2.1. Catalyst Characterization
The polymeric matrix has -CONH
2 groups that can be protonated in the presence of a strong acid such as tungstophosphoric acid, allowing the electrostatic interaction of these groups and the anions [H
3−xPW
12O
40]
x−(where 1 < x ≤ 3). It has been reported that H
+ transfer from [H
3PW
12O
40] to the amine group, resulting in an electrostatic bond between -NH
3+ and [H
3−xPW
12O
40]
x−, is responsible for the efficient immobilization of the heteropolyanion [
33,
34].
The absence of W in the water solution C (obtained from the chloride elimination step) shows that TPA leaching is negligible due to the aforementioned strong interaction.
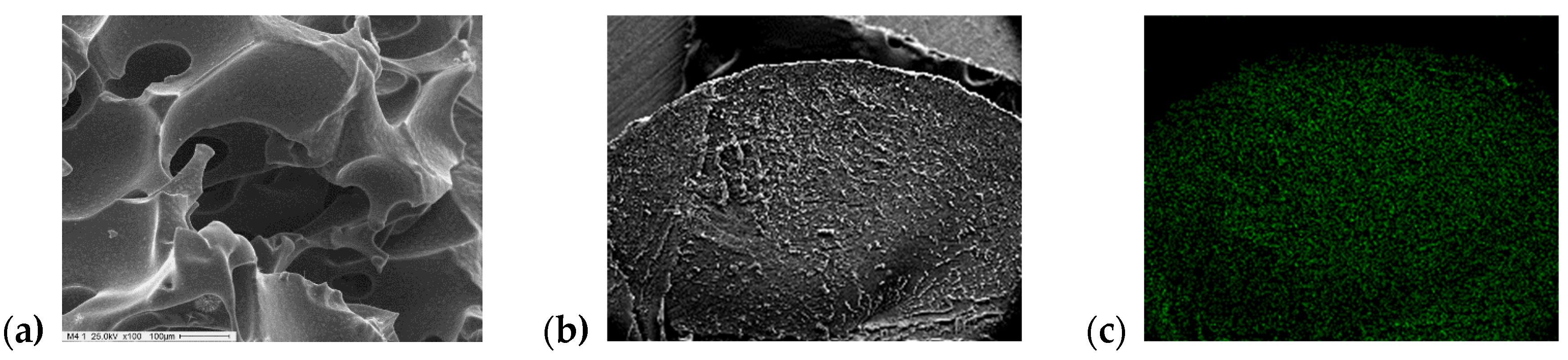
According to SEM micrographs (
Figure 1), PLMTPAXX/YY samples present a sponge-like structure formed by a network of cross-linked channels. The tungsten microanalysis performed by EDAX showed that TPA content along the diameter of the spheres is almost uniform, with a slight increase on their surface.
The SBET values of the PLMTPA60/40T100, PLMTPA40/60T100, and PLMTPA20/80T100 materials are lower than 10 m2/g (6, 4 and 3 m2/g, respectively) and in the same range as those of the bulk TPA and PLM (9 and 2 m2/g, respectively).
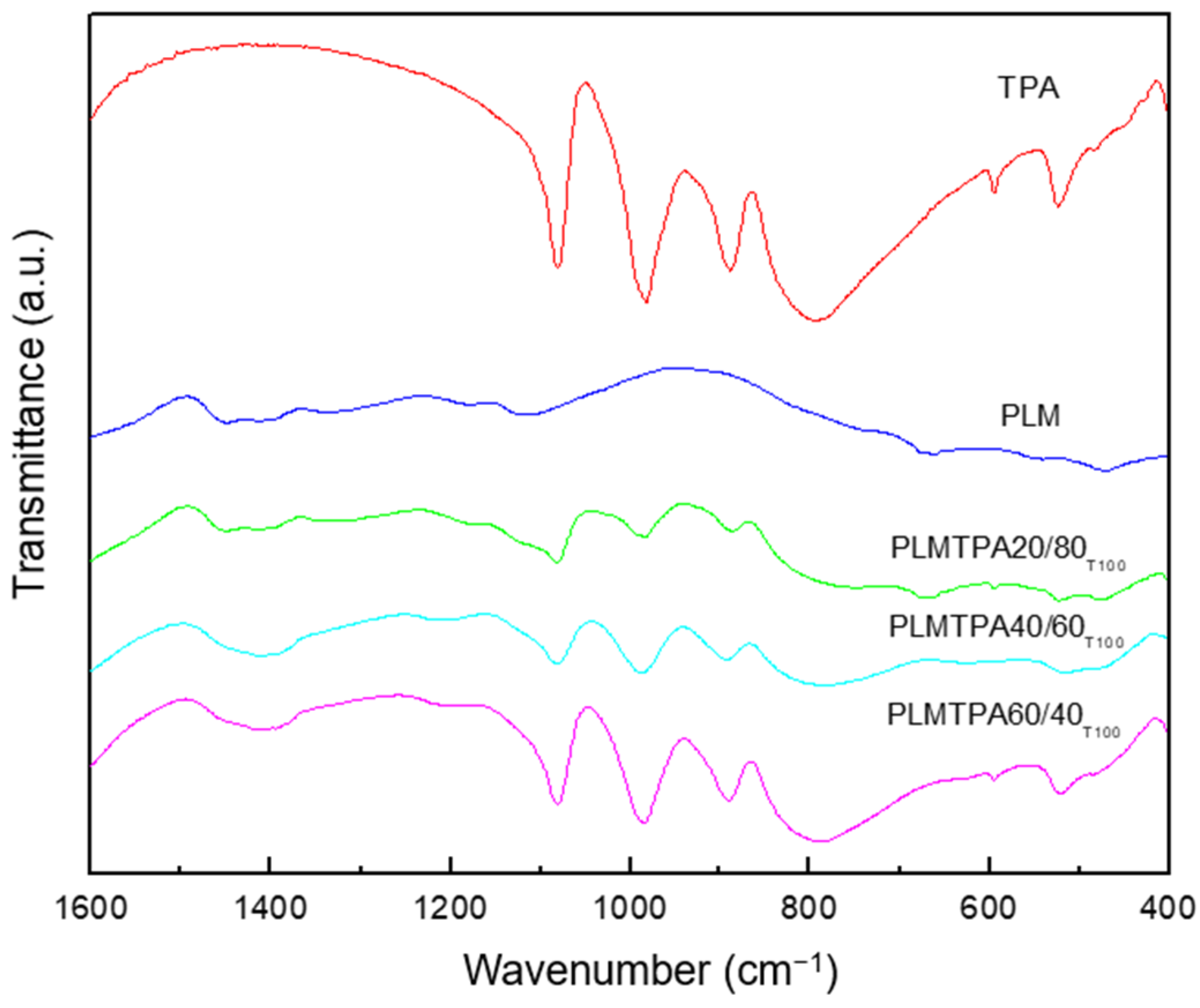
TPA FT-IR spectrum (
Figure 2) shows the bands assigned to the stretching vibrations P-O
a (1081 cm
−1), W-O
d (982 cm
−1), W-O
c-W (888 cm
−1), W-O
c-W (793 cm
−1), and to the bending vibration O
a-P-O
a (595 cm
−1) [
35] (see the subscript meaning in
supplementary materials).
The bands assigned to the stretching vibrations W-Oc-W, W-Ob-W, and P-Oa in the spectrum of PLMTPA60/40
T100, PLMTPA40/60
T100, and PLMTPA20/80
T100 samples overlap the characteristic bands of PLM (
Figure 2). Their intensity increases with the increment of the sample TPA content, and it confirms the presence of [PW
12O
40]
3− anion as the main species. The FT-IR spectra of samples treated at 200 and 300 °C display similar characteristics. However, in the samples calcined at a higher temperature, the relative intensity of the TPA main bands slightly increases due to PLM chemical transformation and partial decomposition (
Figure S2).
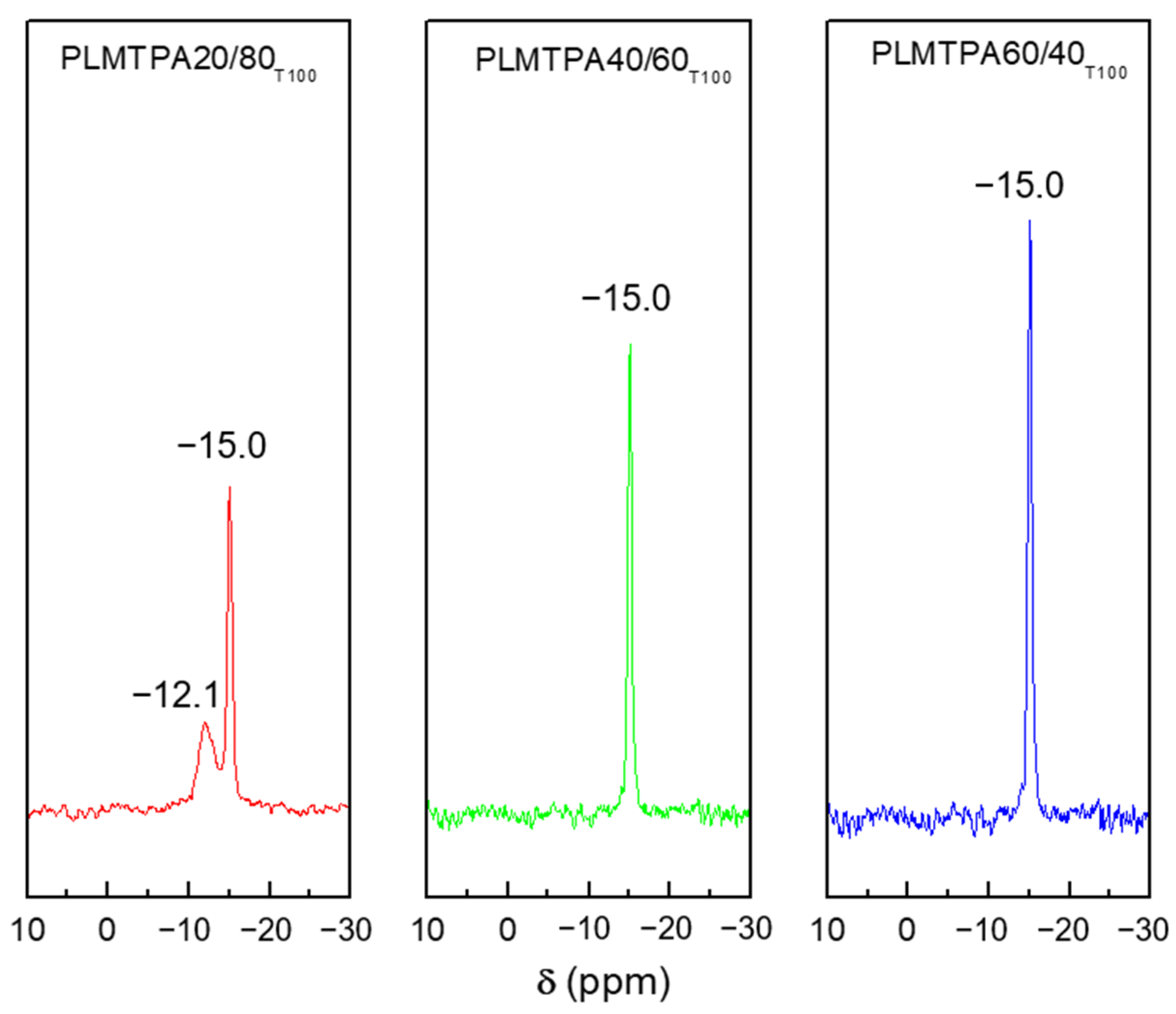
The
31P MAS-NMR spectra of PLMTPA20/80
T100, PLMTPA40/60
T100 and PLMTPA60/40
T100 samples (
Figure 3) display a narrow band with a maximum at around −15.0 ppm, attributed to the [PW
12O
40]
3− anion. The PLMTPA20/80 spectrum also shows a wider and less intense band at −12.1 ppm, assigned to the [P
2W
21O
71]
6− dimeric species [
36,
37].
Based on the FT-IR and
31P MAS-NMR results, we can conclude that the [PW
12O
40]
3− anion is the main species in the PLMTPA samples. However, the anion was partially transformed into [P
2W
21O
71]
6− dimeric species during the synthesis due to the limited stability range of the [PW
12O
40]
3− anion in solution [
38]. No significant changes in the
31P MAS-NMR spectra of the samples treated at 200 and 300 °C were found. These results agree with reports showing that an appreciable transformation of [PW
12O
40]
3− into [P
2W
21O
71]
6− takes place at 400 °C or higher temperatures [
39].
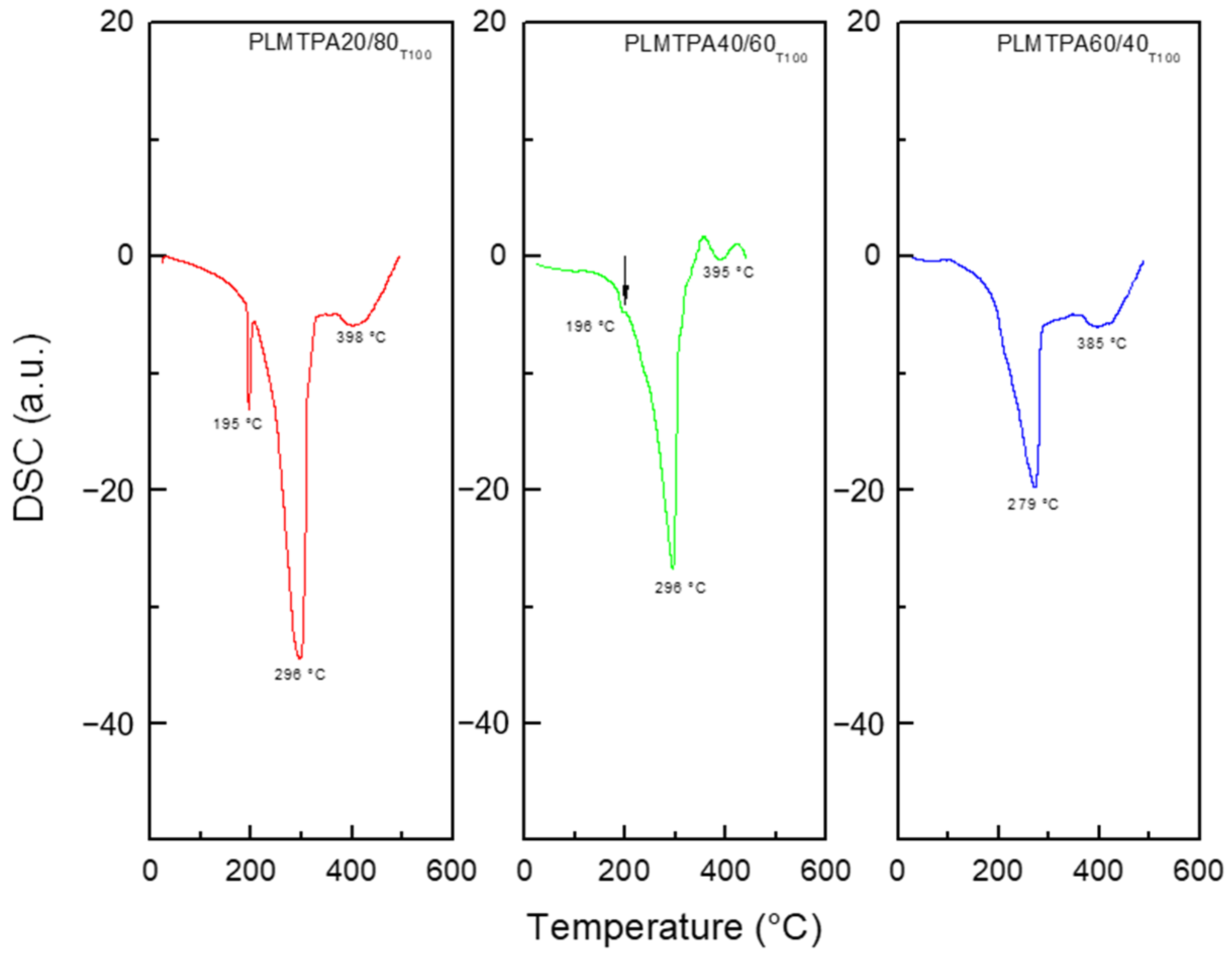
The DSC diagram of dried PLM (
Figure 4) exhibits three endothermic peaks at 199, 300 and 400 °C, which are assigned to the glass transition (T
g) and polymer decomposition [
40,
41]. Above T
g and up to 350 °C, the intra- and intermolecular imidization of the amido groups (with the release of NH
3) and the formation of nitrile groups from the amido group dehydration take place. At temperatures above 350 °C, imide decomposition (with the release of CO
2 and H
2O) and the rupture of the polymeric chain (with the formation of long hydrocarbon chains) occur. According to the TGA analysis, the PLM degradation takes place in two steps between 250 and 350 °C and 350 and 460 °C. The weight loss associated with these steps was 22% and 64%, respectively. TGA diagrams of PLMTPA20/80
T100, PLMTPA40/60
T100 and PLMTPA60/40
T100 samples exhibit the two aforementioned degradation steps. The weight loss ascribed to these steps decreases in parallel with the increment of TPA content in the materials (see
Table S1 in supplementary materials).
The DSC diagrams of the PLMTPA20/80
T100 and PLMTPA40/60
T100 samples (
Figure 4) present the three endothermic peaks mentioned above at slightly lower temperatures. In the case of the PLMTPA40/60
T100 material, the peak assigned to the glass transition is barely visible. For the PLMTPA60/40
T100 samples, the glass transition peak is not present, and the other two endothermic peaks appear at lower temperatures (279 and 385 °C). These results show that PLMTPAXX/YY
T100 materials do not undergo any remarkable chemical changes up to 200 °C.
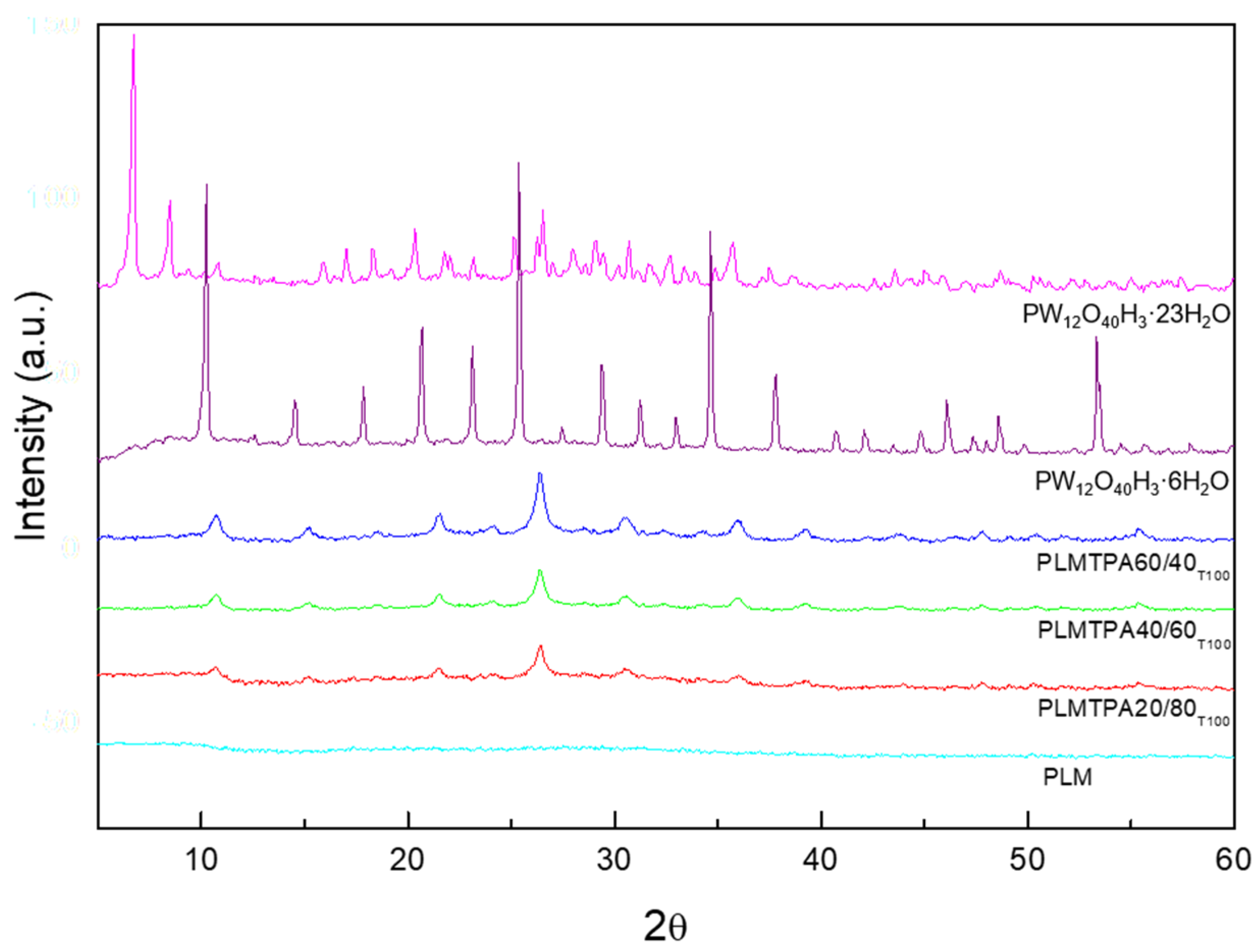
The XRD diagrams of the PLMTPA20/80
T100, PLMTPA40/60
T100 and PLMTPA60/40
T100 samples (
Figure 5) show a new set of broad- and low-intensity peaks overlapping the XRD pattern of bulk PLM; however, they resemble neither those of the H
3PW
12O
40 (JCPDS No. 50-0657) nor those of its more common hydrates (H
3PW
12O
40·6H
2O and H
3PW
12O
40·23H
2O) [
42]. The intensity of these peaks slightly increases as the TPA content rises. These results suggest that most of the TPA is present in the polymeric matrix as a noncrystalline phase. On the other hand, the small TPA crystals seem to be strongly immobilized in the PLM matrix, since they were not removed during the washing with water (three times for 24 h each) performed to eliminate the chloride. The XRD diagrams of samples treated at higher temperatures present similar characteristics.
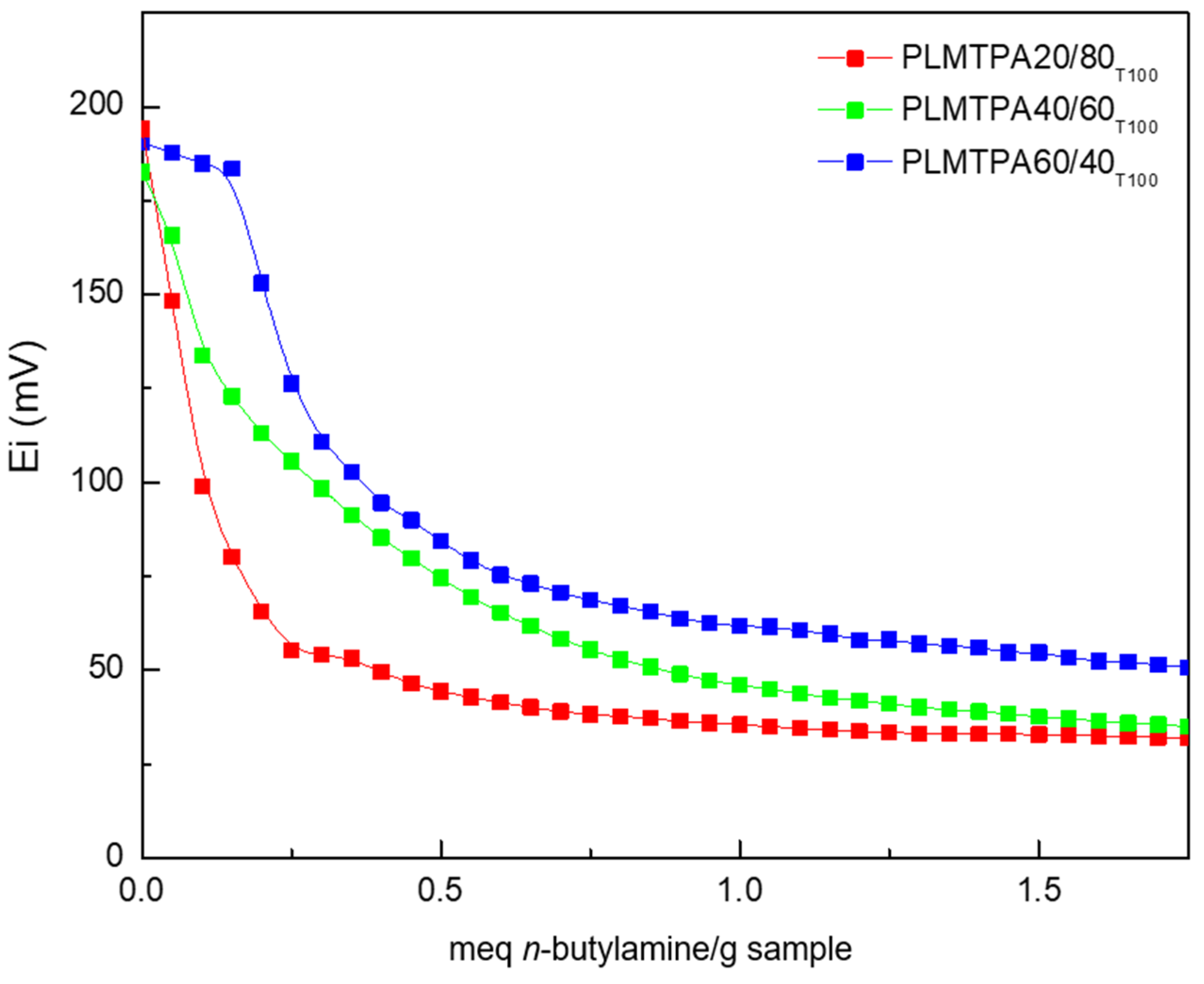
The acidity properties of the PLMTPAXX/YY materials, estimated from the potentiometric titration with
n-butylamine curves (
Figure 6), are listed in
Table 1. According to the potentiometric results, all the materials exhibit Ei values higher than 100 mV [
43], assigned to the presence of very strong acid sites (see the classification scale in
supplementary materials).
The acid strength of PLMTPA20/80
T100 (Ei = 194 mV), PLMTPA40/60
T100 (Ei = 182 mV), and PLMTPA60/40
T100 (Ei = 191 mV) samples is higher than that of PLM (Ei = −44 mV) but lower than that of bulk TPA (Ei = 620 mV) [
44]. The lower acid strength of the PLMTPAXX/YY materials compared to bulk TPA could be due to the fact that the protons in TPA are present as H
+(H
2O)
2 species (hydrated protons), whereas in the PLMTPA samples they interact with the nitrogen of -CONH
2 groups.
The number of acid sites (N
S) estimated as the area under the curve increases in the following order: PLMTPA20/80
T100 > PLMTPA40/60
T100 > PLMTPA60/40
T100 (84, 146, and 201 meq
n-butylamine/g, respectively) in parallel with the increment of TPA content. On the other hand, the thermal treatment of the samples at higher temperatures decreased both Ei and Ns (
Figure S3), because of the structural and chemical modification of the PLM matrix.
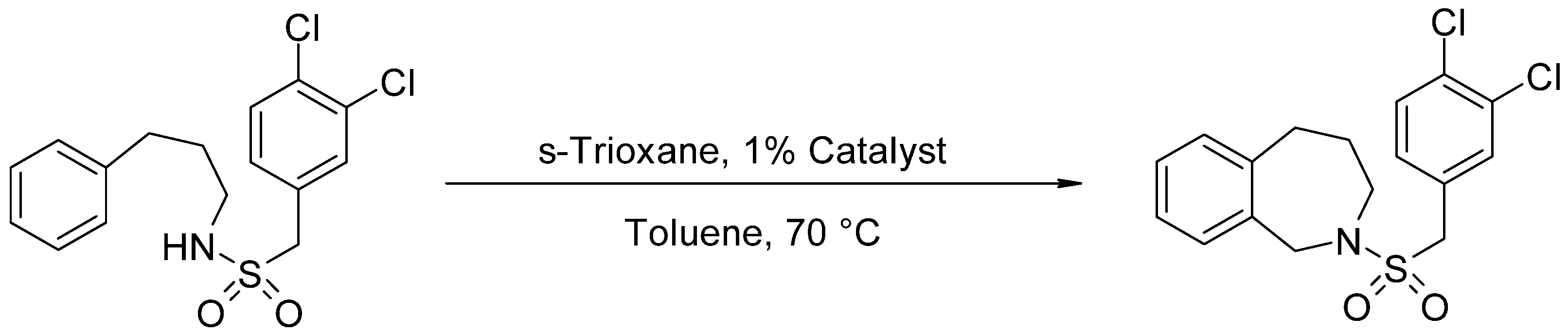
2.2. Catalytic Test for N-(3,4-Dichlorobenzylsulfonyl)-2,3,4,5-Tetrahydro-1H-Benzo[c]azepine Synthesis
The most suitable catalyst was identified using the test reaction between
N-phenylpropyl-3,4-dichlorobenzylsulfonamide and s-trioxane as substrate (see
Scheme 1), with toluene as solvent, at 70 °C for 3 h to obtain
N-(3,4-dichlorobenzylsulfonyl)-2,3,4,5-tetrahydro-1H-benzo[c]azepine.
First, we performed a blank experiment without the catalyst, and no reaction was observed (
Table 1, entry 1). Likewise, no reaction was observed when the support (PLM) was used, under the same conditions (
Table 1, entry 2). Subsequently, the PLMTPA materials were tested, showing very good conversion and selectivity of the desired product (
Table 1, entries 3–5). In these three experiments, very good product yields were obtained for PLMTPA20/80
T100, PLMTPA40/60
T100 and PLMTPA60/40
T100 samples (48%, 62% and 83%, respectively), indicating the need to use a catalyst with acidic properties to produce
N-(3,4-dichlorobenzylsulfonyl)-2,3,4,5-tetrahydro-1
H-benzo[
c]azepine. According to the acidity measurements of the PLMTPA samples obtained by potentiometric titration, the Ns in the catalysts increases as follows: PLMTPA20/80
T100 < PLMTPA40/60
T100 < PLMTPA60/40
T100. The
N-(3,4-dichlorobenzylsulfonyl)-2,3,4,5-tetrahydro-1
H-benzo[
c]azepine yield increases in the same way, suggesting that the acidity strongly affects the catalytic activity of the PLMTPA catalysts. The yields obtained for the samples calcined at 200 and 300 °C (
Table 1, entries 6 and 7) confirm that the drop in Ns and acid strength leads to a decrease in
N-(3,4-dichlorobenzylsulfonyl)-2,3,4,5-tetrahydro-1
H-benzo[
c]azepine production. On the other hand, the yield obtained using bulk TPA in the same reaction conditions was lower (41%) than those achieved using the PLMTPA20/80
T100, PLMTPA40/60
T100, and PLMTPA60/40
T100 catalysts. Consequently, the TPA inclusion in the polymer matrix (mainly present as a noncrystalline phase) increases the
N-(3,4-dichlorobenzylsulfonyl)-2,3,4,5-tetrahydro-1
H-benzo[
c]azepine yields and facilitates the catalyst recovery (by simple centrifugation and filtration) from the reaction medium for its subsequent reuse. On the contrary, the recovery of bulk TPA was more difficult, because it must be washed several times with the reaction solvent to eliminate product traces covering its surface.
To evaluate the influence of the solvent nature, the reaction was conducted using benzene, chloroform, dichloromethane, and hexane instead of toluene (
Table 2). All solvents were selected due to TPA insolubility in them.
Similar results were obtained using aromatic hydrocarbons such as toluene and benzene (
Table 2, entries 1 and 2: 83% and 80%, respectively). The use of chlorinated solvents gives lower yields (
Table 2, entries 3 and 4: 68% and 52%, respectively). Finally, hexane gives a poor yield (
Table 2, entry 5: 35%) due to the very low solubility of the substrates in the reaction medium. Therefore, toluene was chosen as solvent for the next experiments because, although benzene gives comparable results, it is classified as a carcinogen, which increases the risk of cancer and other illnesses.
To evaluate the influence of reaction temperature, five temperatures (25, 50, 70, 90, and 110 °C) were checked in identical reaction conditions (see
Table 3). No product formation was observed at room temperature (25 °C) (
Table 3, entry 1). A temperature increase to 70 °C leads to higher
N-(3,4-dichlorobenzylsulfonyl)-2,3,4,5-tetrahydro-1
H-2-benzo[
c]azepine yields. The maximum yield for a reaction time of 3 h at 70 °C is 83% (
Table 3, entry 3). The catalytic performance in the reaction drops considerably when raising the temperature to 90 and 100 °C (
Table 3, entries 4 and 5: 69% and 51%, respectively). In these cases, the formation of several secondary products, which were detected by TLC (and were not identified), was observed. Based on these results, 70 °C was the reaction temperature chosen for the next experiments.
Table 4 shows the results obtained under the already optimized reaction conditions (toluene as solvent and 70 °C temperature) as a function of time (0.5, 2, 3, 4, and 6 h). The reaction yields increased with time up to 3 h (
Table 4, entry 3) and then remained at a constant value.
Table 5 displays the catalyst amount effect on the yield of
N-(3,4-dichlorobenzylsulfonyl)-2,3,4,5-tetrahydro-1
H-benzo[
c]azepine. The reaction yields increased from 69% to 83% when the catalyst amount increased from 0.5% to 1% (
Table 5, entries 1 and 2). There were no substantial changes by raising the catalyst amount up to 2% (
Table 5, entries 3 and 4).
The PLMTPA60/40
100 reuse was evaluated under optimized reaction conditions: substrate, 0.5 mmol;
s-trioxane, 1.5 mmol; toluene, 2 mL; catalyst, 1% mmol; temperature, 70 °C, and reaction time, 3 h. After each run, the catalyst was filtered, washed with three portions of toluene, dried under vacuum at 25 °C for 24 h, and reused. Each reuse test is presented in
Table 6, showing that the catalyst can be reused for six cycles without appreciable loss of its catalytic activity.
The possible TPA leaching from the catalyst into the reaction media was evaluated as follows: a sample of PLMTPA60/40100 catalyst (the one with the highest TPA content) was refluxed in toluene (3 mL) for 6 h, filtered in vacuum, and dried to constant weight. The activity of the PLMTPA60/40100 catalyst was evaluated, being similar to that evaluated with the fresh catalyst (82%, 70 °C, 3 h). The refluxed toluene was used as solvent for attempting the N-(3,4-dichlorobenzylsulfonyl)-2,3,4,5-tetrahydro-1H-benzo[c]azepine synthesis without adding the catalyst. After 6 h at 70 °C, no significant amounts of the product were detected, and the substrate was recovered quantitatively from the reaction media.
Finally, another experiment consisted of withdrawing the catalyst after 1 h of reaction; the reaction was followed for two more hours, and the reaction yields were evaluated. The reaction yields at 1 and 3 h were practically the same (38% and 40%, respectively), indicating that the synthesis reaction takes place in a heterogeneous phase.
Using the best reaction conditions found for
N-(3,4-dichlorobenzylsulfonyl)-2,3,4,5-tetrahydro-1
H-benzo[
c]azepine synthesis (
N-aralkylsulfonamide, 0.5 mmol;
s-trioxane, 1.5 mmol; toluene, 2 mL; catalyst, PLMTPA60/40
100, 1% mmol; and 70 °C, 3 h), various benzo[
c]azepines, isoquinolines, and one benzo[
c]azocine were prepared. The methodology is not appropriate to prepare analogous rings of five, eight, and nine members, because of the low yield. However, it is suitable for the preparation of heterocycles with six and seven members (
Table 7).
Table 8 shows comparative results for the
N-(3,4-dichlorobenzylsulfonyl)-2,3,4,5-tetrahydro-1
H-benzo[
c]azepine synthesis using different acid catalysts. Better results were obtained using heterogeneous catalysts based on HPAs (
Table 8, entries 3–5), probably due to the milder conditions than the methanesulphonic acid/trifluoracetic acid and methanesulphonic/acetic anhydride media (
Table 8, entries 1 and 2).
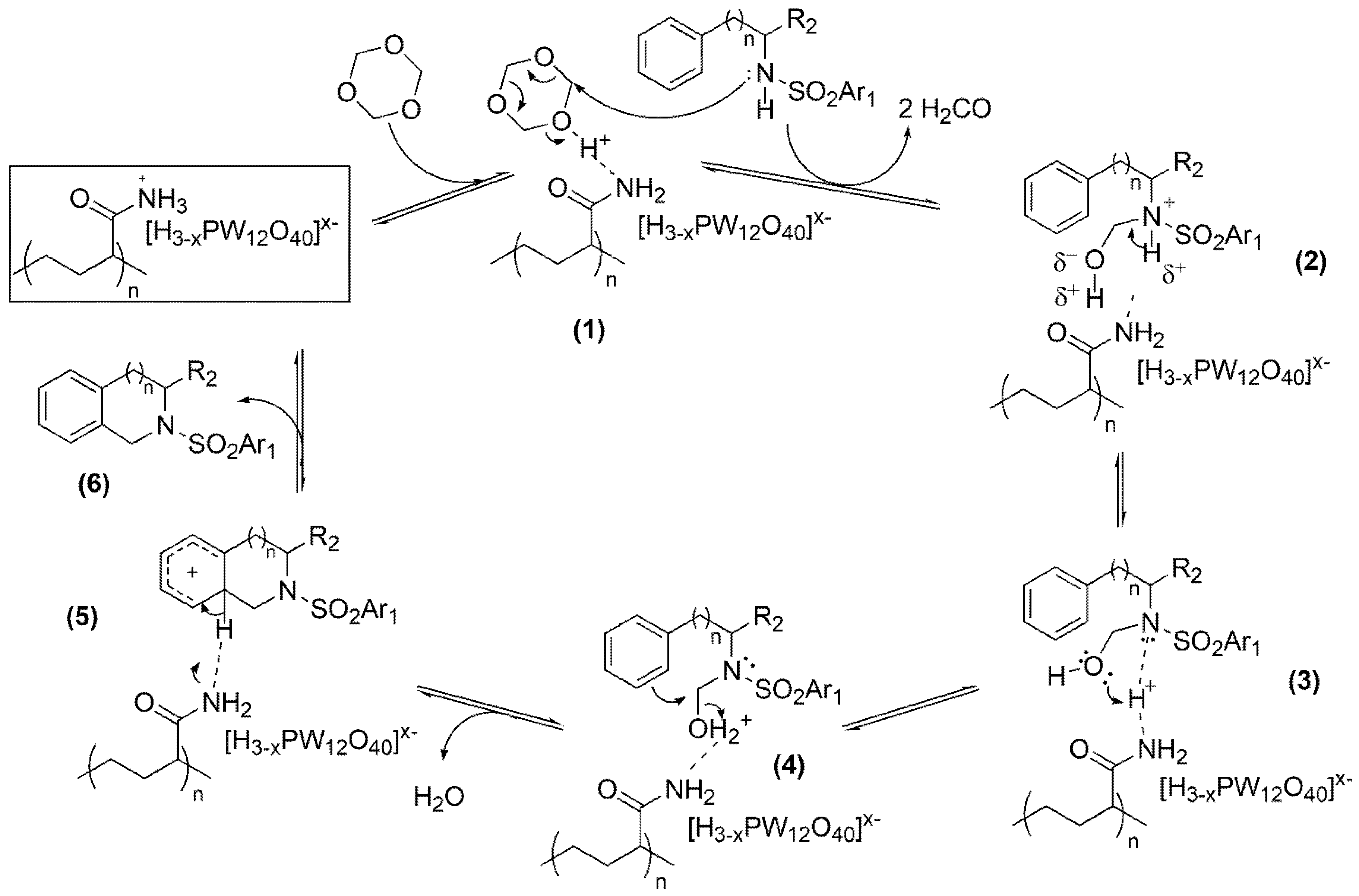
2.3. Mechanism of 2,3,4,5-Tetrahydro-1H-benzo[c]azepine and Analogous Ring Synthesis
A plausible mechanism for the synthesis of 2,3,4,5-tetrahydro-1
H-benzo[
c]azepines and analogous rings is described. This reaction involves the formation of a benzazepine or a homolog in the nitrogen ring from a sulfonamide and formaldehyde formed in situ from
s-trioxane. The reaction is catalyzed by a strong acid. The proposed mechanism corresponds to the reaction in which the catalyst is a supported HPA (
Scheme 2).
Initially, the catalyst coordinates with an s-trioxane molecule forming the adduct (1), which undergoes nucleophilic attack by the sulfonamide, simultaneously with the protonation of s-trioxane, resulting in the alkylation of the N atom by a hydroxymethyl and the formation of an ammonium ion (2).
At the acidic catalytic site, the conjugate base of the catalyst is generated momentarily, which stabilizes the ion by forming a bifurcated hydrogen bridge to both acidic hydrogens. A transfer of a proton protonates the catalyst again, which is coordinated to the hydroxymethylsulfonamide (3). A new proton transfer forms oxonium (4) from (3). Oxonium (4) is again coordinated with the conjugate base of the catalyst and undergoes an intramolecular electrophilic aromatic substitution while removing a molecule of water. Intermediate (5) is finally deprotonated, forming benzo[c]azepine (or analogous ring) (6) and regenerating the catalyst.
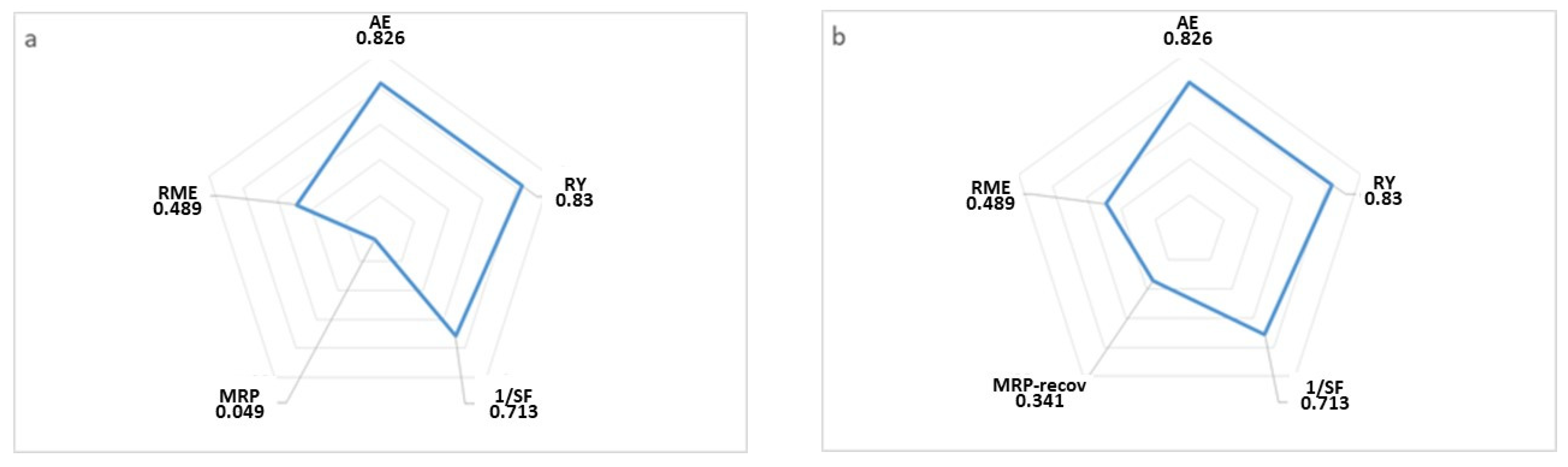
Since Barry Trost introduced the atom economy concept in 1991 [
45] and Paul Anastas and John Warner defined the 12 principles of green chemistry [
46], several groups, including ours, have started to consider the best ways to adopt this working approach. One way of verifying how close the synthesis method used is to an ideal process, from the green chemistry point of view, is using some of the green parameters defined by John Andraos and coworkers in several publications [
47,
48,
49] and representing them in a polygon graphic. In our case, we decided to represent five of these parameters, atom economy (AE), reaction yield (RY), stoichiometry factor (SF), material recovery parameter (MRP), and reaction mass efficiency (RME), in a radial pentagon (
Figure 7a,b). For comparative purposes, we studied the green parameters on the same reaction with (
Figure 7b) and without (
Figure 7a) solvent and catalyst recovery. We noted that the change was in the MRP parameter, which increased approximately ten times when we recovered the solvent and catalyst.


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}













