An Overview of the Photocatalytic Water Splitting over Suspended Particles


Abstract
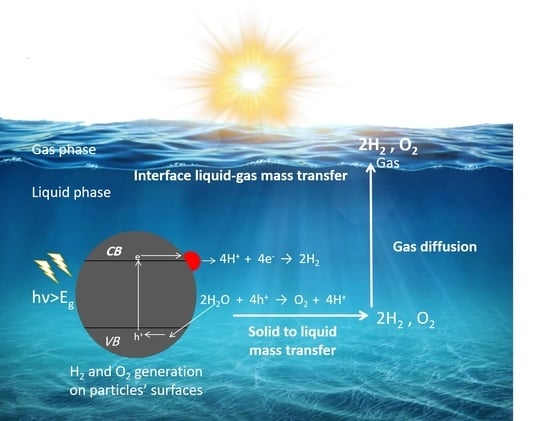
:
1. Introduction
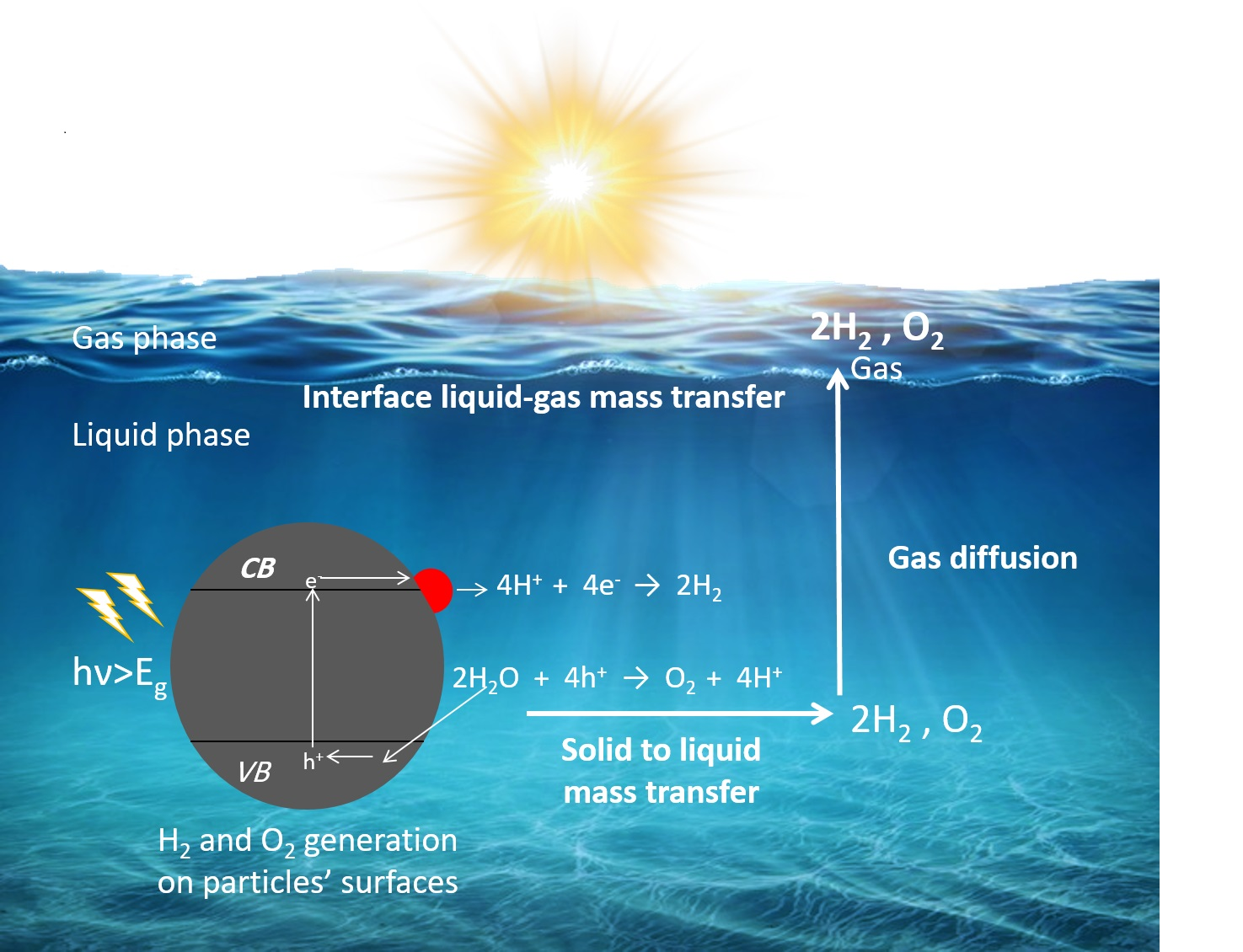
2. Fundamental Processes in Photocatalytic Overall Water Splitting
3. Design and Synthesis of Particulate Photocatalytic Systems
4. Improving Light Absorption
4.1. UV Light Photocatalysts
4.2. Visible Light Photocatalysts
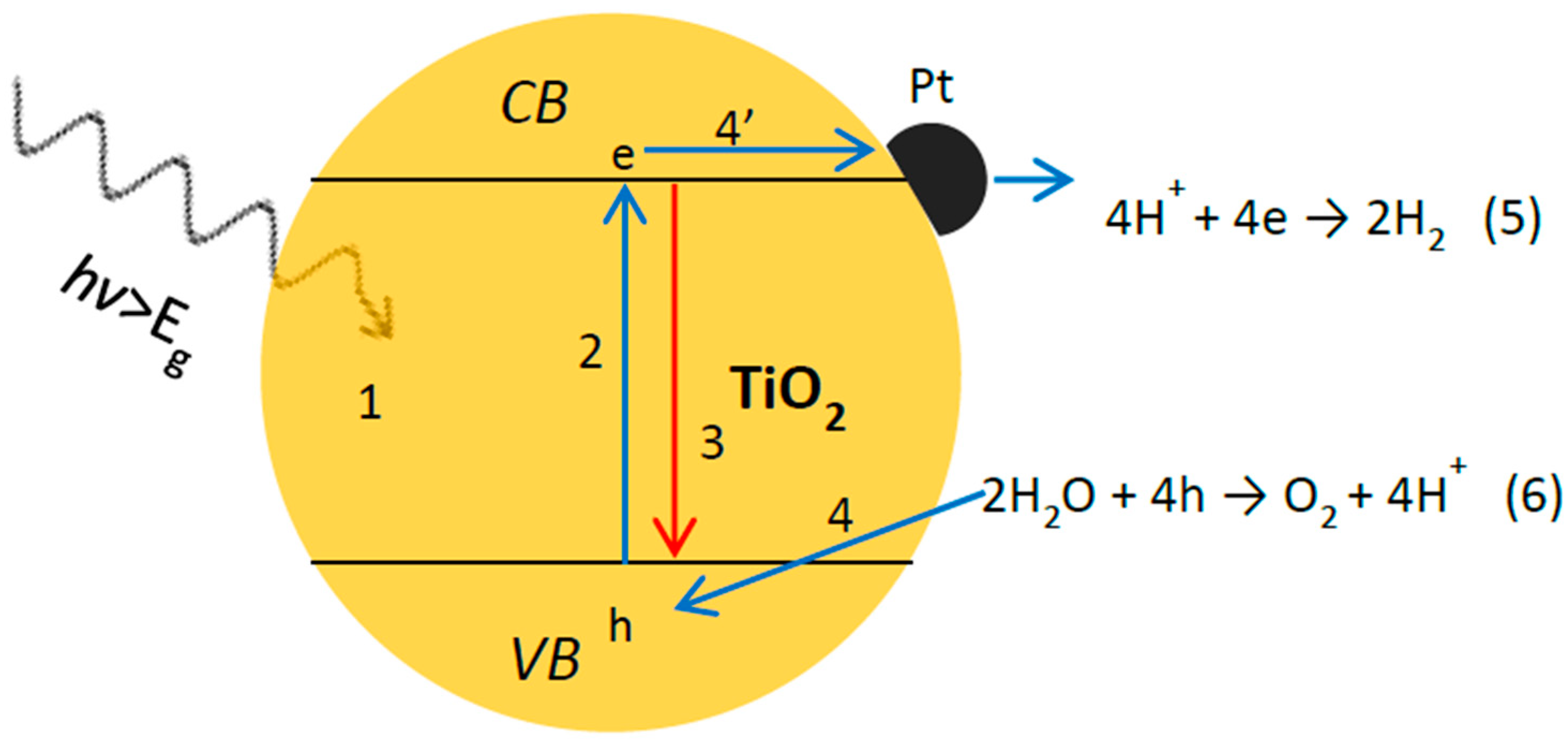
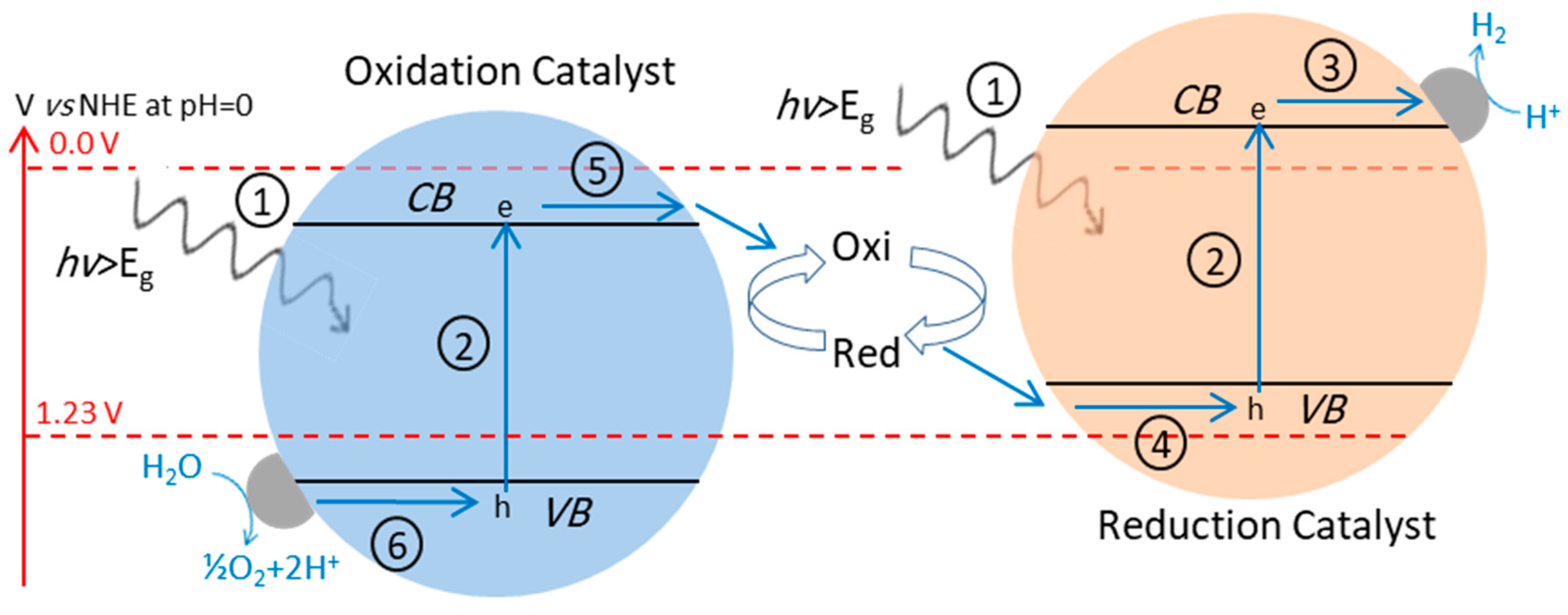
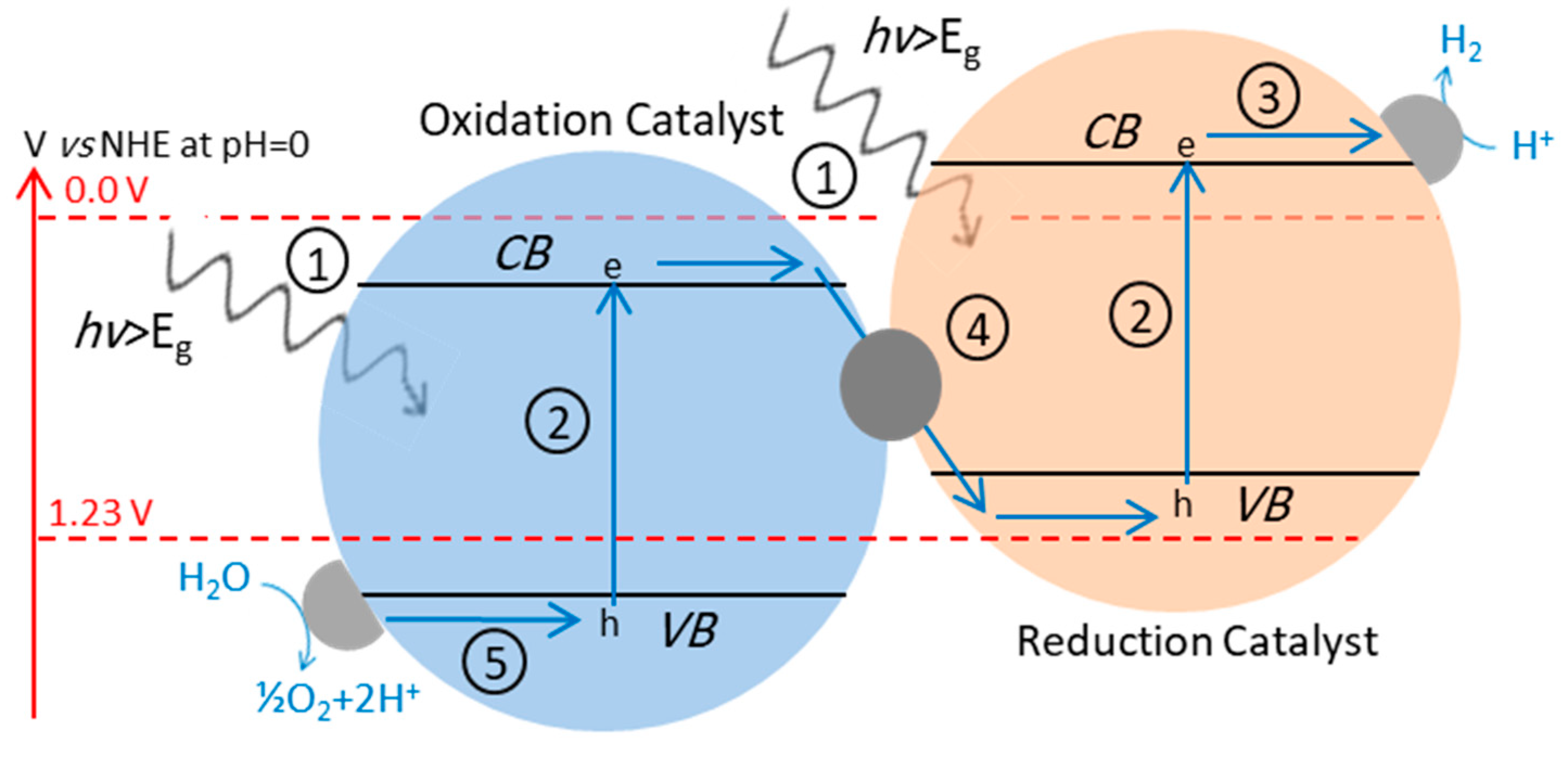
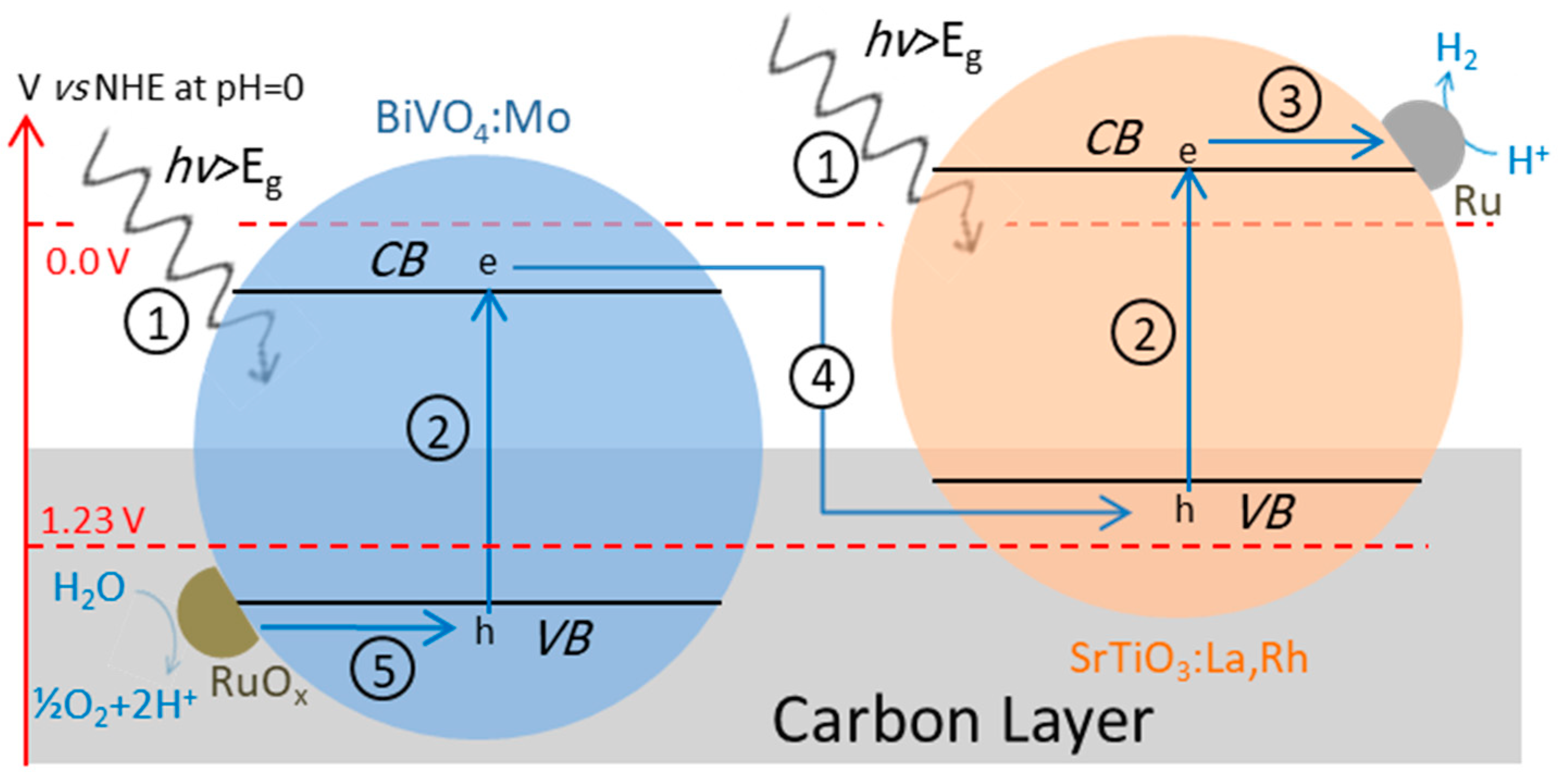
4.3. Z-Scheme: A Two-Step Approach
4.3.1. Z-Scheme with Aqueous Redox Mediator

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| H2 Photocatalyst (wt. % Unless Indicated) | O2 Photocatalyst (wt.%) | Mediator | H2 Rate (mmolh−1) | O2 Rate (mmolh−1) | AQY (%) | Ref. |
|---|---|---|---|---|---|---|
| Pt/SrTiO3:Rh | BiVO4 | Fe3+/Fe2+ | 15 | 7.2 | 0.4 at 420 nm | [110] |
| Pt/SrTiO3:Cr/Ta (Pt = 0.3, Cr, Ta = 4.0 mol% each) | PtOx/WO3 (Pt = 0.5) | IO3−/I− | 16 | 8 | 1 at 420 nm | [111] |
| Pt/TaON (Pt = 0.3) | PtOx/WO3 (Pt = 0.5) | IO3−/I−_ | 16.5 | 8 | 0.5 at 420 nm | [112] |
| Pt/ZrO2/TaON (Pt = 1.0, Zn/Ta = 0.1) | PtOx/WO3 (Pt = 0.5) | IO3−/I−_ | 52 | 27 | 6.3 at 420 nm | [109] |
| Ru/SrTiO3:Rh (Ru = 1.0, Sr:Ti:Rh = 1.1:0.98:0.02) | PtOx/WO3 (Pt = 0.3) | Fe3+/Fe2+ | 88 | 44 | 4.2 at 420 nm | [40] |
| Pt/MgTa2O6−xNy/TaON (Pt = 0.4, Mg/Ta = 0.2) | PtOx/WO3 (Pt = 0.45) | IO3−/I− | 108 | 55 | 6.8 at 420 nm | [110] |
4.3.2. Z-Scheme with Solid-State Electron Mediator
5. Improving Efficiency
5.1. Charge Recombination
5.2. Back-Reaction (2 H2+O2 → 2 H2O)
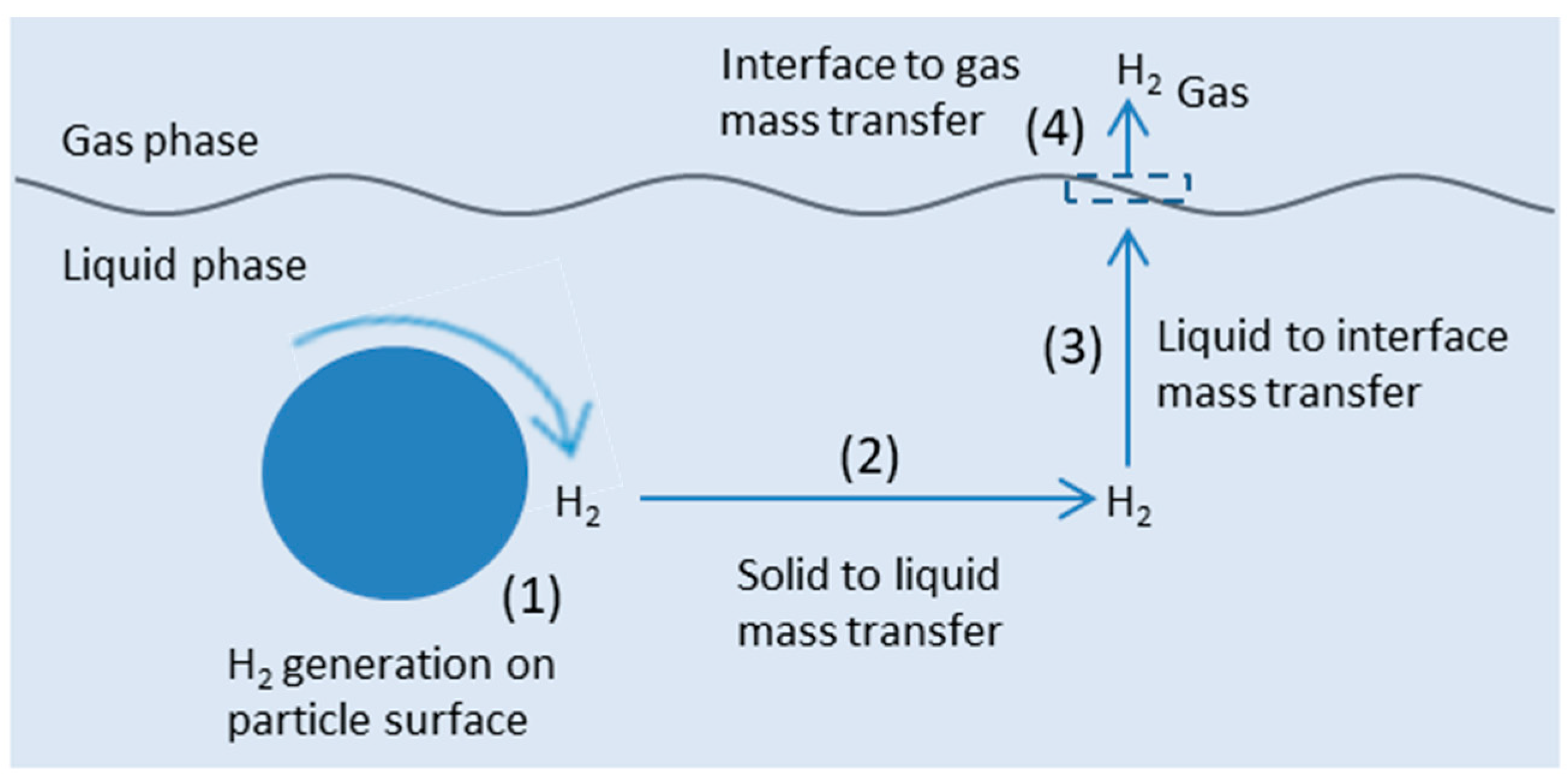
5.3. Mass Transfer Limitations
6. More Recent Overall Water Splitting Systems
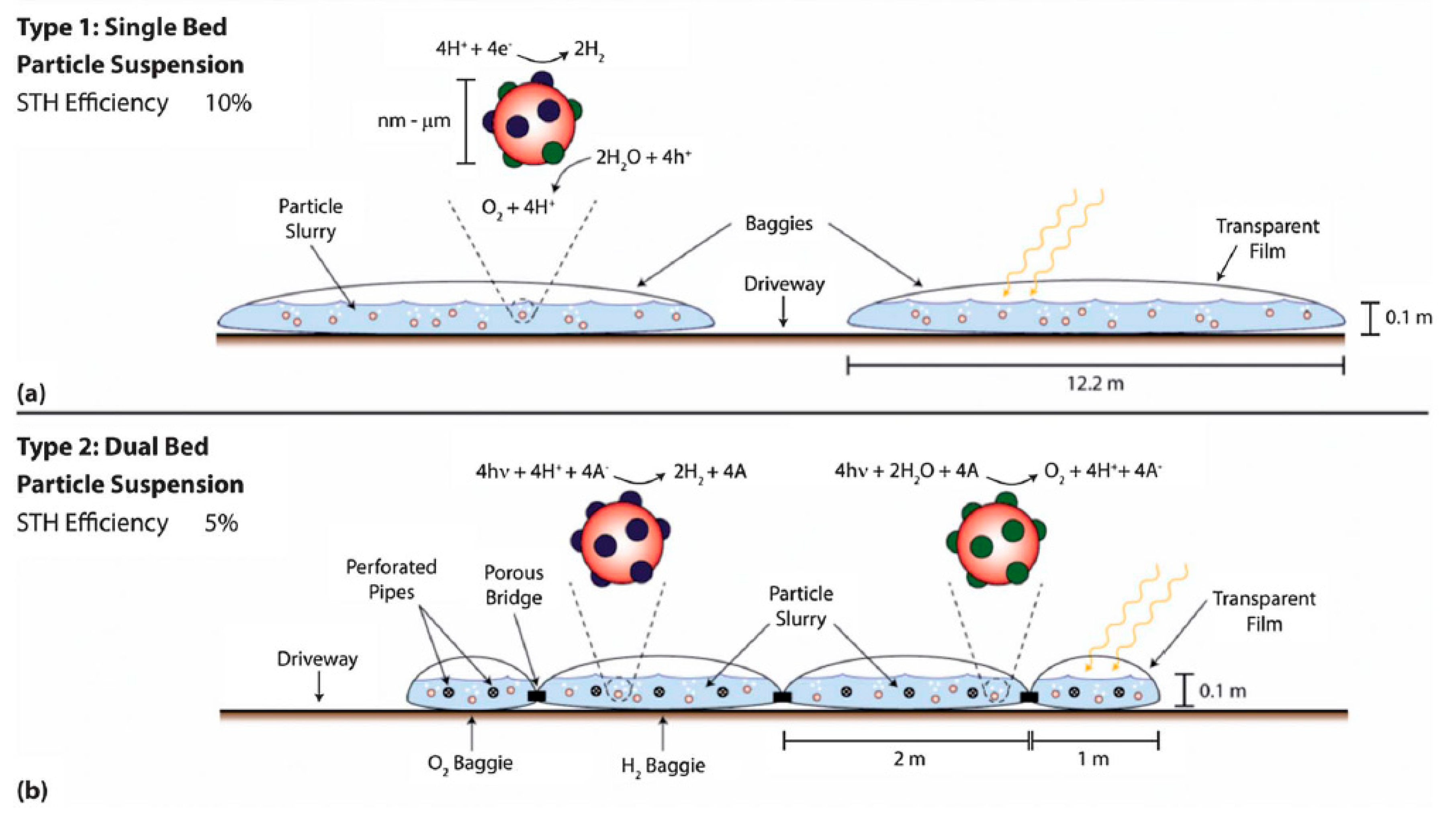
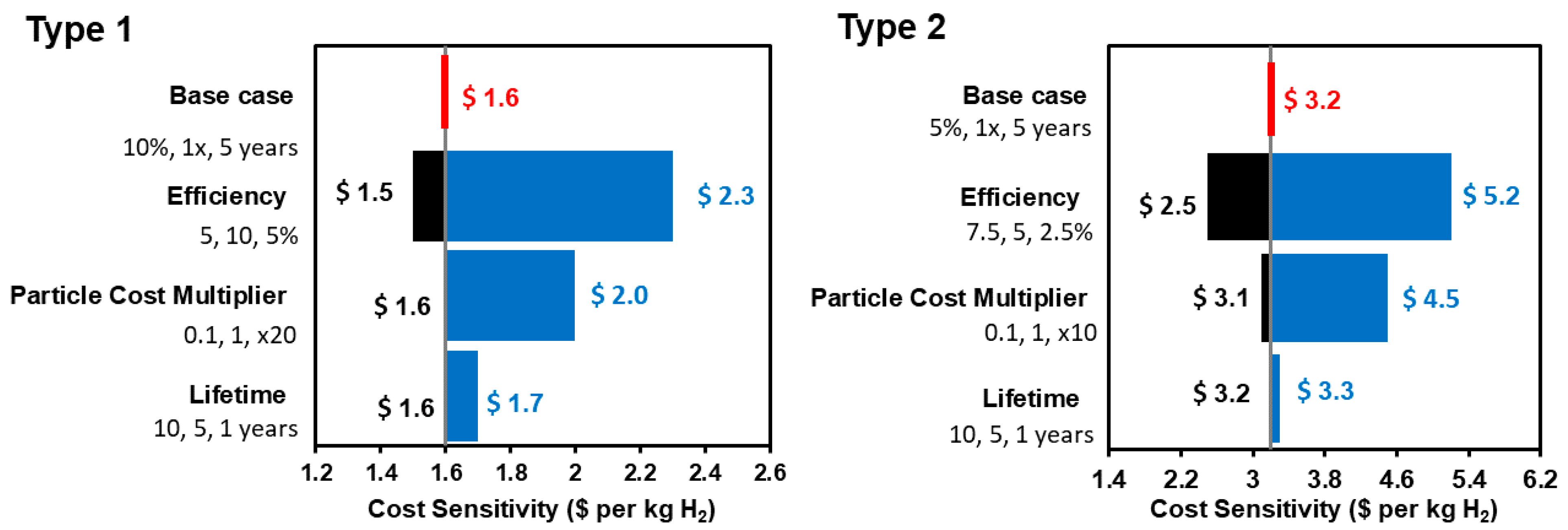
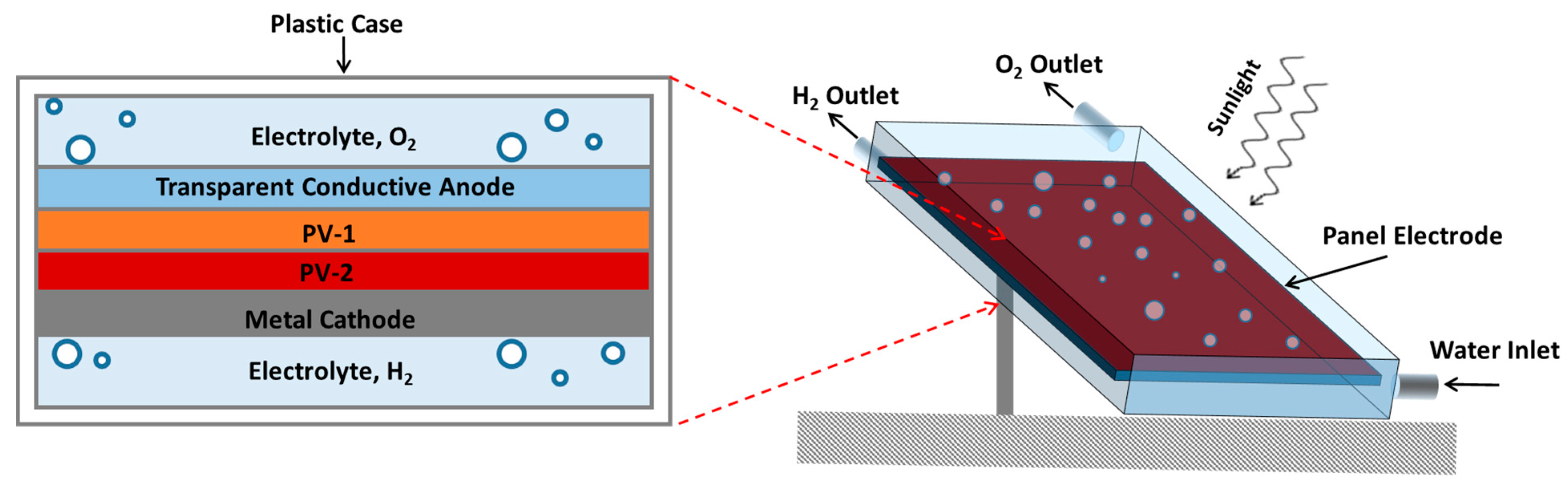
7. Reactor Design and Cost of Hydrogen
8. Conclusions and Future Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Siegmund, P.; Abermann, J.; Baddour, O.; Canadell, P.; Cazenave, A.; Derksen, C.; Garreau, A.; Howell, S.; Huss, H.; Isensee, K.; et al. World Meteorological Organization, The Global Climate in 2015–2019. In The Global Climate in 2015–2019: Climate Change Accelerates; WMO Publication Board: Geneva, Switzerland, 2019. [Google Scholar]
- Morton, O. A New Day Dawning? Silicon Valley Sunrise; Nature Publishing Group: London, UK, 2006.
- Barber, J. Photosynthetic energy conversion: Natural and artificial. Chem. Soc. Rev. 2009, 38, 185–196. [Google Scholar] [CrossRef]
- Lewis, N.S. Toward Cost-Effective Solar Energy Use. Science 2007, 315, 798–801. [Google Scholar] [CrossRef] [Green Version]
- Grätzel, M. Photoelectrochemical cells. Nature 2001, 414, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Erisman, J.W.; Sutton, M.A.; Galloway, J.; Klimont, Z.; Winiwarter, W. How a century of ammonia synthesis changed the world. Nat. Geosci. 2008, 1, 636–639. [Google Scholar] [CrossRef]
- Centi, G.; Perathoner, S. Opportunities and prospects in the chemical recycling of carbon dioxide to fuels. Catal. Today 2009, 148, 191–205. [Google Scholar] [CrossRef]
- Aasberg-Petersen, K.; Dybkjær, I.; Ovesen, C.; Schjødt, N.; Sehested, J.; Thomsen, S. Natural gas to synthesis gas–catalysts and catalytic processes. J. Nat. Gas Sci. Eng. 2011, 3, 423–459. [Google Scholar] [CrossRef]
- Nadeem, M.A.; Idriss, H. Effect of pH, temperature, and low light flux on the performance (16% STH) of coupled triple junction solar cell to water electrolysis. J. Power Sources 2020, 459, 228074. [Google Scholar] [CrossRef]
- Bashir, S.M.; Nadeem, M.A.; Al-Oufi, M.; Al-Hakami, M.; Isimjan, T.T.; Idriss, H. Sixteen Percent Solar-to-Hydrogen Efficiency Using a Power-Matched Alkaline Electrolyzer and a High Concentrated Solar Cell: Effect of Operating Parameters. ACS Omega 2020, 5, 10510–10518. [Google Scholar] [CrossRef] [PubMed]
- James, B.D.; Baum, G.N.; Perez, J.; Baum, K.N.J.D.r. Technoeconomic Analysis of Photoelectrochemical (PEC) Hydrogen Production. Office of Energy Efficiency and Renewable Energy: Arlington, Virginia, 2009. [Google Scholar]
- Shaner, M.R.; Atwater, H.A.; Lewis, N.S.; McFarland, E.W.J.E.; Science, E. A comparative technoeconomic analysis of renewable hydrogen production using solar energy. Energy Environ. Sci. 2016, 9, 2354–2371. [Google Scholar] [CrossRef] [Green Version]
- Pinaud, B.A.; Benck, J.D.; Seitz, L.C.; Forman, A.J.; Chen, Z.; Deutsch, T.G.; James, B.D.; Baum, K.N.; Baum, G.N.; Ardo, S.; et al. Technical and economic feasibility of centralized facilities for solar hydrogen production via photocatalysis and photoelectrochemistry. Energy Environ. Sci. 2013, 6, 1983–2002. [Google Scholar] [CrossRef] [Green Version]
- Fabian, D.M.; Hu, S.; Singh, N.; Houle, F.A.; Hisatomi, T.; Domen, K.; Osterloh, F.E.; Ardo, S. Particle suspension reactors and materials for solar-driven water splitting. Energy Environ. Sci. 2015, 8, 2825–2850. [Google Scholar] [CrossRef] [Green Version]
- Goto, Y.; Hisatomi, T.; Wang, Q.; Higashi, T.; Ishikiriyama, K.; Maeda, T.; Sakata, Y.; Okunaka, S.; Tokudome, H.; Katayama, M.; et al. A Particulate Photocatalyst Water-Splitting Panel for Large-Scale Solar Hydrogen Generation. Joule 2018, 2, 509–520. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Hisatomi, T.; Suzuki, Y.; Pan, Z.; Seo, J.; Katayama, M.; Minegishi, T.; Nishiyama, H.; Takata, T.; Seki, K.; et al. Particulate Photocatalyst Sheets Based on Carbon Conductor Layer for Efficient Z-Scheme Pure-Water Splitting at Ambient Pressure. J. Am. Chem. Soc. 2017, 139, 1675–1683. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, Y.; Hisatomi, T.; Nakabayashi, M.; Shibata, N.; Kubota, J.; Domen, K. Z-scheme water splitting using particulate semiconductors immobilized onto metal layers for efficient electron relay. J. Catal. 2015, 328, 308–315. [Google Scholar] [CrossRef]
- Pan, Z.; Hisatomi, T.; Wang, Q.; Chen, S.; Iwase, A.; Nakabayashi, M.; Shibata, N.; Takata, T.; Katayama, M.; Minegishi, T.; et al. Photoreduced Graphene Oxide as a Conductive Binder to Improve the Water Splitting Activity of Photocatalyst Sheets. Adv. Funct. Mater. 2016, 26, 7011–7019. [Google Scholar] [CrossRef]
- Minegishi, T.; Nishimura, N.; Kubota, J.; Domen, K. Photoelectrochemical properties of LaTiO2N electrodes prepared by particle transfer for sunlight-driven water splitting. Chem. Sci. 2013, 4, 1120–1124. [Google Scholar] [CrossRef]
- Fujishima, A.; Honda, K. Electrochemical photolysis of water at a semiconductor electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef]
- Wagner, F.; Somorjai, G. Photocatalytic hydrogen production from water on Pt-free SrTiO3 in alkali hydroxide solutions. Nature 1980, 285, 559–560. [Google Scholar] [CrossRef]
- Yamaguti, K.; Sato, S. Photolysis of water over metallized powdered titanium dioxide. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1985, 81, 1237–1246. [Google Scholar] [CrossRef]
- Kudo, A.; Domen, K.; Maruya, K.-i.; Onishi, T. Photocatalytic activities of TiO2 loaded with NiO. Chem. Phys. Lett. 1987, 133, 517–519. [Google Scholar] [CrossRef]
- Sayama, K.; Arakawa, H. Effect of carbonate salt addition on the photocatalytic decomposition of liquid water over Pt–TiO2 catalyst. J. Chem. Soc. Faraday Trans. 1997, 93, 1647–1654. [Google Scholar] [CrossRef]
- Tabata, S.; Nishida, H.; Masaki, Y.; Tabata, K. Stoichiometric photocatalytic decomposition of pure water in Pt/TiO2 aqueous suspension system. Catal. Lett. 1995, 34, 245–249. [Google Scholar] [CrossRef]
- Moon, S.-C.; Mametsuka, H.; Suzuki, E.; Anpo, M. Stoichiometric decomposition of pure water over Pt-loaded Ti/B binary oxide under UV-irradiation. Chem. Lett. 1998, 27, 117–118. [Google Scholar] [CrossRef]
- Sayama, K.; Arakawa, H. Effect of Na2CO3 addition on photocatalytic decomposition of liquid water over various semiconductor catalysis. J. Photochem. Photobiol. A Chem. 1994, 77, 243–247. [Google Scholar] [CrossRef]
- Bard, A.J. Photoelectrochemistry. Science 1980, 207, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Nadeem, M.A.; Al-Oufi, M.; Wahab, A.K.; Anjum, D.; Idriss, H. Hydrogen Production on Ag-Pd/TiO2 Bimetallic Catalysts: Is there a Combined Effect of Surface Plasmon Resonance with Schottky Mechanism on the Photo-Catalytic Activity? ChemistrySelect 2017, 2, 2754–2762. [Google Scholar] [CrossRef]
- Khan, M.A.; Al-Oufi, M.; Toseef, A.; Nadeem, M.A.; Idriss, H. Comparing the Reaction Rates of Plasmonic (Gold) and Non-Plasmonic (Palladium) Metal Particles in Photocatalytic Hydrogen Production. Catal. Lett. 2018, 148, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Nadeem, A.; Muir, J.M.; Waterhouse, G.W.; Idriss, H. Hydrogen Photo-Production from Ethanol on TiO2: A Surface Science and Catalysis Study; SPIE Bellingham: Washington, DC, USA, 2011; Volume 8109. [Google Scholar]
- Wahab, A.K.; Nadeem, M.A.; Idriss, H. Hydrogen Production During Ethylene Glycol Photoreactions Over Ag-Pd/TiO2 at Different Partial Pressures of Oxygen. Front. Chem. 2019, 7. [Google Scholar] [CrossRef]
- Nadeem, A.; Waterhouse, G.I.; Metson, J.; Idriss, H. Hydrogen Production by Photoreaction of Ethanol Over Au/TiO2 Anatase: The Effect of TiO2 Particle Size; SPIE Bellingham: Washington, DC, USA, 2010; Volume 7770. [Google Scholar]
- Hussain, E.; Majeed, I.; Nadeem, M.A.; Iqbal, A.; Chen, Y.; Choucair, M.; Jin, R.; Nadeem, M.A. Remarkable effect of BaO on photocatalytic H2 evolution from water splitting via TiO2(P25) supported palladium nanoparticles. J. Environ. Chem. Eng. 2019, 7, 102729. [Google Scholar] [CrossRef]
- Domen, K.; Naito, S.; Soma, M.; Onishi, T.; Tamaru, K. Photocatalytic decomposition of water vapour on an NiO–SrTiO3 catalyst. J. Chem. Soc. Chem. Commun. 1980, 12, 543–544. [Google Scholar] [CrossRef]
- Domen, K.; Kudo, A.; Onishi, T.; Kosugi, N.; Kuroda, H. Photocatalytic decomposition of water into H2 and O2 over NiO-SrTiO3 powder. 1. Structure of the catalyst. J. Phys. Chem. 1986, 90, 292–295. [Google Scholar] [CrossRef]
- Pan, C.; Takata, T.; Nakabayashi, M.; Matsumoto, T.; Shibata, N.; Ikuhara, Y.; Domen, K. A complex perovskite type oxynitride: The first photocatalyst for water splitting operable at up to 600 nm. Angew. Chem. Int. Ed. 2015, 54, 2955–2959. [Google Scholar] [CrossRef] [PubMed]
- Ohno, T.; Bai, L.; Hisatomi, T.; Maeda, K.; Domen, K. Photocatalytic water splitting using modified GaN:ZnO solid solution under visible light: Long-time operation and regeneration of activity. J. Am. Chem. Soc. 2012, 134, 8254–8259. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Teramura, K.; Takata, T.; Hara, M.; Saito, N.; Toda, K.; Inoue, Y.; Kobayashi, H.; Domen, K. Overall Water Splitting on (Ga1-xZnx)(N1-xOx) Solid Solution Photocatalyst: Relationship between Physical Properties and Photocatalytic Activity. J. Phys. Chem. B 2005, 109, 20504–20510. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Sasaki, Y.; Shirakura, N.; Kudo, A. Synthesis of highly active rhodium-doped SrTiO3 powders in Z-scheme systems for visible-light-driven photocatalytic overall water splitting. J. Mater. Chem. A 2013, 1, 12327–12333. [Google Scholar] [CrossRef]
- Kudo, A. Z-scheme photocatalyst systems for water splitting under visible light irradiation. MRS Bull. 2011, 36, 32–38. [Google Scholar] [CrossRef]
- Fujito, H.; Kunioku, H.; Kato, D.; Suzuki, H.; Higashi, M.; Kageyama, H.; Abe, R. Layered Perovskite Oxychloride Bi4NbO8Cl: A Stable Visible Light Responsive Photocatalyst for Water Splitting. J. Am. Chem. Soc. 2016, 138, 2082–2085. [Google Scholar] [CrossRef]
- Tabata, M.; Maeda, K.; Higashi, M.; Lu, D.; Takata, T.; Abe, R.; Domen, K. Modified Ta3N5 Powder as a Photocatalyst for O2 Evolution in a Two-Step Water Splitting System with an Iodate/Iodide Shuttle Redox Mediator under Visible Light. Langmuir 2010, 26, 9161–9165. [Google Scholar] [CrossRef]
- Wu, X.; Zhao, J.; Wang, L.; Han, M.; Zhang, M.; Wang, H.; Huang, H.; Liu, Y.; Kang, Z. Carbon dots as solid-state electron mediator for BiVO4/CDs/CdS Z-scheme photocatalyst working under visible light. Appl. Catal. B Environ. 2017, 206, 501–509. [Google Scholar] [CrossRef]
- Srinivasan, N.; Sakai, E.; Miyauchi, M. Balanced Excitation between Two Semiconductors in Bulk Heterojunction Z-Scheme System for Overall Water Splitting. ACS Catal. 2016, 6, 2197–2200. [Google Scholar] [CrossRef]
- Kobayashi, R.; Takashima, T.; Tanigawa, S.; Takeuchi, S.; Ohtani, B.; Irie, H. A heterojunction photocatalyst composed of zinc rhodium oxide, single crystal-derived bismuth vanadium oxide, and silver for overall pure-water splitting under visible light up to 740 nm. Phys. Chem. Chem. Phys. 2016, 18, 27754–27760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Hisatomi, T.; Ma, S.S.K.; Li, Y.; Domen, K. Core/Shell Structured La- and Rh-Codoped SrTiO3 as a Hydrogen Evolution Photocatalyst in Z-Scheme Overall Water Splitting under Visible Light Irradiation. Chem. Mater. 2014, 26, 4144–4150. [Google Scholar] [CrossRef]
- Iwase, A.; Ng, Y.H.; Ishiguro, Y.; Kudo, A.; Amal, R. Reduced Graphene Oxide as a Solid-State Electron Mediator in Z-Scheme Photocatalytic Water Splitting under Visible Light. J. Am. Chem. Soc. 2011, 133, 11054–11057. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Hwang, D.W.; Kim, H.G.; Bae, S.W.; Lee, J.S.; Li, W.; Oh, S.H. Highly efficient overall water splitting through optimization of preparation and operation conditions of layered perovskite photocatalysts. Top. Catal. 2005, 35, 295–303. [Google Scholar] [CrossRef]
- Asai, R.; Nemoto, H.; Jia, Q.; Saito, K.; Iwase, A.; Kudo, A. A visible light responsive rhodium and antimony-codoped SrTiO3 powdered photocatalyst loaded with an IrO2 cocatalyst for solar water splitting. Chem. Commun. 2014, 50, 2543–2546. [Google Scholar] [CrossRef]
- Kato, H.; Kudo, A. Water splitting into H2 and O2 on alkali tantalate photocatalysts ATaO3 (A = Li, Na, and K). J. Phys. Chem. B 2001, 105, 4285–4292. [Google Scholar] [CrossRef]
- Jo, W.J.; Kang, H.J.; Kong, K.-J.; Lee, Y.S.; Park, H.; Lee, Y.; Buonassisi, T.; Gleason, K.K.; Lee, J.S. Phase transition-induced band edge engineering of BiVO4 to split pure water under visible light. Proc. Natl. Acad. Sci. USA 2015, 112, 13774–13778. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Yuan, J.; Shangguan, W.; Teraoka, Y. Visible-light-responding BiYWO6 solid solution for stoichiometric photocatalytic water splitting. J. Phys. Chem. C 2008, 112, 8521–8523. [Google Scholar] [CrossRef]
- Chiang, T.H.; Lyu, H.; Hisatomi, T.; Goto, Y.; Takata, T.; Katayama, M.; Minegishi, T.; Domen, K. Efficient photocatalytic water splitting using al-doped SrTiO3 coloaded with molybdenum oxide and rhodium–chromium oxide. ACS Catal. 2018, 8, 2782–2788. [Google Scholar] [CrossRef]
- Liao, L.; Zhang, Q.; Su, Z.; Zhao, Z.; Wang, Y.; Li, Y.; Lu, X.; Wei, D.; Feng, G.; Yu, Q.; et al. Efficient solar water-splitting using a nanocrystalline CoO photocatalyst. Nat. Nanotechnol. 2014, 9, 69–73. [Google Scholar] [CrossRef]
- Sakata, Y.; Matsuda, Y.; Yanagida, T.; Hirata, K.; Imamura, H.; Teramura, K. Effect of metal ion addition in a Ni supported Ga2O3 photocatalyst on the photocatalytic overall splitting of H2O. Catal. Lett. 2008, 125, 22–26. [Google Scholar] [CrossRef]
- Sakata, Y.; Hayashi, T.; Yasunaga, R.; Yanaga, N.; Imamura, H. Remarkably high apparent quantum yield of the overall photocatalytic H2O splitting achieved by utilizing Zn ion added Ga2O3 prepared using dilute CaCl2 solution. Chem. Commun. 2015, 51, 12935–12938. [Google Scholar] [CrossRef] [PubMed]
- Sato, J.; Saito, N.; Yamada, Y.; Maeda, K.; Takata, T.; Kondo, J.N.; Hara, M.; Kobayashi, H.; Domen, K.; Inoue, Y. RuO2-Loaded β-Ge3N4 as a Non-Oxide Photocatalyst for Overall Water Splitting. J. Am. Chem. Soc. 2005, 127, 4150–4151. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Takata, T.; Domen, K. Overall water splitting on the transition-metal oxynitride photocatalyst LaMg1/3Ta2/3O2N over a large portion of the visible-light spectrum. Chem. A Eur. J. 2016, 22, 1854–1862. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Lu, D.; Domen, K. Direct Water Splitting into Hydrogen and Oxygen under Visible Light by using Modified TaON Photocatalysts with d0 Electronic Configuration. Chem. A Eur. J. 2013, 19, 4986–4991. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Teramura, K.; Domen, K. Effect of post-calcination on photocatalytic activity of (Ga1-xZnx)(N1−xOx) solid solution for overall water splitting under visible light. J. Catal. 2008, 254, 198–204. [Google Scholar] [CrossRef]
- Kibria, M.G.; Nguyen, H.P.; Cui, K.; Zhao, S.; Liu, D.; Guo, H.; Trudeau, M.L.; Paradis, S.; Hakima, A.-R.; Mi, Z. One-step overall water splitting under visible light using multiband InGaN/GaN nanowire heterostructures. ACS Nano 2013, 7, 7886–7893. [Google Scholar] [CrossRef]
- Xu, J.; Pan, C.; Takata, T.; Domen, K. Photocatalytic overall water splitting on the perovskite-type transition metal oxynitride CaTaO2N under visible light irradiation. Chem. Commun. 2015, 51, 7191–7194. [Google Scholar] [CrossRef]
- Pan, C.; Takata, T.; Kumamoto, K.; Ma, S.S.K.; Ueda, K.; Minegishi, T.; Nakabayashi, M.; Matsumoto, T.; Shibata, N.; Ikuhara, Y. Band engineering of perovskite-type transition metal oxynitrides for photocatalytic overall water splitting. J. Mater. Chem. A 2016, 4, 4544–4552. [Google Scholar] [CrossRef]
- Zhang, G.; Lan, Z.-A.; Lin, L.; Lin, S.; Wang, X. Overall water splitting by Pt/g-C3N4 photocatalysts without using sacrificial agents. Chem. Sci. 2016, 7, 3062–3066. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Liu, Y.; Liu, N.; Han, Y.; Zhang, X.; Huang, H.; Lifshitz, Y.; Lee, S.-T.; Zhong, J.; Kang, Z. Metal-free efficient photocatalyst for stable visible water splitting via a two-electron pathway. Science 2015, 347, 970–974. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Takata, T.; Domen, K. Particulate photocatalysts for overall water splitting. Nat. Rev. Mater. 2017, 2, 17050. [Google Scholar] [CrossRef]
- Kato, H.; Asakura, K.; Kudo, A. Highly efficient water splitting into H2 and O2 over lanthanum-doped NaTaO3 photocatalysts with high crystallinity and surface nanostructure. J. Am. Chem. Soc. 2003, 125, 3082–3089. [Google Scholar] [CrossRef] [PubMed]
- Ham, Y.; Hisatomi, T.; Goto, Y.; Moriya, Y.; Sakata, Y.; Yamakata, A.; Kubota, J.; Domen, K. Flux-mediated doping of SrTiO3 photocatalysts for efficient overall water splitting. J. Mater. Chem. A 2016, 4, 3027–3033. [Google Scholar] [CrossRef] [Green Version]
- Ikarashi, K.; Sato, J.; Kobayashi, H.; Saito, N.; Nishiyama, H.; Inoue, Y. Photocatalysis for water decomposition by RuO2-dispersed ZnGa2O4 with d10 configuration. J. Phys. Chem. B 2002, 106, 9048–9053. [Google Scholar] [CrossRef]
- Sato, J.; Saito, N.; Nishiyama, H.; Inoue, Y. New photocatalyst group for water decomposition of RuO2-loaded p-block metal (In, Sn, and Sb) oxides with d10 configuration. J. Phys. Chem. B 2001, 105, 6061–6063. [Google Scholar] [CrossRef]
- Inoue, Y. Photocatalytic water splitting by RuO2-loaded metal oxides and nitrides with d0-and d10-related electronic configurations. Energy Environ. Sci. 2009, 2, 364–386. [Google Scholar] [CrossRef]
- Kudo, A.; Miseki, Y. Heterogeneous photocatalyst materials for water splitting. Chem. Soc. Rev. 2009, 38, 253–278. [Google Scholar] [CrossRef]
- Ajmal, A.; Majeed, I.; Malik, R.N.; Idriss, H.; Nadeem, M.A. Principles and mechanisms of photocatalytic dye degradation on TiO2 based photocatalysts: A comparative overview. RSC Adv. 2014, 4, 37003–37026. [Google Scholar] [CrossRef]
- Schneider, J.; Matsuoka, M.; Takeuchi, M.; Zhang, J.; Horiuchi, Y.; Anpo, M.; Bahnemann, D.W. Understanding TiO2 photocatalysis: Mechanisms and materials. Chem. Rev. 2014, 114, 9919–9986. [Google Scholar] [CrossRef]
- Wang, M.; Li, Z.; Wu, Y.; Ma, J.; Lu, G. Inhibition of hydrogen and oxygen reverse recombination reaction over Pt/TiO2 by F− ions and its impact on the photocatalytic hydrogen formation. J. Catal. 2017, 353, 162–170. [Google Scholar] [CrossRef]
- Kitano, M.; Matsuoka, M.; Ueshima, M.; Anpo, M. Recent developments in titanium oxide-based photocatalysts. Appl. Catal. A Gen. 2007, 325, 1–14. [Google Scholar] [CrossRef]
- Gaya, U.I.; Abdullah, A.H. Heterogeneous photocatalytic degradation of organic contaminants over titanium dioxide: A review of fundamentals, progress and problems. J. Photochem. Photobiol. C Photochem. Rev. 2008, 9, 1–12. [Google Scholar] [CrossRef]
- Maeda, K.; Domen, K. New non-oxide photocatalysts designed for overall water splitting under visible light. J. Phys. Chem. C 2007, 111, 7851–7861. [Google Scholar] [CrossRef]
- Kamata, K.; Maeda, K.; Lu, D.; Kako, Y.; Domen, K. Synthesis and photocatalytic activity of gallium–zinc–indium mixed oxynitride for hydrogen and oxygen evolution under visible light. Chem. Phys. Lett. 2009, 470, 90–94. [Google Scholar] [CrossRef]
- Inoue, Y.; Niiyama, T.; Asai, Y.; Sato, K. Stable photocatalytic activity of BaTi4O9 combined with ruthenium oxide for decomposition of water. J. Chem. Soc. Chem. Commun. 1992, 7, 579–580. [Google Scholar] [CrossRef]
- Iwase, A.; Kato, H.; Kudo, A. Nanosized Au particles as an efficient cocatalyst for photocatalytic overall water splitting. Catal. Lett. 2006, 108, 7–10. [Google Scholar] [CrossRef]
- Murdoch, M.; Waterhouse, G.; Nadeem, M.; Metson, J.; Keane, M.; Howe, R.; Llorca, J.; Idriss, H. The effect of gold loading and particle size on photocatalytic hydrogen production from ethanol over Au/TiO2 nanoparticles. Nat. Chem. 2011, 3, 489–492. [Google Scholar] [CrossRef]
- Majeed, I.; Nadeem, M.A.; Hussain, E.; Waterhouse, G.I.N.; Badshah, A.; Iqbal, A.; Nadeem, M.A.; Idriss, H. On the Synergism between Cu and Ni for Photocatalytic Hydrogen Production and their Potential as Substitutes of Noble Metals. ChemCatChem 2016, 8, 3146–3155. [Google Scholar] [CrossRef]
- Li, R.; Han, H.; Zhang, F.; Wang, D.; Li, C. Highly efficient photocatalysts constructed by rational assembly of dual-cocatalysts separately on different facets of BiVO4. Energy Environ. Sci. 2014, 7, 1369–1376. [Google Scholar] [CrossRef]
- Alsabban, M.M.; Yang, X.; Wahyudi, W.; Fu, J.-H.; Hedhili, M.N.; Ming, J.; Yang, C.-W.; Nadeem, M.A.; Idriss, H.; Lai, Z.; et al. Design and Mechanistic Study of Highly Durable Carbon-Coated Cobalt Diphosphide Core–Shell Nanostructure Electrocatalysts for the Efficient and Stable Oxygen Evolution Reaction. ACS Appl. Mater. Interfaces 2019, 11, 20752–20761. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Ji, Z.; Zou, W.; Gu, X.; Deng, Y.; Gao, F.; Tang, C.; Dong, L. In situ loading transition metal oxide clusters on TiO2 nanosheets as co-catalysts for exceptional high photoactivity. ACS Catal. 2013, 3, 2052–2061. [Google Scholar] [CrossRef]
- Maeda, K.; Wang, X.; Nishihara, Y.; Lu, D.; Antonietti, M.; Domen, K. Photocatalytic activities of graphitic carbon nitride powder for water reduction and oxidation under visible light. J. Phys. Chem. C 2009, 113, 4940–4947. [Google Scholar] [CrossRef]
- Youngblood, W.J.; Lee, S.-H.A.; Kobayashi, Y.; Hernandez-Pagan, E.A.; Hoertz, P.G.; Moore, T.A.; Moore, A.L.; Gust, D.; Mallouk, T.E. Photoassisted overall water splitting in a visible light-absorbing dye-sensitized photoelectrochemical cell. J. Am. Chem. Soc. 2009, 131, 926–927. [Google Scholar] [CrossRef]
- Kim, H.; Hwang, D.; Kim, Y.; Lee, J. Highly donor-doped (110) layered perovskite materials as novel photocatalysts for overall water splitting. J. Chem. Soc. Chem. Commun. 1999, 12, 1077–1078. [Google Scholar] [CrossRef] [Green Version]
- Sayama, K.; Arakawa, H.; Domen, K. Photocatalytic water splitting on nickel intercalated A4TaxNb6-xO17 (A = K, Rb). Catal. Today 1996, 28, 175–182. [Google Scholar] [CrossRef]
- Laurent-Applegate, L.; Roques, S. Biological Actions of Infrared Radiation. In Sensing, Signaling and Cell Adaptation; Storey, K.B., Storey, J.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2002; pp. 233–242. [Google Scholar]
- Maeda, K.; Takata, T.; Hara, M.; Saito, N.; Inoue, Y.; Kobayashi, H.; Domen, K. GaN:ZnO Solid Solution as a Photocatalyst for Visible-Light-Driven Overall Water Splitting. J. Am. Chem. Soc. 2005, 127, 8286–8287. [Google Scholar] [CrossRef]
- Wang, Z.; Inoue, Y.; Hisatomi, T.; Ishikawa, R.; Wang, Q.; Takata, T.; Chen, S.; Shibata, N.; Ikuhara, Y.; Domen, K. Overall water splitting by Ta3N5 nanorod single crystals grown on the edges of KTaO3 particles. Nat. Catal. 2018, 1, 756–763. [Google Scholar] [CrossRef]
- Murthy, D.H.K.; Matsuzaki, H.; Wang, Z.; Suzuki, Y.; Hisatomi, T.; Seki, K.; Inoue, Y.; Domen, K.; Furube, A. Origin of the overall water splitting activity of Ta3N5 revealed by ultrafast transient absorption spectroscopy. Chem. Sci. 2019, 10, 5353–5362. [Google Scholar] [CrossRef] [Green Version]
- Kudo, A. Recent progress in the development of visible light-driven powdered photocatalysts for water splitting. Int. J. Hydrog. Energy 2007, 32, 2673–2678. [Google Scholar] [CrossRef]
- Konta, R.; Ishii, T.; Kato, H.; Kudo, A. Photocatalytic Activities of Noble Metal Ion Doped SrTiO3 under Visible Light Irradiation. J. Phys. Chem. B 2004, 108, 8992–8995. [Google Scholar] [CrossRef]
- Park, K.-W.; Kolpak, A.M. Understanding photocatalytic overall water splitting on CoO nanoparticles: Effects of facets, surface stoichiometry, and the CoO/water interface. J. Catal. 2018, 365, 115–124. [Google Scholar] [CrossRef]
- Wang, X.; Maeda, K.; Thomas, A.; Takanabe, K.; Xin, G.; Carlsson, J.M.; Domen, K.; Antonietti, M. A metal-free polymeric photocatalyst for hydrogen production from water under visible light. Nat. Mater. 2009, 8, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.J.; Reardon, P.J.T.; Moniz, S.J.A.; Tang, J. Visible Light-Driven Pure Water Splitting by a Nature-Inspired Organic Semiconductor-Based System. J. Am. Chem. Soc. 2014, 136, 12568–12571. [Google Scholar] [CrossRef] [PubMed]
- Ng, B.J.; Putri, L.K.; Kong, X.Y.; Teh, Y.W.; Pasbakhsh, P.; Chai, S.P. Z-Scheme Photocatalytic Systems for Solar Water Splitting. Adv. Sci. (Weinh. Baden-Wurtt. Ger.) 2020, 7, 1903171. [Google Scholar] [CrossRef] [PubMed]
- Abe, R.; Sayama, K.; Domen, K.; Arakawa, H. A new type of water splitting system composed of two different TiO2 photocatalysts (anatase, rutile) and a IO3−/I− shuttle redox mediator. Chem. Phys. Lett. 2001, 344, 339–344. [Google Scholar] [CrossRef]
- Ryu, A. Development of a New System for Photocatalytic Water Splitting into H2 and O2 under Visible Light Irradiation. Bull. Chem. Soc. Jpn. 2011, 84, 1000–1030. [Google Scholar] [CrossRef] [Green Version]
- Ma, G.; Chen, S.; Kuang, Y.; Akiyama, S.; Hisatomi, T.; Nakabayashi, M.; Shibata, N.; Katayama, M.; Minegishi, T.; Domen, K. Visible Light-Driven Z-Scheme Water Splitting Using Oxysulfide H2 Evolution Photocatalysts. J. Phys. Chem. Lett. 2016, 7, 3892–3896. [Google Scholar] [CrossRef]
- Qi, Y.; Chen, S.; Li, M.; Ding, Q.; Li, Z.; Cui, J.; Dong, B.; Zhang, F.; Li, C. Achievement of visible-light-driven Z-scheme overall water splitting using barium-modified Ta3N5 as a H2-evolving photocatalyst. Chem. Sci. 2017, 8, 437–443. [Google Scholar] [CrossRef] [Green Version]
- Kato, T.; Hakari, Y.; Ikeda, S.; Jia, Q.; Iwase, A.; Kudo, A. Utilization of Metal Sulfide Material of (CuGa)1–xZn2xS2 Solid Solution with Visible Light Response in Photocatalytic and Photoelectrochemical Solar Water Splitting Systems. J. Phys. Chem. Lett. 2015, 6, 1042–1047. [Google Scholar] [CrossRef]
- Chen, S.; Qi, Y.; Hisatomi, T.; Ding, Q.; Asai, T.; Li, Z.; Ma, S.S.K.; Zhang, F.; Domen, K.; Li, C. Efficient Visible-Light-Driven Z-Scheme Overall Water Splitting Using a MgTa2O6−xNy /TaON Heterostructure Photocatalyst for H2 Evolution. Angew. Chem. Int. Ed. 2015, 54, 8498–8501. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Chen, J.; Li, C.; Tian, W. Achieving solar overall water splitting with hybrid photosystems of photosystem II and artificial photocatalysts. Nat. Commun. 2014, 5, 4647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, K.; Higashi, M.; Lu, D.; Abe, R.; Domen, K. Efficient nonsacrificial water splitting through two-step photoexcitation by visible light using a modified oxynitride as a hydrogen evolution photocatalyst. J. Am. Chem. Soc. 2010, 132, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Hori, M.; Konta, R.; Shimodaira, Y.; Kudo, A. Construction of Z-scheme type heterogeneous photocatalysis systems for water splitting into H2 and O2 under visible light irradiation. Chem. Lett. 2004, 33, 1348–1349. [Google Scholar] [CrossRef]
- Abe, R.; Sayama, K.; Sugihara, H. Development of new photocatalytic water splitting into H2 and O2 using two different semiconductor photocatalysts and a shuttle redox mediator IO3-/I. J. Phys. Chem. B 2005, 109, 16052–16061. [Google Scholar] [CrossRef] [PubMed]
- Abe, R.; Higashi, M.; Domen, K. Overall Water Splitting under Visible Light through a Two-Step Photoexcitation between TaON and WO3 in the Presence of an Iodate–Iodide Shuttle Redox Mediator. ChemSusChem 2011, 4, 228–237. [Google Scholar] [CrossRef]
- Tsuji, K.; Tomita, O.; Higashi, M.; Abe, R. Manganese-Substituted Polyoxometalate as an Effective Shuttle Redox Mediator in Z-Scheme Water Splitting under Visible Light. ChemSusChem 2016, 9, 2201–2208. [Google Scholar] [CrossRef]
- Sasaki, Y.; Kato, H.; Kudo, A. [Co(bpy)3]3+/2+ and [Co(phen)3]3+/2+ Electron Mediators for Overall Water Splitting under Sunlight Irradiation Using Z-Scheme Photocatalyst System. J. Am. Chem. Soc. 2013, 135, 5441–5449. [Google Scholar] [CrossRef]
- Zhao, W.; Maeda, K.; Zhang, F.; Hisatomi, T.; Domen, K. Effect of post-treatments on the photocatalytic activity of Sm2Ti2S2O5 for the hydrogen evolution reaction. Phys. Chem. Chem. Phys. 2014, 16, 12051–12056. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, Y.; Nemoto, H.; Saito, K.; Kudo, A. Solar Water Splitting Using Powdered Photocatalysts Driven by Z-Schematic Interparticle Electron Transfer without an Electron Mediator. J. Phys. Chem. C 2009, 113, 17536–17542. [Google Scholar] [CrossRef]
- Tada, H.; Mitsui, T.; Kiyonaga, T.; Akita, T.; Tanaka, K. All-solid-state Z-scheme in CdS–Au–TiO2 three-component nanojunction system. Nat. Mater. 2006, 5, 782–786. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.G.; Jeong, E.D.; Borse, P.H.; Jeon, S.; Yong, K.; Lee, J.S.; Li, W.; Oh, S.H. Photocatalytic Ohmic layered nanocomposite for efficient utilization of visible light photons. Appl. Phys. Lett. 2006, 89, 064103. [Google Scholar] [CrossRef] [Green Version]
- Bai, S.; Jiang, J.; Zhang, Q.; Xiong, Y. Steering charge kinetics in photocatalysis: Intersection of materials syntheses, characterization techniques and theoretical simulations. Chem. Soc. Rev. 2015, 44, 2893–2939. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Song, M.; Wang, H.; Zhang, X.; Sui, N.; Zhang, Q.; Colvin, V.L.; Yu, W.W. Latest progress in constructing solid-state Z scheme photocatalysts for water splitting. Nanoscale 2019, 11, 11071–11082. [Google Scholar] [CrossRef] [PubMed]
- Hisatomi, T.; Takanabe, K.; Domen, K. Photocatalytic Water-Splitting Reaction from Catalytic and Kinetic Perspectives. Catal. Lett. 2015, 145, 95–108. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Durrant, J.R.; Klug, D.R. Mechanism of Photocatalytic Water Splitting in TiO2. Reaction of Water with Photoholes, Importance of Charge Carrier Dynamics, and Evidence for Four-Hole Chemistry. J. Am. Chem. Soc. 2008, 130, 13885–13891. [Google Scholar] [CrossRef]
- Scott, M.; Nadeem, A.M.; Waterhouse, G.I.W.; Idriss, H. Hydrogen Production from Ethanol. Comparing Thermal Catalytic Reactions to Photo-catalytic Reactions. MRS Proc. 2011, 1326, mrss11-1326-f1307-1307. [Google Scholar] [CrossRef]
- Leytner, S.; Hupp, J.T. Evaluation of the energetics of electron trap states at the nanocrystalline titanium dioxide/aqueous solution interface via time-resolved photoacoustic spectroscopy. Chem. Phys. Lett. 2000, 330, 231–236. [Google Scholar] [CrossRef]
- Majeed, I.; Nadeem, M.A.; Al-Oufi, M.; Nadeem, M.A.; Waterhouse, G.I.N.; Badshah, A.; Metson, J.B.; Idriss, H. On the role of metal particle size and surface coverage for photo-catalytic hydrogen production: A case study of the Au/CdS system. Appl. Catal. B Environ. 2016, 182, 266–276. [Google Scholar] [CrossRef]
- Nadeem, M.A.; Murdoch, M.; Waterhouse, G.I.N.; Metson, J.B.; Keane, M.A.; Llorca, J.; Idriss, H. Photoreaction of ethanol on Au/TiO2 anatase: Comparing the micro to nanoparticle size activities of the support for hydrogen production. J. Photochem. Photobiol. A Chem. 2010, 216, 250–255. [Google Scholar] [CrossRef]
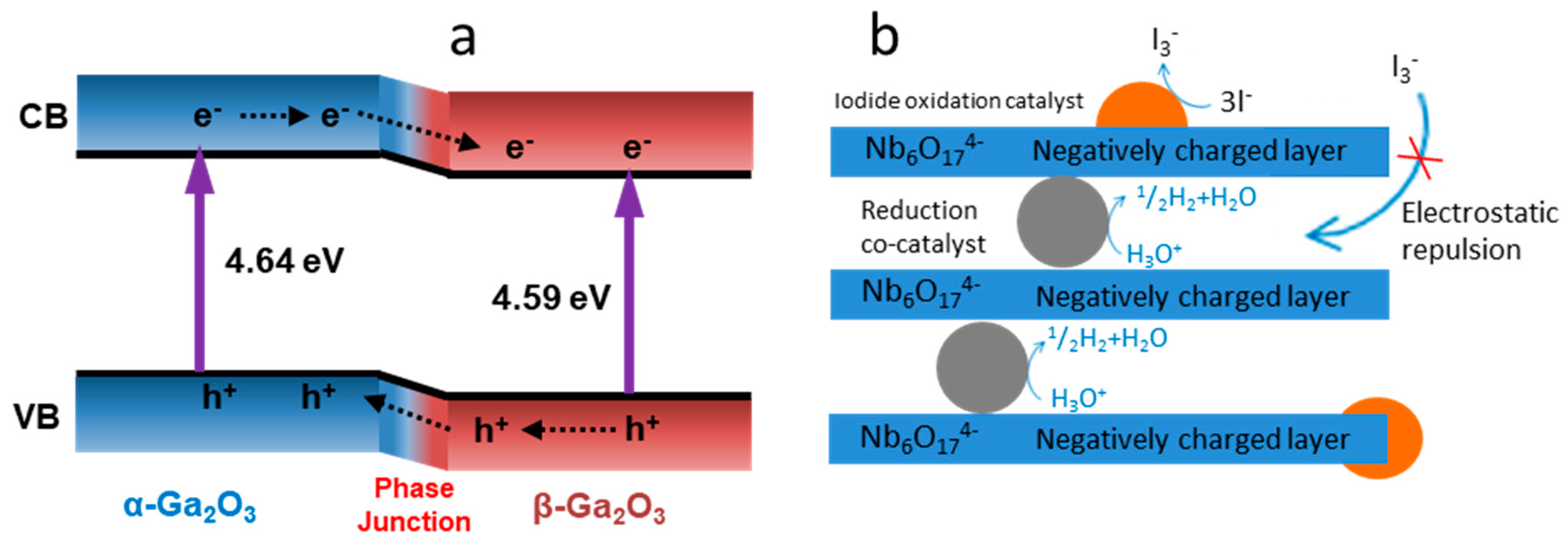
- Wang, X.; Xu, Q.; Li, M.; Shen, S.; Wang, X.; Wang, Y.; Feng, Z.; Shi, J.; Han, H.; Li, C. Photocatalytic overall water splitting promoted by an α–β phase junction on Ga2O3. Angew. Chem. Int. Ed. 2012, 51, 13089–13092. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Peng, Y.-K.; Hu, L.; Zheng, J.; Prabhakaran, D.; Wu, S.; Puchtler, T.J.; Li, M.; Wong, K.-Y.; Taylor, R.A.; et al. Photocatalytic water splitting by N-TiO2 on MgO (111) with exceptional quantum efficiencies at elevated temperatures. Nat. Commun. 2019, 10, 4421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Ren, B.; Chang, S.; Mi, W.; He, J.; Wang, W. Achieving effective control of the photocatalytic performance for CoFe2O4/MoS2 heterojunction via exerting external magnetic fields. Mater. Lett. 2020, 260, 126979. [Google Scholar] [CrossRef]
- Gao, W.; Lu, J.; Zhang, S.; Zhang, X.; Wang, Z.; Qin, W.; Wang, J.; Zhou, W.; Liu, H.; Sang, Y. Suppressing Photoinduced Charge Recombination via the Lorentz Force in a Photocatalytic System. Adv. Sci. 2019, 6, 1901244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Pei, Q.; Wang, R.; Zhou, Y.; Zhang, Z.; Cao, Q.; Wang, D.; Mi, W.; Du, Y. Enhanced Photocatalytic Performance through Magnetic Field Boosting Carrier Transport. ACS Nano 2018, 12, 3351–3359. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yates, J.T. Band Bending in Semiconductors: Chemical and Physical Consequences at Surfaces and Interfaces. Chem. Rev. 2012, 112, 5520–5551. [Google Scholar] [CrossRef]
- Yoshida, Y.; Matsuoka, M.; Moon, S.; Mametsuka, H.; Suzuki, E.; Anpo, M. Photocatalytic decomposition of liquid-water on the Pt-loaded TiO2 catalysts: Effects of the oxidation states of Pt species on the photocatalytic reactivity and the rate of the back reaction. Res. Chem. Intermed. 2000, 26, 567–574. [Google Scholar] [CrossRef]
- Sanap, K.K.; Varma, S.; Dalavi, D.; Patil, P.; Waghmode, S.; Bharadwaj, S. Variation in noble metal morphology and its impact on functioning of hydrogen mitigation catalyst. Int. J. Hydrog. Energy 2011, 36, 10455–10467. [Google Scholar] [CrossRef]
- Berto, T.F.; Sanwald, K.E.; Byers, J.P.; Browning, N.D.; Gutiérrez, O.Y.; Lercher, J.A. Enabling overall water splitting on photocatalysts by CO-covered noble metal co-catalysts. J. Phys. Chem. Lett. 2016, 7, 4358–4362. [Google Scholar] [CrossRef]
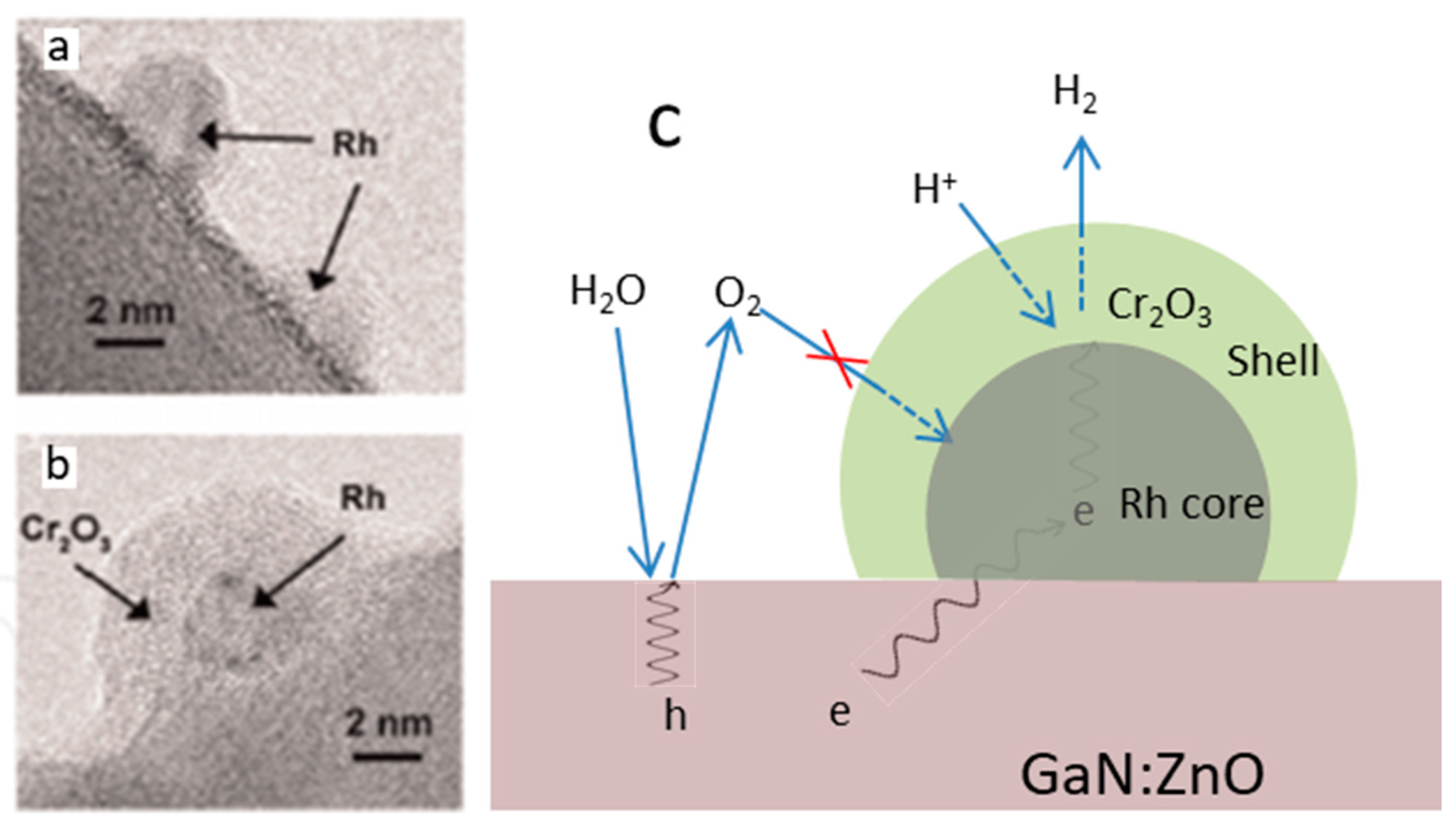
- Dionigi, F.; Vesborg, P.C.; Pedersen, T.; Hansen, O.; Dahl, S.; Xiong, A.; Maeda, K.; Domen, K.; Chorkendorff, I. Suppression of the water splitting back reaction on GaN:ZnO photocatalysts loaded with core/shell cocatalysts, investigated using a μ-reactor. J. Catal. 2012, 292, 26–31. [Google Scholar] [CrossRef]
- Yoshida, M.; Takanabe, K.; Maeda, K.; Ishikawa, A.; Kubota, J.; Sakata, Y.; Ikezawa, Y.; Domen, K. Role and function of noble-metal/Cr-layer core/shell structure cocatalysts for photocatalytic overall water splitting studied by model electrodes. J. Phys. Chem. C 2009, 113, 10151–10157. [Google Scholar] [CrossRef]
- Takata, T.; Pan, C.; Nakabayashi, M.; Shibata, N.; Domen, K. Fabrication of a core–shell-type photocatalyst via photodeposition of group IV and V transition metal oxyhydroxides: An effective surface modification method for overall water splitting. J. Am. Chem. Soc. 2015, 137, 9627–9634. [Google Scholar] [CrossRef] [PubMed]
- Abe, R.; Sayama, K.; Arakawa, H. Significant effect of iodide addition on water splitting into H2 and O2 over Pt-loaded TiO2 photocatalyst: Suppression of backward reaction. Chem. Phys. Lett. 2003, 371, 360–364. [Google Scholar] [CrossRef]
- Nadeem, M.; Idriss, H. Photo-thermal reactions of ethanol over Ag/TiO2 catalysts. The role of silver plasmon resonance in the reaction kinetics. Chem. Commun. 2018, 54, 5197–5200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Hisatomi, T.; Jia, Q.; Tokudome, H.; Zhong, M.; Wang, C.; Pan, Z.; Takata, T.; Nakabayashi, M.; Shibata, N.; et al. Scalable water splitting on particulate photocatalyst sheets with a solar-to-hydrogen energy conversion efficiency exceeding 1%. Nat. Mater. 2016, 15, 611–615. [Google Scholar] [CrossRef] [PubMed]
- Standard Test Methods for Measurement of Electrical Performance and Spectral Response of Nonconcentrator Multijunction Photovoltaic Cells and Modules. In E2236-10(2019); ASTM International: West Conshohocken, PA, USA, 2019.
- Ipek, B.; Uner, D. Artificial photosynthesis from a chemical engineering perspective. In Artificial Photosynthesis; IntechOpen: London, UK, 2012; pp. 13–36. [Google Scholar]
- Skillen, N.; Adams, M.; McCullagh, C.; Ryu, S.Y.; Fina, F.; Hoffmann, M.R.; Irvine, J.T.S.; Robertson, P.K.J. application of a novel fluidised photo reactor under UV–Visible and natural solar irradiation in the photocatalytic generation of hydrogen. Chem. Eng. J. 2016, 286, 610–621. [Google Scholar] [CrossRef] [Green Version]
- Reilly, K.; Wilkinson, D.P.; Taghipour, F. Photocatalytic water splitting in a fluidized bed system: Computational modeling and experimental studies. Appl. Energy 2018, 222, 423–436. [Google Scholar] [CrossRef]
- Chen, D.; Li, F.; Ray, A.K. Effect of mass transfer and catalyst layer thickness on photocatalytic reaction. AIChE J. 2000, 46, 1034–1045. [Google Scholar] [CrossRef]
- Wang, Q.; Hisatomi, T.; Katayama, M.; Takata, T.; Minegishi, T.; Kudo, A.; Yamada, T.; Domen, K. Particulate photocatalyst sheets for Z-scheme water splitting: Advantages over powder suspension and photoelectrochemical systems and future challenges. Faraday Discuss. 2017, 197, 491–504. [Google Scholar] [CrossRef]
- Paramelle, D.; Sadovoy, A.; Gorelik, S.; Free, P.; Hobley, J.; Fernig, D.G. A rapid method to estimate the concentration of citrate capped silver nanoparticles from UV-visible light spectra. Analyst 2014, 139, 4855–4861. [Google Scholar] [CrossRef]
- Huang, X.; El-Sayed, M.A. Gold nanoparticles: Optical properties and implementations in cancer diagnosis and photothermal therapy. J. Adv. Res. 2010, 1, 13–28. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.A.; Sinatra, L.; Oufi, M.; Bakr, O.M.; Idriss, H. Evidence of Plasmonic Induced Photocatalytic Hydrogen Production on Pd/TiO2 Upon Deposition on Thin Films of Gold. Catal. Lett. 2017, 147, 811–820. [Google Scholar] [CrossRef] [Green Version]
- Majeed, I.; Nadeem, M.A.; Hussain, E.; Badshah, A.; Gilani, R.; Nadeem, M.A. Effect of deposition method on metal loading and photocatalytic activity of Au/CdS for hydrogen production in water electrolyte mixture. Int. J. Hydrog. Energy 2017, 42, 3006–3018. [Google Scholar] [CrossRef]
- Mubeen, S.; Lee, J.; Singh, N.; Krämer, S.; Stucky, G.D.; Moskovits, M. An autonomous photosynthetic device in which all charge carriers derive from surface plasmons. Nat. Nanotechnol. 2013, 8, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Wolff, C.M.; Frischmann, P.D.; Schulze, M.; Bohn, B.J.; Wein, R.; Livadas, P.; Carlson, M.T.; Jäckel, F.; Feldmann, J.; Würthner, F.; et al. All-in-one visible-light-driven water splitting by combining nanoparticulate and molecular co-catalysts on CdS nanorods. Nat. Energy 2018, 3, 862–869. [Google Scholar] [CrossRef]
- Lyu, H.; Hisatomi, T.; Goto, Y.; Yoshida, M.; Higashi, T.; Katayama, M.; Takata, T.; Minegishi, T.; Nishiyama, H.; Yamada, T.; et al. An Al-doped SrTiO3 photocatalyst maintaining sunlight-driven overall water splitting activity for over 1000 h of constant illumination. Chem. Sci. 2019, 10, 3196–3201. [Google Scholar] [CrossRef] [Green Version]
- Takata, T.; Jiang, J.; Sakata, Y.; Nakabayashi, M.; Shibata, N.; Nandal, V.; Seki, K.; Hisatomi, T.; Domen, K. Photocatalytic water splitting with a quantum efficiency of almost unity. Nature 2020, 581, 411–414. [Google Scholar] [CrossRef]
- Alsayegh, S.; Johnson, J.; Ohs, B.; Lohaus, J.; Wessling, M.J.I.J.o.H.E. Systematic optimization of H2 recovery from water splitting process using membranes and N2 diluent. Int. J. Hydrog. Energy 2017, 42, 6000–6011. [Google Scholar] [CrossRef]
- Alsayegh, S.; Johnson, J.; Wei, X.; Ohs, B.; Lohaus, J.; Wessling, M. CO2 aided H2 recovery from water splitting processes. Int. J. Hydrog. Energy 2017, 42, 21793–21805. [Google Scholar] [CrossRef]
- Kibria, M.G.; Chowdhury, F.A.; Zhao, S.; AlOtaibi, B.; Trudeau, M.L.; Guo, H.; Mi, Z. Visible light-driven efficient overall water splitting using p-type metal-nitride nanowire arrays. Nat. Commun. 2015, 6, 6797. [Google Scholar] [CrossRef] [Green Version]
- Takata, T.; Pan, C.; Domen, K. Design and Development of Oxynitride Photocatalysts for Overall Water Splitting under Visible Light Irradiation. ChemElectroChem 2016, 3, 31–37. [Google Scholar] [CrossRef]
- Zhang, C.; Chen, C.; Dong, H.; Shen, J.-R.; Dau, H.; Zhao, J. A synthetic Mn4Ca-cluster mimicking the oxygen-evolving center of photosynthesis. Science 2015, 348, 690–693. [Google Scholar] [CrossRef] [PubMed]
- Kanan, M.W.; Nocera, D.G. In Situ Formation of an Oxygen-Evolving Catalyst in Neutral Water Containing Phosphate and Co2+. Science 2008, 321, 1072–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, A.; Teramura, K.; Hosokawa, S.; Kominami, H.; Tanaka, T. Visible light-induced water splitting in an aqueous suspension of a plasmonic Au/TiO2 photocatalyst with metal co-catalysts. Chem. Sci. 2017, 8, 2574–2580. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Fan, F.; Chen, R.; An, H.; Feng, Z.; Li, C. Direct Imaging of Highly Anisotropic Photogenerated Charge Separations on Different Facets of a Single BiVO4 Photocatalyst. Angew. Chem. Int. Ed. 2015, 54, 9111–9114. [Google Scholar] [CrossRef]










| Photocatalysts | Preparation Method | Examples | Cocatalyst Loading | Examples | ||||
|---|---|---|---|---|---|---|---|---|
| Metal oxides | → | Molten salt (Flux) Solid state reactions Ammonia precipitation | → | SrTiO3:Al, SrTiO3:Rh,Sb, La2Ti2O7:Ba, NaTaO3, Ga2O3:Zn, BiYWO6, Bi1−xInxV1−xMoxO4 | → | Impregnation Photodeposition | → | NiOx, CoOx, IrO2, RuO2, Rh2−yCryO3 |
| Calcination under controlled atmosphere | → | MxOy | ||||||
| Metal (oxy) nitrides | → | Thermal nitridation of metal oxides using NH3 | → | Ge3N4, TaON:ZrO2, (Zn0.18Ga0.82)(N0.82O0.18), LaMg1/3Ta2/3O2N, CaTaO2N, Ta3N5, LaScxTa1−xO1−2xN2−2x, GaN:Mg/InGaN:Mg | → | Impregnation Photodeposition | → | RuO2, Rh2−yCryO3 |
| Metal-free photocatalysts | → | Thermal polymerization Electrochemical | → | g-C3N4, C-dot/g-C3N4 | ||||
| Semiconductor | Metal/Metal Oxide (wt.%) | Eg (eV) | H2 Rate (mmolh−1) | O2 Rate (mmolh−1) | AQY (%) | Ref. |
|---|---|---|---|---|---|---|
| La2Ti2O7:Ba(8.0 mol %) | Ni (2.0) | 3.2 | 5 | 2.5 | 50 (not given) | [49] |
| SrTiO3:Al(0.1 mol %) | RhxCryO3 (Rh = 0.1, Cr = 0.1) | 3.2 | 1.4 | 0.7 | 56 at 365 nm | [15,54] |
| SrTiO3:Al(0.1 mol %) | MoOy/RhCrOx (Mo = 0.03, Rh = 0.1, Cr = 0.1) | 3.2 | 1.8 | 0.9 | 69 at 365 nm | [54] |
| SrTiO3:Rh,Sb(0.5 & 2.0 wt.%) | IrO2 (3.0) | 3.2 | 4.4 | 1.9 | 0.1 at 420 nm | [50] |
| NaTaO3 | NiO (0.05) | 4.0 | 3.4 | 1.6 | 20 at 270 nm | [51] |
| Ga2O3:Zn(1.0 mol %) | Ni (1.0) | 4.4 | 4.1 | 2.2 | 20 at 270 nm | [56] |
| Ga2O3:Zn(3.0 mol %) | RhxCryO3 (Rh = 0.5, Cr = 1.5) | 4.4 | 3.2 | 1.6 | 71 at 254 nm | [57] |
| Ge3N4 | RuO2 (1.0) | 3.8 | 0.2 | 0.1 | 9 at 300 nm | [58] |
| Semiconductor | Metal Oxide (wt.% Unless Indicated) | Eg (eV) | H2 Rate (µmolh−1) | O2 Rate (µmolh−1) | AQY (%) | Ref. |
|---|---|---|---|---|---|---|
| Bi1−xInxV1−xMoxO4 | RuO2(3.0) | 2.5 | 17 | 7.8 | 3.2 at 420 nm | [52] |
| BiYWO6 | RuO2(1.0) | 2.7 | 4.1 | 1.8 | 0.17 at 420 nm | [53] |
| LaMg1/3Ta2/3O2N | RhCrOx (Rh = 0.5 Cr = 0.5) | - | 22 | 11 | 0.18 at 440 nm | [59] |
| TaON:ZrO2 (Zr/Ta = 0.1) | RuOx/Cr2O3/IrO2 (Ru = 3.0, Cr = 2.5, Ir/Ta = 0.04) | 2.5 | 6.7 × 10−3 | 2.3 × 10−3 | <0.1 at 420 nm | [60] |
| CoO | - | 2.6 | 1785 | 848 | 5% (STH) | [55] |
| (Zn0.18Ga0.82) (N0.82O0.18) | Rh2−yCryO3 (Rh = 2.5, Cr = 2.0) | 2.64 | 927 | 460 | 5.9 at 420 nm | [61] |
| GaN:Mg/InGaN (grown using MBE) | Rh/Cr2O3 (Not applicable) | 2.22 | 38 | 21 | 12.3 at 400 nm | [62] |
| CaTaO2N | RhCrOy (Rh = 0.5, Cr = 0.5) | 2.43 | 14 × 10−2 | 7 × 10−2 | 0.003 at 440 nm | [63] |
| LaSc0.5Ta0.5O2N | RhCrOy (Rh = 0.5, Cr = 0.5) | 2.1 | 2.4 × 10−3 | 1.2 × 10−3 | - | [64] |
| Ta3N5/KTaO3 (Ta3N5 = 1.4 wt.%) | Rh/Cr2O3 (Rh = 0.002, Cr = 0.004) | 2.1 | 6 × 10−3 | 3 × 10−3 | 0.25 at 400 nm | [94] |
| g-C3N4 | CoOx (1.0) | 2.8 | 8.5 | 3.5 | 0.3 at 405 nm | [65] |
| C3N4/C-dots | - | 2.74 | 46 | - | 16 at 420 nm | [66] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nadeem, M.A.; Khan, M.A.; Ziani, A.A.; Idriss, H. An Overview of the Photocatalytic Water Splitting over Suspended Particles. Catalysts 2021, 11, 60. https://doi.org/10.3390/catal11010060
Nadeem MA, Khan MA, Ziani AA, Idriss H. An Overview of the Photocatalytic Water Splitting over Suspended Particles. Catalysts. 2021; 11(1):60. https://doi.org/10.3390/catal11010060
Chicago/Turabian StyleNadeem, Muhammad Amtiaz, Mohd Adnan Khan, Ahmed Abdeslam Ziani, and Hicham Idriss. 2021. "An Overview of the Photocatalytic Water Splitting over Suspended Particles" Catalysts 11, no. 1: 60. https://doi.org/10.3390/catal11010060