
Study on the Mechanism of SO2 Poisoning of MnOx/PG for Lower Temperature SCR by Simple Washing Regeneration
by

Xianlong Zhang
1,2,*, 
Shiwen Liu
2,
Kang Ma
2,
Yazhong Chen
2,
Shi Jin
2,
Xinyu Wang
2 and
Xueping Wu
2,* 1
Anhui Province Key Laboratory of Advanced Catalytic Materials and Reaction Engineering, Hefei University of Technology, Hefei 230009, China
2
School of Chemistry and Chemical Engineering, Hefei University of Technology, Hefei 230009, China
*
Authors to whom correspondence should be addressed.
Catalysts 2021, 11(11), 1360; https://doi.org/10.3390/catal11111360
Submission received: 28 October 2021
/
Revised: 8 November 2021
/
Accepted: 10 November 2021
/
Published: 12 November 2021
(This article belongs to the Special Issue Frontiers in Catalytic Emission Control)
Abstract
:Manganese oxide-supported palygorskite (MnOx/PG) catalysts are considered highly efficient for low-temperature SCR of NOx. However, the MnOx/PG catalyst tends to be poisoned by SO2. The effect of SO2 on activity of the SO2-pretreated poisoning catalysts under ammonia-free conditions was explored. It was determined that the MnOx/PG catalyst tends to be considerably deactivated by SO2 in the absence of ammonia and that water-washed regeneration can completely recover activity of the deactivated catalyst. Based on these results and characterizations of the catalysts, a reasonable mechanism for the deactivation of MnOx/PG catalyst by SO2 was proposed in this study. SO2 easily oxidized to SO3 on the surface of the catalyst, leading to the formation of polysulfuric acid, wrapping of the active component and blocking the micropores. The deactivation of the MnOx/PG catalyst is initially caused by the formation of polysulfuric rather than the deposition of ammonia sulfate, which occurs later.
1. Introduction
NOx is a series of reactive gases, including NO2, NO, and N2O. Considerable NOx emissions due to human activities cause significant environmental problems, such as acid rain, the ozone hole, and photochemical smog [1,2,3,4]. Currently, selective catalytic reduction using NH3 (NH3-SCR) technology is widely used in thermal power plants, industrial boilers, and other fixed-source flue gas-control [5], owing to its high efficiency and low cost. A catalyst is essential for NH3-SCR technology. Metal oxide catalysts are widely used in NH3-SCR for their excellent denitrification performance and availability. Currently, V2O5/TiO2 and V2O5-WO3/TiO2 catalysts are widely used. Because they need a relatively high reaction temperature of above 300 °C, so the process is usually performed in the gas upstream [6]. An SCR denitrification device is commonly located in front of the dust removal and desulfurization device, which results in the catalyst being easily inactivated by the poisoning of SO2 and dust. Therefore, low-temperature SCR technology is attractive. Owing to its advantages of low energy consumption and low impact of SO2 and dust, SCR technology has gradually attracted the attention of researchers [7,8,9,10].
To date, a series of low-temperature metal oxide catalysts (e.g., cerium (Ce), cobalt (Co), copper (Cu), iron (Fe), manganese (Mn), molybdenum (Mo), nickel (Ni), and vanadium (V)) have been studied. Of these, manganese oxide (MnOx) has various oxidation states, a high valence state, and characteristic crystal form, and it shows excellent NO conversion and N2 selectivity. Pena et al. [11] showed that MnOx/TiO2 had the highest activity compared to Co, Cr, Cu, Fe, Mn, Ni, and V oxides supported on TiO2 at a low temperature. Among the characteristic crystal forms of MnOx, the active components of the catalysts MnO2 and Mn2O3 show the highest NO conversion and N2 selectivity [12]. However, residual sulfur dioxide after desulfurization has a strong poisoning effect on metal oxidation catalysts at low temperatures. Thus, it is important to clarify the poisoning mechanism of the catalyst. There are different opinions about the poisoning mechanism of SO2. Kijlstra et al. [13] speculated that the main reason for the decrease in the activity of the MnOx/Al2O3 catalyst after passing SO2 was the sulfation of the active center atom, Mn, to form manganese sulfate. Luo et al. [14] believed that an SO2-induced decrease in the MnOx/MWCNTs catalyst’s activity was attributed, in part, to the formation of ammonium sulfate on the catalyst’s surface that blocks the catalyst’s pores and is also attributed, in part, to the sulfation of the active center atom, Mn, to form MnSO4. Qi and Yang [15] thought that the main reason for MnOx/TiO2 deactivation is the competitive adsorption of SO2 and NO. This deactivation is reversible, and the activity of the catalyst can be completely restored after removing SO2.
The structural formula of palygorskite (PG) is Mg5Si8O20(OH)2(OH2)4·4H2O. In our previous study, MnOx/PG showed a high NOx conversion rate at a low temperature window of 100–300 °C [16]. Because of its easily available raw materials, abundant channel structure, and large specific surface area, MnOx/PG has a good industrialization prospect. However, its NOx conversion rate is easily reduced under low concentrations of SO2. Currently, as there are no unanimous conclusions on the poisoning mechanism of Mn-based catalysts, our group explored the poisoning mechanism of MnOx/PG. Zhang et al. [17] determined the poisoning mechanism of MnOx/PG catalysts by simulating the conditions of poisoning and the recovery of thermal regeneration. They thought that the reason for the deactivation of the catalyst was the collaborative effect of ammonium sulfate and metal sulfate and that the deposition of ammonium sulfate was the main reason for the deactivation of the catalyst at low temperatures. The addition of Ce enables MnOx/PG to resist sulfur [18]. The abovementioned researchers believed that the fundamental reason for the resistance to Ce is to inhibit MnSO4 production. As a transition metal, Ce easily combines with SOx to form stable Ce2(SO4)3, which protects the sulfation of Mn. The formation of MnSO4 is an important reason for the deactivation of the catalyst above 150 °C. However, there is no clear conclusion about the deactivation mechanism of the Mn8/PG catalysts. Therefore, it is necessary to explore the cause of low-temperature SCR catalyst inactivation caused by SO2 so as to further find a solution to SCR-catalyst sulfide poisoning. In addition to ammonium sulfate salts and ammonium sulfite salts, the effect of metal sulfate salts on the deactivation of catalysts is controversial. In this study, the reasons why SO2 poisoning leads to a decrease in the catalytic activity and water-washed regeneration leads to the recovery of activity were studied under ammonia-free conditions, combined with specific surface-area and pore-size determination (BET), elemental analysis (EA), X-ray diffraction (XRD), temperature-programmed reduction (TPR), temperature-programmed desorption (TPD), and temperature-programmed surface reaction (TPSR). After discussing the mechanisms of SO2-caused poisoning and water-washed regeneration in depth, we proposed a possible explanation for the catalyst deactivation by SO2 and regeneration of the deactivation catalyst by a water-washing process.
2. Results and Discussion
2.1. Elemental Analysis, Specific Surface-Area and Pore-Volume Determination, and X-ray Diffraction
To explore the composition of soluble sulfur compounds and their effect on the physical environment of the catalyst surface, elemental and BET analysis tests were performed. Table 1 shows that the S content of the Mn8/PG-S catalyst is significantly higher than that of fresh the Mn8/PG catalyst. A negligible portion of sulfur was observed in the Mn8/PG catalyst due to the measurement accuracy of the method or impurities in the instrument. The S content on the catalyst surface was significantly decreased after water-washed regeneration, which indicates that the accumulation and removal of S from the catalyst were the key factors for the deactivation and regeneration of the catalyst.
The BET results show that the specific surface area and pore volume of the catalyst decreased after the SO2-pretreated poisoning at 250 °C; the specific surface area and pore volume decreased by 24% and 3%, respectively. Clearly, the formation of sulfur blocked pores and reduced the specific surface area, which was an important factor that inhibited the SCR activity of the Mn8/PG-S catalyst. The specific surface area and pore volume of the Mn8/PG-S-W catalyst considerably increased compared to that of the Mn8/PG-S catalyst. Thus, the SCR activity of the Mn8/PG-S-W catalyst recovered even more than that of fresh samples. This suggests that sulfur compounds on the catalyst were efficiently removed by the water-washing process, which resulted in a large extent of recovery of specific surface area. A small portion of sulfur was observed in the Mn8/PG catalyst due to the measurement accuracy of the method or impurities in the instrument because no sulfur-containing precursors were used during the preparation of the catalyst.
To explore the changes in the crystal shape of the catalyst before and after the SO2-pretreated poisoning and water-washed regeneration, XRD tests were performed on the three catalysts. The XRD patterns of the three catalysts are shown in Figure 1. All of the catalysts obtained by different treatments maintained the primary structure of palygorskite.
The main forms of manganese oxide on the surfaces of Mn8/PG catalysts were MnO2 (i.e., 37.6°, 42.8°, 57.0°) [PDF#30-0820] and Mn2O3 [PDF#10-0069]. It is easier to form amorphous Mn2O3 than MnO2; thus, it is difficult to observe Mn2O3 by XRD. The intensity of the MnO2 diffraction peak in the catalyst was weakened after Mn8/PG catalysts were SO2-pretreated, and the intensity of the diffraction peak of the MnO2 catalyst was restored after the water-washed treatment. The abovementioned results were obtained because sulfur compounds, which were formed on the surface of the catalyst, covered MnO2 after the treatment in the SO2 atmosphere, and the characteristic diffraction peak intensity of MnO2 was weakened. The water-washed regeneration can remove sulfur compounds and restore the intensity of the diffraction peaks of MnO2.
2.2. Scanning Electron Microscopy
According to the abovementioned analysis, it is believed that the presence of sulfur compounds likely causes a significant change in the morphology of the catalyst; thus, SEM analysis was performed to investigate morphological changes of the three catalysts. Figure 2a shows that the Mn8/PG catalyst has a one-dimensional rod-shaped structure with individual rods being uniformly dispersed. These properties are considered to be essential to obtain superior catalyst activity. For the Mn8/PG-S catalyst (Figure 2b), a clear agglomeration of catalysts was observed. A paste-like compound wrapped the catalyst’s surface. For the Mn8/PG-S-W catalyst (Figure 2c), the individual rods were highly dispersed, and the paste-like compound on the rods was clearly removed. The appearance of the Mn8/PG-S-W catalyst was similar to that of the Mn8/PG catalyst. Combined with the BET analysis, it is suggested that the paste-like compound, which is formed during the sulfur treatment, causes a decrease in specific surface area and catalytic deactivation.
2.3. In Situ DRIFT
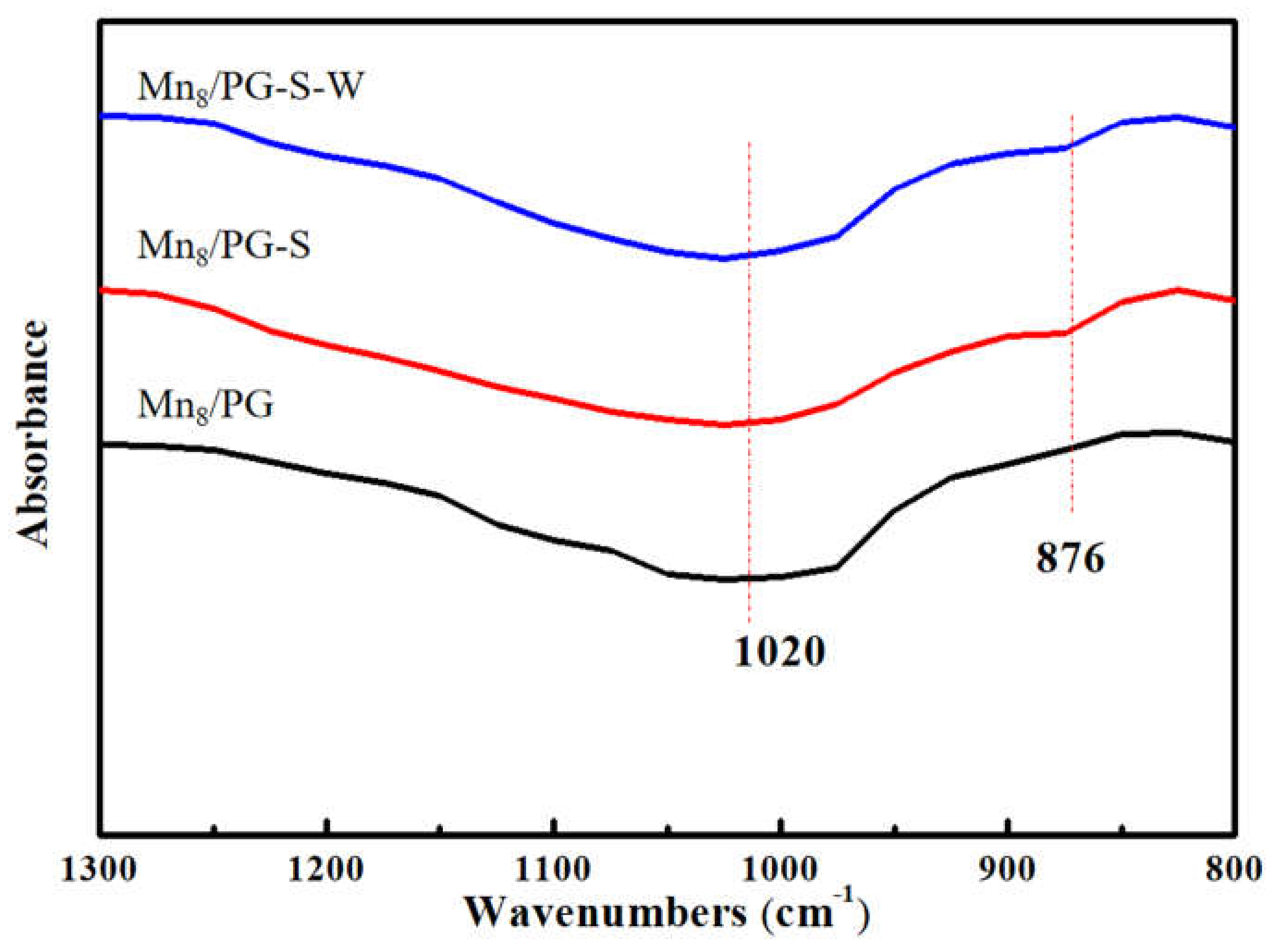
To further study the internal functional groups of the catalyst after the SO2-pretreated poisoning and water-washed regeneration, the three catalysts were evaluated by FT-IR spectroscopy, as shown in Figure 3. Two peaks can be seen in the wavelength range of 1300–800 cm−1. All the samples show the same broad peak at 1020 cm−1, which is attributed to the anti-symmetrical vibration of quartz Si–O–Si, which is contributed by the PG support. The peak vibration at 876 cm−1 is attributed to the S–O stretching vibration that was observed on Mn8/PG-S and Mn8/PG-S-W samples [19]. There was no change detected in the 1020 cm−1 peak intensity for the three catalysts, which indicates that Mn8/PG-S poisoning and Mn8/PG-S-W regeneration did not affect the carrier, with SiO2 as the main component of the catalyst. However, the peak intensity of S–O at 876 cm−1 considerably changed for the three samples. For the Mn8/PG-S catalyst, there was a clear peak at that frequency, which indicates that sulfur compounds formed on the surface of the catalysts. After water-washed regeneration, the peak at that frequency became weak, which indicates that some of the sulfur compounds were washed away, and some metal sulfate salts remained on the catalyst surface.
On the basis of the abovementioned tests and characterization analyses, it can be suggested that the deactivation of the catalyst after the SO2 pretreatment is due to the formation of soluble sulfur compounds rather than MnSO4. Those sulfur compounds can be easily removed by water washing to completely restore the SCR activity of the catalyst. According to reports by Seong et al. [20], at a higher SO42− concentration, the isolated sulfate will be converted into a polynuclear sulfate type, which is larger and will wrap around the active component. It is reasonable to suggest that the soluble sulfur species is actually an SO42− polymer.
2.4. Atomic Absorption Spectroscopy and X-ray Fluorescence Spectroscopy
Because of the compositional characteristics of the palygorskite carrier (magnesium aluminum silicate), the sample contains a large amount of O, Si, Mg, and Al elements. By comparing the Mn content of the three catalysts, we determined that the Mn content does not considerably change (as shown in Table 2) and that the overall growth trend is attributed to the water-washing process, which washes away a part of the carrier.
Combined with the atomic absorption spectroscopy analysis, shown in Table 3, we determined that there are small amounts of Mn and Mg in the water-washed solution of the Mn8/PG catalyst because of the incompletely decomposed precursor Mn(NO3)2 and soluble magnesium salt on the carrier. A small amount of Mn was observed in the water-washed solutions of the Mn8/PG-S catalyst that was washed once and thrice; this is attributed to the fact that SO3 reacts with active manganese and eventually produces MnSO4, which is not strongly bonded to the carrier. The Mn element that is present during the SO2-pretreatment process is converted into the content of lost manganese. It can be determined that the amount of manganese removed during the SO2 pretreatment process is considerably small (approximately 1% of manganese), which suggests that Mn is not lost during the SO2 pretreatment process. The SO2 pretreatment process does not wash away manganese present on the catalyst, which is an important prerequisite for the recovery of the catalyst activity. To further determine the loss of Mn during the SO2 pretreatment process, we excluded other factors that may affect the experimental results. Thus, a simulation experiment with pure MnO2 was performed. MnO2 was used as a SO2 pretreatment with the same process as for Mn8/PG. It was determined that MnO2 had the same trend of activity as that of the Mn8/PG-S catalyst. The analysis of the water-washed solution of SO2-pretreated MnO2 showed that the loss of manganese was in the amount of 0.002%, which can be neglected. The elimination of deactivation of the catalyst via sulfation is not the main cause during the SO2 pretreatment process.
To investigate the acidity and alkalinity of the three catalysts, the pH of the water-washed solution of the three catalysts was determined. The pH of the water-washed solution of the Mn8/PG catalyst was 7.56, which is neutral. The pH of the water-washed solution of the Mn8/PG-S catalyst was 5.3, which was weakly acidic. The pH of the water-washed solution of the Mn8/PG-S-W catalyst was 7.23. Clearly, compared to the fresh and water-washed samples, the Mn8/PG-S catalyst was more covered by soluble sulfur compounds. It can be concluded that the removal of acidic sulfur compounds during the SO2 pretreatment process is the main cause of the recovery of the deactivated catalyst.
2.5. Effect of SO2 Pretreatment and Water Washing on the Activity of Mn8/PG Catalysts
There are two reasons for the deactivation of metal oxides by SO2: the deposition of ammonium sulfate salt on the catalyst and the sulfation of active sites. To eliminate the effect of ammonium sulfate on the activity of the catalyst, Mn8/PG was SO2-pretreated under ammonia-free conditions to investigate the effect of metal sulfates on catalytic activity.
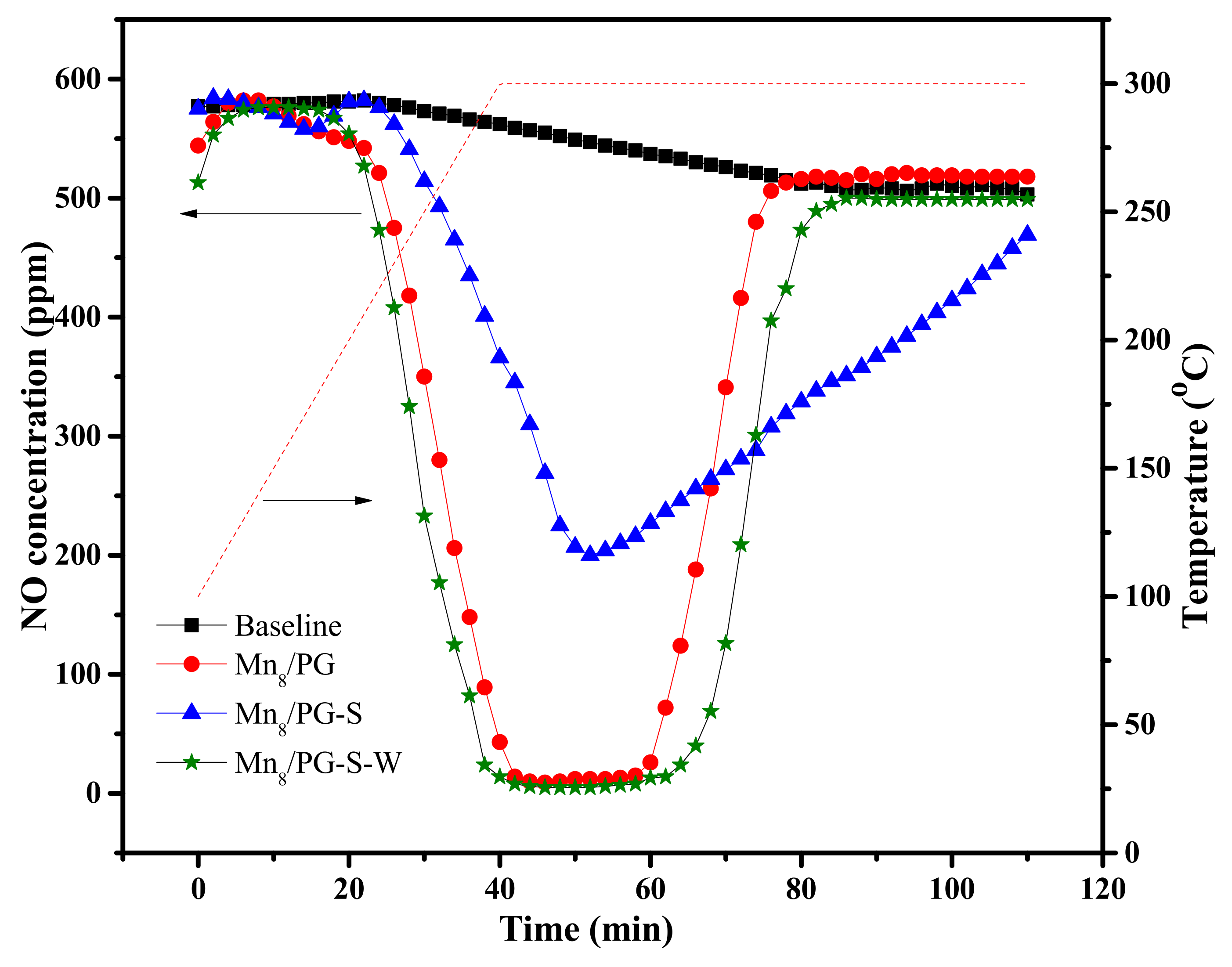
Figure 4 shows the NO conversion of different Mn8/PG catalysts in the studied temperature range. The NO conversion of Mn8/PG-S was significantly inhibited compared with Mn8/PG, indicating that the activity of catalysts was remarkably affected by SO2 pretreatment. The activity of the catalyst was considerably restored after Mn8/PG-S catalysts were water-washed, and the NO conversion was even slightly higher than that of the Mn8/PG catalyst. The results show that SO2 pretreatment causes irreversible deactivation in the low-temperature range of the catalyst, which is consistent with the results of the research on the reversible and irreversible deactivation of the Cu-CHA NH3-SCR catalyst by Hammershøia et al. [21]. The NH3-SCR activity of the Mn8/PG-S catalyst considerably increased from 250 °C to 300 °C. This occurs because the oxidation efficiency of manganese increases with an increase in temperature, which directly leads to an increase in the SCR reaction rate per unit of time.
The NO conversion of the Mn8/PG-S catalyst is considerably inhibited compared with that of the Mn8/PG catalyst in the low-temperature range, indicating that the formation of ammonium sulfate on the catalyst surface is not the main cause of catalyst deactivation. The catalyst was possibly deactivated before the formation of ammonium sulfate. We tentatively concluded that the stratification of ammonium sulfate was not the main reason for the deactivation of the Mn8/PG catalyst.
2.6. X-ray Photoelectron Spectroscopy (XPS)
The S atom concentration of the Mn8/PG-S catalyst is 3.18%, whereas the S atom concentration of the Mn8/PG-S-W catalyst is 0.67% (Table 4). Combined with the result shown in Figure 5, it was determined that the denitration activity of the Mn8/PG-S catalyst was restored to that of the fresh catalyst by washing with water. This observation suggests that the removal of S atoms during the water-washing process is essential for recovering the catalyst’s denitration activity.
Figure 5a shows the XPS spectrum of the S 2p orbital of the Mn8/PG-S catalyst. The S element mainly exists in the forms of S4+ and S6+ in the Mn8/PG-S catalyst. The peak at 168.9 eV is attributed to SO32−, which contributes to the sulfurous-acid generation by the SO2 and H2O adsorption on the surface of the catalyst. The peak at 169.7 eV is attributed to SO42−, which contributes to the sulfuric-acid generation by SO3 and H2O on the surface of the catalysts and surface-generated MnSO4 [22,23]. The S peak can be divided into S6+ and S4+. According to the calculated area of the two peaks, it was determined that the proportions of S6+ and S4+ are 49% and 51%, respectively. Compared to the catalytic reaction without a sulfur atmosphere, more S4+ was oxidized to S6+ due to the presence of MnO2 (Mn4+) and SO2 when the catalytic reaction proceeded in the sulfur-rich atmosphere [24]. Thus, sulfur was primarily present as a sulfuric acid and MnSO4 on the surface of the catalyst. After water-washed regeneration, most S was washed away. The XPS spectrum of the S 2p orbital of the washed regenerated catalyst cannot be observed because of the extremely low content of S.
The XPS spectra of the Mn 2p orbitals for the three catalysts are shown in Figure 5b. The Mn 2p 3/2 peak of the Mn8/PG catalyst can be divided into two peaks: MnO2 (643.1 eV) and Mn2O3 (641.6 eV) [25]. Based on the calculation of the peak area, the proportions of MnO2 and Mn2O3 are 68.2% and 38.8%, respectively. The peak of Mn 2p3/2 can be divided into three peaks, which correspond to MnO2 (643 eV), Mn2O3 (641.8 eV), and manganese sulfate (644.9 eV), owing to the sulfur-containing atmosphere [26]. The proportions of the abovementioned compounds were 25.2%, 38.6%, and 36.2%, respectively. The presence of MnO2 and manganese sulfate in the Mn8/PG-S catalyst indicate that the surface high-valence Mn is reduced to Mn2+ by S, and then S and Mn combine to form stable MnSO4 on the surface of Mn8/PG-S catalysts. After splitting and fitting the orbital peak of the Mn8/PG-S-W catalyst Mn 2p, the peak of Mn2+ (MnSO4) still exists in the Mn8/PG-S-W catalyst. However, the activity of the catalyst was significantly restored, which suggests that the main cause of catalyst deactivation may not be MnSO4.
2.7. H2-Temperature-Programmed Reduction (H2-TPR)
The redox capacity of manganese-based catalysts is a key factor in the catalytic reduction of NH3-SCR [27]. The reduction temperature can be used to evaluate the redox ability of catalysts. The low reduction temperature leads to the stronger redox ability of the catalysts [28]. The redox properties of the three catalysts were investigated by performing the H2-TPR analysis in Figure 6 and measuring the produced H2 concentration during the temperature increase from 50 °C to 800 °C. The profile of H2 consumption on each catalyst, along with the temperature rising in the H2-TPR experiment, is shown in Figure 6.
For fresh samples, there are two prominent reduction peaks (307 °C and 456 °C), as shown in the H2-TPR analysis. The two peaks correspond to the reduction of Mn4+ and Mn3+, respectively [29,30]. In the case of the Mn8/PG-S catalyst, the reduction peak at 412 °C (Mn4+) was clearly weakened, and a new reduction peak at 647 °C (Mn2+) appeared. During the SO2 pretreatment process of the Mn8/PG catalyst, Mn4+ and Mn3+ were reduced to Mn2+ by SO2, S4+ was oxidized to S6+, and S6+ was present on the surface of catalysts in the form of SO42−. A shift in the reduction peak of the Mn8/PG catalyst was caused by the sulfur compounds that are formed on the catalyst and that suppress the oxidizing activity of MnOx, which is in agreement with the XPS result. In the case of the Mn8/PG-S-W catalyst, the reduction peaks appeared at 390 °C and 501 °C. The intensity of the reduction peak of the Mn8/PG-S-W catalyst Mn4+ could not be completely restored to that of Mn8/PG catalyst because a part of Mn8/PG catalyst Mn4+ was converted to Mn2+, which could not be reoxidized to Mn4+ by the water-washing process. The water-washing process removes the sulfur species from the sulfated catalyst, thereby restoring the redox capacity of the catalyst. This conclusion further proves that sulfur species are an important cause for catalyst deactivation.
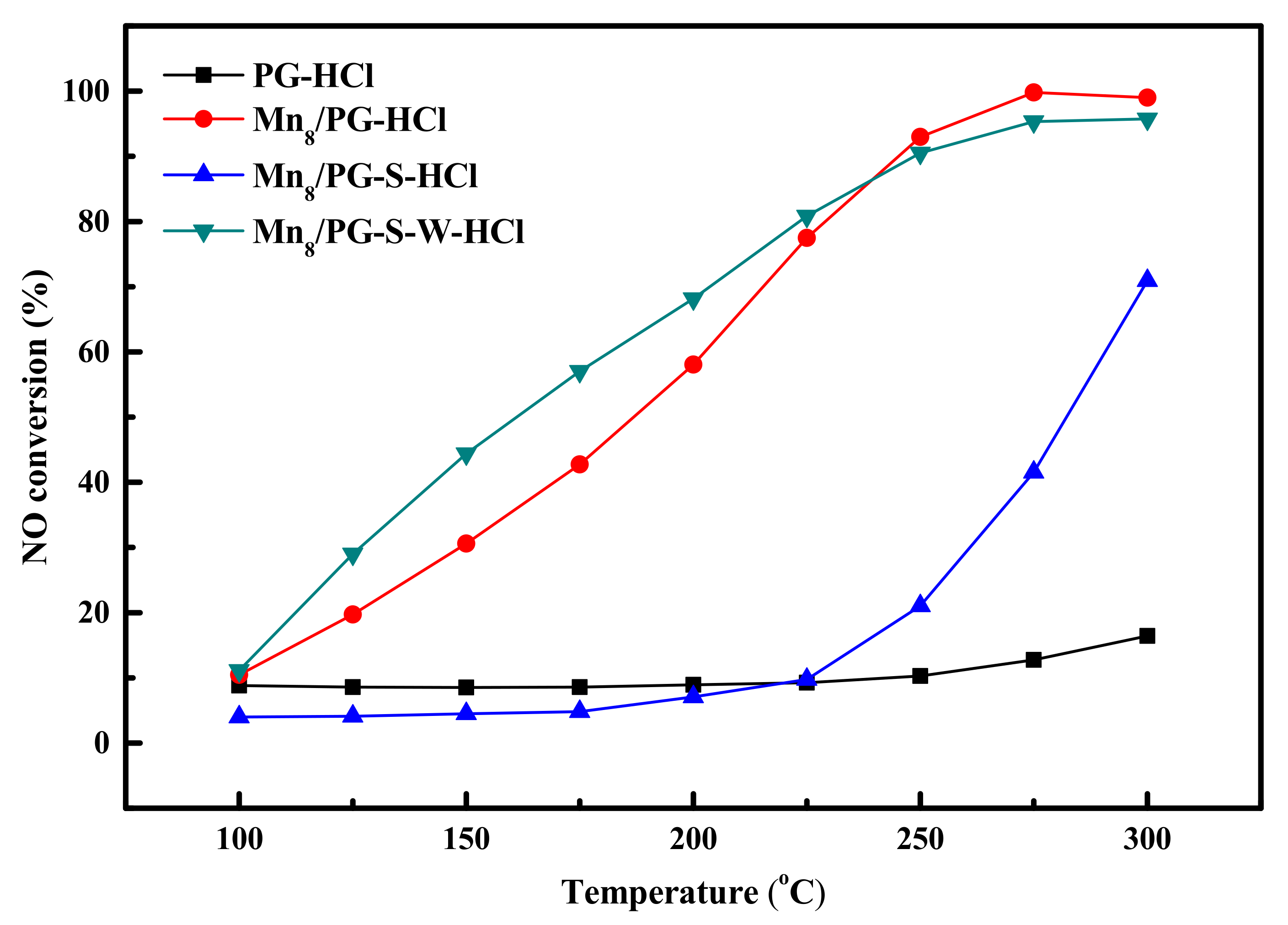
2.8. Activity Test on Catalysts Treated by HCl
The results of atomic absorption spectroscopy showed that a small amount of Mg2+ exists in the water-washed solution of different catalysts. To eliminate the potential influence of sulfurization of elements from the carrier, we pretreated the PG carrier with 1.2 mol/L HCl to make sure the metal oxides in the carrier were completely removed. Then, manganese oxide was reloaded by the wetness co-impregnation method to prepare different Mn8/PG-HCl catalysts, and the results are shown in Figure 7. Because PG contains large amounts of alkali earth metal oxides (e.g., MgO and Al2O3) that can contribute to a lower SCR activity, PG may be sulfurized by SO2 to block the surface of the carrier. After pretreatment with an HCl solution, PG was considerably inactive throughout the low-temperature range, which indicates that alkali earth metal oxides, which provided activity in the carrier, were considerably removed. When MnOx was reloaded, the activity of the Mn8/PG-HCl catalyst was almost the same as that of Mn8/PG, which indicates that Mn, as an active component, provided the main denitrification activity in the lower temperature range. Mn8/PG-S-HCl and Mn8/PG-S-W-HCl catalysts showed a trend that was similar to that which occurred after the Mn8/PG treatment, which indicates deactivation due to the Mn8/PG sulfurization of alkali earth metal from the carrier. However, it is debatable whether the deactivation of the catalyst was caused by the formation of sulfur compounds from the active component.
2.9. SCR Activity Test of MnSO4-Doped PG
To evaluate the potential deactivation of the catalyst by the formation of MnSO4, the PG carrier was doped with MnSO4 and then tested for SCR. Figure 8 shows the NO conversion of PG; MnSO4 (1%)/PG and MnSO4 (8%)/PG were compared. The experimental results show that PG has a certain catalytic activity, which is attributed to the existence of oxygen-containing functional groups, which are an active component and can promote the redox process [17]. After the loading of 1% MnSO4 (simulates SO2-pretreated poisoning with S content during the elemental analysis), a considerable increase in the SCR activity was observed. However, for MnSO4 (8%)/PG (assuming all active components of MnOx are completely deactivated), a clear decrease in the SCR activity appeared. This result shows that Mn2+ has a certain activity, and a small amount of MnSO4 is easily distributed on the catalyst, which is favorable for catalytic activity. When the content of MnSO4 is 8%, the MnSO4 bulk phase is large. Thus, an excessively high MnSO4 content is not advantageous for the uniform dispersion on the catalyst, aggregation tends to easily occur, and the activity of PG decreases. This result further confirms that the small amount of MnSO4 that is generated during the SO2 pretreatment process is not the main reason for the deactivation of the catalyst.
2.10. SO2 Adsorption and TPD
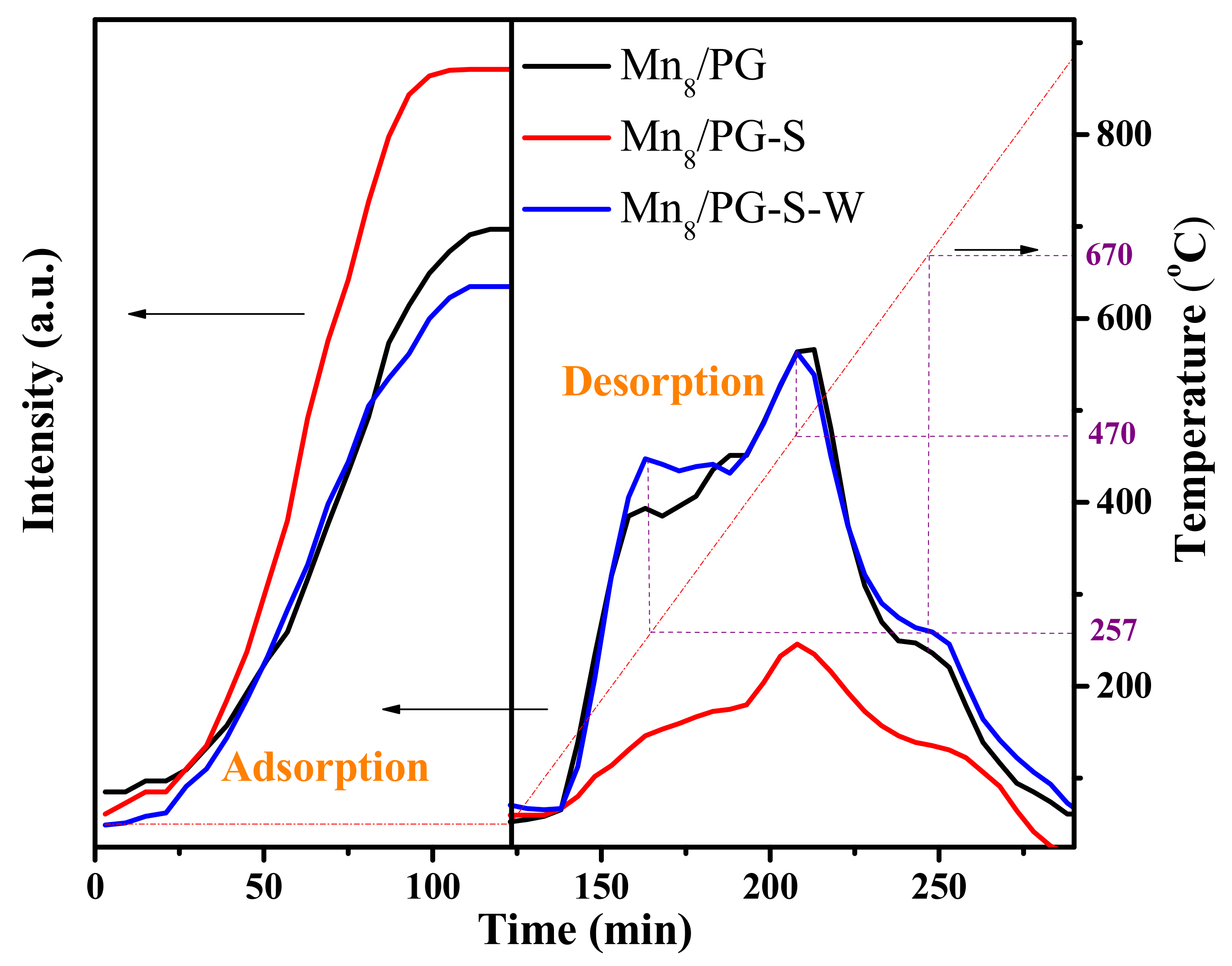
Considering that the oxidation of SO2 is frequently induced by the adsorption of SO2 on catalysts, the performance of SO2 adsorption on Mn8/PG, Mn8/PG-S, and Mn8/PG-S-W catalysts was measured. Each sample was exposed to an SO2-containing flowing gas for 100 min until equilibrium. The TPD of SO2 for each sample from 50 °C to 1000 °C was also performed. The profiles of SO2 adsorption equilibria and SO2 TPD for the three catalysts are compared in Figure 9.
As shown in Figure 9, SO2 adsorption equilibrium profiles obtained at 50 °C for 100 min vary significantly. For the Mn8/PG-S catalyst, SO2 penetrates immediately and reaches the equilibrium. However, 6 and 12 min diffusion time was observed for the Mn8/PG-S-W and Mn8/PG catalysts, respectively. This implies that the Mn8/PG-S catalyst has almost no SO2 adsorption capacity, and the water-washing process can restore a part of its SO2 adsorption capacity. By calculating the corresponding area of each curve, the SO2 adsorption capacity of each catalyst can be compared more directly. The SO2 adsorption capacities of the Mn8/PG, Mn8/PG-S and Mn8/PG-S-W catalysts are 1.34 × 10−4, 4.14 × 10−5, and 6.7 × 10−5 mol·g−1, respectively. Adsorbed SO2 is oxidized to SO3 by MnOx during the adsorption process. Figure 6 shows that there is a difference between the SO2 adsorption equilibrium concentration and the inlet concentration, and the difference between the SO2 equilibrium concentration and the inlet concentration (400 ppm) is believed to be attributed to SO2 oxidizing to SO3. The SO2 oxidation rates of the Mn8/PG, Mn8/PG-S, and Mn8/PG-S-W catalysts are 6.25%, 1.25%, and 2%, respectively. SO2 is continuously adsorbed and oxidized by the active component. A part of SO2 forms MnSO4 with the active component, and the other part forms a removable sulfur compound. These removable sulfur compounds are responsible for the decreased SO2 adsorption and oxidation capacity. Removable sulfur compounds that cover the active component can be removed by the water-washing process, thereby recovering the adsorption capacity of the catalyst. Because the water-washing process does not wash away MnSO4, the oxidizing capability is not significantly recovered. Most of the soluble sulfur compounds can be removed by the water-washing process; thus, the adsorption capacity of the catalyst is recovered. Because MnSO4 is still present after the water-washing process (the presence of SO42− may inhibit the adsorption of SO2), the adsorption and oxidation capacities of the catalyst are not fully recovered.
For direct comparison with SO2 adsorption, the SO2-TPD profiles of each catalyst are shown in Figure 9. The figure shows that there is no SO2 desorption peak for the Mn8/PG catalyst. A considerable SO2 desorption peak was obtained on the Mn8/PG-S catalyst in the temperature range of 600 °C–800 °C. This may be attributed to the joint desorption action of a soluble sulfur compound and MnSO4. The desorption peak of the water-washed regenerated catalyst was observed at 700 °C–800 °C, and the peak was weaker than that of the sulfur Mn8/PG-S sample. This desorption peak may be attributed to the decomposition of MnSO4, which remained after water washing. By comparing the SO2 desorption peak areas of the Mn8/PG-S and Mn8/PG-S-W catalysts, it can be observed that the soluble sulfur compound is the main product of SO2, which is also the main reason for the deactivation of the Mn8/PG-S catalyst.
2.11. NH3 Adsorption and TPD
It is widely believed that the adsorption and activation of NH3 on the catalyst surface is the first important step in the SCR reaction, according to either E–R or L–H mechanism [31]. In general, an increase in the acidity of the catalyst surface is favorable for the adsorption of NH3 and SCR activity. The NH3 adsorption ability of each sample was measured, followed by the TPD experiments. During the adsorption, the catalyst was exposed to a gas mixture containing 600 ppm of NH3 balanced by N2 at 50 °C for more than 100 min until equilibrium. Then, after a sweeping with N2, TPD profiles were also recorded, as shown in Figure 10.
From the comparison of the NH3 adsorption equilibrium curves for the three catalysts, it was observed that the diffusion time of the Mn8/PG-S sample was 4 min faster than that of the fresh sample. The area enclosed by the NH3 curve of the Mn8/PG-S catalyst was significantly smaller than that of the fresh sample during the process of adsorption equilibrium, which indicates that the NH3 adsorption on the Mn8/PG-S catalyst was considerably inhibited. However, the diffusion time and the area enclosed by the NH3 curve of the Mn8/PG-S-W catalyst were found to be slightly greater than those of fresh samples, which indicates that the NH3 adsorption of the catalyst was efficiently recovered during the removal of sulfate species from the poisoned catalyst.
Figure 10 shows the NH3 TPD curve after 125 min of adsorption. There are several clear desorption peaks of NH3 in the range of 50–900 °C: a weak acidic site desorption peak at 132–380 °C, a moderate acidic site desorption peak at 380–640 °C, and a strong acidic site desorption peak at 640–775 °C. The desorption peak of the Mn8/PG-S catalyst is lower than that of the Mn8/PG catalyst due to the reduction of stored NH3 during the adsorption. However, the temperature corresponding to the desorption peak did not change, which indicates that the binding position of NH3 on the catalyst did not change. The amount of desorption of NH3 on the water-washed regeneration catalyst was slightly higher than that of the Mn8/PG catalyst, which indicates that the water-washing process can remove the SO42− polymer, which inhibits the adsorption of NH3.
The NH3 adsorption-desorption curve confirms that the formation of ammonium sulfates is not the main cause of the decrease in catalytic activity. The deactivation of the catalyst occurred before the possible formation of ammonium sulfates. Based on the result that the NH3 adsorption of the catalyst was considerably inhibited by the SO2 pretreatment of the catalyst, it can be speculated that the formation of an SO42− polymer caused the pore blockage of the catalyst and the occupation of active sites for NH3 adsorption.
2.12. NO Adsorption and TPD
Currently, some researchers believe that the SCR reaction follows the L–H mechanism, and the NO adsorption behavior in the L–H mechanism considerably affects the SCR reaction [32,33]. To investigate the effect of soluble sulfur compounds (SO42− polymer) on the adsorption and desorption of NO on the catalyst, we performed the adsorption and desorption analyses of NO on the three catalysts. During the absorption period, the catalyst was placed in an atmosphere containing 600 ppm of NO with N2 as the carrier gas at 50 °C, and the outlet NO concentration is shown in Figure 11.
The adsorption equilibrium curves for the three catalysts at 50 °C before 80 min are shown in Figure 11. The diffusion time of the Mn8/PG-S catalyst by NO was 50 min faster than that of the Mn8/PG catalyst. The enclosed area of the Mn8/PG-S catalyst was significantly smaller than that of the Mn8/PG catalyst during the adsorption period, which indicates that the capacity of stored NO was considerably affected by the generated sulfur compounds. The adsorption equilibrium time of the Mn8/PG-S-W catalyst is approximately the same as that of the Mn8/PG catalyst. However, the amount of adsorbed NO is slightly lower than that of the Mn8/PG catalyst, which may be due to the inhibition of NO adsorption by the residual SO42−.
There are two distinct desorption peaks for fresh Mn8/PG. The first desorption peak appears at 250–320 °C. The desorption peak is attributed to the decomposition of the NO adsorption product, which is mainly involved in SCR. It may be formed by the decomposition of bridging nitrate salts [34]. The second peak belongs to the high-temperature peak at approximately 400 °C. It may be formed by the oxidation of NO to form nitrite salts and nitrate salts, which are decomposed and desorbed at high temperatures [35]. Because the decomposition temperature of this nitrate is considerably high, it cannot easily participate in the SCR reaction at low temperatures. When the catalyst is reacted in a sulfur-containing atmosphere, the low- and high-temperature peaks are both attenuated. The NO storage is reduced during the adsorption period due to the presence of sulfur compounds. The water-washed regeneration process can remove the SO42− polymer from the catalyst surface, but MnSO4 still exists. The presence of MnSO4 inhibits the activation of NO; thus, the peak intensity does not recover at 400 °C.
2.13. Temperature-Programmed Surface Reaction (TPSR)
To explore the effect of sulfuric acid and sulfurous acid on the surface environment of the catalyst, a further investigation was conducted using the TPSR, as shown in Figure 12.
As a result, the concentration of NO was quickly increased to an inlet concentration of 570 ppm at 4 min for the Mn8/PG catalyst without NH3 adsorption. When the temperature of the system increased above 230 °C, the NO concentration gradually decreased to a low concentration. After 80 min, the NO concentration did not reach the inlet concentration due to the consumption of the NO heat reaction under high temperature conditions.
For the Mn8/PG catalyst with NH3 adsorption, there is a clear inverted peak in the TPSR curve. The larger NO depletion peak at 20–80 min is attributed to the consumption of the NO surface reaction with adsorbed NH3. The TPSR curve of the Mn8/PG-S catalyst also shows a distinct consumption peak (1.94 × 10−4 mol·g−1), but the depletion peak attributed to the adsorbed NH3 reaction at the same time period was lower than the Mn8/PG consumption peak (2.26 × 10−4 mol·g−1). The presence of soluble sulfur compounds inhibits the adsorption of NH3 at certain specific acidic sites, which are the most critical active sites during low-temperature SCR reactions. The TPSR curve for the Mn8/PG-S-W catalyst shows the same consumption peak (2.69 × 10−4 mol·g−1) as that of the Mn8/PG catalyst in the same time period because the water-washing process can remove the SO42− polymer at the key active sites in the low-temperature SCR reaction. Meanwhile, it was observed that the amount of activated adsorption of NH3 by the Mn8/PG-S-W catalyst is slightly higher than that of the Mn8/PG catalyst, which may be an important reason why the activity of the Mn8/PG-S-W catalyst is slightly higher than that of the Mn8/PG catalyst.
2.14. Proposed SO2-Pretreated Poisoning and Water-Washed Regeneration Mechanism Model
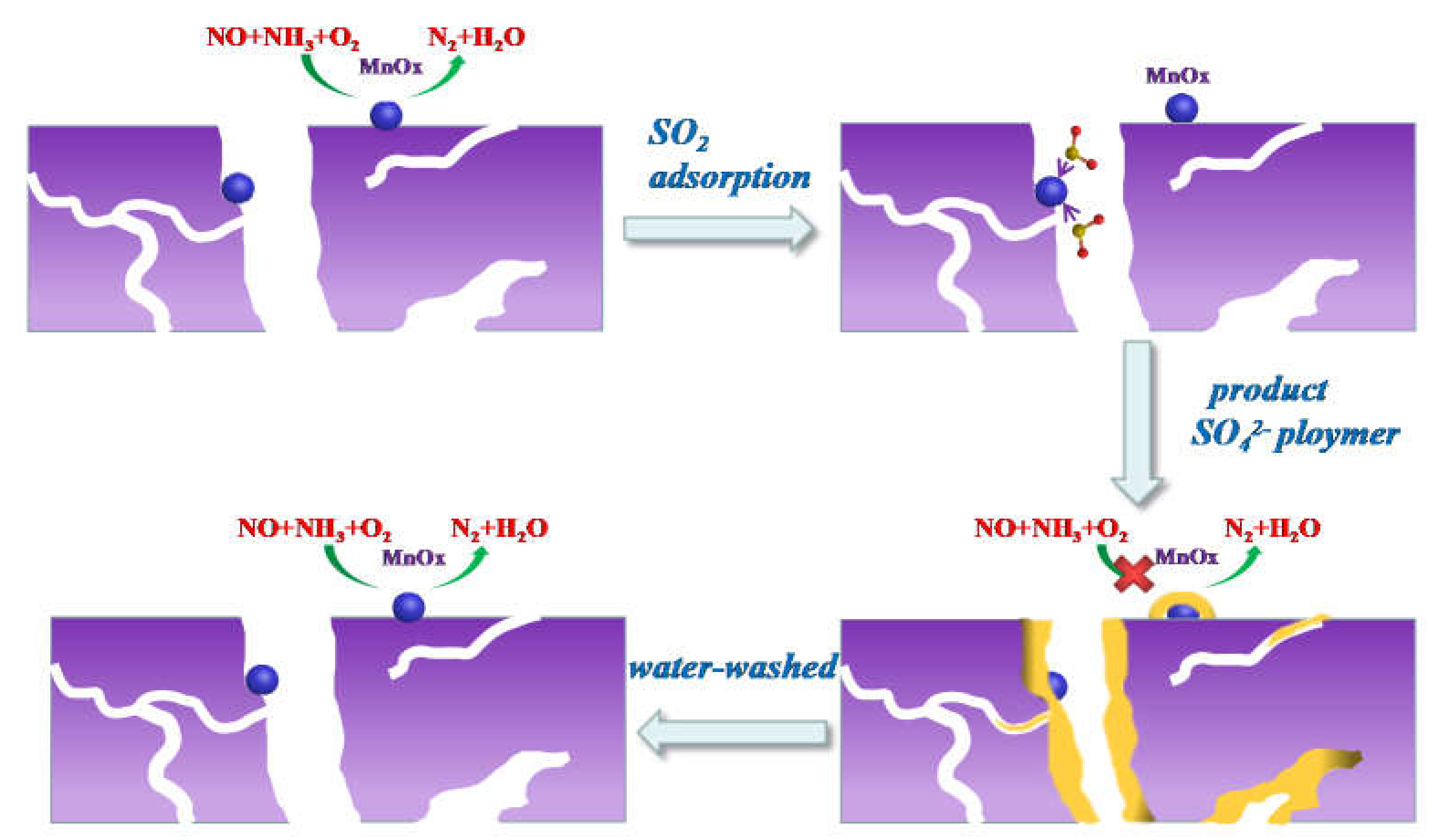
To better illustrate and summarize the abovementioned characterization analysis, in Figure 13, we propose a mechanism model for SO2-pretreated poisoning and water-washed regeneration.
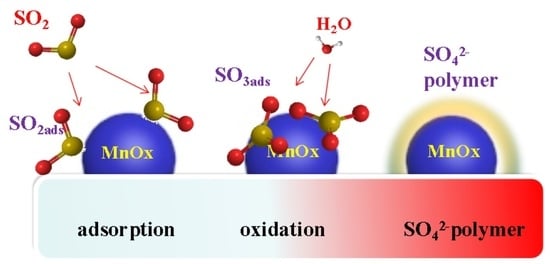
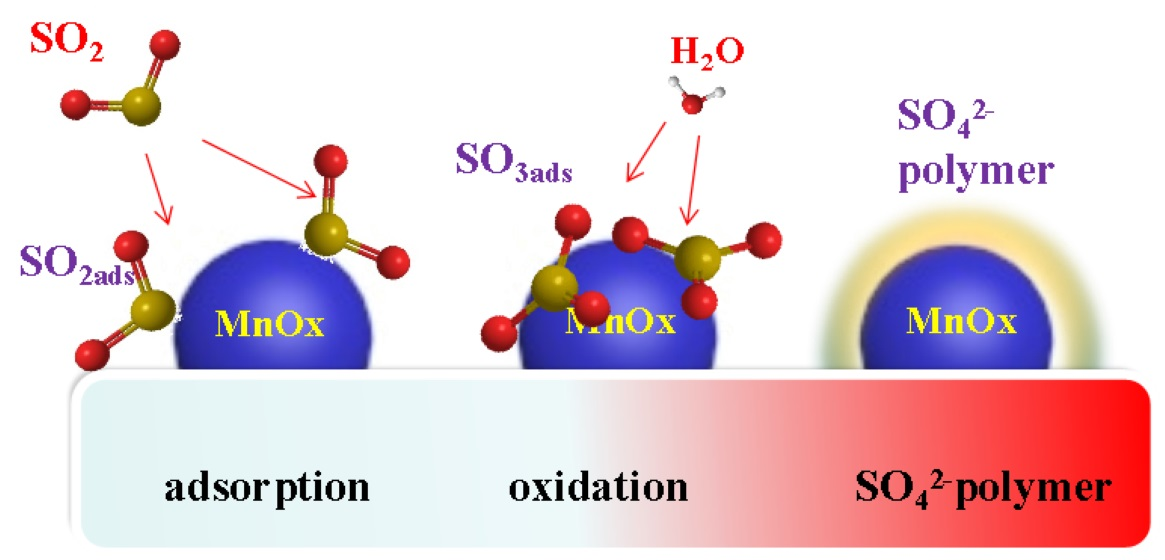
The process of SO2-pretreated poisoning of the catalyst is as follows. First, SO2 adsorbs on the active component, MnOx, of the Mn8/PG catalyst. Then, SO2 is oxidized by the active component to SO3 and adsorbed on the surface of the active component. When the concentration of SO42− on the active site reaches a certain level, sulfate is converted into an SO42− polymer. Thus, the specific surface area of the catalyst is decreased, which corresponds to a reduction in the adsorption and activation of NH3 and NO.
The mechanism of water-washed regeneration is as follows. The water-washing process can easily wash away the SO42− polymer from the active component. Thus, the catalytic activity of the active components can be restored.
3. Experimental
3.1. Materials
The palygorskite, supplied by Xinzhou New Material Factory (Anhui, China), was ground and sieved through 0.42–0.84 mm mesh. The chemical composition of the palygorskite was 69.44 wt.% SiO2, 11.84 wt.% Al2O3, 5.14 wt.% Fe2O3, 12.16 wt.% MgO, 0.43 wt.% TiO2, 0.5 wt.% K2O, 0.21 wt.% CaO, and 0.28 wt.% others. The surface area, pore volume, and pore size of palygorskite were 228.5 m2·g−1, 0.41 cc·g−1, 2.8 nm, respectively. All used chemicals, including manganese nitrate, manganese sulfate and hydrochloric acid, were analytical grade and purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, SCR, China) Simulated flue gas provided by Nanjing Shangyuan Industrial Gas Plant, (Nanjing, China). MnO2 is produced by calcination of manganese nitrate.
3.2. Catalyst Preparation
Fresh Mn8/PG catalyst: The catalysts were prepared by the wetness co-impregnation loading of active species. Palygorskite, as a support, was pore-volume impregnated by a mixed aqueous solution of manganese nitrate as a precursor. The mass ratios of manganese to palygorskite were 8 wt%. The catalysts were initially dried at 50 °C for 6 h, followed by drying at 110 °C for 12 h. The dried catalysts were ground and sieved through 20–40 mm meshes and subsequently calcined in air at 300 °C for 3 h. Thus, the obtained catalysts were designated as Mn8/PG, where “8” refers to the mass percentage of each element.
SO2-pretreated catalyst: A total of 2-g fresh Mn8/PG catalyst was continuously ventilated under the following reaction conditions: 400 ppm SO2, 3 vol% O2, N2 as an equilibrium gas, total flow rate of 400 mL·min−1, and reaction temperature of 250 °C for 5 h until SO2 adsorption was saturated. The sample was labeled as Mn8/PG-S.
Water-washed catalyst: A total of 2-g Mn8/PG-S catalyst was added to an Erlenmeyer flask containing 50-mL deionized water; the mixture was placed in an SHA-B-type water bath, stirred at a constant temperature (25 °C) for 10 min, and then dried at 105 °C to obtain a water-washed catalyst. The sample was labeled as Mn8/PG-S-W.
Multi-cycles of SO2 pretreatment followed by water washing of the catalyst were also carried out to evaluate the potential weight loss of manganese species. Concrete steps are as follows: first, in the absence of NH3, SO2 was piped in, the catalyst was presulfurized, again according to the process of the catalyst presulfurization water experiment, and dried after a bath. The catalyst was then washed in SO2, secondary vulcanization processing took place, followed by water processing, the cycle. The samples were labeled as Mn8/PG-W-x, where “x” means the number of cycles.
MnSO4 (x)/PG catalyst: MnSO4 was impregnated with the equal volume method. Firstly, 10 g PG and a certain amount of MnSO4 were weighed, and a certain amount of deionized water was added to MnSO4 to be stirred and dissolved. Then, manganese sulfate solution was poured into PG, stirred for 15 min, and allowed to stand for 24 h to obtain the MnSO4 (X)/PG catalyst. The mass of manganese sulfate was calculated according to the load ratio, X.
3.3. Catalytic Activity Test
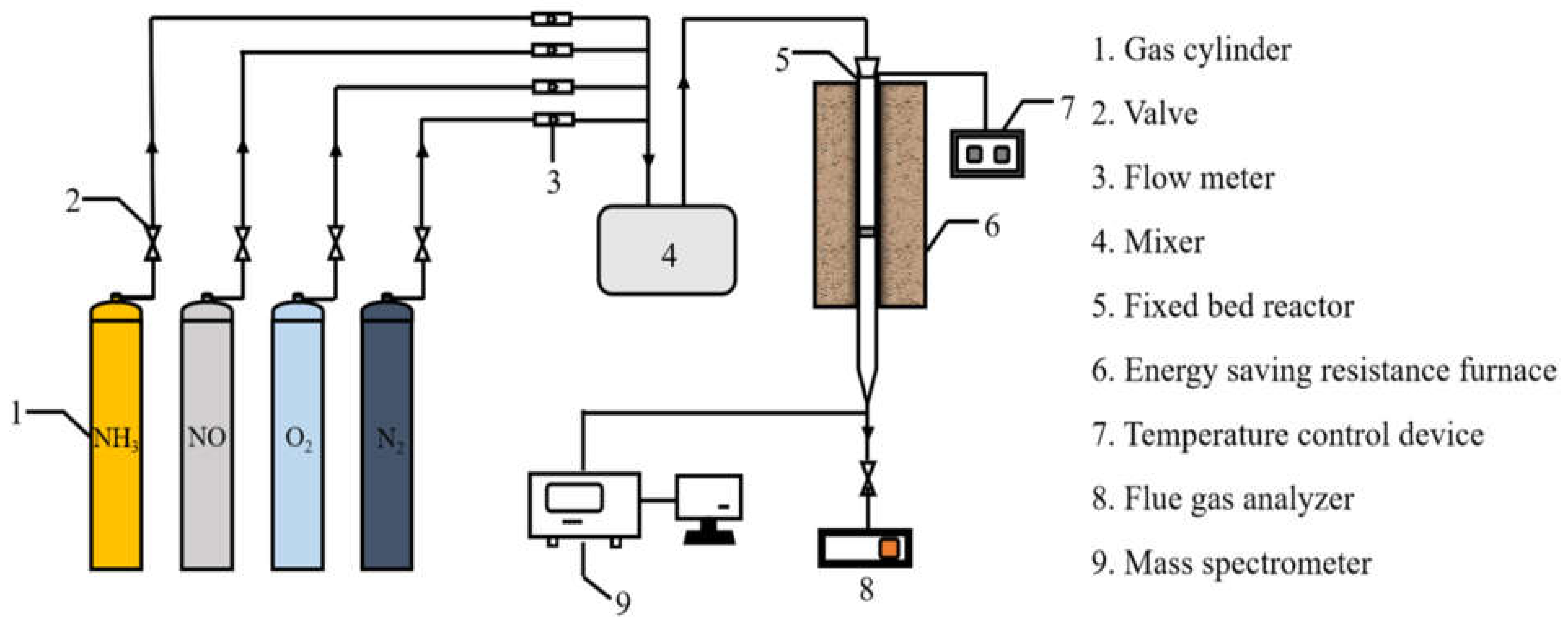
Catalytic performance of the catalysts for NH3-SCR was evaluated in a fixed-bed quartz tube (with an inner diameter of 15 mm and a height of 80 mm) continuous-flow reactor containing 3 mL of the catalysts. The evaluation device consists of three parts: simulated flue-gas region, fixed-bed reactor, and analysis region. The device composition is shown in Figure 14. The typical feed-gas composition was as follows: 600 ppm of NO, 600 ppm of NH3, 400 ppm of SO2, 3% O2, and the balance of N2. The concentrations of NO and SO2 in the inlet and outlet streams of the reactor were monitored online by an NO analyzer (Testo 350XL, Testo SE & Co. KGaA, Lenzkirch, Germany) and an SO2 (MRU-OPTIMA7) analyzer (Testo SE & Co. KGaA, Germany), respectively. All data were collected after 40 min when the SCR reaction reached a steady state.
The denitrification performance of catalysts was based on the NO conversion (XNO). The calculation formula is as follows:
where [NOx]in is the inlet concentration of NO and [NOx]in is the outlet concentration of NO.
3.4. Catalyst Characterization
The surface area of the catalyst was measured using a Quantachrome Nove (2200 ev) analyzer (Anton Paar Co. Ltd, Graz, Austria). The microscopic surface composition of the catalysts was analyzed using a new Hitachi SU8020 high-resolution field-emission scanning electron microscopy (SEM) instrument (Carl Zeiss AG, Oberkochen, Germany). The powder XRD measurement was performed on a Rigaku D/MAx2500V system (PANalytical B.V., Almelo, Netherlands) with a Cu Kα radiation. The XPS analysis was performed on an ESCALAB 250-type electron energy spectrometer (Thermo Fisher Scientific, Waltham, America) with Al Kα, hν = 1486.6 eV and a power of 150 W. The X-ray fluorescence (XRF) spectroscopic analysis was performed on an XRF-1800 X-ray fluorescence spectrometer (Shimadzu, Kyoto, Janpan), Rh target, a maximum power of 4 kW, with a minimum area of 0.5 mm.
The NH3 isothermal adsorption and temperature-programmed desorption measurement of ammonia (NH3-TPD) catalysts was performed in fixed quartz reaction tubes. A total of 0.5-g catalysts was loaded into a quartz tube, pretreated at 110 °C for 3 h in N2 flow (100 mL·min−1), and cooled to 50 °C before the measurement. The adsorption of NH3 was performed at 50 °C. After adsorption saturation, the catalysts were heated at a rate of 5 °C·min−1, from 50 °C to 900 °C under a constant N2 flow of 200 mL·min−1. The desorbed amount of NH3 was monitored online by mass spectrometry (Hiden QIC-200, Beijing Enghead Analysis Technology Co., Ltd, Beijing, China). SO2 and NO isothermal adsorption and TPD experiments were consistent with NH3-TPD experiments.
Hydrogen temperature-programmed reduction (H2-TPR) experiments were performed using a homemade H2-TPR device (Tianjin Xianquan Instrument Co. Ltd, Tianjin, China). A total of 50.0-mg catalyst with a particle diameter of 0.38–0.83 mm was placed into the device, a 5% H2–Ar gas mixture was passed, and the gas-flow rate was set at 20 mL·min−1. The baseline was purged at 50 °C until the baseline stabilized and switched to the analytical state. Then, the temperature was increased from 50 °C to 850 °C at a ramping rate of 10 °C·min−1. Hydrogen concentration was measured using a thermal conductivity cell (TCD detector, Tianjin Xianquan Instrument Co. Ltd, Tianjin, China) to produce a TPR spectrum. The detection temperature was 80 °C, and the bridge flow was 60 mA.
Temperature-programmed surface reactions (TPSR) were carried out to study the reactivity of catalyst-pre-adsorbed ammonia with gaseous NO. The catalysts (0.5 g) were adsorbed with NH3 at 50 °C, then reacted with simulated flue gas containing 600 ppm NO/Ar and 3% O2 from 50 °C to 300 °C at a rate of 5 °C·min−1, and the total gas-flow rate was 350 mL·min−1. Finally, the gas concentrations of NO were measured by a flue-gas analyzer (Testo350-XL, Testo SE & Co. KGaA, Germany).
4. Conclusions
In this study, the deactivation of MnOx/PG catalysts by SO2 for low-temperature SCR was investigated. The catalyst was pretreated in an SO2-containing ammonia-free gas to avoid a reaction between SO2 and NH3, which helps explore the reasons for the deactivation of the catalyst by SO2. Interestingly, the catalyst was determined to be considerably deactivated only by SO2 and can be easily recovered by simple washing with water. The results suggest that the poisoning species is neither ammonia sulfates nor manganese sulfates but something similar to polysulfuric acid that coats active manganese oxides. The effects of Mn8/PG SO2-pretreated poisoning and water-washed regeneration on the SCR reaction were studied. In addition, fresh SO2-pretreated poisoning and water-washed regeneration catalysts were characterized in detail. A possible SO2-poisoning and regeneration mechanism was proposed. The obtained conclusions are as follows.
- The stratification of the ammonium sulfate salt on the surface and the sulfation of the active component (MnSO4 formed) is not the main cause of catalyst deactivation. The main reason for the catalyst deactivation caused by SO2-pretreated poisoning is the SO42− polymer. This polymer coats the active component and affects the adsorption and activation of NH3 and NO.
- The recovery mechanism of water-washed regeneration is as follows. The SO42− polymer that coats the active component can be removed by the water-washing process to restore the catalytic activity of the active component; the water-washing process does not cause a loss of the active component.
- MnSO4 produced during vulcanization cannot be removed by washing. Therefore, MnSO4 is not the main cause of catalyst inactivation. The loading of a small amount of MnSO4 on the PG does not result in a decrease in the catalytic activity. Instead, the abovementioned process can increase the denitrification activity of PG in the low-temperature range.
Author Contributions
Conceptualization, X.Z. and X.W. (Xueping Wu); funding acquisition, X.W. (Xueping Wu); investigation, S.J. and X.W. (Xinyu Wang); formal analysis, S.J. and K.M.; project administration, X.Z. and Y.C.; validation, Y.C. and X.W.; writing—original draft, K.M. and S.L.; writing—review and editing, X.Z., S.L., and X.W. (Xinyu Wang). All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Not applicable.
Acknowledgments
This study was financially supported, in part, by the National Natural Science Foundation of China (No. 51872070).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Amini, H.; Taghavi-Shahri, S.M.; Henderson, S.; Hosseini, V.; Hassankhany, H.; Naderi, M.; Ahadi, S.; Schindler, C. Annual and seasonal spatial models for nitrogen oxides in Tehran, Iran. Sci. Rep. 2016, 6, 32970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, A.; Burrows, J.P.; Nüss, H.; Graier, C.; Niemeier, U. Increase in tropospheric nitrogen dioxide over China observed from space. Nature 2005, 437, 129–132. [Google Scholar] [CrossRef] [PubMed]
- Li, W.J.; Li, T.Y.; Wey, M.Y. Preferred enhancement of fast-SCR by Mn/CeSiOx catalyst: Study on Ce/Si promotion and shape dependence. Chem. Eng. J. 2021, 403, 126317. [Google Scholar] [CrossRef]
- Zhu, J.J.; Thomas, A. Perovskite-type mixed oxides as catalytic material for NO removal. Appl. Catal. B 2009, 92, 225–233. [Google Scholar] [CrossRef]
- Beale, A.M.; Lezcano-Gonzalez, I.; Maunula, T. Development and characterization of thermally stable supported V–W–TiO2 catalysts for mobile NH3–SCR applications. Catal. Struct. React. 2014, 1, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Qi, G.; Yang, R.T. A superior catalyst for low-temperature NO reduction with NH3. Chem. Commun. 2003, 34, 848–849. [Google Scholar] [CrossRef] [PubMed]
- Pieterse, J.A.Z.; van den Brink, R.W.; Booneveld, S.; de Bruijn, F.A. Durability of ZSM5-supported Co-Pd catalysts in the reduction of NOx with methane. Appl. Catal. B 2002, 39, 167–179. [Google Scholar] [CrossRef]
- Notoya, F.; Su, C.; Sasaoka, E.; Nojima, S. Effect of SO2 on the Low-Temperature Selective Catalytic Reduction of Nitric Oxide with Ammonia over TiO2, ZrO2, and Al2O3. Ind. Eng. Chem. Res. 2001, 40, 3732–3739. [Google Scholar] [CrossRef]
- Huang, J.; Tong, Z.; Yan, H. Selective catalytic reduction of NO with NH3 at low temperatures over iron and manganese oxides supported on mesoporous silica. Appl. Catal. B 2008, 78, 309–314. [Google Scholar] [CrossRef]
- Lin, T.; Zhang, Q.L.; Li, W.; Gong, M.C.; Xing, Y.X.; Chen, Y.Q. Monolith manganese-based catalyst supported on ZrO2-TiO2 for NH3-SCR reaction at low temperature. Acta. Phys-Chim. Sin. 2008, 24, 1127–1131. [Google Scholar] [CrossRef]
- Peña, D.A.; Uphade, B.S.; Smirniotis, P.G. TiO2-supported metal oxide catalysts for low-temperature selective catalytic reduction of NO with NH3: I. Evaluation and characterization of first row transition metals. J. Catal. 2004, 221, 421–431. [Google Scholar] [CrossRef]
- Kapteijn, F.; Singoredjon, L.; Andreini, A.; Moulijn, J.A. Activity and selectivity of pure manganese oxides in the selective catalytic reduction of nitric oxide with ammonia. Appl. Catal. B 1994, 3, 173–189. [Google Scholar] [CrossRef]
- Kijlstra, W.S.; Biervliet, M.; Poels, E.K.; Bliek, A. Deactivation by SO2 of MnOx/Al2O3 catalysts used for the selective catalytic reduction of NO with NH3 at low temperatures. Appl. Catal. B 1998, 16, 327–337. [Google Scholar] [CrossRef]
- Luo, H.C.; Huang, B.C.; Fu, M.L.; Wu, J.L.; Ye, D.Q. SO2 Deactivation mechanism of MnOx/MWCNTs catalyst for low-temperature selective catalytic reduction of NOx by ammonia. Acta Phys-Chim. Sin. 2012, 28, 2175–2182. [Google Scholar]
- Qi, G.; Yang, R.T. Low-temperature selective catalytic reduction of NO with NH3 over iron and manganese oxides supported on titania. Appl. Catal. B 2003, 44, 217–225. [Google Scholar] [CrossRef]
- Li, J.H.; Zhang, X.L.; Chen, T.H. Characterization and ammonia adsorption-desorption of palygorskite- sup-ported manganese oxide as a low-temperature selective catalytic reduction catalyst. Chin. J. Catal. 2010, 31, 454–460. [Google Scholar]
- Zhang, C.P.; Zhang, X.L.; Wu, X.P. The mechanism of SO2 influence on the denitration of MnOx/PG catalysts at low temperature. J. Environ. Sci. 2013, 33, 2686–2693. [Google Scholar]
- Zhang, L.F.; Zhang, X.L.; Lv, S.S.; Wu, X.P. Promoted performance of a MnOx/PG catalyst for low-temperature SCR against SO2 poisoning by addition of cerium oxide. RSC Adv. 2015, 5, 82952–82959. [Google Scholar] [CrossRef]
- Hammershøi, P.S.; Jangjou, Y.; Epling, W.S. Reversible and irreversible deactivation of Cu-CHA NH3-SCR catalysts by SO2 and SO3. Appl. Catal. B 2018, 226, 38–45. [Google Scholar] [CrossRef] [Green Version]
- Román, E.; Segovia, J.D.; Martín-Gago, J.A.; Comtet, G.; Hellner, L. Study of the interaction of SO2 with TiO2 (110) surface. Vacuum 1997, 48, 597. [Google Scholar] [CrossRef]
- Romano, E.J.; Schulz, K.H. A XPS investigation of SO2 adsorption on ceria–zirconia mixed-metal oxides. Appl. Surf. Sci. 2005, 246, 262–270. [Google Scholar] [CrossRef]
- Shen, B.X.; Liu, T. Deactivation of MnOx-CeOx/ACF catalysts for low-temperature NH3-SCR in the presence of SO2. Acta Phys-Chim. Sin. 2010, 26, 3009. [Google Scholar]
- Wang, L.S.; Huang, B.C.; Su, Y.X.; Zhou, G.Y.; Wang, K.L.; Luo, H.C. Manganese oxides supported on multi-walled carbon nanotubes for selective catalytic reduction of NO with NH3: Catalytic activity and characterization. Chem. Eng. J. 2012, 192, 232–241. [Google Scholar] [CrossRef]
- Romeo, M.; Bak, K.; Fallah, J.E. XPS Study of the Reduction of Cerium Dioxide. Surf. Interface Anal. 1993, 20, 508–512. [Google Scholar] [CrossRef]
- Andreoli, S.; Deorsola, F.A.; Galletti, C.; Pirone, R. Nanostructured MnOx catalysts for low-temperature NOx SCR. Chem. Eng. J. 2015, 278, 174–182. [Google Scholar] [CrossRef]
- Yu, C.; Dong, L.; Feng, C.; Liu, X.; Huang, B. Low-temperature SCR of NOx by NH3 over MnOx/SAPO-34 prepared by two different methods: A comparative study. Environ. Technol. Lett. 2017, 38, 1030–1042. [Google Scholar] [CrossRef]
- Jin, R.B.; Liu, Y.; Wang, Y.; Cen, W.L.; Wu, Z.B.; Wang, H.Q.; Weng, X.L. The role of cerium in the improved SO2 tolerance for NO reduction with NH3 over Mn-Ce/TiO2 catalyst at low temperature. Appl. Catal. B 2014, 148, 582–588. [Google Scholar] [CrossRef]
- Liu, J.; Zhao, Z.; Wang, J.Q.; Xu, C.M.; Duan, A.J.; Jiang, G.Y.; Yang, Q. The highly active catalysts of nanometricCeO2-supported cobalt oxides for soot combustion. Appl. Catal. B 2008, 84, 185–195. [Google Scholar] [CrossRef]
- Zhang, L.; Li, L.L.; Cao, Y.; Yao, X.J. Getting insight into the influence of SO2 on TiO2/CeO2 for the selective catalytic reduction of NO by NH3. Appl. Catal. B 2015, 165, 589–598. [Google Scholar] [CrossRef]
- Jung, S.M.; Grange, P. Characterization and reactivity of pure TiO2-SO42− SCR catalyst: Influence of SO42- content. Catal. Today 2000, 59, 305–312. [Google Scholar] [CrossRef]
- Liao, P.W.; Yang, L.; Wang, F.; Hu, Y.F.; Sheng, Z.Y. Performance study for low-temperature SCR catalysts based on Mn-Ce prepared by different methods. Acta Chim. Sinica 2011, 69, 2723–2728. [Google Scholar]
- Yang, J.; Li, Z.F.; Yang, C.L.; Ma, Y.Y.; Li, Y.Y.; Zhang, Q.; Song, K.; Cui, J.X. Significant promoting effect of La doping on the wide temperature NH3-SCR performance of Ce and Cu modified ZSM-5 catalysts. J. Solid State Chem. 2021, 305, 122700. [Google Scholar] [CrossRef]
- Li, M.; Guo, R.; Hu, C.; Sun, P.; Pan, W.; Liu, S.; Sun, X.; Liu, S.; Liu, J. The enhanced resistance to K deactivation of Ce/TiO2 catalyst for NH3-SCR reaction by the modification with P. Appl. Surf. Sci. 2018, 436, 814–822. [Google Scholar] [CrossRef]
- Yang, N.Z.; Guo, R.T.; Tian, Y.; Pan, W.G.; Chen, Q.L.; Wang, Q.S.; Lu, C.Z.; Wang, S.X. The enhanced performance of ceria by HF treatment for selective catalytic reduction of NO with NH3. Fuel 2016, 179, 305–311. [Google Scholar] [CrossRef]
- Macleod, N.; Cropley, R.; Lambert, R.M. Efficient reduction of NOx by H2 under oxygen-rich conditions over Pd/TiO2 catalysts: An in situ DRIFTS study. Catal. Lett. 2003, 86, 69–75. [Google Scholar] [CrossRef]
Figure 1.
X-ray diffraction patterns for the catalysts.

Figure 2.
Scanning electron microscopic images of different catalysts: (a) Mn8/PG, (b) Mn8/PG-S, and (c) Mn8/PG-S-W.
Figure 2.
Scanning electron microscopic images of different catalysts: (a) Mn8/PG, (b) Mn8/PG-S, and (c) Mn8/PG-S-W.


Figure 3.
DRIFT spectra of different catalysts.

Figure 4.
Selective catalytic reduction activity of different Mn8/PG catalysts. Reaction conditions: [NH3] = [NO] = 600 ppm, [O2] = 3 vol%, N2 balance, GHSV = 7000 h−1, total flow rate = 350 mL min−1.
Figure 4.
Selective catalytic reduction activity of different Mn8/PG catalysts. Reaction conditions: [NH3] = [NO] = 600 ppm, [O2] = 3 vol%, N2 balance, GHSV = 7000 h−1, total flow rate = 350 mL min−1.

Figure 5.
S 2p (a) and Mn 2p (b) XPS spectra of two different catalysts.

Figure 6.
H2-temperature-programmed reduction of different catalysts.

Figure 7.
Activity of different HCl-treated, PG-loaded Mn catalysts. Reaction conditions: [NH3] = [NO] = 600 ppm, [O2] = 3 vol%, Ar balance, GHSV = 7000 h−1, total flow rate = 350 mL min−1.
Figure 7.
Activity of different HCl-treated, PG-loaded Mn catalysts. Reaction conditions: [NH3] = [NO] = 600 ppm, [O2] = 3 vol%, Ar balance, GHSV = 7000 h−1, total flow rate = 350 mL min−1.

Figure 8.
Activity of different PG catalysts. Reaction conditions: [NH3] = [NO] = 600 ppm, [O2] = 3 vol%, Ar balance, GHSV = 7000 h−1, total flow rate = 350 mL min−1.
Figure 8.
Activity of different PG catalysts. Reaction conditions: [NH3] = [NO] = 600 ppm, [O2] = 3 vol%, Ar balance, GHSV = 7000 h−1, total flow rate = 350 mL min−1.

Figure 9.
The SO2 emission profiles of isothermal adsorption and following temperature-programmed desorption (TPD). Reaction conditions: 0.5 g of the catalyst, 400 ppm of SO2, GHSV = 16,000 h−1.
Figure 9.
The SO2 emission profiles of isothermal adsorption and following temperature-programmed desorption (TPD). Reaction conditions: 0.5 g of the catalyst, 400 ppm of SO2, GHSV = 16,000 h−1.

Figure 10.
The NH3 emission profiles of isothermal adsorption and following temperature-programmed desorption (TPD). Reaction conditions: 0.5 g of the catalyst, 600 ppm of NO, GHSV = 16,000 h−1.
Figure 10.
The NH3 emission profiles of isothermal adsorption and following temperature-programmed desorption (TPD). Reaction conditions: 0.5 g of the catalyst, 600 ppm of NO, GHSV = 16,000 h−1.

Figure 11.
The NO emission profiles of isothermal adsorption and following temperature-programmed desorption (TPD). Reaction conditions: 0.5 g of the catalyst, 600 ppm of NO, GHSV = 16,000 h−1.
Figure 11.
The NO emission profiles of isothermal adsorption and following temperature-programmed desorption (TPD). Reaction conditions: 0.5 g of the catalyst, 600 ppm of NO, GHSV = 16,000 h−1.

Figure 12.
NH3 adsorption and desorption curve for different MnO8/PG catalysts. Reaction conditions: 600 ppm of NO, 3% O2, Ar as a balance, total flue rate: 350 mL·min−1, GHSV: 7000 h−1, heating rate (5 °C·min−1).
Figure 12.
NH3 adsorption and desorption curve for different MnO8/PG catalysts. Reaction conditions: 600 ppm of NO, 3% O2, Ar as a balance, total flue rate: 350 mL·min−1, GHSV: 7000 h−1, heating rate (5 °C·min−1).

Figure 13.
Proposed sulfur-treated poisoning and water-washed regeneration mechanism model.

Figure 14.
The self-designed fixed-bed catalysis system for SCR reaction and detection.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
S content, specific surface area and pore volume of the three catalysts.
| Catalysts | S Content (wt%) | SBET (m2/g) | Vp (cm3/g) |
|---|---|---|---|
| Mn8/PG | 0.025 | 121.44 | 0.436 |
| Mn8/PG-S | 0.736 | 92.76 | 0.421 |
| Mn8/PG-S-W | 0.123 | 115.21 | 0.432 |
Table 2.
Content of elements in different catalysts.
| Element Content (wt%) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Catalysts | Si | O | Mn | Mg | Al | Fe | C | K | Ca | Ti | S | R |
| Mn8/PG | 36.2 | 30.8 | 8.32 | 8.51 | 5.44 | 4.70 | 1.79 | 1.03 | 1.42 | 0.82 | 0.08 | 0.91 |
| Mn8/PG-W-1 | 35.5 | 33.4 | 8.65 | 7.43 | 5.25 | 4.21 | 2.63 | 0.85 | 0.82 | 0.54 | 0.42 | 0.28 |
| Mn8/PG-W-2 | 36.4 | 33 | 9.18 | 7.46 | 4.86 | 3.63 | 3.13 | 0.86 | 0.45 | 0.48 | 0.34 | 0.24 |
| Mn8/PG-W-3 | 36 | 33.7 | 8.65 | 7.44 | 4.97 | 3.56 | 2.85 | 0.87 | 0.27 | 0.48 | 0.33 | 0.84 |
| Mn8/PG-W-4 | 36.8 | 33.8 | 9.29 | 7.24 | 4.92 | 3.84 | 3.10 | 0.86 | 0.17 | 0.49 | 0.30 | 0.22 |
Table 3.
Elements’ content in different water-washed solutions of catalysts.
| Catalysts | Mn | Mg | PH |
|---|---|---|---|
| Mn8/PG | 2.1 mg/L | 27.7 mg/L | 7.56 |
| Mn8/PG-W-1 | 4.6 mg/L | 56.2 mg/L | 5.30 |
| Mn8/PG-W-3 | 10.8 mg/L | 38 mg/L | 7.23 |
| MnO2-W-1 | 0.24 mg/L | - | 6.5 |
Table 4.
XPS results for surface Mn atomic percentage.
| Catalysts | Surface Atomic Concentration/% | |||||
|---|---|---|---|---|---|---|
| Mn | S | O | ||||
| Mn4+ | Mn3+ | Mn2+ | ||||
| Mn8/PG | 1.29 | 68.2% | 38.8% | - | - | 98.71 |
| Mn8/PG-S | 1.07 | 25.2% | 38.6% | 36.2% | 3.18 | 95.75 |
| Mn8/PG-S-W | 1.44 | 32.7% | 43.7% | 23.6% | 0.67 | 97.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhang, X.; Liu, S.; Ma, K.; Chen, Y.; Jin, S.; Wang, X.; Wu, X. Study on the Mechanism of SO2 Poisoning of MnOx/PG for Lower Temperature SCR by Simple Washing Regeneration. Catalysts 2021, 11, 1360. https://doi.org/10.3390/catal11111360
AMA Style
Zhang X, Liu S, Ma K, Chen Y, Jin S, Wang X, Wu X. Study on the Mechanism of SO2 Poisoning of MnOx/PG for Lower Temperature SCR by Simple Washing Regeneration. Catalysts. 2021; 11(11):1360. https://doi.org/10.3390/catal11111360
Chicago/Turabian StyleZhang, Xianlong, Shiwen Liu, Kang Ma, Yazhong Chen, Shi Jin, Xinyu Wang, and Xueping Wu. 2021. "Study on the Mechanism of SO2 Poisoning of MnOx/PG for Lower Temperature SCR by Simple Washing Regeneration" Catalysts 11, no. 11: 1360. https://doi.org/10.3390/catal11111360
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.