
A Review of CeO2 Supported Catalysts for CO2 Reduction to CO through the Reverse Water Gas Shift Reaction



Abstract
:1. Introduction
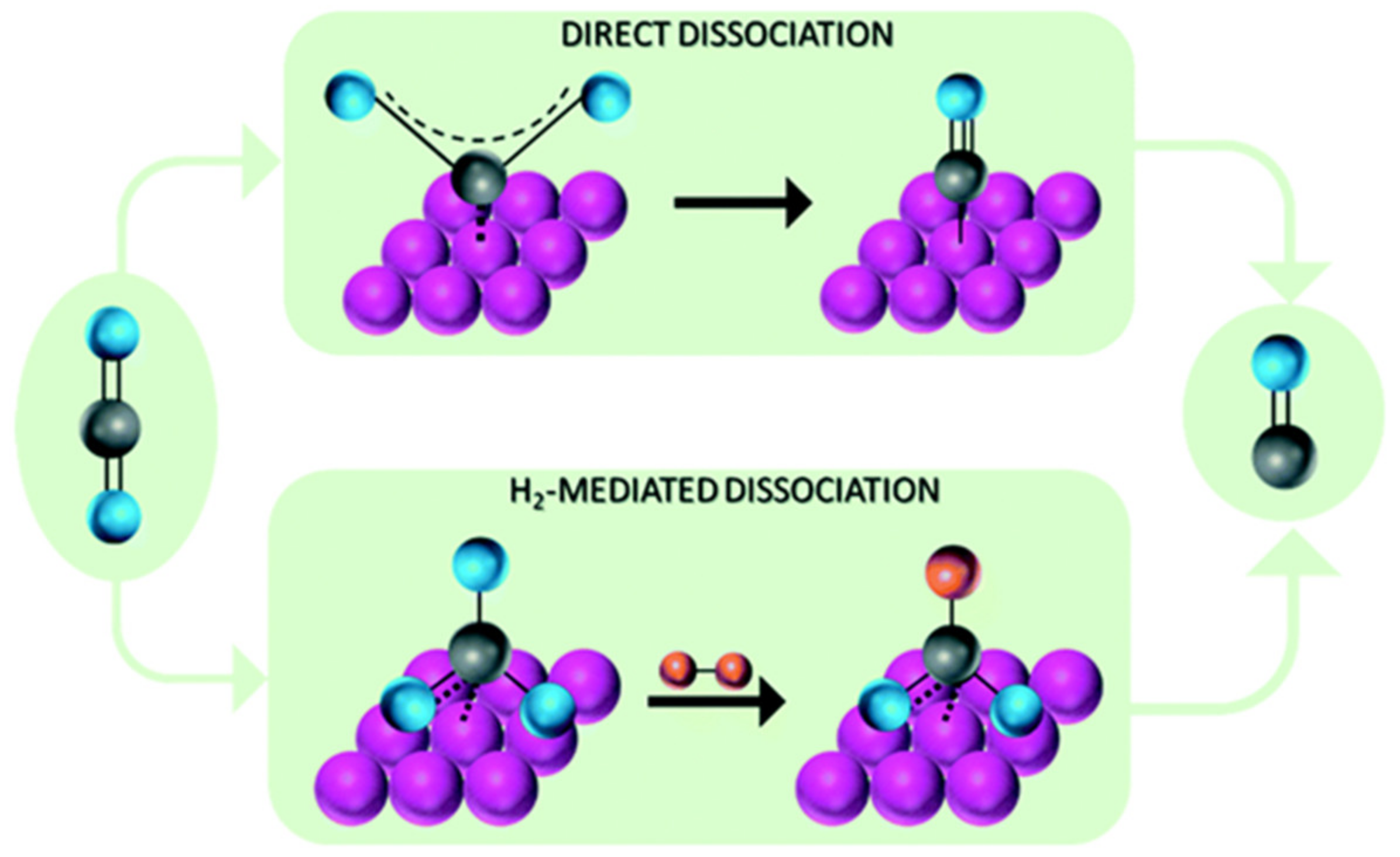
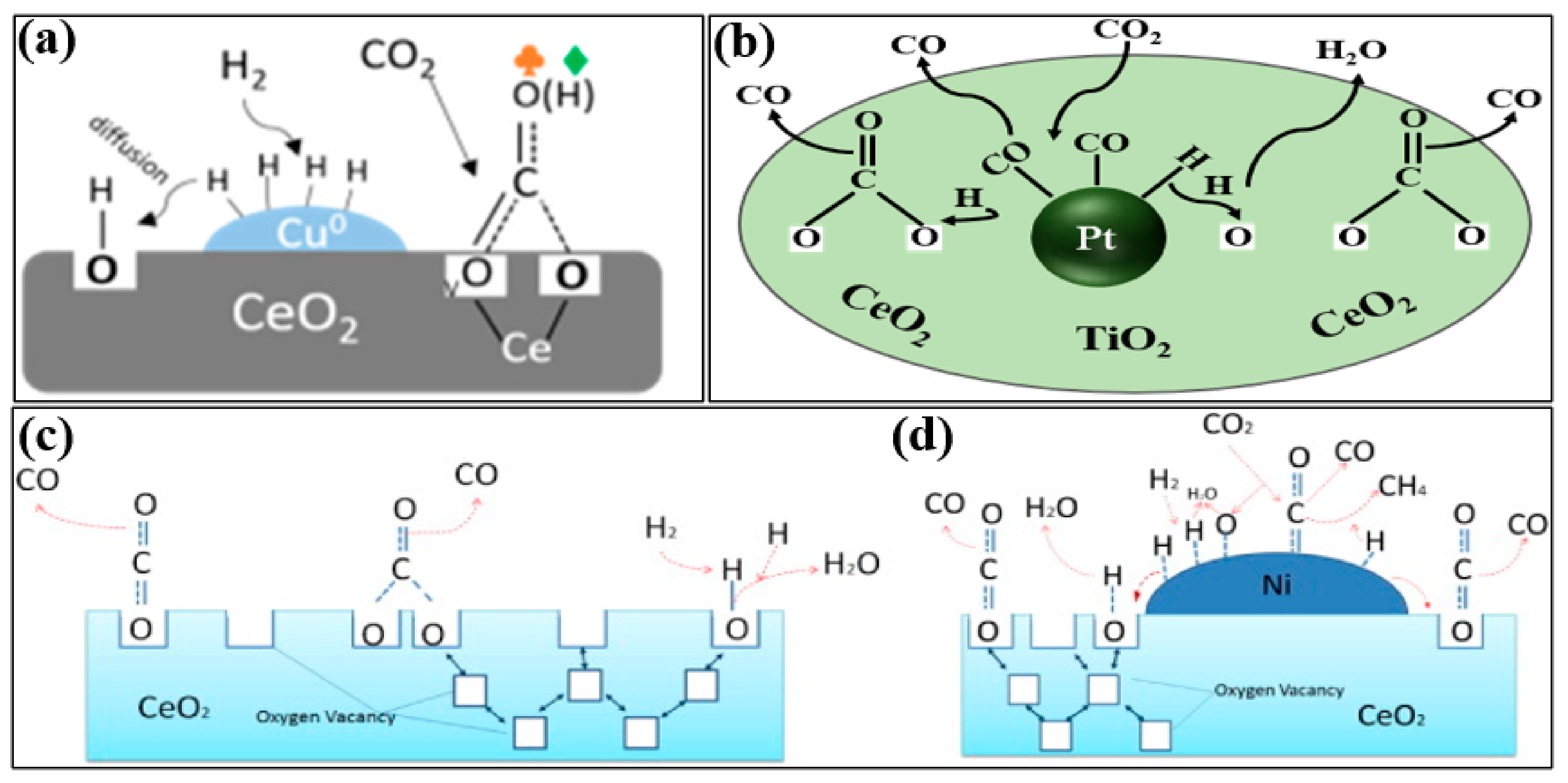
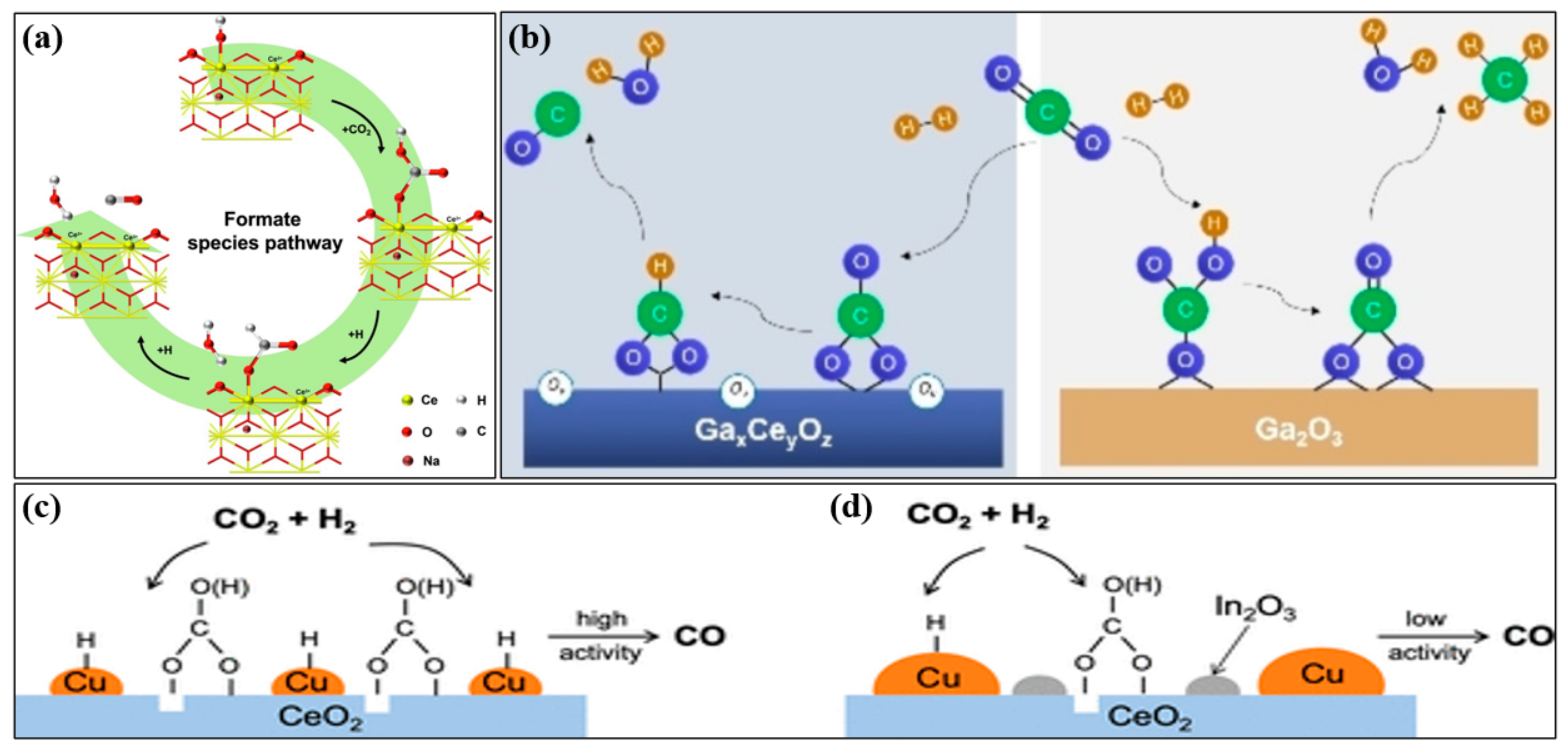
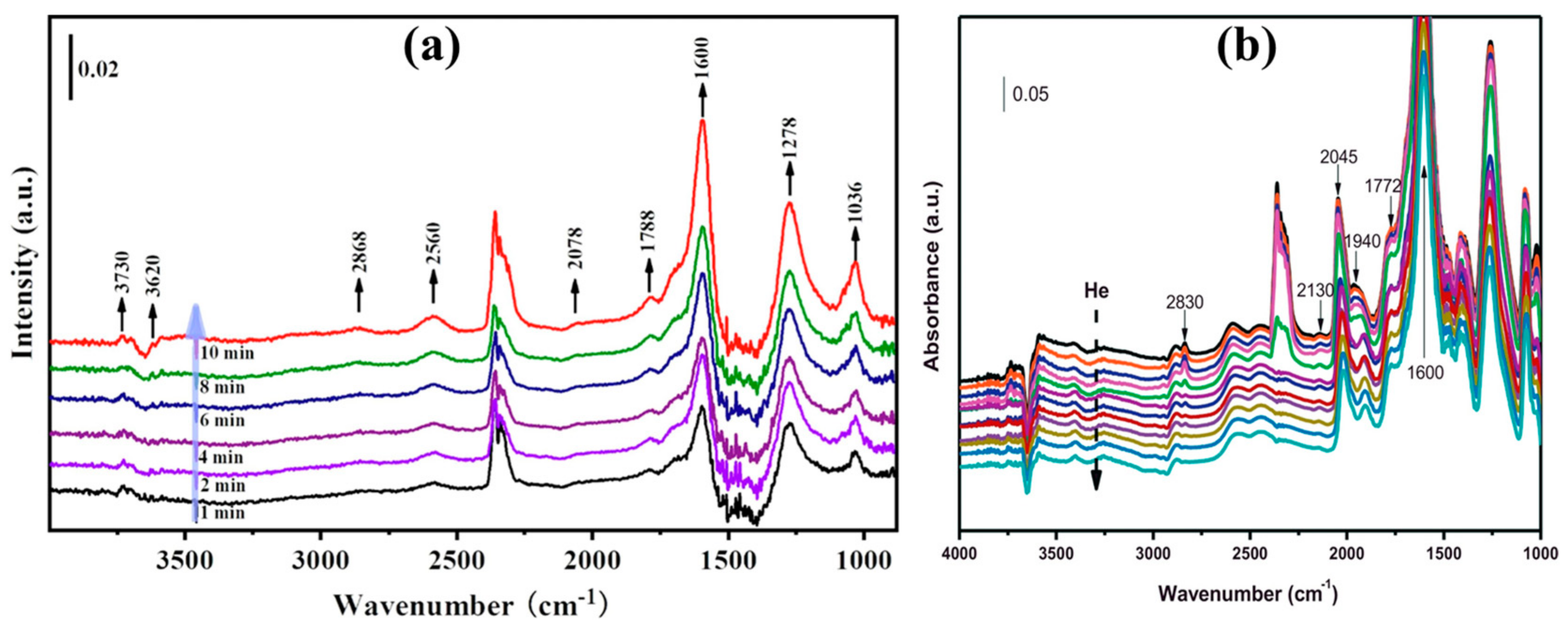
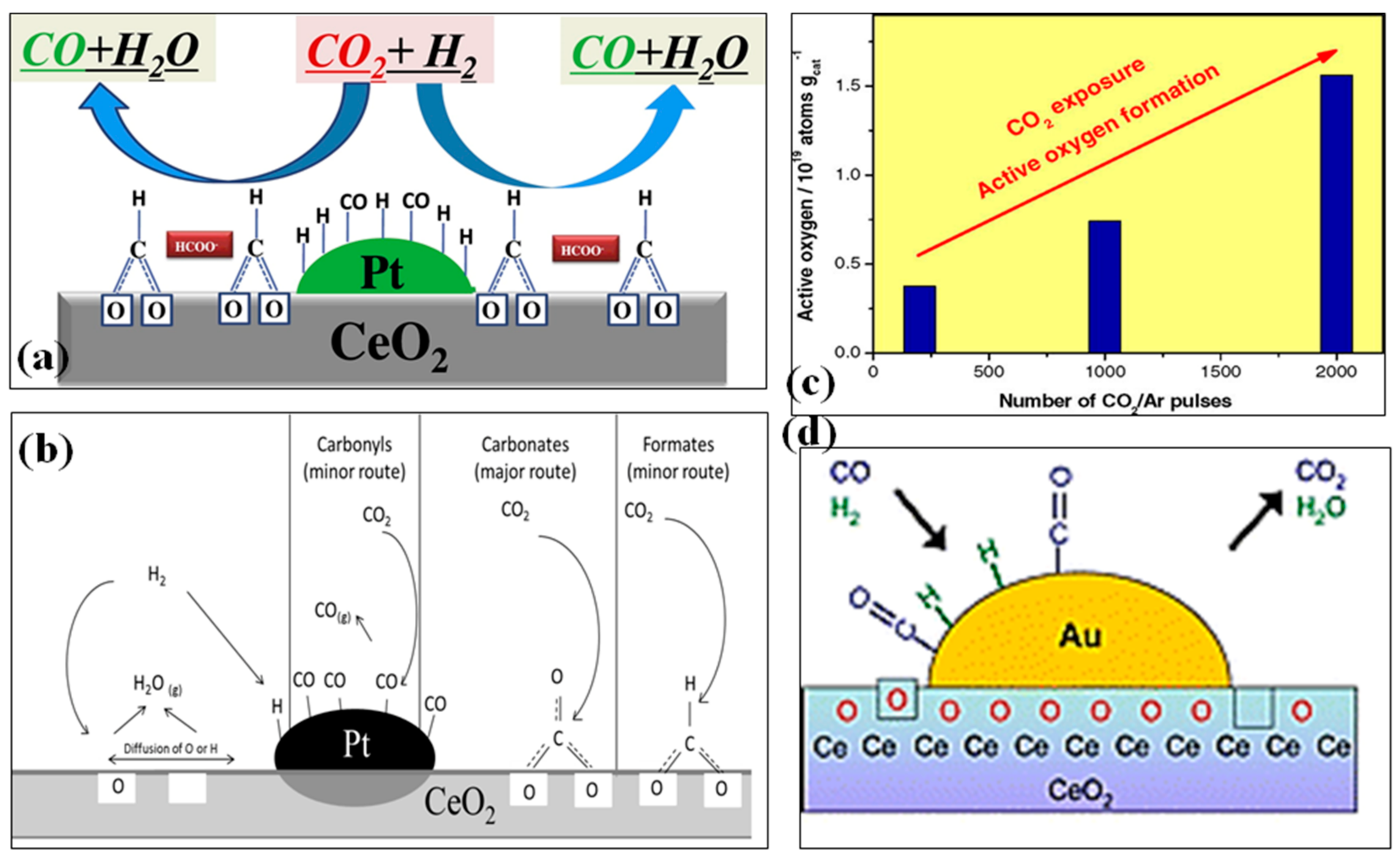
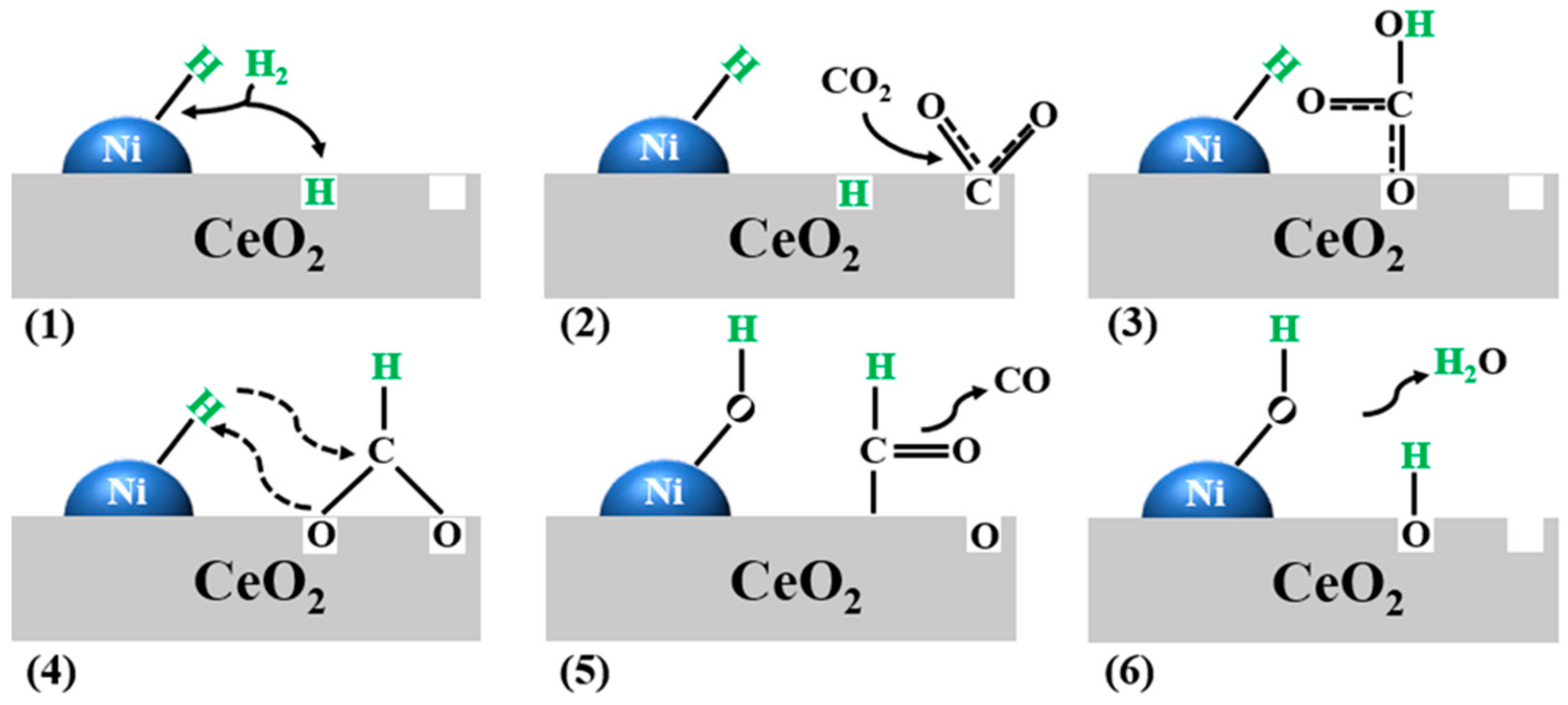
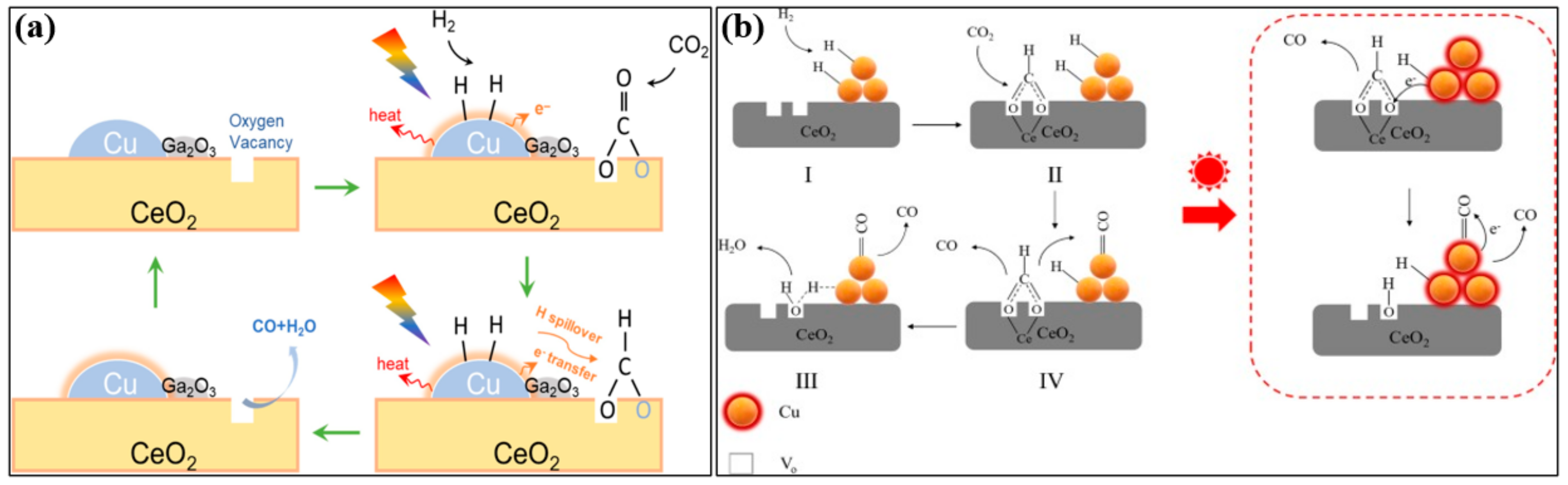
2. Mechanism




3. Kinetics of RWGS Reaction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | RWGS Reaction Rate | Assumption | Ref. |
|---|---|---|---|
| Cu-CeO2/ZrO2 |
| [107] | |
| Ni-Al2O3 |
| [110] | |
| CuO-ZnO/Al2O3 |
| [105] | |
| Pt-TiO2 Pt-Al2O3 |
| [79] | |
| Pt-Al2O3 |
| [115] | |
| FeMo-Al2O3 |
| [116] | |
| ALE-Cu/SiO2 |
| [117] | |
| Fe-K/γ-Al2O3 |
| [118] | |
| Fe/K@γ-Al2O3 |
| [120] |
4. Effect of Different Parameters on Catalyst Performance in RWGS Reaction
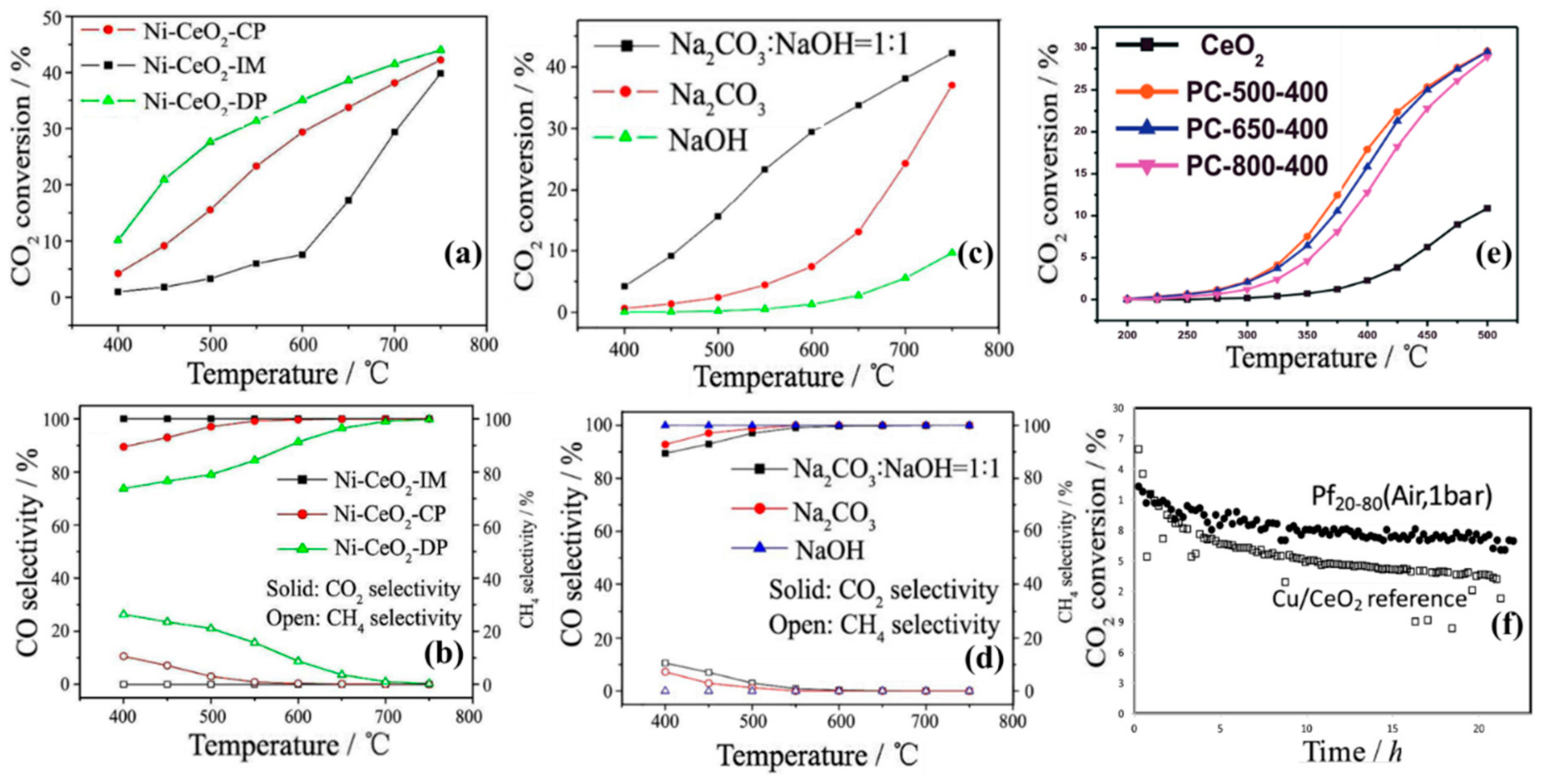
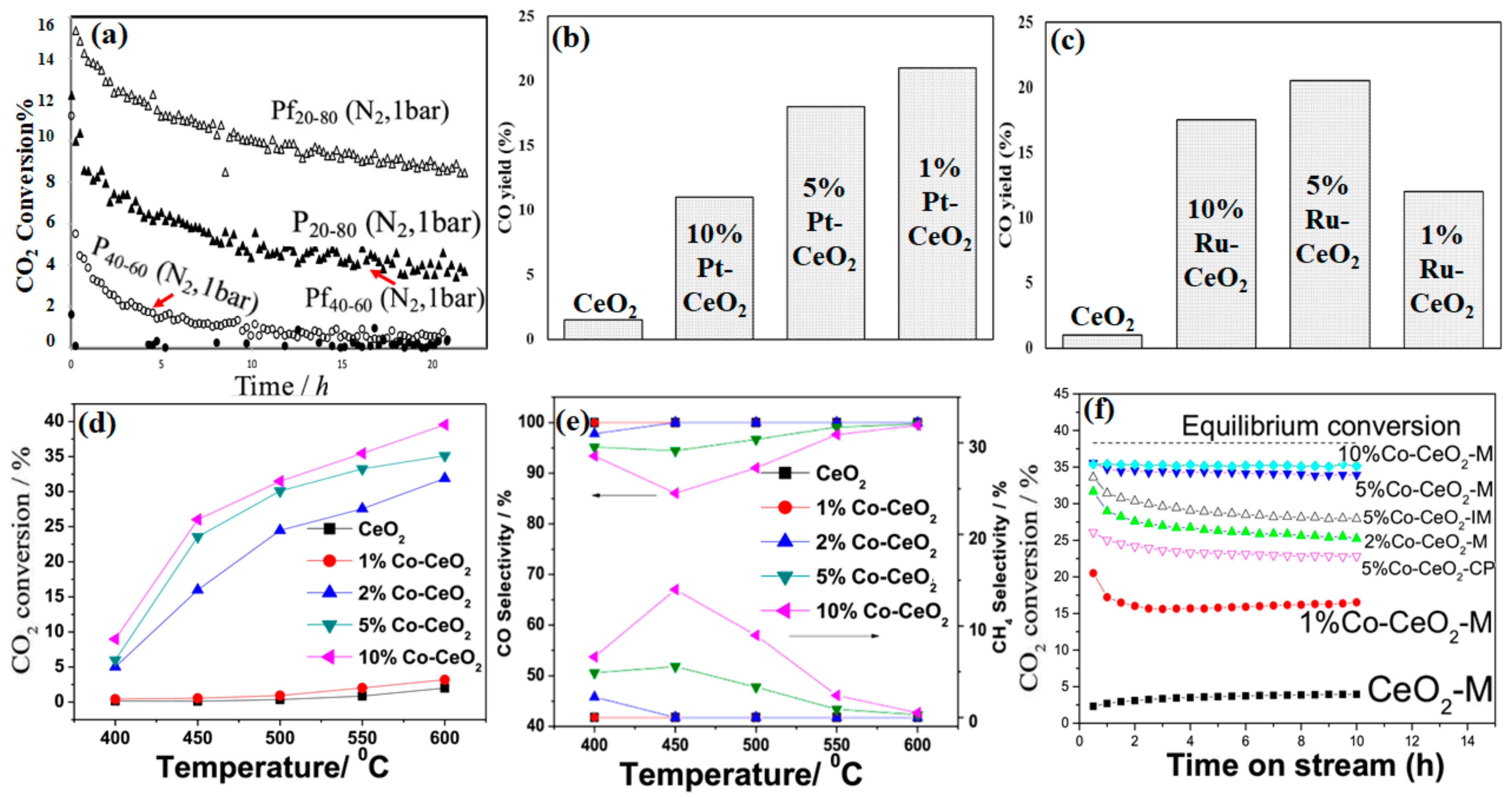
4.1. Preparation Methods
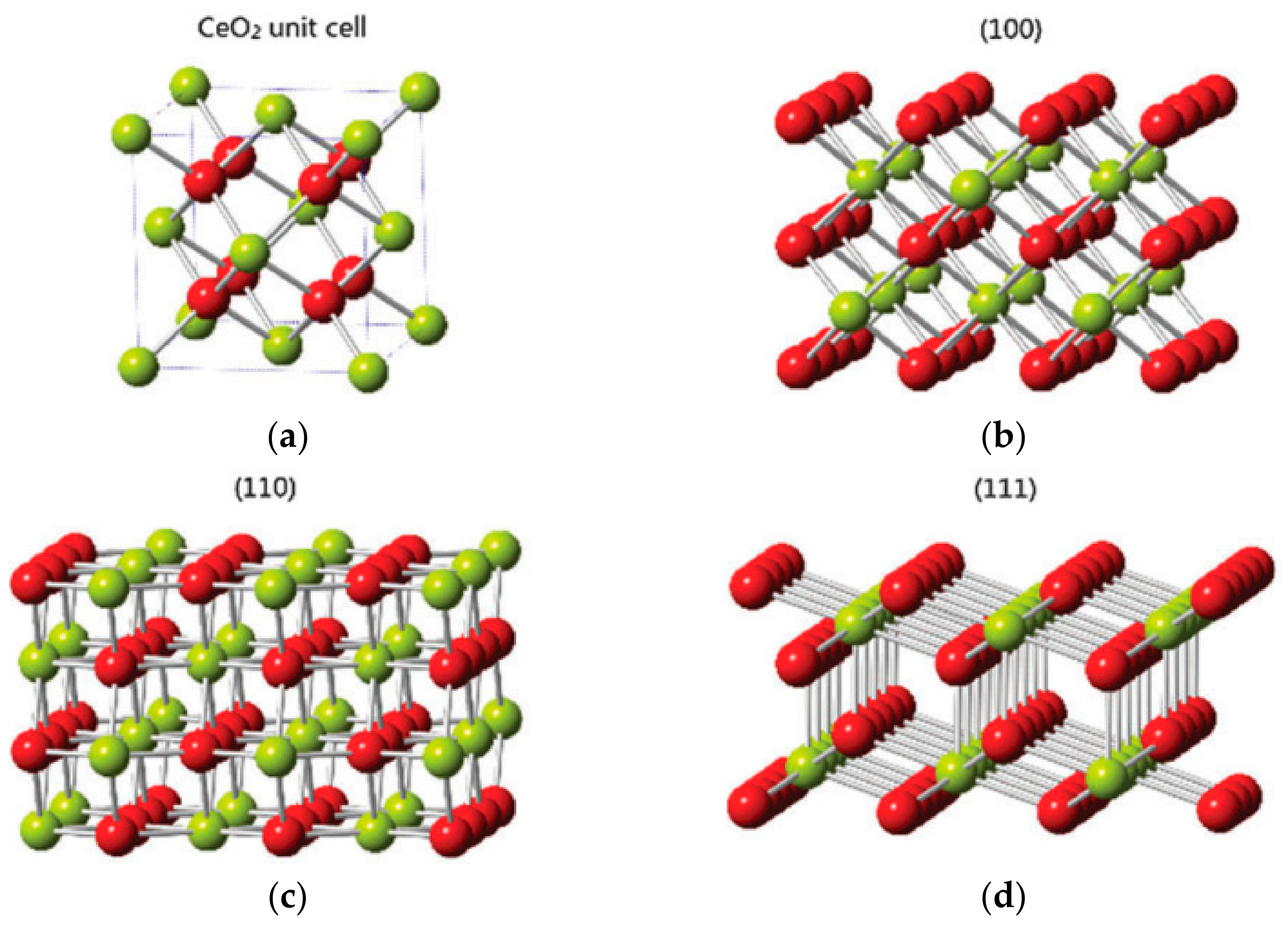
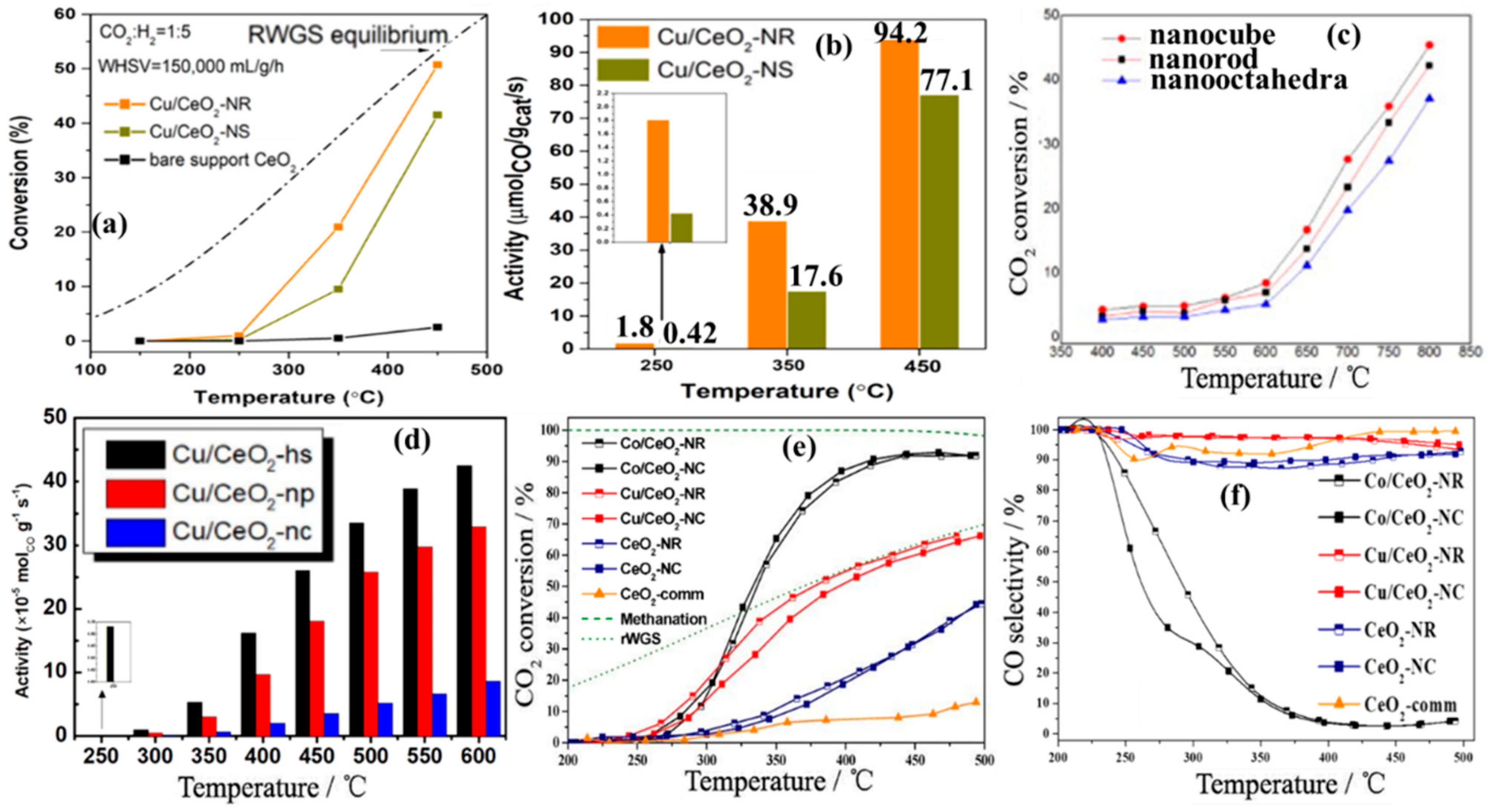
4.2. Shape and Crystal Face Effect
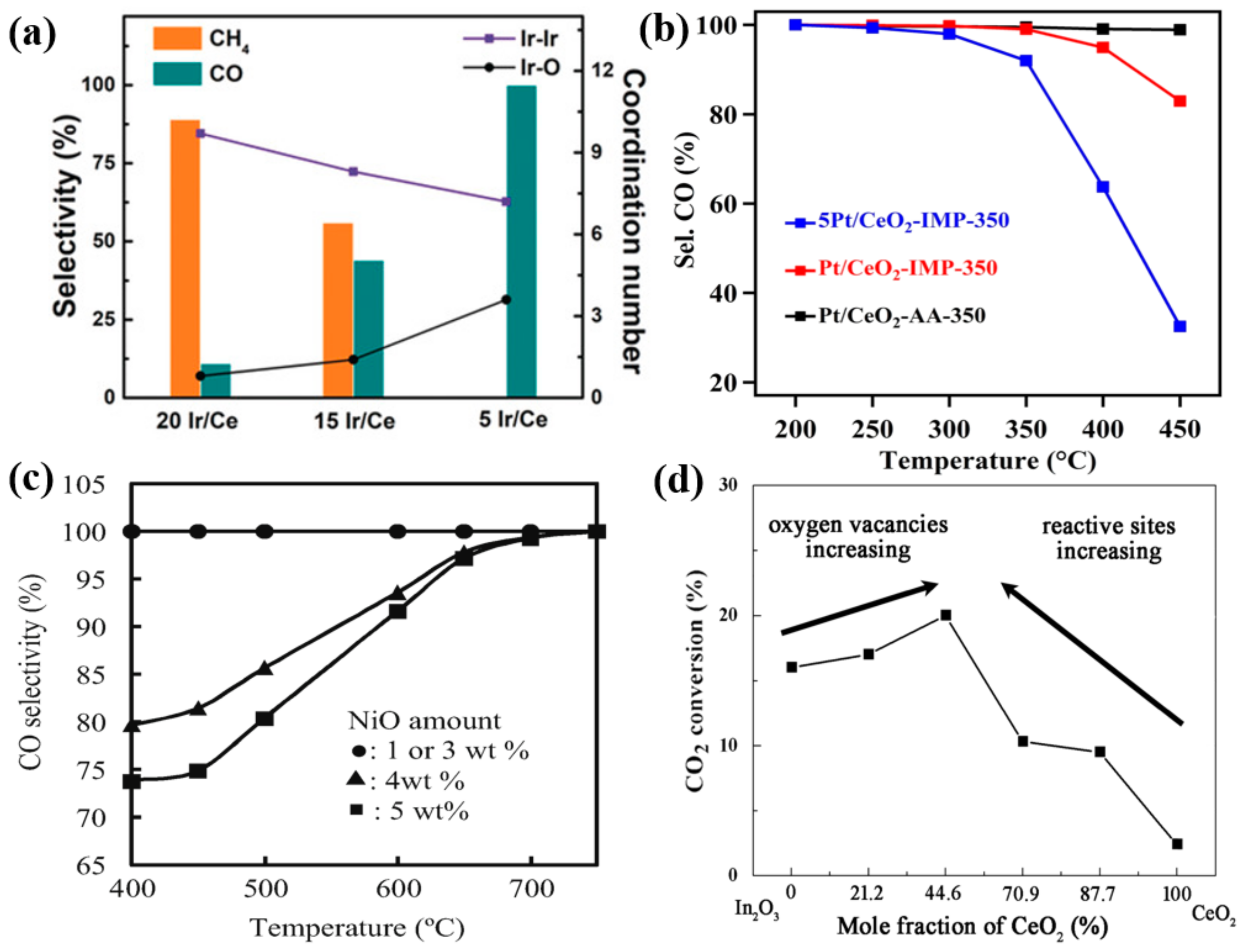
4.3. Metal–Support Interactions

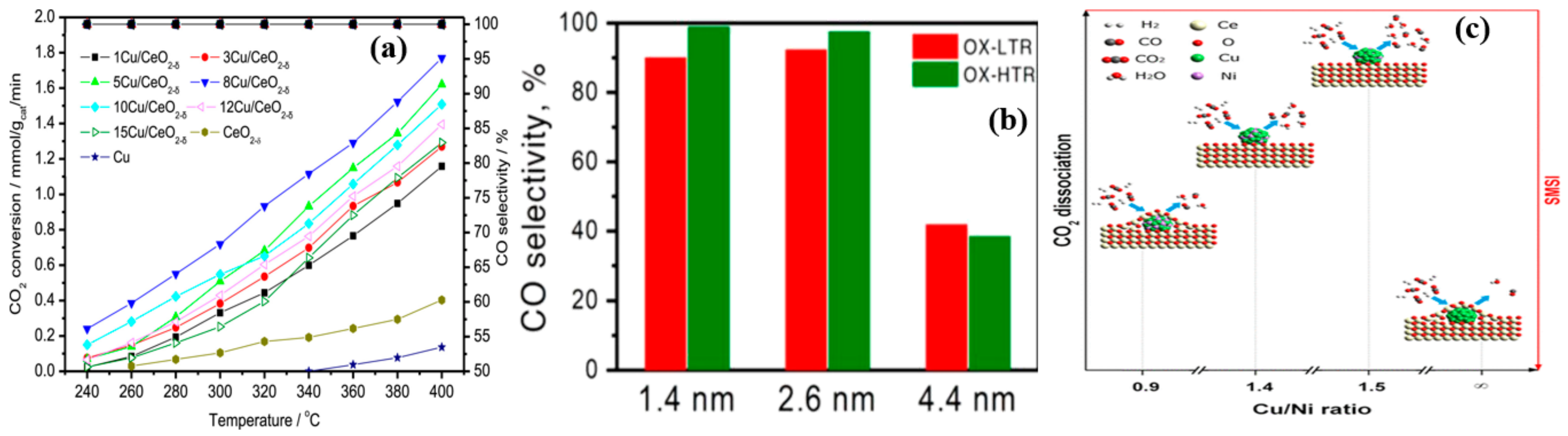
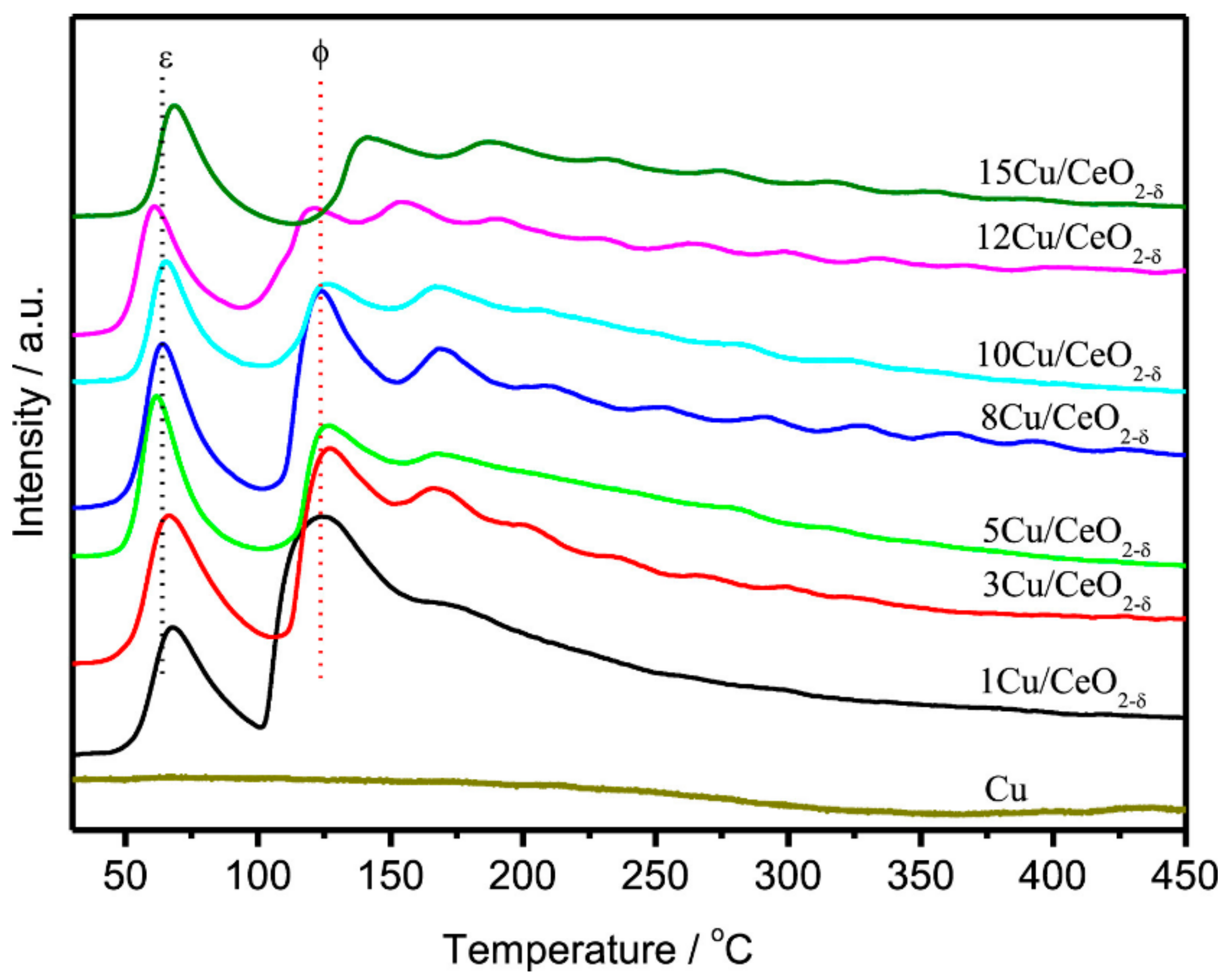
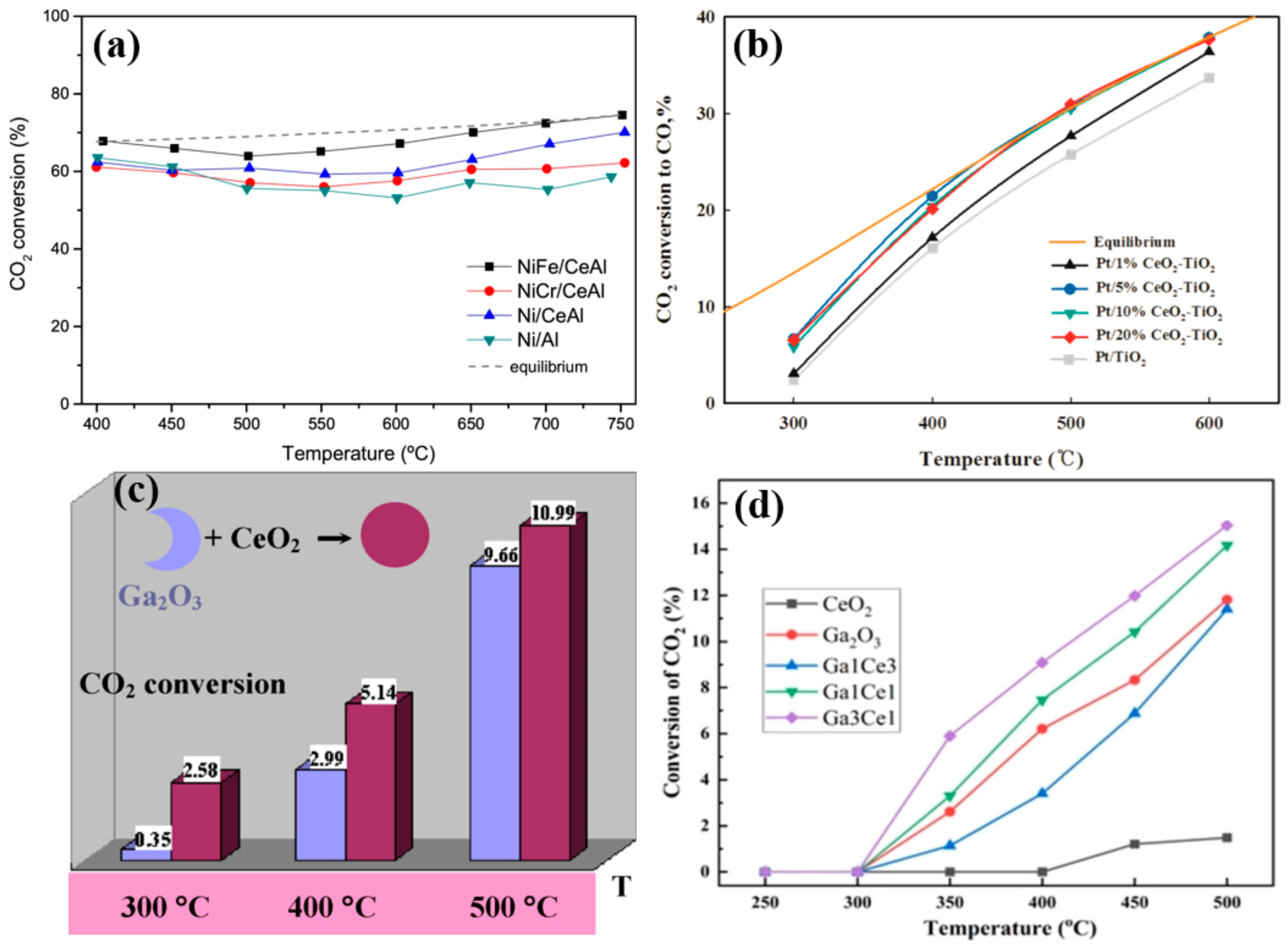
4.4. Active Metal Loading
4.5. Metal Size Effect
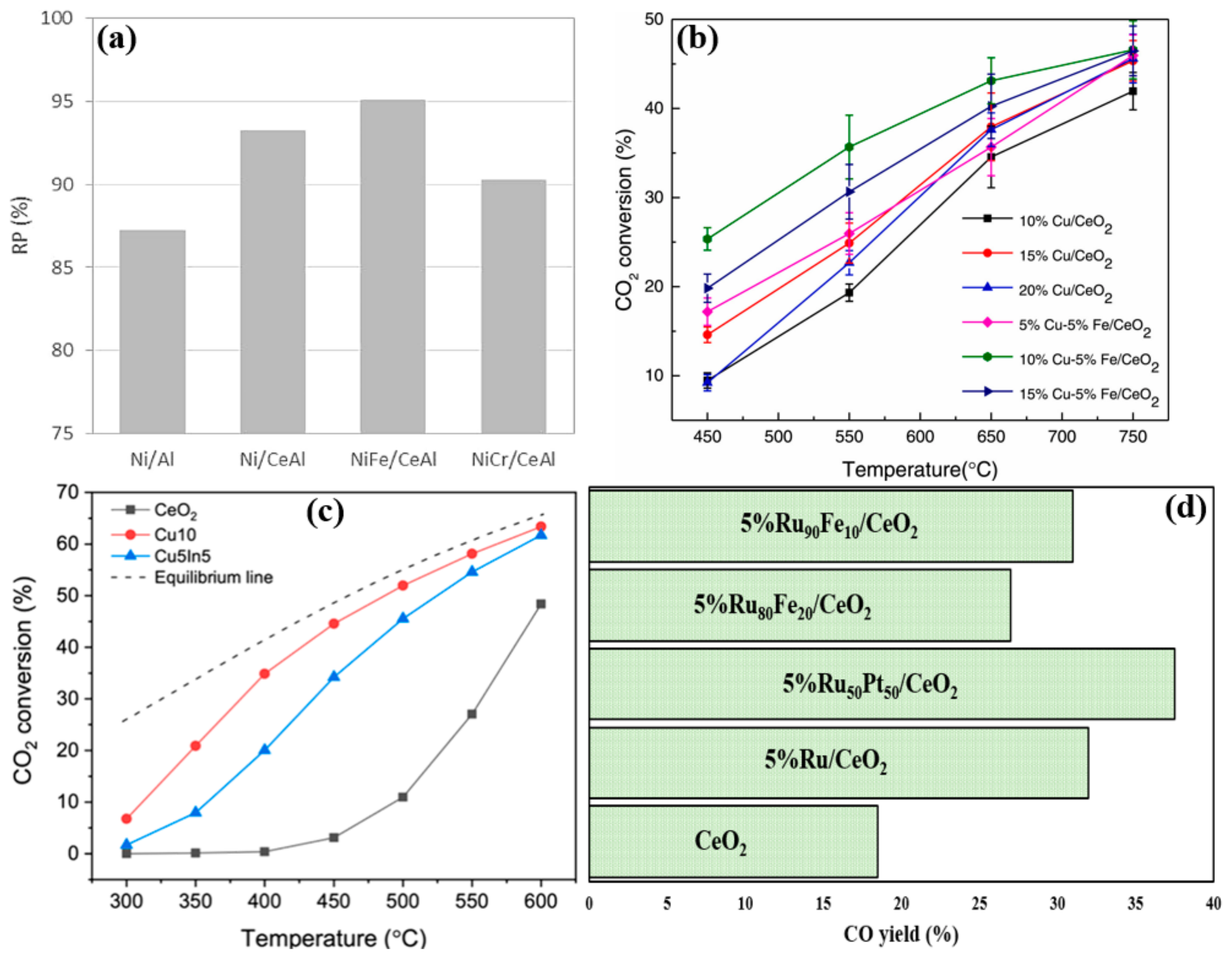
4.6. Effect of Adding CeO2 as a Reducible Transition Metal Oxide Promoter
4.7. Bimetallic Effect
5. Summary and Outlook
- -
- CeO2 supports are renowned for their acid-base properties, and high oxygen mobility and stability, which improve RWGS reaction activity when added to a suitable active metal catalytic system.
- -
- Surface defects (Ce3+ and oxygen vacancies) enable metal particles to attach well to the ceria support, resulting in increased metal dispersion.
- -
- A Ce4+/Ce3+ redox pair with strong activity can significantly improve CO2 dissociative activation.
- -
- Sintering processes and coke formation may be hampered by a strong metal–support interaction (SMSI) leading to an enhancement in catalyst stability.
- -
- CeO2 can be produced by a variety of methods to provide suitable design and technical features generating appropriate metal–support interactions that can further be tuned by synthesis methods and catalyst pretreatment techniques.
- -
- CeO2 support could influence the reaction mechanism by changing the adsorption energy of key intermediates with MSI.
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ABC | Artificial Bee Colony |
| ALE | Atomic Layer Epitaxy |
| BET | Brunauer–Emmett–Teller |
| CA | Complex |
| CP | Co-Precipitation |
| DE | Differential Evolution |
| DFT | Density Functional Theory |
| DP | Deposition-Precipitation |
| DRIFTS | Diffuse Reflectance Infrared Fourier-Transform Spectroscopy |
| EDX | Energy Dispersive X-ray |
| FCC | Face-Centered Cubic |
| FTS | Fischer–Tropsch Synthesis |
| HT | Hard-template |
| HTR | High Temperature Reduction |
| IM | Impregnation |
| LHHW | Langmuir–Hinshelwood–Hougen–Watson |
| LN | Liquid Nitrogen |
| LSPR | Localized Surface Plasmon Resonance |
| LTR | Low Temperature Reduction |
| M | Mesoporous |
| MOF | Metal Organic Framework |
| MSI | Metal–Support Interaction |
| NAP | Near Ambient Pressure |
| NC | Nanocube |
| NO | Nanooctahedra |
| NR | Nanorod |
| NS | Nanospheres |
| OSC | Oxygen Storage Capacity |
| OX | Oxidation |
| P | Pyrolysis |
| PC | Precipitation |
| PF | Flash Pyrolized |
| RDS | Rate-Determining Step |
| RM | Reverse Microemulsion |
| RP | Reduction Percentage |
| RWGS | Reverse Water Gas Shift |
| SEM | Scanning Electron Microscopy |
| SMSI | Strong Metal–Support Interaction |
| TEM | Transmission Electron Microscopy |
| TPR | Temperature Programmed Reduction |
| WGS | Water Gas Shift |
| WI | Wet Impregnation |
| XPS | X-ray Photoelectron Spectroscopy |
| XRD | X-ray Diffraction |
References
- Yang, X.; Su, X.; Chen, X.; Duan, H.; Liang, B.; Liu, Q.; Liu, X.; Ren, Y.; Huang, Y.; Zhang, T. Promotion effects of potassium on the activity and selectivity of Pt/zeolite catalysts for reverse water gas shift reaction. Appl. Catal. B Environ. 2017, 216, 95–105. [Google Scholar] [CrossRef]
- Ebrahimi, P.; Kumar, A.; Khraisheh, M. Thermodynamic assessment of effect of ammonia, hydrazine and urea on water gas shift reaction. Int. J. Hydrogen Energy 2020, 47, 3237–3247. [Google Scholar] [CrossRef]
- Sasol ECOFT and Deutsche Aircraft join forces to accelerate power-to-liquid (PTL) aiming for carbon-neutral flight. Focus Catal. 2022, 2022, 4. [CrossRef]
- Liu, X.; Pajares, A.; Matienzo, D.D.C.; de la Piscina, P.R.; Homs, N. Preparation and characterization of bulk MoXC catalysts and their use in the reverse water-gas shift reaction. Catal. Today 2020, 356, 384–389. [Google Scholar] [CrossRef]
- Centi, G.; Perathoner, S. Opportunities and prospects in the chemical recycling of carbon dioxide to fuels. Catal. Today 2009, 148, 191–205. [Google Scholar] [CrossRef]
- Aresta, M. Carbon Dioxide as Chemical Feedstock; John Wiley & Sons: Hoboken, NJ, USA, 2010. [Google Scholar]
- Ueckerdt, F.; Bauer, C.; Dirnaichner, A.; Everall, J.; Sacchi, R.; Luderer, G. Potential and risks of hydrogen-based e-fuels in climate change mitigation. Nat. Clim. Chang. 2021, 11, 384–393. [Google Scholar] [CrossRef]
- Olah, G.A.; Goeppert, A.; Prakash, G.S. Chemical recycling of carbon dioxide to methanol and dimethyl ether: From greenhouse gas to renewable, environmentally carbon neutral fuels and synthetic hydrocarbons. J. Org. Chem. 2009, 74, 487–498. [Google Scholar] [CrossRef]
- Sterner, M. Bioenergy and Renewable Power Methane in Integrated 100% Renewable Energy Systems. Limiting Global Warming by Transforming Energy Systems: Limiting Global Warming by Transforming Energy Systems; Kassel University Press GmbH: Hesse, Germany, 2009; Volume 14. [Google Scholar]
- Pastor-Pérez, L.; Baibars, F.; Le Sache, E.; Arellano-Garcia, H.; Gu, S.; Reina, T.R. CO2 valorisation via reverse water-gas shift reaction using advanced Cs doped Fe-Cu/Al2O3 catalysts. J. CO2 Util. 2017, 21, 423–428. [Google Scholar] [CrossRef]
- Galván, C.Á.; Schumann, J.; Behrens, M.; Fierro, J.L.G.; Schlögl, R.; Frei, E. Reverse water-gas shift reaction at the Cu/ZnO interface: Influence of the Cu/Zn ratio on structure-activity correlations. Appl. Catal. B Environ. 2016, 195, 104–111. [Google Scholar] [CrossRef]
- Sheehan, S.W. Electrochemical methane production from CO2 for orbital and interplanetary refueling. Iscience 2021, 24, 102230. [Google Scholar] [CrossRef]
- Xu, X.D.; Moulijn, J.A. Mitigation of CO2 by chemical conversion: Plausible chemical reactions and promising products. Energy Fuels 1996, 10, 305–325. [Google Scholar]
- Kwak, J.H.; Kovarik, L.; Szanyi, J.N. Heterogeneous catalysis on atomically dispersed supported metals: CO2 reduction on multifunctional Pd catalysts. ACS Catal. 2013, 3, 2094–2100. [Google Scholar] [CrossRef]
- Yasuda, T.; Uchiage, E.; Fujitani, T.; Tominaga, K.-I.; Nishida, M. Reverse water gas shift reaction using supported ionic liquid phase catalysts. Appl. Catal. B Environ. 2018, 232, 299–305. [Google Scholar] [CrossRef]
- Wender, I. Reactions of synthesis gas. Fuel Process. Technol. 1996, 48, 189–297. [Google Scholar] [CrossRef]
- ExxonMobil unveils tech for methanol-to-SAF. Focus Catal. 2022, 2022, 6. [CrossRef]
- Kumar, A.; Mohammed, A.A.; Saad, M.A.; Al-Marri, M.J. Effect of nickel on combustion synthesized copper/fumed-SiO2 catalyst for selective reduction of CO2 to CO. Int. J. Energy Res. 2022, 46, 441–451. [Google Scholar] [CrossRef]
- De Miranda, P.E.V. Chapter 5.3.3—Application of Hydrogen by Use of Chemical Reactions of Hydrogen and Carbon Dioxide. In Science and Engineering of Hydrogen-Based Energy Technologies; de Miranda, P.E.V., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 279–289. [Google Scholar]
- Stangeland, K.; Kalai, D.; Li, H.; Yu, Z. CO2 methanation: The effect of catalysts and reaction conditions. Energy Procedia 2017, 105, 2022–2027. [Google Scholar] [CrossRef]
- Nityashree, N.; Price, C.; Pastor-Perez, L.; Manohara, G.; Garcia, S.; Maroto-Valer, M.M.; Reina, T. Carbon stabilised saponite supported transition metal-alloy catalysts for chemical CO2 utilisation via reverse water-gas shift reaction. Appl. Catal. B Environ. 2020, 261, 118241. [Google Scholar] [CrossRef]
- Dias, Y.R.; Perez-Lopez, O.W. Carbon dioxide methanation over Ni-Cu/SiO2 catalysts. Energy Convers. Manage. 2020, 203, 112214. [Google Scholar] [CrossRef]
- Elsernagawy, O.Y.; Hoadley, A.; Patel, J.; Bhatelia, T.; Lim, S.; Haque, N. Thermo-economic analysis of reverse water-gas shift process with different temperatures for green methanol production as a hydrogen carrier. J. CO2 Util. 2020, 41, 101280. [Google Scholar] [CrossRef]
- Kaiser, P.; Unde, R.B.; Kern, C.; Jess, A. Production of liquid hydrocarbons with CO2 as carbon source based on reverse water-gas shift and Fischer-Tropsch synthesis. Chem. Ing. Tech. 2013, 85, 489–499. [Google Scholar] [CrossRef]
- York, A.P.; Xiao, T.C.; Green, M.L.; Claridge, J.B. Methane oxyforming for synthesis gas production. Catal. Rev. 2007, 49, 511–560. [Google Scholar] [CrossRef]
- Unde, R.B. Kinetics and Reaction Engineering Aspects of Syngas Production by the Heterogeneously Catalysed Reverse Water Gas Shift Reaction; Universitaet Bayreuth: Bayreuth, Germany, 2012. [Google Scholar]
- Wu, H.; Chang, Y.; Wu, J.; Lin, J.; Lin, I.; Chen, C. Methanation of CO2 and reverse water gas shift reactions on Ni/SiO2 catalysts: The influence of particle size on selectivity and reaction pathway. Catal. Sci. Technol. 2015, 5, 4154–4163. [Google Scholar] [CrossRef]
- Ebrahimi, P.; Kumar, A.; Khraisheh, M. Combustion synthesis of copper ceria solid solution for CO2 conversion to CO via reverse water gas shift reaction. Int. J. Hydrogen Energy 2022, in press. [CrossRef]
- Ammal, S.C.; Heyden, A. Origin of the unique activity of Pt/TiO2 catalysts for the water–gas shift reaction. J. Catal. 2013, 306, 78–90. [Google Scholar] [CrossRef]
- Ebrahimi, P.; Kumar, A.; Khraisheh, M. A review of recent advances in water-gas shift catalysis for hydrogen production. Emergent Mater. 2020, 3, 881–917. [Google Scholar] [CrossRef]
- Boaro, M.; Colussi, S.; Trovarelli, A. Ceria-based materials in hydrogenation and reforming reactions for CO2 valorization. Front. Chem. 2019, 7, 28. [Google Scholar] [CrossRef]
- Ebrahimi, P.; Kumar, A.; Khraisheh, M. Analysis of combustion synthesis method for Cu/CeO2 synthesis by integrating thermodynamics and design of experiments approach. Results Eng. 2022, 15, 100574. [Google Scholar] [CrossRef]
- Ishito, N.; Hara, K.; Nakajima, K.; Fukuoka, A. Selective synthesis of carbon monoxide via formates in reverse water–gas shift reaction over alumina-supported gold catalyst. J. Energy Chem. 2016, 25, 306–310. [Google Scholar] [CrossRef]
- Trovarelli, A. Catalytic properties of ceria and CeO2-containing materials. Catal. Rev. 1996, 38, 439–520. [Google Scholar] [CrossRef]
- Yeung, C.M.; Tsang, S.C. A study of co-precipitated bimetallic gold catalysts for water–gas shift reaction. Catal. Commun. 2008, 9, 1551–1557. [Google Scholar]
- Chang, K.; Zhang, H.; Cheng, M.-J.; Lu, Q. Application of ceria in CO2 conversion catalysis. ACS Catal. 2019, 10, 613–631. [Google Scholar] [CrossRef]
- Castaño, M.G.; Reina, T.R.; Ivanova, S.; Centeno, M.; Odriozola, J.A. Pt vs. Au in water–gas shift reaction. J. Catal. 2014, 314, 1–9. [Google Scholar] [CrossRef]
- Moreira, M.N.; Ribeiro, A.M.; Cunha, A.F.; Rodrigues, A.E.; Zabilskiy, M.; Djinović, P.; Pintar, A. Copper based materials for water-gas shift equilibrium displacement. Appl. Catal. B Environ. 2016, 189, 199–209. [Google Scholar] [CrossRef]
- Porosoff, M.D.; Chen, J.G. Trends in the catalytic reduction of CO2 by hydrogen over supported monometallic and bimetallic catalysts. J. Catal. 2013, 301, 30–37. [Google Scholar] [CrossRef]
- Younis, A.; Chu, D.; Li, S. Cerium oxide nanostructures and their applications. Funct. Nanomater. 2016, 3, 53–68. [Google Scholar]
- Yao, S.; Xu, W.; Johnston-Peck, A.; Zhao, F.; Liu, Z.; Luo, S.; Senanayake, S.; Martínez-Arias, A.; Liu, W.; Rodriguez, J. Morphological effects of the nanostructured ceria support on the activity and stability of CuO/CeO2 catalysts for the water-gas shift reaction. PCCP 2014, 16, 17183–17195. [Google Scholar] [CrossRef]
- Sayle, D.C.; Maicaneanu, S.A.; Watson, G.W. Atomistic models for CeO2 (111), (110), and (100) nanoparticles, supported on yttrium-stabilized zirconia. J. Am. Chem. Soc. 2002, 124, 11429–11439. [Google Scholar] [CrossRef]
- Jiang, Y.; Adams, J.B.; Van Schilfgaarde, M. Density-functional calculation of CeO2 surfaces and prediction of effects of oxygen partial pressure and temperature on stabilities. J. Chem. Phys. 2005, 123, 064701. [Google Scholar] [CrossRef]
- Chen, Y.; Hu, P.; Lee, M.-H.; Wang, H. Au on (1 1 1) and (1 1 0) surfaces of CeO2: A density-functional theory study. Surf. Sci. 2008, 602, 1736–1741. [Google Scholar] [CrossRef]
- Sayle, T.X.; Cantoni, M.; Bhatta, U.M.; Parker, S.C.; Hall, S.R.; Möbus, G.; Molinari, M.; Reid, D.; Seal, S.; Sayle, D.C. Strain and architecture-tuned reactivity in ceria nanostructures; enhanced catalytic oxidation of CO to CO2. Chem. Mater. 2012, 24, 1811–1821. [Google Scholar] [CrossRef]
- Liu, X.; Zhou, K.; Wang, L.; Wang, B.; Li, Y. Oxygen vacancy clusters promoting reducibility and activity of ceria nanorods. J. Am. Chem. Soc. 2009, 131, 3140–3141. [Google Scholar] [CrossRef] [PubMed]
- Si, R.; Flytzani-Stephanopoulos, M. Shape and crystal-plane effects of nanoscale ceria on the activity of Au-CeO2 catalysts for the water–gas shift reaction. Angew. Chem. 2008, 120, 2926–2929. [Google Scholar] [CrossRef]
- Stubenrauch, J.; Brosha, E.; Vohs, J. Reaction of carboxylic acids on CeO2 (111) and CeO2 (100). Catal. Today 1996, 28, 431–441. [Google Scholar] [CrossRef]
- Mei, D. First-principles characterization of formate and carboxyl adsorption on stoichiometric CeO2 (111) and CeO2 (110) surfaces. J. Energy Chem. 2013, 22, 524–532. [Google Scholar] [CrossRef]
- Senanayake, S.D.; Mullins, D.R. Redox pathways for HCOOH decomposition over CeO2 surfaces. J. Phys. Chem. C 2008, 112, 9744–9752. [Google Scholar] [CrossRef]
- Zhou, G.; Dai, B.; Xie, H.; Zhang, G.; Xiong, K.; Zheng, X. CeCu composite catalyst for CO synthesis by reverse water–gas shift reaction: Effect of Ce/Cu mole ratio. J. CO2 Util. 2017, 21, 292–301. [Google Scholar] [CrossRef]
- Reddy, B.M.; Thrimurthulu, G.; Katta, L. Design of efficient CexM1− xO2− δ (M= Zr, Hf, Tb and Pr) nanosized model solid solutions for CO oxidation. Catal. Lett. 2011, 141, 572–581. [Google Scholar] [CrossRef]
- Lu, B.; Kawamoto, K. Preparation of mesoporous CeO2 and monodispersed NiO particles in CeO2, and enhanced selectivity of NiO/CeO2 for reverse water gas shift reaction. Mater. Res. Bull. 2014, 53, 70–78. [Google Scholar] [CrossRef]
- Wang, L.; Liu, H.; Chen, Y.; Yang, S. Reverse water–gas shift reaction over co-precipitated Co–CeO2 catalysts: Effect of Co content on selectivity and carbon formation. Int. J. Hydrogen Energy 2017, 42, 3682–3689. [Google Scholar] [CrossRef]
- Aitbekova, A.; Wu, L.; Wrasman, C.J.; Boubnov, A.; Hoffman, A.S.; Goodman, E.D.; Bare, S.R.; Cargnello, M. Low-temperature restructuring of CeO2-supported Ru nanoparticles determines selectivity in CO2 catalytic reduction. J. Am. Chem. Soc. 2018, 140, 13736–13745. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Sherman, B.J.; Lo, C.S. Carbon dioxide activation and dissociation on ceria (110): A density functional theory study. J. Chem. Phys. 2013, 138, 014702. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Wang, W.; Wei, S.; Guo, C.; Shao, Y.; Zhang, M.; Deng, Z.; Zhu, H.; Guo, W. Initial reduction of CO2 on perfect and O-defective CeO2 (111) surfaces: Towards CO or COOH? RSC Adv. 2015, 5, 97528–97535. [Google Scholar] [CrossRef]
- Wu, K.; Sun, L.D.; Yan, C.H. Recent Progress in Well-Controlled Synthesis of Ceria-Based Nanocatalysts towards Enhanced Catalytic Performance. Adv. Energy Mater. 2016, 6, 1600501. [Google Scholar] [CrossRef]
- Trovarelli, A.; Llorca, J. Ceria catalysts at nanoscale: How do crystal shapes shape catalysis? ACS Catal. 2017, 7, 4716–4735. [Google Scholar] [CrossRef]
- Ma, Y.; Gao, W.; Zhang, Z.; Zhang, S.; Tian, Z.; Liu, Y.; Ho, J.C.; Qu, Y. Regulating the surface of nanoceria and its applications in heterogeneous catalysis. Surf. Sci. Rep. 2018, 73, 1–36. [Google Scholar] [CrossRef]
- Saeidi, S.; Najari, S.; Fazlollahi, F.; Nikoo, M.K.; Sefidkon, F.; Klemeš, J.J.; Baxter, L.L. Mechanisms and kinetics of CO2 hydrogenation to value-added products: A detailed review on current status and future trends. Renew. Sustain. Energy Rev. 2017, 80, 1292–1311. [Google Scholar] [CrossRef]
- Wang, W.; Wang, S.; Ma, X.; Gong, J. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 2011, 40, 3703–3727. [Google Scholar] [CrossRef]
- Porosoff, M.D.; Yan, B.; Chen, J.G. Catalytic reduction of CO2 by H2 for synthesis of CO, methanol and hydrocarbons: Challenges and opportunities. Energy Environ. Sci. 2016, 9, 62–73. [Google Scholar] [CrossRef]
- Kattel, S.; Liu, P.; Chen, J.G. Tuning selectivity of CO2 hydrogenation reactions at the metal/oxide interface. J. Am. Chem. Soc. 2017, 139, 9739–9754. [Google Scholar] [CrossRef]
- Su, X.; Yang, X.; Zhao, B.; Huang, Y. Designing of highly selective and high-temperature endurable RWGS heterogeneous catalysts: Recent advances and the future directions. J. Energy Chem. 2017, 26, 854–867. [Google Scholar] [CrossRef]
- Trovarelli, A. Catalysis by Ceria and Related Materials; World Scientific: Singapore, 2002; Volume 2. [Google Scholar]
- Montini, T.; Melchionna, M.; Monai, M.; Fornasiero, P. Fundamentals and catalytic applications of CeO2-based materials. Chem. Rev. 2016, 116, 5987–6041. [Google Scholar] [CrossRef]
- Xie, S.; Wang, Z.; Cheng, F.; Zhang, P.; Mai, W.; Tong, Y. Ceria and ceria-based nanostructured materials for photoenergy applications. Nano Energy 2017, 34, 313–337. [Google Scholar] [CrossRef]
- Devaiah, D.; Reddy, L.H.; Park, S.-E.; Reddy, B.M. Ceria–zirconia mixed oxides: Synthetic methods and applications. Catal. Rev. 2018, 60, 177–277. [Google Scholar] [CrossRef]
- Su, X.; Xu, J.; Liang, B.; Duan, H.; Hou, B.; Huang, Y. Catalytic carbon dioxide hydrogenation to methane: A review of recent studies. J. Energy Chem. 2016, 25, 553–565. [Google Scholar] [CrossRef]
- Frontera, P.; Macario, A.; Ferraro, M.; Antonucci, P. Supported catalysts for CO2 methanation: A review. Catalysts 2017, 7, 59. [Google Scholar] [CrossRef]
- Zhang, M.; Zijlstra, B.; Filot, I.A.; Li, F.; Wang, H.; Li, J.; Hensen, E.J. A theoretical study of the reverse water-gas shift reaction on Ni (111) and Ni (311) surfaces. Can. J. Chem. Eng. 2020, 98, 740–748. [Google Scholar] [CrossRef]
- Fornero, E.L.; Chiavassa, D.L.; Bonivardi, A.L.; Baltanás, M.A. Transient analysis of the reverse water gas shift reaction on Cu/ZrO2 and Ga2O3/Cu/ZrO2 catalysts. J. CO2 Util. 2017, 22, 289–298. [Google Scholar] [CrossRef]
- Zhu, M.; Ge, Q.; Zhu, X. Catalytic reduction of CO2 to CO via reverse water gas shift reaction: Recent advances in the design of active and selective supported metal catalysts. Trans. Tianjin Univ. 2020, 26, 172–187. [Google Scholar] [CrossRef]
- Lin, W.; Stocker, K.M.; Schatz, G.C. Mechanisms of hydrogen-assisted CO2 reduction on nickel. J. Am. Chem. Soc. 2017, 139, 4663–4666. [Google Scholar] [CrossRef]
- González-Castaño, M.; Dorneanu, B.; Arellano-García, H. The reverse water gas shift reaction: A process systems engineering perspective. React. Chem. Eng. 2021, 6, 954–976. [Google Scholar]
- Kim, K.-J.; Lee, Y.-L.; Na, H.-S.; Ahn, S.-Y.; Shim, J.-O.; Jeon, B.-H.; Roh, H.-S. Efficient waste to energy conversion based on CO-CeO2 catalyzed water-gas shift reaction. Catalysts 2020, 10, 420. [Google Scholar]
- Mathew, T.; Saju, S.; Raveendran, S.N. Survey of Heterogeneous Catalysts for the CO2 Reduction to CO via Reverse Water Gas Shift. In Engineering Solutions for CO2 Conversion; Reina, T.R., Arellano-Garcia, H., Odriozola, J.A., Eds.; Wiley: Hoboken, NJ, USA, 2021; Chapter 12; pp. 281–316. [Google Scholar]
- Kim, S.S.; Lee, H.H.; Hong, S.C. A study on the effect of support’s reducibility on the reverse water-gas shift reaction over Pt catalysts. Appl. Catal. A Gen. 2012, 423, 100–107. [Google Scholar]
- Wang, L.-C.; Khazaneh, M.T.; Widmann, D.; Behm, R.J. TAP reactor studies of the oxidizing capability of CO2 on a Au/CeO2 catalyst—A first step toward identifying a redox mechanism in the Reverse Water–Gas Shift reaction. J. Catal. 2013, 302, 20–30. [Google Scholar]
- Lin, L.; Yao, S.; Liu, Z.; Zhang, F.; Li, N.; Vovchok, D.; Martinez-Arias, A.; Castañeda, R.; Lin, J.; Senanayake, S.D. In situ characterization of Cu/CeO2 nanocatalysts for CO2 hydrogenation: Morphological effects of nanostructured ceria on the catalytic activity. J. Phys. Chem. C 2018, 122, 12934–12943. [Google Scholar]
- Lee, S.M.; Eom, H.; Kim, S.S. A study on the effect of CeO2 addition to a Pt/TiO2 catalyst on the reverse water gas shift reaction. Environ. Technol. 2021, 42, 182–192. [Google Scholar]
- Liu, Y.; Li, Z.; Xu, H.; Han, Y. Reverse water–gas shift reaction over ceria nanocube synthesized by hydrothermal method. Catal. Commun. 2016, 76, 1–6. [Google Scholar]
- Goguet, A.; Meunier, F.C.; Tibiletti, D.; Breen, J.P.; Burch, R. Spectrokinetic investigation of reverse water-gas-shift reaction intermediates over a Pt/CeO2 catalyst. J. Phys. Chem. B 2004, 108, 20240–20246. [Google Scholar]
- Zhu, X.; Qu, X.; Li, X.; Liu, J.; Liu, J.; Zhu, B.; Shi, C. Selective reduction of carbon dioxide to carbon monoxide over Au/CeO2 catalyst and identification of reaction intermediate. Chin. J. Catal. 2016, 37, 2053–2058. [Google Scholar]
- Lu, B.; Zhang, T.; Zhang, L.; Xu, Y.; Zhang, Z.; Wu, F.; Li, X.; Luo, C. Promotion effects of oxygen vacancies on activity of Na-doped CeO2 catalysts for reverse water gas shift reaction. Appl. Surf. Sci. 2022, 587, 152881. [Google Scholar]
- Wang, M.; Shen, M.; Jin, X.; Tian, J.; Li, M.; Zhou, Y.; Zhang, L.; Li, Y.; Shi, J. Oxygen vacancy generation and stabilization in CeO2–x by Cu introduction with improved CO2 photocatalytic reduction activity. ACS Catal. 2019, 9, 4573–4581. [Google Scholar] [CrossRef]
- Zhang, Y.; Liang, L.; Chen, Z.; Wen, J.; Zhong, W.; Zou, S.; Fu, M.; Chen, L.; Ye, D. Highly efficient Cu/CeO2-hollow nanospheres catalyst for the reverse water-gas shift reaction: Investigation on the role of oxygen vacancies through in situ UV-Raman and DRIFTS. Appl. Surf. Sci. 2020, 516, 146035. [Google Scholar] [CrossRef]
- Jacobs, G.; Davis, B.H. Reverse water-gas shift reaction: Steady state isotope switching study of the reverse water-gas shift reaction using in situ DRIFTS and a Pt/ceria catalyst. Appl. Catal. A Gen. 2005, 284, 31–38. [Google Scholar] [CrossRef]
- Dai, H.; Zhang, A.; Xiong, S.; Xiao, X.; Zhou, C.; Pan, Y. The Catalytic Performance of Ga2O3−CeO2 Composite Oxides over Reverse Water Gas Shift Reaction. ChemCatChem 2022, 14, e202200049. [Google Scholar] [CrossRef]
- Li, M.; My Pham, T.H.; Ko, Y.; Zhao, K.; Zhong, L.; Luo, W.; Züttel, A. Support-Dependent Cu–In Bimetallic Catalysts for Tailoring the Activity of Reverse Water Gas Shift Reaction. ACS Sustain. Chem. Eng. 2022, 10, 1524–1535. [Google Scholar] [CrossRef]
- Meunier, F.C.; Tibiletti, D.; Goguet, A.; Reid, D.; Burch, R. On the reactivity of carbonate species on a Pt/CeO2 catalyst under various reaction atmospheres: Application of the isotopic exchange technique. Appl. Catal. A Gen. 2005, 289, 104–112. [Google Scholar] [CrossRef]
- Kalamaras, C.M.; Americanou, S.; Efstathiou, A.M. “Redox” vs “associative formate with–OH group regeneration” WGS reaction mechanism on Pt/CeO2: Effect of platinum particle size. J. Catal. 2011, 279, 287–300. [Google Scholar] [CrossRef]
- Chen, X.; Su, X.; Liang, B.; Yang, X.; Ren, X.; Duan, H.; Huang, Y.; Zhang, T. Identification of relevant active sites and a mechanism study for reverse water gas shift reaction over Pt/CeO2 catalysts. J. Energy Chem. 2016, 25, 1051–1057. [Google Scholar] [CrossRef]
- Juarez, R.; Parker, S.F.; Concepcion, P.; Corma, A.; Garcia, H. Heterolytic and heterotopic dissociation of hydrogen on ceria-supported gold nanoparticles. Combined inelastic neutron scattering and FT-IR spectroscopic study on the nature and reactivity of surface hydrogen species. Chem. Sci. 2010, 1, 731–738. [Google Scholar] [CrossRef]
- Wang, L.; Widmann, D.; Behm, R.J. Reactive removal of surface oxygen by H2, CO and CO/H2 on a Au/CeO2 catalyst and its relevance to the preferential CO oxidation (PROX) and reverse water gas shift (RWGS) reaction. Catal. Sci. Technol. 2015, 5, 925–941. [Google Scholar] [CrossRef]
- Shen, H.; Dong, Y.; Yang, S.; He, Y.; Wang, Q.; Cao, Y.; Wang, W.; Wang, T.; Zhang, Q.; Zhang, H. Identifying the roles of Ce3+−OH and Ce−H in the reverse water-gas shift reaction over highly active Ni-doped CeO2 catalyst. Nano Res. 2022, 15, 5831–5841. [Google Scholar] [CrossRef]
- Deng, B.; Song, H.; Peng, K.; Li, Q.; Ye, J. Metal-organic framework-derived Ga-Cu/CeO2 catalyst for highly efficient photothermal catalytic CO2 reduction. Appl. Catal. B Environ. 2021, 298, 120519. [Google Scholar] [CrossRef]
- Zhao, B.; Pan, Y.-X.; Liu, C.-J. The promotion effect of CeO2 on CO2 adsorption and hydrogenation over Ga2O3. Catal. Today 2012, 194, 60–64. [Google Scholar] [CrossRef]
- Yang, Z.; Zeng, M.; Wang, K.; Yue, X.; Chen, X.; Dai, W.; Fu, X. Visible light-assisted thermal catalytic reverse water gas reaction over Cu-CeO2: The synergistic of hot electrons and oxygen vacancies induced by LSPR effect. Fuel 2022, 315, 123186. [Google Scholar] [CrossRef]
- Wolf, A.; Jess, A.; Kern, C. Syngas Production via Reverse Water-Gas Shift Reaction over a Ni-Al2O3 Catalyst: Catalyst Stability, Reaction Kinetics, and Modeling. Chem. Eng. Technol. 2016, 39, 1040–1048. [Google Scholar] [CrossRef]
- Pekridis, G.; Kalimeri, K.; Kaklidis, N.; Vakouftsi, E.; Iliopoulou, E.; Athanasiou, C.; Marnellos, G. Study of the reverse water gas shift (RWGS) reaction over Pt in a solid oxide fuel cell (SOFC) operating under open and closed-circuit conditions. Catal. Today 2007, 127, 337–346. [Google Scholar] [CrossRef]
- Spencer, M. On the activation energies of the forward and reverse water-gas shift reaction. Catal. Lett. 1995, 32, 9–13. [Google Scholar] [CrossRef]
- Ernst, K.-H.; Campbell, C.T.; Moretti, G. Kinetics of the reverse water-gas shift reaction over Cu (110). J. Catal. 1992, 134, 66–74. [Google Scholar] [CrossRef]
- Ginés, M.; Marchi, A.; Apesteguía, C. Kinetic study of the reverse water-gas shift reaction over CuO/ZnO/Al2O3 catalysts. Appl. Catal. A Gen. 1997, 154, 155–171. [Google Scholar] [CrossRef]
- Osaki, T.; Narita, N.; Horiuchi, T.; Sugiyama, T.; Masuda, H.; Suzuki, K. Kinetics of reverse water gas shift (RWGS) reaction on metal disulfide catalysts. J. Mol. Catal. A Chem. 1997, 125, 63–71. [Google Scholar] [CrossRef]
- Poto, S.; van Berkel, D.V.; Gallucci, F.; d’Angelo, M.F.N. Kinetic modelling of the methanol synthesis from CO2 and H2 over a CuO/CeO2/ZrO2 catalyst: The role of CO2 and CO hydrogenation. Chem. Eng. J. 2022, 435, 134946. [Google Scholar] [CrossRef]
- Henkel, T. Modellierung Von Reaktion und Stofftransport in Geformten Katalysatoren am Beispiel der Methanolsynthese. Ph.D. Thesis, Technische Universität München, München, Germany, 2011. [Google Scholar]
- Graaf, G.; Stamhuis, E.; Beenackers, A. Kinetics of low-pressure methanol synthesis. Chem. Eng. Sci. 1988, 43, 3185–3195. [Google Scholar] [CrossRef]
- Lalinde, J.A.H.; Roongruangsree, P.; Ilsemann, J.; Baeumer, M.; Kopyscinski, J. CO2 methanation and reverse water gas shift reaction. Kinetic study based on in situ spatially-resolved measurements. Chem. Eng. J. 2020, 390, 124629. [Google Scholar] [CrossRef]
- Kopyscinski, J.; Schildhauer, T.J.; Vogel, F.; Biollaz, S.M.; Wokaun, A. Applying spatially resolved concentration and temperature measurements in a catalytic plate reactor for the kinetic study of CO methanation. J. Catal. 2010, 271, 262–279. [Google Scholar] [CrossRef]
- Daza, Y.A.; Kuhn, J.N. CO2 conversion by reverse water gas shift catalysis: Comparison of catalysts, mechanisms and their consequences for CO2 conversion to liquid fuels. RSC Adv. 2016, 6, 49675–49691. [Google Scholar] [CrossRef]
- Campbell, C.T.; Ernst, K.-H. Forward and Reverse Water—Gas Shift Reactions on Model Copper Catalysts: Kinetics and Elementary Steps; ACS Publications: Washington, DC, USA, 1992. [Google Scholar]
- Kim, S.S.; Lee, H.H.; Hong, S.C. The effect of the morphological characteristics of TiO2 supports on the reverse water–gas shift reaction over Pt/TiO2 catalysts. Appl. Catal. B Environ. 2012, 119, 100–108. [Google Scholar] [CrossRef]
- Jadhav, S.G.; Vaidya, P.D.; Bhanage, B.M.; Joshi, J.B. Kinetics of reverse water-gas shift reaction over Pt/Al2O3 catalyst. Can. J. Chem. Eng. 2016, 94, 101–106. [Google Scholar] [CrossRef]
- Ghodoosi, F.; Khosravi-Nikou, M.R.; Shariati, A. Mathematical Modeling of Reverse Water-Gas Shift Reaction in a Fixed-Bed Reactor. Chem. Eng. Technol. 2017, 40, 598–607. [Google Scholar] [CrossRef]
- Chen, C.S.; Wu, J.H.; Lai, T.W. Carbon dioxide hydrogenation on Cu nanoparticles. J. Phys. Chem. C 2010, 114, 15021–15028. [Google Scholar] [CrossRef]
- Najari, S.; Gróf, G.; Saeidi, S.; Gallucci, F. Modeling and optimization of hydrogenation of CO2: Estimation of kinetic parameters via Artificial Bee Colony (ABC) and Differential Evolution (DE) algorithms. Int. J. Hydrogen Energy 2019, 44, 4630–4649. [Google Scholar] [CrossRef]
- Najari, S.; Saeidi, S.; Gróf, G.; Keil, F.J.; Rodrigues, A.E. Kinetic parameters estimation via dragonfly algorithm (DA) and comparison of cylindrical and spherical reactors performance for CO2 hydrogenation to hydrocarbons. Energy Convers. Manage. 2020, 226, 113550. [Google Scholar] [CrossRef]
- Brübach, L.; Hodonj, D.; Pfeifer, P. Kinetic Analysis of CO2 Hydrogenation to Long-Chain Hydrocarbons on a Supported Iron Catalyst. Ind. Eng. Chem. Res. 2022, 61, 1644–1654. [Google Scholar] [CrossRef]
- Luhui, W.; Hui, L.; Yuan, L.; Ying, C.; Shuqing, Y. Influence of preparation method on performance of Ni-CeO2 catalysts for reverse water-gas shift reaction. J. Rare Earths 2013, 31, 559–564. [Google Scholar]
- Li, Y.; Zhang, B.; Tang, X.; Xu, Y.; Shen, W. Hydrogen production from methane decomposition over Ni/CeO2 catalysts. Catal. Commun. 2006, 7, 380–386. [Google Scholar] [CrossRef]
- Yisup, N.; Cao, Y.; Feng, W.-L.; Dai, W.-L.; Fan, K.-N. Catalytic oxidation of methane over novel Ce–Ni–O mixed oxide catalysts prepared by oxalate gel-coprecipitation. Catal. Lett. 2005, 99, 207–213. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, A.; Li, L.; Wang, X.; Su, D.; Zhang, T. “Ir-in-ceria”: A highly selective catalyst for preferential CO oxidation. J. Catal. 2008, 255, 144–152. [Google Scholar] [CrossRef]
- Li, L.; Song, L.; Wang, H.; Chen, C.; She, Y.; Zhan, Y.; Lin, X.; Zheng, Q. Water-gas shift reaction over CuO/CeO2 catalysts: Effect of CeO2 supports previously prepared by precipitation with different precipitants. Int. J. Hydrogen Energy 2011, 36, 8839–8849. [Google Scholar] [CrossRef]
- Yongyi, H.; Qibiao, L.; Yongzhao, W.; Yongxiang, Z. Selective catalytic dehydration of 1, 4-butanediol to 3-buten-1-ol over CeO2 with different morphology. Chin. J. Catal. 2010, 31, 619–622. [Google Scholar]
- Dong, X.-F.; Zou, H.-B.; Lin, W.-M. Effect of preparation conditions of CuO–CeO2–ZrO2 catalyst on CO removal from hydrogen-rich gas. Int. J. Hydrogen Energy 2006, 31, 2337–2344. [Google Scholar] [CrossRef]
- Xue, L.; Zhang, C.; He, H.; Teraoka, Y. Promotion effect of residual K on the decomposition of N2O over cobalt–cerium mixed oxide catalyst. Catal. Today 2007, 126, 449–455. [Google Scholar] [CrossRef]
- Mei, Y.; Meisheng, C.; Zhang, N.; Zhiqi, L.; Huang, X. Characterization of CeO2-ZrO2 mixed oxides prepared by two different co-precipitation methods. J. Rare Earths 2013, 31, 251–256. [Google Scholar]
- Luhui, W.; Hui, L.; Yuan, L.; Ying, C.; Shuqing, Y. Effect of precipitants on Ni-CeO2 catalysts prepared by a co-precipitation method for the reverse water-gas shift reaction. J. Rare Earths 2013, 31, 969–974. [Google Scholar]
- Dai, B.; Cao, S.; Xie, H.; Zhou, G.; Chen, S. Reduction of CO2 to CO via reverse water-gas shift reaction over CeO2 catalyst. Korean J. Chem. Eng. 2018, 35, 421–427. [Google Scholar] [CrossRef]
- Ronda-Lloret, M.; Rico-Francés, S.; Sepúlveda-Escribano, A.; Ramos-Fernandez, E.V. CuOx/CeO2 catalyst derived from metal organic framework for reverse water-gas shift reaction. Appl. Catal. A: Gen. 2018, 562, 28–36. [Google Scholar] [CrossRef]
- Kim, T.K.; Lee, K.J.; Cheon, J.Y.; Lee, J.H.; Joo, S.H.; Moon, H.R. Nanoporous metal oxides with tunable and nanocrystalline frameworks via conversion of metal–organic frameworks. J. Am. Chem. Soc. 2013, 135, 8940–8946. [Google Scholar] [CrossRef]
- Liu, L.; Fan, F.; Bai, M.; Xue, F.; Ma, X.; Jiang, Z.; Fang, T. Mechanistic study of methanol synthesis from CO2 hydrogenation on Rh-doped Cu (111) surfaces. Mol. Catal. 2019, 466, 26–36. [Google Scholar] [CrossRef]
- Lykaki, M.; Pachatouridou, E.; Carabineiro, S.A.; Iliopoulou, E.; Andriopoulou, C.; Kallithrakas-Kontos, N.; Boghosian, S.; Konsolakis, M. Ceria nanoparticles shape effects on the structural defects and surface chemistry: Implications in CO oxidation by Cu/CeO2 catalysts. Appl. Catal. B: Environ. 2018, 230, 18–28. [Google Scholar] [CrossRef]
- Zheng, X.; Li, Y.; Zhang, L.; Shen, L.; Xiao, Y.; Zhang, Y.; Au, C.; Jiang, L. Insight into the effect of morphology on catalytic performance of porous CeO2 nanocrystals for H2S selective oxidation. Appl. Catal. B Environ. 2019, 252, 98–110. [Google Scholar] [CrossRef]
- Kovacevic, M.; Mojet, B.L.; van Ommen, J.G.; Lefferts, L. Effects of morphology of cerium oxide catalysts for reverse water gas shift reaction. Catal. Lett. 2016, 146, 770–777. [Google Scholar] [CrossRef]
- Esch, F.; Fabris, S.; Zhou, L.; Montini, T.; Africh, C.; Fornasiero, P.; Comelli, G.; Rosei, R. Electron localization determines defect formation on ceria substrates. Science 2005, 309, 752–755. [Google Scholar] [CrossRef]
- Skorodumova, N.; Baudin, M.; Hermansson, K. Surface properties of CeO2 from first principles. Phys. Rev. B 2004, 69, 075401. [Google Scholar] [CrossRef]
- Konsolakis, M.; Lykaki, M.; Stefa, S.; Carabineiro, S.A.; Varvoutis, G.; Papista, E.; Marnellos, G.E. CO2 hydrogenation over nanoceria-supported transition metal catalysts: Role of ceria morphology (nanorods versus nanocubes) and active phase nature (Co versus Cu). Nanomaterials 2019, 9, 1739. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-S.; Chung, S.-H.; Yoo, C.-J.; Lee, M.S.; Cho, I.-H.; Lee, D.-W.; Lee, K.-Y. Catalytic reduction of nitrate in water over Pd–Cu/TiO2 catalyst: Effect of the strong metal-support interaction (SMSI) on the catalytic activity. Appl. Catal. B Environ. 2013, 142, 354–361. [Google Scholar] [CrossRef]
- Liotta, L.; Longo, A.; Macaluso, A.; Martorana, A.; Pantaleo, G.; Venezia, A.; Deganello, G. Influence of the SMSI effect on the catalytic activity of a Pt (1%)/Ce0.6Zr0.4O2 catalyst: SAXS, XRD, XPS and TPR investigations. Appl. Catal. B Environ. 2004, 48, 133–149. [Google Scholar] [CrossRef]
- Bertella, F.; Concepción, P.; Martínez, A. The impact of support surface area on the SMSI decoration effect and catalytic performance for Fischer-Tropsch synthesis of Co-Ru/TiO2-anatase catalysts. Catal. Today 2017, 296, 170–180. [Google Scholar] [CrossRef]
- Tauster, S.; Fung, S.; Garten, R.L. Strong metal-support interactions. Group 8 noble metals supported on titanium dioxide. J. Am. Chem. Soc. 1978, 100, 170–175. [Google Scholar] [CrossRef]
- Hosokawa, S.; Taniguchi, M.; Utani, K.; Kanai, H.; Imamura, S. Affinity order among noble metals and CeO2. Appl. Catal. A: Gen. 2005, 289, 115–120. [Google Scholar] [CrossRef]
- Ivanova, A.; Slavinskaya, E.; Gulyaev, R.; Zaikovskii, V.; Stonkus, O.; Danilova, I.; Plyasova, L.; Polukhina, I.; Boronin, A. Metal–support interactions in Pt/Al2O3 and Pd/Al2O3 catalysts for CO oxidation. Appl. Catal. B Environ. 2010, 97, 57–71. [Google Scholar] [CrossRef]
- Strobel, R.; Pratsinis, S.E. Flame synthesis of supported platinum group metals for catalysis and sensors. Platin. Met. Rev. 2009, 53, 11–20. [Google Scholar] [CrossRef]
- Zou, H.; Dong, X.; Lin, W. Selective CO oxidation in hydrogen-rich gas over CuO/CeO2 catalysts. Appl. Surf. Sci. 2006, 253, 2893–2898. [Google Scholar] [CrossRef]
- Zhou, G.; Xie, F.; Deng, L.; Zhang, G.; Xie, H. Supported mesoporous Cu/CeO2-δ catalyst for CO2 reverse water–gas shift reaction to syngas. Int. J. Hydrogen Energy 2020, 45, 11380–11393. [Google Scholar] [CrossRef]
- Tauster, S. Strong metal-support interactions. Acc. Chem. Res. 1987, 20, 389–394. [Google Scholar] [CrossRef]
- Matte, L.P.; Kilian, A.S.; Luza, L.; Alves, M.C.; Morais, J.; Baptista, D.L.; Dupont, J.; Bernardi, F. Influence of the CeO2 support on the reduction properties of Cu/CeO2 and Ni/CeO2 nanoparticles. J. Phys. Chem. C 2015, 119, 26459–26470. [Google Scholar] [CrossRef]
- Thill, A.S.; Kilian, A.S.; Bernardi, F. Key role played by metallic nanoparticles on the ceria reduction. J. Phys. Chem. C 2017, 121, 25323–25332. [Google Scholar] [CrossRef]
- Figueiredo, W.T.; Escudero, C.; Perez-Dieste, V.; Ospina, C.A.; Bernardi, F. Determining the Surface Atomic Population of Cux Ni1–x/CeO2 (0 < x ≤ 1) Nanoparticles during the Reverse Water–Gas Shift (RWGS) Reaction. J. Phys. Chem. C 2020, 124, 16868–16878. [Google Scholar]
- Einakchi, R. Metal Nanoparticles Over Active Ionic-Conductive Supports for the Reverse Water Gas Shift Reaction. Master’s Dissertation, Université d’Ottawa/University of Ottawa, Ottawa, ON, Canada, 2016. [Google Scholar]
- Wang, L.; Liu, H. Mesoporous Co-CeO2 catalyst prepared by colloidal solution combustion method for reverse water-gas shift reaction. Catal. Today 2018, 316, 155–161. [Google Scholar] [CrossRef]
- Jurković, D.L.; Pohar, A.; Dasireddy, V.D.; Likozar, B. Effect of copper-based catalyst support on reverse water-gas shift reaction (RWGS) activity for CO2 reduction. Chem. Eng. Technol. 2017, 40, 973–980. [Google Scholar] [CrossRef]
- Bouarab, R.; Akdim, O.; Auroux, A.; Cherifi, O.; Mirodatos, C. Effect of MgO additive on catalytic properties of Co/SiO2 in the dry reforming of methane. Appl. Catal. A Gen. 2004, 264, 161–168. [Google Scholar] [CrossRef]
- Pino, L.; Vita, A.; Cipitì, F.; Laganà, M.; Recupero, V. Hydrogen production by methane tri-reforming process over Ni–ceria catalysts: Effect of La-doping. Appl. Catal. B Environ. 2011, 104, 64–73. [Google Scholar] [CrossRef]
- Chen, X.; Su, X.; Duan, H.; Liang, B.; Huang, Y.; Zhang, T. Catalytic performance of the Pt/TiO2 catalysts in reverse water gas shift reaction: Controlled product selectivity and a mechanism study. Catal. Today 2017, 281, 312–318. [Google Scholar] [CrossRef]
- Liu, L.; Corma, A. Metal catalysts for heterogeneous catalysis: From single atoms to nanoclusters and nanoparticles. Chem. Rev. 2018, 118, 4981–5079. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Xu, Y.; Chen, Y.; Li, W.; Lin, L.; Li, M.; Deng, Y.; Wang, X.; Ge, B.; Yang, C. Tuning the selectivity of catalytic carbon dioxide hydrogenation over iridium/cerium oxide catalysts with a strong metal–support interaction. Angew. Chem. 2017, 129, 10901–10905. [Google Scholar] [CrossRef]
- Chen, X.; Chen, Y.; Song, C.; Ji, P.; Wang, N.; Wang, W.; Cui, L. Recent advances in supported metal catalysts and oxide catalysts for the reverse water-gas shift reaction. Front. Chem. 2020, 8, 709. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Wang, M.; Ma, P.; Zheng, Y.; Chen, J.; Li, H.; Zhang, X.; Zheng, K.; Kuang, Q.; Xie, Z.-X. Atomically dispersed Pt/CeO2 catalyst with superior CO selectivity in reverse water gas shift reaction. Appl. Catal. B Environ. 2021, 291, 120101. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, Y.; Wang, Z.; Yan, J.-M.; Ge, Q.; Liu, C.-J. Reverse water gas shift over In 2O3–CeO2 catalysts. Catal. Today 2016, 259, 402–408. [Google Scholar] [CrossRef]
- Ro, I.; Sener, C.; Stadelman, T.M.; Ball, M.R.; Venegas, J.M.; Burt, S.P.; Hermans, I.; Dumesic, J.A.; Huber, G.W. Measurement of intrinsic catalytic activity of Pt monometallic and Pt-MoOx interfacial sites over visible light enhanced PtMoOx/SiO2 catalyst in reverse water gas shift reaction. J. Catal. 2016, 344, 784–794. [Google Scholar] [CrossRef]
- Pettigrew, D.; Trimm, D.; Cant, N. The effects of rare earth oxides on the reverse water-gas shift reaction on palladium/alumina. Catal. Lett. 1994, 28, 313–319. [Google Scholar] [CrossRef]
- Yang, L.; Pastor-Pérez, L.; Gu, S.; Sepúlveda-Escribano, A.; Reina, T. Highly efficient Ni/CeO2-Al2O3 catalysts for CO2 upgrading via reverse water-gas shift: Effect of selected transition metal promoters. Appl. Catal. B Environ. 2018, 232, 464–471. [Google Scholar] [CrossRef]
- Dorner, R.W.; Hardy, D.R.; Williams, F.W.; Willauer, H.D. Effects of ceria-doping on a CO2 hydrogenation iron–manganese catalyst. Catal. Commun. 2010, 11, 816–819. [Google Scholar] [CrossRef]
- Galvita, V.V.; Poelman, H.; Bliznuk, V.; Detavernier, C.; Marin, G.B. CeO2-modified Fe2O3 for CO2 utilization via chemical looping. Ind. Eng. Chem. Res. 2013, 52, 8416–8426. [Google Scholar] [CrossRef]
- Yan, B.; Zhao, B.; Kattel, S.; Wu, Q.; Yao, S.; Su, D.; Chen, J.G. Tuning CO2 hydrogenation selectivity via metal-oxide interfacial sites. J. Catal. 2019, 374, 60–71. [Google Scholar] [CrossRef]
- Rahmani, F.; Haghighi, M.; Estifaee, P. Synthesis and characterization of Pt/Al2O3–CeO2 nanocatalyst used for toluene abatement from waste gas streams at low temperature: Conventional vs. plasma–ultrasound hybrid synthesis methods. Microporous Mesoporous Mater. 2014, 185, 213–223. [Google Scholar] [CrossRef]
- Zonetti, P.C.; Letichevsky, S.; Gaspar, A.B.; Sousa-Aguiar, E.F.; Appel, L.G. The NixCe0. 75Zr0.25−xO2 solid solution and the RWGS. Appl. Catal. A Gen. 2014, 475, 48–54. [Google Scholar] [CrossRef]
- Wenzel, M.; Rihko-Struckmann, L.; Sundmacher, K. Continuous production of CO from CO2 by RWGS chemical looping in fixed and fluidized bed reactors. Chem. Eng. J. 2018, 336, 278–296. [Google Scholar] [CrossRef]
- Yang, L.; Pastor-Pérez, L.; Villora-Picó, J.J.; Gu, S.; Sepúlveda-Escribano, A.; Reina, T.R. CO2 valorisation via reverse water-gas shift reaction using promoted Fe/CeO2-Al2O3 catalysts: Showcasing the potential of advanced catalysts to explore new processes design. Appl. Catal. A Gen. 2020, 593, 117442. [Google Scholar] [CrossRef]
- Ray, K.; Sengupta, S.; Deo, G. Reforming and cracking of CH4 over Al2O3 supported Ni, Ni-Fe and Ni-Co catalysts. Fuel Proces. Technol. 2017, 156, 195–203. [Google Scholar] [CrossRef]
- Theofanidis, S.A.; Galvita, V.V.; Poelman, H.; Marin, G.B. Enhanced carbon-resistant dry reforming Fe-Ni catalyst: Role of Fe. Acs Catal. 2015, 5, 3028–3039. [Google Scholar] [CrossRef]
- Chen, L.; Wu, D.; Wang, C.; Ji, M.; Wu, Z. Study on Cu-Fe/CeO2 bimetallic catalyst for reverse water gas shift reaction. J. Environ. Chem. Eng. 2021, 9, 105183. [Google Scholar] [CrossRef]
- Goguet, A.; Meunier, F.; Breen, J.; Burch, R.; Petch, M.; Ghenciu, A.F. Study of the origin of the deactivation of a Pt/CeO2 catalyst during reverse water gas shift (RWGS) reaction. J. Catal. 2004, 226, 382–392. [Google Scholar] [CrossRef]
- Kattel, S.; Yu, W.; Yang, X.; Yan, B.; Huang, Y.; Wan, W.; Liu, P.; Chen, J.G. CO2 Hydrogenation over Oxide-Supported PtCo Catalysts: The Role of the Oxide Support in Determining the Product Selectivity. Angew. Chem. Int. Ed. 2016, 55, 7968–7973. [Google Scholar] [CrossRef]
- Panaritis, C.; Edake, M.; Couillard, M.; Einakchi, R.; Baranova, E.A. Insight towards the role of ceria-based supports for reverse water gas shift reaction over RuFe nanoparticles. J. CO2 Util. 2018, 26, 350–358. [Google Scholar] [CrossRef]
- Le Saché, E.; Pastor-Perez, L.; Haycock, B.J.; Villora-Picó, J.J.; Sepulveda-Escribano, A.; Reina, T.R. Switchable catalysts for chemical CO2 recycling: A step forward in the methanation and reverse water–Gas shift reactions. ACS Sustain. Chem. Eng. 2020, 8, 4614–4622. [Google Scholar] [CrossRef]
- Choi, E.J.; Lee, Y.H.; Lee, D.-W.; Moon, D.-J.; Lee, K.-Y. Hydrogenation of CO2 to methanol over Pd–Cu/CeO2 catalysts. Mol. Catal. 2017, 434, 146–153. [Google Scholar] [CrossRef]
- Porosoff, M.D.; Yang, X.; Boscoboinik, J.A.; Chen, J.G. Molybdenum carbide as alternative catalysts to precious metals for highly selective reduction of CO2 to CO. Angew. Chem. Int. Ed. 2014, 53, 6705–6709. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-C.; Pang, S.H.; Sulmonetti, T.P.; Su, W.-N.; Lee, J.-F.; Hwang, B.-J.; Jones, C.W. Synergy between ceria oxygen vacancies and Cu nanoparticles facilitates the catalytic conversion of CO2 to CO under mild conditions. ACS Catal. 2018, 8, 12056–12066. [Google Scholar] [CrossRef]
- Dai, B.; Zhou, G.; Ge, S.; Xie, H.; Jiao, Z.; Zhang, G.; Xiong, K. CO2 reverse water-gas shift reaction on mesoporous M-CeO2 catalysts. Can. J. Chem. Eng. 2017, 95, 634–642. [Google Scholar] [CrossRef]
- Vovchok, D.; Zhang, C.; Hwang, S.; Jiao, L.; Zhang, F.; Liu, Z.; Senanayake, S.D.; Rodriguez, J.A. Deciphering dynamic structural and mechanistic complexity in Cu/CeO2/ZSM-5 catalysts for the reverse water-gas shift reaction. ACS Catal. 2020, 10, 10216–10228. [Google Scholar] [CrossRef]
- Wang, L.; Liu, H.; Chen, Y.; Zhang, R.; Yang, S. K-Promoted Co–CeO2 Catalyst for the Reverse Water–Gas Shift Reaction. Chem. Lett. 2013, 42, 682–683. [Google Scholar] [CrossRef]
- Luhui, W.; Zhang, S.; Yuan, L. Reverse water gas shift reaction over Co-precipitated Ni-CeO2 catalysts. J. Rare Earths 2008, 26, 66–70. [Google Scholar]
- Sun, F.-M.; Yan, C.-F.; Guo, C.-Q.; Huang, S.-L. Ni/Ce–Zr–O catalyst for high CO2 conversion during reverse water gas shift reaction (RWGS). Int. J. Hydrogen Energy 2015, 40, 15985–15993. [Google Scholar] [CrossRef]
- Belekar, R. Suppression of coke formation during reverse water-gas shift reaction for CO2 conversion using highly active Ni/Al2O3-CeO2 catalyst material. Phys. Lett. A 2021, 395, 127206. [Google Scholar] [CrossRef]












| Active Metal | Mechanism | Ref. |
|---|---|---|
| Cu | Redox or Associative | [81,91,100] |
| Cu5In5 | Associative | [91] |
| Ni | Redox | [83] |
| Ga | Associative | [90] |
| Pt | Redox and/or Associative | [84,94] |
| Au | Redox and/or Associative | [80,96] |
| 10Ga5Cu | Associative | [98] |
| Ce | Redox | [83] |
| Catalyst | Synthesizing Method | Temperature (°C) | Conversion (%) | Selectivity (%) | Ref. |
|---|---|---|---|---|---|
| Pt-CeO2 | Co-Precipitation | 300 | 6.7 | - | [94] |
| 2%Pt-CeO2 | Commercial | 290 | 21.7 | ~100 | [178] |
| 1 wt.%Pt-CeO2 | Polyol | 500 | ~24 | ~100 | [154] |
| 3.2%PtCo-CeO2 | Incipient Wetness Impregnation | 300 | 9.1 | 92.3 | [179] |
| 5 wt.%Ru-CeO2 | Polyol | 500 | ~25 | ~100 | [154] |
| 5%Ru-CeO2 | Polyol | 350 | ~16 | ~31 | [180] |
| Ru50Pt50-CeO2 | Polyol | 500 | ~28 | ~100 | [154] |
| RuNi-CeZ | Wet Impregnation | 350 | 53 | 93 | [181] |
| 5%Ru/Sm-CeO2 | Polyol | 350 | ~16 | ~69 | [180] |
| FeNi-CeZr | Wet Impregnation | 350 | 13 | 60 | [181] |
| PtCo-CeO2 | Incipient Wetness Impregnation | 300 | 9.1 | 92.31 | [179] |
| 0.5Pd10Cu-CeO2 | Precipitation–Impregnation | 270 | 12 | - | [182] |
| 1Pd10Cu-CeO2 | Precipitation–Impregnation | 270 | 17.8 | - | [182] |
| 2Pd10Cu-CeO2 | Precipitation–Impregnation | 270 | 11.3 | - | [182] |
| PdNi-CeO2 | Incipient Wetness Impregnation | 300 | 2.5 | 37.5 | [183] |
| 10Cu-CeO2 | Precipitation-Impregnation | 270 | 6.4 | - | [182] |
| Cu-CeO2 | Space-Confined | 300 | ~18 | ~100 | [184] |
| CeO2-NC | Hydrothermal–Incipient Wetness Impregnation | 700 | 27.8 | ~100 | [83] |
| CeO2-NR | Hydrothermal–Incipient Wetness Impregnation | 700 | 23.8 | ~100 | [83] |
| CeO2-NO | Hydrothermal–Incipient Wetness Impregnation | 700 | 19.8 | ~100 | [83] |
| CeO2-HT | Hard-Template | 580 | 15.9 | ~100 | [131] |
| Cu/CeO2 | Wet Impregnation | 380 | 52 | 95 | [140] |
| CuCeOx | Hard Template | 400 | 33 | ~100 | [51] |
| CuOx-CeO2 | Wet Impregnation | 400 | 10 | ~100 | [132] |
| 1 wt.%Cu-CeO2 | Combustion | 600 | ~70 | ~100 | [28] |
| 5.60 wt.%Cu-CeO2-hs | Hydrothermal–Impregnation | 600 | ~50 | ~100 | [88] |
| 5 wt.%Cu-CeO2-nr | Hydrothermal–incipient wetness impregnation | 450 | ~50 | - | [81] |
| 5 wt.%Cu-CeO2-ns | Microemulsion-incipient wetness impregnation | 450 | ~40 | - | [81] |
| 0.25 mole%Cu-CeO2 | Hard Template–Impregnation | 400 | 31.34 | 100 | [185] |
| 5Cu/48CeO2/ZSM | Physical Mixing | 600 | ~68 | 100 | [186] |
| Fe-CeO2 | Hard Template–Impregnation | 340 | 3.3 | ~100 | [185] |
| Mn-CeO2 | Hard Template–Impregnation | 340 | 3.3 | ~100 | [128] |
| Co-CeO2 | Hard Template–Impregnation | 340 | 9.3 | ~100 | [128] |
| 10%Co-CeO2 | Colloidal Solution Combustion | 300 | 3.8 | 39.4 | [155] |
| 10Cu5Fe-CeO2 | Impregnation | 750 | 42 | 100 | [177] |
| 5Cu5In-CeO2 | Impregnation | 500 | 45 | 100 | [91] |
| 10Cu-CeO2 | Impregnation | 500 | 50 | 100 | [91] |
| 10 wt.%Co-CeO2 | Co-precipitation–Impregnation | 500 | 28 | ~91 | [187] |
| 1%K10% Co-CeO2 | Co-precipitation–Impregnation | 500 | 31 | 100 | [187] |
| l%Ni-CeO2 | Co-precipitation | 600 | ~35 | 100 | [188] |
| 1%Ni-CeO2 | Co-precipitation | 400 | ~4.5 | ~90 | [130] |
| Ni-CeO2 | Co-precipitation | 700 | 37.5 | ~100 | [121] |
| Ni-CeO2 | Deposition–Precipitation | 700 | 41.7 | ~100 | [121] |
| Ni-CeO2 | Impregnation | 700 | 29.2 | ~100 | [121] |
| Ni-CeZrOx | Impregnation | 700 | 46.1 | 97.3 | [189] |
| Ni/CeO2-Al2O3 | Wet Impregnation | 750 | 59 | 94 | [167] |
| Ni-CeZrOx | Precipitation-Co-precipitation | 550 | 48 | 87.5 | [172] |
| 1%NiO-CeO2/SBA-15 | Calcination | 450 | ~2.5 | 100 | [53] |
| 6%Ni/Al2O3-CeO2 | Combustion | 750 | ~63 | ~90 | [190] |
| RuFe-CeO2 | Polyol | 800 | 47.5 | ~100 | [180] |
| 0.7%Ir-CeO2 | Adsorption–Precipitation | 300 | 2.9 | >99 | [161] |
| 5%Ir-CeO2 | Adsorption–Precipitation | 300 | 6.8 | >99 | [161] |
| 20%Ir-CeO2 | Adsorption–Precipitation | 300 | 8.8 | 12 | [161] |
| In2O3-CeO2 | Co-precipitation | 500 | 20.4 | ~100 | [164] |
| Ga2O3-CeO2 | Physical Mixing | 400 | 5.14 | ~100 | [99] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ebrahimi, P.; Kumar, A.; Khraisheh, M. A Review of CeO2 Supported Catalysts for CO2 Reduction to CO through the Reverse Water Gas Shift Reaction. Catalysts 2022, 12, 1101. https://doi.org/10.3390/catal12101101
Ebrahimi P, Kumar A, Khraisheh M. A Review of CeO2 Supported Catalysts for CO2 Reduction to CO through the Reverse Water Gas Shift Reaction. Catalysts. 2022; 12(10):1101. https://doi.org/10.3390/catal12101101
Chicago/Turabian StyleEbrahimi, Parisa, Anand Kumar, and Majeda Khraisheh. 2022. "A Review of CeO2 Supported Catalysts for CO2 Reduction to CO through the Reverse Water Gas Shift Reaction" Catalysts 12, no. 10: 1101. https://doi.org/10.3390/catal12101101