solvent was removed at reduced pressure to afford the crude product and was purified by silica gel column chromatography, using PE/EA (9:1) as an eluent, to remove the undesired low polar fractions, followed by increasing the polarity of the eluent gradually until 8:2 to obtain the desired compound 28 as an orange powder (0.4 g, 69%) (m.p. 227 °C). 1H-NMR (850 MHz, CDCl3) δ ppm: 10.16 (s, 1H, CHO), 8.41 (d, 1H, J = 0.85 Hz, ArH), 8.32 (d, 1H, J = 1.7 Hz, ArH), 8.21 (d, 1H, J = 7.65 Hz, ArH), 8.14 (d, 1H, J = 8.5 Hz, ArH), 8.06 (d, 2H, J = 7.65 Hz, ArH), 7.91 (d, 2H, J = 7.65 Hz, ArH), 7.82 (t, 1H, J = 7.65 Hz, ArH), 7.74–7.76 (m, 2H, ArH), 7.72 (dd, 1H, J = 8.5, 1.7 Hz, ArH), 7.63 (d, 2H, J = 7.65 Hz, ArH), 7.44 (d, 1H, J = 8.5 Hz, ArH), 7.39 (d, 1H, J = 8.5 Hz, ArH), 7.31 (d, 2H, J = 7.65 Hz, ArH), 7.14 (d, 1H, J = 4.25 Hz, ArH), 6.75 (d, 1H, J = 4.25 Hz, ArH), 4.16–4.21 (m, 2H, N-CH2), 2.44 (s, 3H, CH3), 2.09 (sept, 1H, J = 6.8 Hz, CH), 1.36–1.46 (m, 4H, 2CH2), 1.26–1.35 (m, 4H, 2CH2), 0.94 (t, 3H, J = 7.65 Hz, CH3), 0.88 (t, 3H, J = 7.65 Hz, CH3). 13C-NMR (213 MHz, CDCl3) δ ppm: 11.05, 14.19, 21.24, 23.20, 24.53, 28.95, 31.14, 39.62, 47.81, 109.63, 109.66, 118.19, 118.83, 122.98, 123.32, 123.59, 124.25, 124.93, 125.72, 127.22, 128.71, 129.06, 129.70, 130.09, 130.26, 130.28, 131.13, 131.93, 132.90, 136.39, 136.85, 139.09, 139.45, 140.00, 140.89, 141.32, 141.55, 145.25, 146.61, 150.73, 151.01, 192.01. IR (cm−1): C-H olefinic 3060, C-H aliphatic 2957, 2857, C-H aldehyde 2724, C=O 1699, C=C 1603, 1537. HRMS (ESI): m/z cacld for C46H42N3OS 684.3049 [M+1]+, found 684.3048.
3.2.24. 2,3-Bis(5-bromothiophen-2-yl)quinoxaline (29)
To a solution of 24 (0.4 g, 1.5 mmol, 1.0 eq) in DMF (10 mL), a solution of NBS (0.6 g, 3.5 mmol, 2.3 eq) in DMF (4 mL) was added in a dropwise manner. It was then stirred for 36 h in the dark at room temperature while monitoring the reaction by TLC. After completion, the reaction was stopped by adding ice. The precipitate was filtered off and washed several times with distilled water to obtain compound 29 as a yellow solid (0.6 g, 94%) (m.p. 136 °C). 1H-NMR (850 MHz, CDCl3) δ ppm: 8.02 (dd, 2H, J = 6.8, 3.4 Hz, ArH), 7.73 (dd, 2H, J = 6.8, 3.4 Hz, ArH), 7.12 (d, 2H, J = 4.25 Hz, ArH), 7.01 (d, 2H, J = 4.25 Hz, ArH). 13C-NMR (213 MHz, CDCl3) δ ppm: 116.93, 128.95, 129.73, 130.69, 130.71, 140.68, 143.00, 145.11. IR (cm−1): C-H olefinic 3076, C=C 1519, 1475. HRMS (ESI): m/z cacld for C16H9Br2N2S2 450.8574 [M+1]+ and 452.8730 [M+3]+, found 450.8576 and 454.8548, respectively.
3.2.25. 4-(5-(3-(5-Bromothiophen-2-yl)quinoxalin-2-yl)thiophen-2-yl)benzaldehyde (30)
Compound 29 (0.3 g, 0.7 mmol, 1.0 eq), 4-formylphenylboronic acid (0.1 g, 0.7 mmol, 1.1 eq), K2CO3 (0.9 g, 6.6 mmol, 10.0 eq), and Pd(PPh3)4 (0.1 g, 0.1 mmol, 0.1 eq) were mixed in dry toluene (30 mL) under an argon atmosphere and a drying system of CaCl2 with stirring at ambient temperature. Then, the reaction mixture was heated for 24 h at 90 °C and monitored by TLC. After the reaction was complete, the solvent was removed under vacuum and extracted by distilled water and DCM (30 mL × 4–5 times). The combined organic extract was washed with brine and dried over anhydrous Na2SO4. Then, the solvent was removed at reduced pressure to afford the crude product and was purified by silica gel column chromatography, using PE/DCM (2:1) as an eluent, to remove the residue of the start and the impurities, followed by increasing the polarity of the eluent gradually until 1:1 to obtain the desired compound 30 as an orange powder (0.1 g, 20%) (m.p. 111 °C). 1H-NMR (850 MHz, CDCl3) δ ppm: 10.03 (s, 1H, CHO), 8.08 (dd, 1H, J = 6.8, 2.55 Hz, ArH), 8.06 (dd, 1H, J = 6.8, 2.55 Hz, ArH), 7.93 (d, 2H, J = 7.65 Hz, ArH), 7.84 (d, 2H, J = 8.5 Hz, ArH), 7.75–7.76 (m, 2H, ArH), 7.42 (d, 1H, J = 3.4 Hz, ArH), 7.38 (d, 1H, J = 4.25 Hz, ArH), 7.16 (d, 1H, J = 4.25 Hz, ArH), 7.02 (d, 1H, J = 3.4 Hz, ArH). 13C-NMR (213 MHz, CDCl3) δ ppm: 117.03, 125.58, 126.33, 128.18, 128.98, 128.99, 129.85, 130.52, 130.69, 130.73, 130.79, 135.74, 139.53, 140.73, 140.79, 142.51, 143.22, 145.42, 145.56, 146.10, 191.55. IR, (cm−1): C-H olefinic 3057, C-H aldehyde 2848, 2732, C=O 1695, C=C 1599, 1565. HRMS (ESI): m/z cacld for C23H14BrN2OS2 476.9731 [M+1]+ and 478.9887 [M+3]+ found 476.9733 and 478.9718, respectively.
3.2.26. 4-(5-(3-(5-(9-(2-Ethylhexyl)-6-p-tolyl-9H-carbazol-3-yl)thiophen-2-yl)quinoxalin-2-yl)thiophen-2-yl)benzaldehyde (31)
Compound 30 (0.1 g, 0.1 mmol, 1.0 eq), 9-(2-ethylhexyl)-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-6-p-tolyl-9H-carbazole 10c (0.1 g, 0.2 mmol, 1.5 eq), K2CO3 (0.3 g, 2.1 mmol, 14.0 eq), and Pd(PPh3)4 (0.01 g, 0.01 mmol, 0.05 eq) were mixed in a degassed aqueous solvent of toluene/EtOH/H2O 2:1:1 (60 mL) under an argon atmosphere with stirring at ambient temperature. Thereafter, the reaction mixture was heated for 3 h at 80 °C and monitored by TLC. After the reaction was complete, the solvent was removed under vacuum and extracted by distilled water and CHCl3 (20 mL × 4–5 times). The combined organic extract was washed with brine and dried over anhydrous Na2SO4. Then, the solvent was removed under reduced pressure to afford the crude product and was purified by silica gel column chromatography, using PE/EA (9:1) as an eluent, to produce compound 31 as an orange powder (0.1 g, 81%) (m.p. 74 °C). 1H-NMR (850 MHz, CDCl3) δ ppm: 10.02 (s, 1H, CHO), 8.45 (s, 1H, ArH), 8.33 (s, 1H, ArH), 8.13 (d, 1H, J = 8.5 Hz, ArH), 8.10 (d, 1H, J = 8.5 Hz, ArH), 7.92 (d, 2H, J = 7.65 Hz, ArH), 7.85 (d, 2H, J = 7.65 Hz, ArH), 7.80 (d, 1H, J = 8.5 Hz, ArH), 7.72–7.76 (m, 3H, ArH), 7.63 (d, 2H, J = 7.65 Hz, ArH), 7.50 (d, 1H, J = 4.25 Hz, ArH), 7.45 (d, 1H, J = 8.5 Hz, ArH), 7.43 (dd, 1H, J = 3.4, 0.85 Hz, ArH), 7.41–7.42 (m, 2H, ArH), 7.31 (dd, 1H, J = 3.4, 0.85 Hz, ArH), 7.30 (d, 2H, J = 7.65 Hz, ArH), 4.16–4.22 (m, 2H, N-CH2), 2.43 (s, 3H, CH3), 2.10 (sept, 1H, J = 6.8 Hz, CH), 1.35–1.46 (m, 4H, 2CH2), 1.26–1.34 (m, 4H, 2CH2), 0.94 (t, 3H, J = 7.65 Hz, CH3), 0.89 (t, 3H, J = 7.65 Hz, CH3). 13C-NMR (213 MHz, CDCl3) δ ppm: 11.05, 14.20, 21.24, 23.20, 24.53, 28.96, 31.14, 39.63, 47.82, 109.65, 109.69, 118.22, 118.85, 122.82, 123.31, 123.62, 124.35, 124.92, 125.65, 125.74, 126.35, 127.22, 128.35, 128.71, 128.94, 129.69, 130.56, 130.68, 131.03, 131.36, 131.95, 132.58, 132.91, 135.71, 136.40, 137.87, 139.09, 139.56, 140.13, 140.18, 140.90, 141.55, 142.26, 145.92, 146.26, 150.85, 191.55. IR (cm−1): C-H olefinic 3058, C-H aliphatic 2956, 2851, C-H aldehyde 2732, C=O 1696, C=C 1600. HRMS (ESI): m/z cacld for C50H44N3OS2 766.2926 [M+1]+, found 766.2927.
3.2.27. 4-(3-(9-(2-ethylhexyl)-6-(p-tolyl)-9H-carbazol-3-yl)quinoxalin-2-yl)benzaldehyde (32)
Compound 25 (0.1 g, 0.4 mmol, 1.0 eq), 9-(2-ethylhexyl)-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-6-p-tolyl-9H-carbazole 10c (0.3 g, 0.2 mmol, 1.5 eq), K2CO3 (0.7 g, 5.2 mmol, 14.0 eq), and Pd(PPh3)4 (0.01 g, 0.01 mmol, 0.05 eq) were mixed in a degassed aqueous solvent of toluene/EtOH/H2O 2:1:1 (40 mL) under an argon atmosphere with stirring at ambient temperature. Then, the reaction mixture was heated at 80 °C for 3 h and monitored by TLC. After the reaction was complete, the solvent was removed under vacuum and extracted by distilled water and DCM (20 mL × 4–5 times). The combined organic extract was washed with brine and dried over anhydrous Na2SO4. Then, the solvent was removed at reduced pressure to afford the crude product and was purified by silica gel column chromatography, using PE/EA (9:1) as an eluent, to remove the undesired low polar fractions, followed by increasing the polarity of the eluent gradually until 8:2 to obtain the desired compound 32 as a yellow powder (0.2 g, 86%) (m.p. 99 °C). 1H-NMR (850 MHz, CDCl3) δ ppm: 10.01 (s, 1H, CHO), 8.49 (s, 1H, ArH), 8.44 (d, 1H, J = 7.65 Hz, ArH), 8.26 (d, 1H, J = 8.5 Hz, ArH), 8.21 (d, 1H, J = 0.85 Hz, ArH), 7.86–7.88 (m, 1H, ArH), 7.83–7.85 (m, 3H, ArH), 7.77 (d, 2H, J = 8.5 Hz, ArH), 7.71 (dd, 1H, J = 8.5, 1.7 Hz, ArH), 7.57 (d, 2H, J = 8.5 Hz, ArH), 7.50 (dd, 1H, J = 8.5, 1.7 Hz, ArH), 7.45 (d, 1H, J = 8.5 Hz, ArH), 7.30 (d, 1H, J = 8.5 Hz, ArH), 7.28 (d, 2H, J = 7.65 Hz, ArH), 4.16–4.21 (m, 2H, N-CH2), 2.42 (s, 3H, CH3), 2.07 (sept, 1H, J = 6.8 Hz, CH), 1.33–1.42 (m, 4H, 2CH2), 1.22–1.32 (m, 4H, 2CH2), 0.92 (t, 3H, J = 7.65 Hz, CH3), 0.86 (t, 3H, J = 7.65 Hz, CH3). 13C-NMR (213 MHz, CDCl3) δ ppm: 11.04, 14.17, 21.23, 23.18, 24.50, 28.88, 31.11, 39.56, 47.83, 109.21, 109.71, 118.99, 122.94, 123.44, 123.51, 125.88, 127.30, 128.19, 128.36, 128.69, 129.36, 129.67, 129.78, 130.46, 130.74, 131.37, 133.27, 136.30, 136.45, 139.08, 140.26, 140.87, 141.05, 142.23, 145.11, 152.68, 153.42, 191.95. IR (cm−1): C-H olefinic 3057, C-H aliphatic 2956, 2857, C-H aldehyde 2731, C=O 1701, C=C 1602, 1477. HRMS (ESI): m/z cacld for C42H40N3O 602.3171 [M+1]+, found 602.3170.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





















