
Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways


{kind=link}
Abstract
:1. Introduction
1.1. Brief History—Discovery
1.2. ERBB Family Members
1.3. EGFR Expression
1.4. Physiological Role of EGFR
1.5. EGFR Structure
1.6. EGFR Mutations
2. Signal Transduction
2.1. Ligand—EGF
2.2. EGFR Dimerization
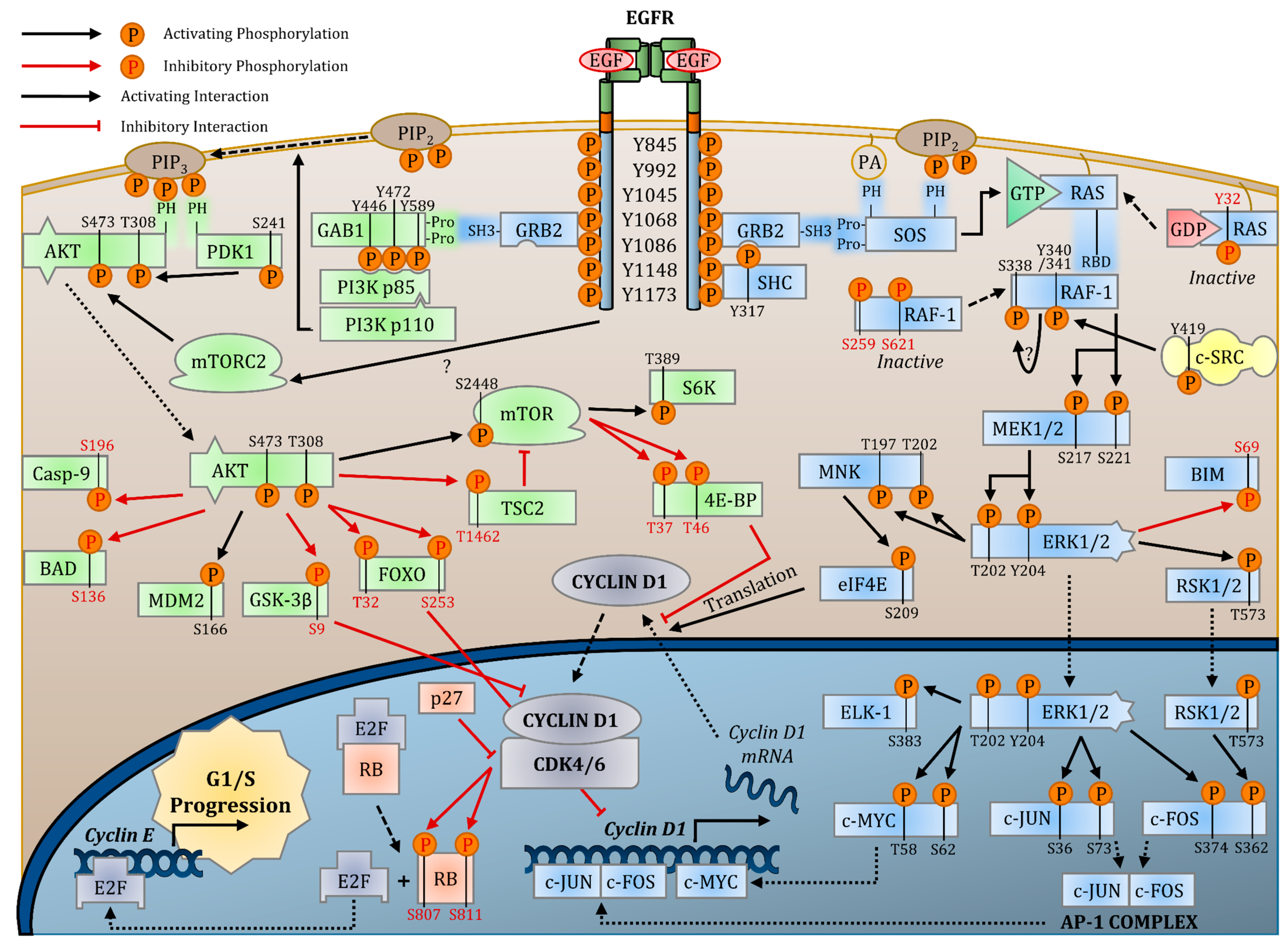
2.3. Signaling Pathways
2.3.1. EGFR Transautophosphorylation
2.3.2. RAS-RAF-MEK-ERK MAPK Pathway
2.3.3. PI3K-AKT-mTOR Pathway
2.3.4. PLC-γ1-PKC Pathway
2.3.5. SRC
2.3.6. Nuclear EGFR Signaling
2.4. EGFR-Targeted Therapies
2.5. Effects of EGFR Signaling on Cell Cycle Progression
2.5.1. EGFR Activation of the CDK4/6-CYCLIN D Complex
2.5.2. EGFR Inhibition of CDKi Activity
3. Conclusions
Acknowledgments
Conflicts of Interest
References
- Pines, G.; Köstler, W.J.; Yarden, Y. Oncogenic mutant forms of EGFR: Lessons in signal transduction and targets for cancer therapy. FEBS Lett. 2010, 584, 2699–2706. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S. Origins of growth factors: NGF and EGF. Ann. N. Y. Acad. Sci. 2004, 1038, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S. Isolation of a mouse submaxillary gland protein accelerating incisor eruption and eyelid opening in the new-born animal. J. Biol. Chem. 1962, 237, 1555–1562. [Google Scholar] [PubMed]
- Cohen, S. The stimulation of epidermal proliferation by a specific protein (EGF). Dev. Biol. 1965, 12, 394–407. [Google Scholar] [CrossRef]
- Cohen, S.; Elliott, G.A. The stimulation of epidermal keratinization by a protein isolated from the submaxillary gland of the mouse. J. Investig. Dermatol. 1963, 40, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Carpenter, G.; King, L. Epidermal growth factor-receptor-protein kinase interactions. Co-purification of receptor and epidermal growth factor-enhanced phosphorylation activity. J. Biol. Chem. 1980, 255, 4834–4842. [Google Scholar] [PubMed]
- Cohen, S.; Ushiro, H.; Stoscheck, C.; Chinkers, M. A native 170,000 epidermal growth factor receptor-kinase complex from shed plasma membrane vesicles. J. Biol. Chem. 1982, 257, 1523–1531. [Google Scholar] [PubMed]
- Carpenter, G.; Lembach, K.J.; Morrison, M.M.; Cohen, S. Characterization of the binding of 125-I-labeled epidermal growth factor to human fibroblasts. J. Biol. Chem. 1975, 250, 4297–4304. [Google Scholar] [PubMed]
- Ushiro, H.; Cohen, S. Identification of phosphotyrosine as a product of epidermal growth factor-activated protein kinase in A-431 cell membranes. J. Biol. Chem. 1980, 255, 8363–8365. [Google Scholar] [PubMed]
- Downward, J.; Yarden, Y.; Mayes, E.; Scrace, G.; Totty, N.; Stockwell, P.; Ullrich, A.; Schlessinger, J.; Waterfield, M.D. Close similarity of epidermal growth factor receptor and v-erb-B oncogene protein sequences. Nature 1984, 307, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, A.; Coussens, L.; Hayflick, J.S.; Dull, T.J.; Gray, A.; Tam, A.W.; Lee, J.; Yarden, Y.; Libermann, T.A.; Schlessinger, J. Human epidermal growth factor receptor cDNA sequence and aberrant expression of the amplified gene in A431 epidermoid carcinoma cells. Nature 1984, 309, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Olofsson, B.; Pizon, V.; Zahraoui, A.; Tavitian, A.; Therwath, A. Structure and expression of the chicken epidermal growth factor receptor gene locus. Eur. J. Biochem. 1986, 160, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Gusterson, B.; Cowley, G.; Smith, J.A.; Ozanne, B. Cellular localisation of human epidermal growth factor receptor. Cell Biol. Int. Rep. 1984, 8, 649–658. [Google Scholar] [CrossRef]
- Cowley, G.P.; Smith, J.A.; Gusterson, B.A. Increased EGF receptors on human squamous carcinoma cell lines. Br. J. Cancer 1986, 53, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Gusterson, B.; Cowley, G.; McIlhinney, J.; Ozanne, B.; Fisher, C.; Reeves, B. Evidence for increased epidermal growth factor receptors in human sarcomas. Int. J. Cancer 1985, 36, 689–693. [Google Scholar] [PubMed]
- Veale, D.; Ashcroft, T.; Marsh, C.; Gibson, G.J.; Harris, A.L. Epidermal growth factor receptors in non-small cell lung cancer. Br. J. Cancer 1987, 55, 513–516. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.J.; Bigner, S.H.; Bigner, D.D.; Kinzler, K.W.; Hamilton, S.R.; Vogelstein, B. Increased expression of the epidermal growth factor receptor gene in malignant gliomas is invariably associated with gene amplification. Proc. Natl. Acad. Sci. USA 1987, 84, 6899–6903. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.; Sainsbury, C.; Needham, G.; Farndon, J.; Malcolm, A.; Harris, A. Epidermal-growth-factor receptor status as predictor of early recurrence of and death from breast cancer. Lancet 1987, 329, 1398–1402. [Google Scholar] [CrossRef]
- Velu, T.; Beguinot, L.; Vass, W.; Willingham, M.; Merlino, G.; Pastan, I.; Lowy, D. Epidermal-growth-factor-dependent transformation by a human EGF receptor proto-oncogene. Science 1987, 238, 1408–1410. [Google Scholar] [CrossRef] [PubMed]
- Di Fiore, P.P.; Pierce, J.H.; Fleming, T.P.; Hazan, R.; Ullrich, A.; King, C.R.; Schlessinger, J.; Aaronson, S.A. Overexpression of the human EGF receptor confers an EGF-dependent transformed phenotype to NIH 3T3 cells. Cell 1987, 51, 1063–1070. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Tzahar, E.; Waterman, H.; Chen, X.; Levkowitz, G.; Karunagaran, D.; Lavi, S.; Ratzkin, B.J.; Yarden, Y. A hierarchical network of interreceptor interactions determines signal transduction by Neu differentiation factor/neuregulin and epidermal growth factor. Mol. Cell. Biol. 1996, 16, 5276–5287. [Google Scholar] [CrossRef] [PubMed]
- Graus-Porta, D.; Beerli, R.R.; Daly, J.M.; Hynes, N.E. ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J. 1997, 16, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- Holbro, T.; Beerli, R.R.; Maurer, F.; Koziczak, M.; Barbas, C.F.; Hynes, N.E. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc. Natl. Acad. Sci. USA 2003, 100, 8933–8938. [Google Scholar] [CrossRef] [PubMed]
- Burgess, A.W.; Cho, H.S.; Eigenbrot, C.; Ferguson, K.M.; Garrett, T.P.J.; Leahy, D.J.; Lemmon, M.A.; Sliwkowski, M.X.; Ward, C.W.; Yokoyama, S.; et al. An Open-and-Shut Case? Recent Insights into the Activation of EGF/ErbB Receptors. Mol. Cell 2003, 12, 541–552. [Google Scholar] [CrossRef]
- Tomas, A.; Futter, C.E.; Eden, E.R. EGF receptor trafficking: Consequences for signaling and cancer. Trends Cell Biol. 2014, 24, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Aroian, R.V.; Koga, M.; Mendel, J.E.; Ohshima, Y.; Sternberg, P.W. The let-23 gene necessary for Caenorhabditis elegans vulval induction encodes a tyrosine kinase of the EGF receptor subfamily. Nature 1990, 348, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Wasserman, J.D.; Freeman, M. Control of EGF receptor activation in Drosophila. Trends Cell Biol. 1997, 7, 431–436. [Google Scholar] [CrossRef]
- Lusk, J.; Lam, V.; Tolwinski, N. Epidermal Growth Factor Pathway Signaling in Drosophila Embryogenesis: Tools for Understanding Cancer. Cancers 2017, 9, 16. [Google Scholar] [CrossRef] [PubMed]
- Davies, R.L.; Grosse, V.A.; Kucherlapati, R.; Bothwell, M. Genetic analysis of epidermal growth factor action: Assignment of human epidermal growth factor receptor gene to chromosome 7. Proc. Natl. Acad. Sci. USA 1980, 77, 4188–4192. [Google Scholar] [CrossRef] [PubMed]
- Kondo, I.; Shimizu, N. Mapping of the human gene for epidermal growth factor receptor (EGFR) on the p13 leads to q22 region of chromosome 7. Cytogenet. Cell Genet. 1983, 35, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, G.; Cohen, S. Epidermal growth factor. Annu. Rev. Biochem. 1979, 48, 193–216. [Google Scholar] [CrossRef] [PubMed]
- Gullick, W.J.; Marsden, J.J.; Whittle, N.; Ward, B.; Bobrow, L.; Waterfield, M.D. Expression of Epidermal Growth Factor Receptors on Human Cervical, Ovarian, and Vulval Carcinomas. Cancer Res. 1986, 46, 285–292. [Google Scholar] [PubMed]
- Clark, A.J.; Ishii, S.; Richert, N.; Merlino, G.T.; Pastan, I. Epidermal growth factor regulates the expression of its own receptor. Proc. Natl. Acad. Sci. USA 1985, 82, 8374–8378. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, R.; Merlino, G.T.; Pastan, I. A transcription factor active on the epidermal growth factor receptor gene. Proc. Natl. Acad. Sci. USA 1988, 85, 5016–5020. [Google Scholar] [CrossRef] [PubMed]
- Haley, J.; Whittle, N.; Bennet, P.; Kinchington, D.; Ullrich, A.; Waterfield, M. The human EGF receptor gene: Structure of the 110 kb locus and identification of sequences regulating its transcription. Oncogene Res. 1987, 1, 375–396. [Google Scholar] [PubMed]
- Johnson, A.C.; Ishii, S.; Jinno, Y.; Pastan, I.; Merlino, G.T. Epidermal growth factor receptor gene promoter. J. Biol. Chem. 1988, 263, 5693–5699. [Google Scholar] [PubMed]
- Johnson, A.C. Activation of epidermal growth factor receptor gene transcription by phorbol 12-myristate 13-acetate is mediated by activator protein 2. J. Biol. Chem. 1996, 271, 3033–3038. [Google Scholar] [PubMed]
- Mialon, A.; Sankinen, M.; Söderström, H.; Junttila, T.T.; Holmström, T.; Koivusalo, R.; Papageorgiou, A.C.; Johnson, R.S.; Hietanen, S.; Elenius, K.; et al. DNA topoisomerase I is a cofactor for c-Jun in the regulation of epidermal growth factor receptor expression and cancer cell proliferation. Mol. Cell. Biol. 2005, 25, 5040–5051. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.A.; Lakshmanan, J. Metabolism and effects of epidermal growth factor and related growth factors in mammals. Endocr. Rev. 1990, 11, 418–442. [Google Scholar] [CrossRef] [PubMed]
- Chia, C.M.; Winston, R.M.; Handyside, A.H. EGF, TGF-alpha and EGFR expression in human preimplantation embryos. Development 1995, 121, 299–307. [Google Scholar] [PubMed]
- Roskoski, R. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef] [PubMed]
- Olayioye, M.A.; Neve, R.M.; Lane, H.A.; Hynes, N.E. The ErbB signaling network: Receptor heterodimerization in development and cancer. EMBO J. 2000, 19, 3159–3167. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.D.; Bork, P. Epidermal growth factor-like modules. Curr. Opin. Struct. Biol. 1993, 3, 385–392. [Google Scholar] [CrossRef]
- Threadgill, D.W.; Dlugosz, A.A.; Hansen, L.A.; Tennenbaum, T.; Lichti, U.; Yee, D.; LaMantia, C.; Mourton, T.; Herrup, K.; Harris, R.C.; et al. Targeted disruption of mouse EGF receptor: Effect of genetic background on mutant phenotype. Science 1995, 269, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.F.; Simon, H.; Chen, H.; Bates, B.; Hung, M.C.; Hauser, C. Requirement for neuregulin receptor erbB2 in neural and cardiac development. Nature 1995, 378, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.K.; Weichun, L.; Hauser, C.; Marchuk, Y.; Getman, D.; Kuo-Fen, L. Rescue of the cardiac defect in erbB2 mutant mice reveals essential roles of erbB2 in peripheral nervous system development. Neuron 1999, 23, 273–283. [Google Scholar] [CrossRef]
- Lin, W.; Sanchez, H.B.; Deerinck, T.; Morris, J.K.; Ellisman, M.; Lee, K.F. Aberrant development of motor axons and neuromuscular synapses in erbB2-deficient mice. Proc. Natl. Acad. Sci. USA 2000, 97, 1299–1304. [Google Scholar] [CrossRef] [PubMed]
- Erickson, S.L.; O’Shea, K.S.; Ghaboosi, N.; Loverro, L.; Frantz, G.; Bauer, M.; Lu, L.H.; Moore, M.W. ErbB3 is required for normal cerebellar and cardiac development: A comparison with ErbB2-and heregulin-deficient mice. Development 1997, 124, 4999–5011. [Google Scholar] [CrossRef] [PubMed]
- Riethmacher, D.; Sonnenberg-Riethmacher, E.; Brinkmann, V.; Yamaai, T.; Lewin, G.R.; Birchmeier, C. Severe neuropathies in mice with targeted mutations in the ErbB3 receptor. Nature 1997, 389, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Golding, J.P.; Trainor, P.; Krumlauf, R.; Gassmann, M. Defects in pathfinding by cranial neural crest cells in mice lacking the Neuregulin receptor ErbB4. Nat. Cell Biol. 2000, 2, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Jones, F.E.; Golding, J.P.; Gassmann, M. ErbB4 signaling during breast and neural development: Novel genetic models reveal unique ErbB4 activities. Cell Cycle 2003, 2, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Gassmann, M.; Casagranda, F.; Orioli, D.; Simon, H.; Lai, C.; Klein, R.; Lemke, G. Aberrant neural and cardiac development in mice lacking the ErbB4 neuregulin receptor. Nature 1995, 378, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Basketter, D.A.; Couchman, J.R.; Rees, D.A. Distribution and number of epidermal growth factor receptors in skin is related to epithelial cell growth. Dev. Biol. 1983, 100, 506–512. [Google Scholar] [CrossRef]
- Zieske, J.D.; Wasson, M. Regional variation in distribution of EGF receptor in developing and adult corneal epithelium. J. Cell Sci. 1993, 106 (Pt 1), 145–152. [Google Scholar] [PubMed]
- Fowler, K.J.; Walker, F.; Alexander, W.; Hibbs, M.L.; Nice, E.C.; Bohmer, R.M.; Mann, G.B.; Thumwood, C.; Maglitto, R.; Danks, J.A. A mutation in the epidermal growth factor receptor in waved-2 mice has a profound effect on receptor biochemistry that results in impaired lactation. Proc. Natl. Acad. Sci. USA 1995, 92, 1465–1469. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Paterson, A.J.; Chin, E.; Nabell, L.M.; Kudlow, J.E. Targeted expression of a dominant negative epidermal growth factor receptor in the mammary gland of transgenic mice inhibits pubertal mammary duct development. Mol. Endocrinol. 1997, 11, 1766–1781. [Google Scholar] [CrossRef] [PubMed]
- Wiesen, J.F.; Young, P.; Werb, Z.; Cunha, G.R. Signaling through the stromal epidermal growth factor receptor is necessary for mammary ductal development. Development 1999, 126, 335–344. [Google Scholar] [PubMed]
- Yamada, M.; Ikeuchi, T.; Hatanaka, H. The neurotrophic action and signalling of epidermal growth factor. Prog. Neurobiol. 1997, 51, 19–37. [Google Scholar] [CrossRef]
- Liu, B.; Neufeld, A.H. Activation of epidermal growth factor receptors in astrocytes: From development to neural injury. J. Neurosci. Res. 2007, 85, 3523–3529. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Pinilla, F.; Knauer, D.J.; Nieto-Sampedro, M. Epidermal growth factor receptor immunoreactivity in rat brain. Development and cellular localization. Brain Res. 1988, 438, 385–390. [Google Scholar] [CrossRef]
- Ciccolini, F.; Mandl, C.; Hölzl-Wenig, G.; Kehlenbach, A.; Hellwig, A. Prospective isolation of late development multipotent precursors whose migration is promoted by EGFR. Dev. Biol. 2005, 284, 112–125. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Sampedro, M.; Gomez-Pinilla, F.; Knauer, D.J.; Broderick, J.T. Epidermal growth factor receptor immunoreactivity in rat brain astrocytes. Response to injury. Neurosci. Lett. 1988, 91, 276–282. [Google Scholar] [CrossRef]
- Carpenter, C.D.; Ingraham, H.A.; Cochet, C.; Walton, G.M.; Lazar, C.S.; Sowadski, J.M.; Rosenfeld, M.G.; Gill, G.N. Structural analysis of the transmembrane domain of the epidermal growth factor receptor. J. Biol. Chem. 1991, 266, 5750–5755. [Google Scholar] [PubMed]
- Morrow, M.R.; Grant, C.W.M. The EGF receptor transmembrane domain: Peptide-peptide interactions in fluid bilayer membranes. Biophys. J. 2000, 79, 2024–2032. [Google Scholar] [CrossRef]
- Cymer, F.; Schneider, D. Transmembrane helix-helix interactions involved in ErbB receptor signaling. Cell Adhes. Migr. 2010, 4, 299–312. [Google Scholar] [CrossRef]
- Tanner, K.G.; Kyte, J. Dimerization of the extracellular domain of the receptor for epidermal growth factor containing the membrane-spanning segment in response to treatment with epidermal growth factor. J. Biol. Chem. 1999, 274, 35985–35990. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.A.; Tynan, J.A.; Hart, K.C.; Meyer, A.N.; Robertson, S.C.; Donoghue, D.J. Rotational coupling of the transmembrane and kinase domains of the Neu receptor tyrosine kinase. Mol. Biol Cell 2000, 11, 3589–3599. [Google Scholar] [CrossRef] [PubMed]
- Walton, G.M.; Chen, W.S.; Rosenfeld, M.G.; Gill, G.N. Analysis of deletions of the carboxyl terminus of the epidermal growth factor receptor reveals self-phosphorylation at tyrosine 992 and enhanced in vivo tyrosine phosphorylation of cell substrates. J. Biol. Chem. 1990, 265, 1750–1754. [Google Scholar] [PubMed]
- Stamos, J.; Sliwkowski, M.X.; Eigenbrot, C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 2002, 277, 46265–46272. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Gureasko, J.; Shen, K.; Cole, P.A.; Kuriyan, J. An Allosteric Mechanism for Activation of the Kinase Domain of Epidermal Growth Factor Receptor. Cell 2006, 125, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Vivanco, I.; Beroukhim, R.; Huang, J.H.Y.; Feng, W.L.; DeBiasi, R.M.; Yoshimoto, K.; King, J.C.; Nghiemphu, P.; Yuza, Y.; et al. Epidermal growth factor receptor activation in glioblastoma through novel missense mutations in the extracellular domain. PLoS Med. 2006, 3, e485. [Google Scholar] [CrossRef] [PubMed]
- Barber, T.D.; Vogelstein, B.; Kinzler, K.W.; Velculescu, V.E. Somatic Mutations of EGFR in Colorectal Cancers and Glioblastomas. N. Engl. J. Med. 2004, 351, 2883. [Google Scholar] [CrossRef] [PubMed]
- Cappuzzo, F.; Finocchiaro, G.; Rossi, E.; Jänne, P.A.; Carnaghi, C.; Calandri, C.; Bencardino, K.; Ligorio, C.; Ciardiello, F.; Pressiani, T.; et al. EGFR FISH assay predicts for response to cetuximab in chemotherapy refractory colorectal cancer patients. Ann. Oncol. 2008, 19, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Fujino, S.; Enokibori, T.; Tezuka, N.; Asada, Y.; Inoue, S.; Kato, H.; Mori, A. A comparison of epidermal growth factor receptor levels and other prognostic parameters in non-small cell lung cancer. Eur. J. Cancer 1996, 32, 2070–2074. [Google Scholar] [CrossRef]
- Endres, N.F.; Barros, T.; Cantor, A.J.; Kuriyan, J. Emerging concepts in the regulation of the EGF receptor and other receptor tyrosine kinases. Trends Biochem. Sci. 2014, 39, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Red Brewer, M.; Yun, C.-H.; Lai, D.; Lemmon, M.A.; Eck, M.J.; Pao, W. Mechanism for activation of mutated epidermal growth factor receptors in lung cancer. Proc. Natl. Acad. Sci. USA 2013, 110, E3595–E3604. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.K.; Cvrljevic, A.N.; Johns, T.G. The epidermal growth factor receptor variant III (EGFRvIII): Where wild things are altered. FEBS J. 2013, 280, 5350–5370. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.H.H.; Furnari, F.B.; Cavenee, W.K.; Bögler, O. Epidermal growth factor receptor signaling intensity determines intracellular protein interactions, ubiquitination, and internalization. Proc. Natl. Acad. Sci. USA 2003, 100, 6505–6510. [Google Scholar] [CrossRef] [PubMed]
- Paez, J.G.; Jänne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Shigematsu, H.; Lin, L.; Takahashi, T.; Nomura, M.; Suzuki, M.; Wistuba, I.I.; Fong, K.M.; Lee, H.; Toyooka, S.; Shimizu, N.; et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J. Natl. Cancer Inst. 2005, 97, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Carey, K.D.; Garton, A.J.; Romero, M.S.; Kahler, J.; Thomson, S.; Ross, S.; Park, F.; Haley, J.D.; Gibson, N.; Sliwkowski, M.X. Kinetic analysis of epidermal growth factor receptor somatic mutant proteins shows increased sensitivity to the epidermal growth factor receptor tyrosine kinase inhibitor, erlotinib. Cancer Res. 2006, 66, 8163–8171. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.H.; Boggon, T.J.; Li, Y.; Woo, M.S.; Greulich, H.; Meyerson, M.; Eck, M.J. Structures of Lung Cancer-Derived EGFR Mutants and Inhibitor Complexes: Mechanism of Activation and Insights into Differential Inhibitor Sensitivity. Cancer Cell 2007, 11, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Petri, E.T.; Halmos, B.; Boggon, T.J. Structure and clinical relevance of the epidermal growth factor receptor in human cancer. J. Clin. Oncol. 2008, 26, 1742–1751. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, H.; Park, E.; Yun, C.-H.; Sng, N.J.; Lucena-Araujo, A.R.; Yeo, W.-L.; Huberman, M.S.; Cohen, D.W.; Nakayama, S.; Ishioka, K.; et al. Structural, biochemical, and clinical characterization of epidermal growth factor receptor (EGFR) exon 20 insertion mutations in lung cancer. Sci. Transl. Med. 2013, 5, 216ra177. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Stiegler, A.L.; Boggon, T.J.; Kobayashi, S.; Halmos, B. EGFR-mutated lung cancer: A paradigm of molecular oncology. Oncotarget 2010, 1, 497–514. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.-H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.-K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef] [PubMed]
- Inukai, M.; Toyooka, S.; Ito, S.; Asano, H.; Ichihara, S.; Soh, J.; Suehisa, H.; Ouchida, M.; Aoe, K.; Aoe, M.; et al. Presence of epidermal growth factor receptor gene T790M mutation as a minor clone in non-small cell lung cancer. Cancer Res. 2006, 66, 7854–7858. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.V.; Blagoev, B.; Gnad, F.; Macek, B.; Kumar, C.; Mortensen, P.; Mann, M. Global, In Vivo, and Site-Specific Phosphorylation Dynamics in Signaling Networks. Cell 2006, 127, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Waters, K.M.; Liu, T.; Quesenberry, R.D.; Willse, A.R.; Bandyopadhyay, S.; Kathmann, L.E.; Weber, T.J.; Smith, R.D.; Wiley, H.S.; Thrall, B.D. Network analysis of epidermal growth factor signaling using integrated genomic, proteomic and phosphorylation data. PLoS ONE 2012, 7, e34515. [Google Scholar] [CrossRef] [PubMed]
- Oda, K.; Matsuoka, Y.; Funahashi, A.; Kitano, H. A comprehensive pathway map of epidermal growth factor receptor signaling. Mol. Syst. Biol. 2005, 1, 2005.0010. [Google Scholar] [CrossRef] [PubMed]
- Conte, A.; Sigismund, S. The Ubiquitin Network in the Control of EGFR Endocytosis and Signaling. Prog. Mol. Biol. Transl. Sci. 2016. [Google Scholar] [CrossRef]
- Sigismund, S.; Woelk, T.; Puri, C.; Maspero, E.; Tacchetti, C.; Transidico, P.; Di Fiore, P.P.; Polo, S. Clathrin-independent endocytosis of ubiquitinated cargos. Proc. Natl. Acad. Sci. USA 2005, 102, 2760–2765. [Google Scholar] [CrossRef] [PubMed]
- Kasselberg, A.G.; Orth, D.N.; Gray, M.E.; Stahlman, M.T. Immunocytochemical localization of human epidermal growth factor/urogastrone in several human tissues. J. Histochem. Cytochem. 1985, 33, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Edwin, F.; Wiepz, G.J.; Singh, R.; Peet, C.R.; Chaturvedi, D.; Bertics, P.J.; Patel, T.B. A historical perspective of the EGF receptor and related systems. Methods Mol. Biol. 2006, 327, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.B.; Harris, R.C. Autocrine, paracrine and juxtacrine signaling by EGFR ligands. Cell Signal. 2005, 17, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Carpenter, G.; Coffey, R.J. EGF receptor ligands: Recent advances. F1000Research 2016, 5, 2270. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; De Luca, A.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; Carotenuto, A.; De Feo, G.; Caponigro, F.; Salomon, D.S. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006, 366, 2–16. [Google Scholar] [CrossRef] [PubMed]
- Carraway, K.L.; Weber, J.L.; Unger, M.J.; Ledesma, J.; Yu, N.; Gassmann, M.; Lai, C. Neuregulin-2, a new ligand of ErbB3/ErbB4-receptor tyrosine kinases. Nature 1997, 387, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Riese, D.J.; Gilbert, W.; Stern, D.F.; McMahan, U.J. Ligands for ErbB-family receptors encoded by a neuregulin-like gene. Nature 1997, 387, 509–512. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.L.; Dunn, E.F.; Harari, P.M. Understanding resistance to EGFR inhibitors-impact on future treatment strategies. Nat. Rev. Clin. Oncol. 2010, 7, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Sliwkowski, M.X.; Mark, M.; Frantz, G.; Akita, R.; Sun, Y.; Hillan, K.; Crowley, C.; Brush, J.; Godowski, P.J. Neuregulin-3 (NRG3): A novel neural tissue-enriched protein that binds and activates ErbB4. Proc. Natl. Acad. Sci. USA 1997, 94, 9562–9567. [Google Scholar] [CrossRef] [PubMed]
- Roepstorff, K.; Grandal, M.V.; Henriksen, L.; Knudsen, S.L.J.; Lerdrup, M.; Grøvdal, L.; Willumsen, B.M.; Van Deurs, B. Differential effects of EGFR ligands on endocytic sorting of the receptor. Traffic 2009, 10, 1115–1127. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, C.; Fambrough, D.; Huard, C.; Diamonti, A.J.; Lander, E.S.; Cantley, L.C.; Carraway, K.L. Growth Factor-specific Signaling Pathway Stimulation and Gene Expression Mediated by ErbB Receptors. J. Biol. Chem. 2001, 276, 22685–22698. [Google Scholar] [CrossRef] [PubMed]
- Ebner, R.; Derynck, R. Epidermal growth factor and transforming growth factor-alpha: Differential intracellular routing and processing of ligand-receptor complexes. Cell Regul. 1991, 2, 599–612. [Google Scholar] [CrossRef] [PubMed]
- French, A.R.; Tadaki, D.K.; Niyogi, S.K.; Lauffenburger, D.A. Intracellular trafficking of epidermal growth factor family ligands is directly influenced by the pH sensitivity of the receptor/ligand interaction. J. Biol. Chem. 1995, 270, 4334–4340. [Google Scholar] [CrossRef] [PubMed]
- Waterman, H.; Sabanai, I.; Geiger, B.; Yarden, Y. Alternative intracellular routing of ErbB receptors may determine signaling potency. J. Biol. Chem. 1998, 273, 13819–13827. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.J.; Gilmore, J.L.; Foley, J.; Lemmon, M.A.; Riese, D.J. Functional selectivity of EGF family peptide growth factors: Implications for cancer. Pharmacol. Ther. 2009, 122, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sporn, M.B.; Roberts, A.B. Autocrine growth factors and cancer. Nature 1985, 313, 745–747. [Google Scholar] [CrossRef] [PubMed]
- De Larco, J.E.; Todaro, G.J. Growth factors from murine sarcoma virus-transformed cells. Proc. Natl. Acad. Sci. USA 1978, 75, 4001–4005. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Schlessinger, J. Epidermal growth factor induces rapid, reversible aggregation of the purified epidermal growth factor receptor. Biochemistry 1987, 26, 1443–1451. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Schlessinger, J. Self-phosphorylation of epidermal growth factor receptor: Evidence for a model of intermolecular allosteric activation. Biochemistry 1987, 26, 1434–1442. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, K.M.; Berger, M.B.; Mendrola, J.M.; Cho, H.S.; Leahy, D.J.; Lemmon, M.A. EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Mol. Cell 2003, 11, 507–517. [Google Scholar] [CrossRef]
- Jura, N.; Endres, N.F.; Engel, K.; Deindl, S.; Das, R.; Lamers, M.H.; Wemmer, D.E.; Zhang, X.; Kuriyan, J. Mechanism for Activation of the EGF Receptor Catalytic Domain by the Juxtamembrane Segment. Cell 2009, 137, 1293–1307. [Google Scholar] [CrossRef] [PubMed]
- Clayton, A.H.A.; Walker, F.; Orchard, S.G.; Henderson, C.; Fuchs, D.; Rothacker, J.; Rothacker, J.; Nice, E.C.; Burgess, A.W. Ligand-induced dimer-tetramer transition during the activation of the cell surface epidermal growth factor receptor-A multidimensional microscopy analysis. J. Biol. Chem. 2005, 280, 30392–30399. [Google Scholar] [CrossRef] [PubMed]
- Gadella, T.W.; Jovin, T.M. Oligomerization of epidermal growth factor receptors on A431 cells studied by time-resolved fluorescence imaging microscopy. A stereochemical model for tyrosine kinase receptor activation. J. Cell Biol. 1995, 129, 1543–1558. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.-H.; Maruyama, I.N. All EGF(ErbB) receptors have preformed homo- and heterodimeric structures in living cells. J. Cell Sci. 2008, 121, 3207–3217. [Google Scholar] [CrossRef] [PubMed]
- Chung, I.; Akita, R.; Vandlen, R.; Toomre, D.; Schlessinger, J.; Mellman, I. Spatial control of EGF receptor activation by reversible dimerization on living cells. Nature 2010, 464, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Bessman, N.J.; Bagchi, A.; Ferguson, K.M.; Lemmon, M.A. Complex Relationship between Ligand Binding and Dimerization in the Epidermal Growth Factor Receptor. Cell Rep. 2014, 9, 1306–1317. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Mi, L.Z.; Schürpf, T.; Walz, T.; Springer, T.A. Mechanisms for kinase-mediated dimerization of the epidermal growth factor receptor. J. Biol. Chem. 2012, 287, 38244–38253. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Hristova, K. Receptor tyrosine kinase transmembrane domains: Function, dimer structure and dimerization energetics. Cell Adhes. Migr. 2010, 4, 249–254. [Google Scholar] [CrossRef]
- Sato, K.-I. Cellular Functions Regulated by Phosphorylation of EGFR on Tyr845. Int. J. Mol. Sci. 2013, 14, 10761–10790. [Google Scholar] [CrossRef] [PubMed]
- Hunter, T.; Ling, N.; Cooper, J.A. Protein kinase C phosphorylation of the EGF receptor at a threonine residue close to the cytoplasmic face of the plasma membrane. Nature 1984, 311, 480–483. [Google Scholar] [CrossRef] [PubMed]
- Pawson, T. Specificity in Signal Transduction: From Phosphotyrosine-SH2 Domain Interactions to Complex Cellular Systems. Cell 2004, 116, 191–203. [Google Scholar] [CrossRef]
- Morrison, D.K. MAP Kinase Pathways. Cold Spring Harb. Perspect. Biol. 2012, 4, a011254. [Google Scholar] [CrossRef] [PubMed]
- Lewis, T.S.; Shapiro, P.S.; Ahn, N.G. Signal Transduction through MAP Kinase Cascades. Adv. Cancer Res. 1998, 74, 49–139. [Google Scholar] [CrossRef] [PubMed]
- Lowenstein, E.J.; Daly, R.J.; Batzer, A.G.; Li, W.; Margolis, B.; Lammers, R.; Ullrich, A.; Skolnik, E.Y.; Bar-Sagi, D.; Schlessinger, J. The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell 1992, 70, 431–442. [Google Scholar] [CrossRef]
- Buday, L.; Downward, J. Epidermal growth factor regulates p21ras through the formation of a complex of receptor, Grb2 adapter protein, and Sos nucleotide exchange factor. Cell 1993, 73, 611–620. [Google Scholar] [CrossRef]
- Jiang, X.; Huang, F.; Marusyk, A.; Sorkin, A. Grb2 regulates internalization of EGF receptors through clathrin-coated pits. Mol. Biol Cell 2003, 14, 858–870. [Google Scholar] [CrossRef] [PubMed]
- Batzer, A.G.; Rotin, D.; Ureña, J.M.; Skolnik, E.Y.; Schlessinger, J. Hierarchy of binding sites for Grb2 and Shc on the epidermal growth factor receptor. Mol. Cell. Biol. 1994, 14, 5192–5201. [Google Scholar] [CrossRef] [PubMed]
- Pelicci, G.; Lanfrancone, L.; Grignani, F.; McGlade, J.; Cavallo, F.; Forni, G.; Nicoletti, I.; Grignani, F.; Pawson, T.; Giuseppe Pelicci, P. A novel transforming protein (SHC) with an SH2 domain is implicated in mitogenic signal transduction. Cell 1992, 70, 93–104. [Google Scholar] [CrossRef]
- Okabayashi, Y.; Kido, Y.; Okutani, T.; Sugimoto, Y.; Sakaguchi, K.; Kasuga, M. Tyrosines 1148 and 1173 of activated human epidermal growth factor receptors are binding sites of Shc in intact cells. J. Biol. Chem. 1994, 269, 18674–18678. [Google Scholar] [PubMed]
- Sakaguchi, K.; Okabayashi, Y.; Kido, Y.; Kimura, S.; Matsumura, Y.; Inushima, K.; Kasuga, M. Shc phosphotyrosine-binding domain dominantly interacts with epidermal growth factor receptors and mediates Ras activation in intact cells. Mol. Endocrinol. 1998, 12, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Salcini, A.E.; McGlade, J.; Pelicci, G.; Nicoletti, I.; Pawson, T.; Pelicci, P.G. Formation of Shc-Grb2 complexes is necessary to induce neoplastic transformation by overexpression of Shc proteins. Oncogene 1994, 9, 2827–2836. [Google Scholar] [PubMed]
- Van der Geer, P.; Wiley, S.; Ka-Man Lai, V.; Olivier, J.P.; Gish, G.D.; Stephens, R.; Kaplan, D.; Shoelson, S.; Pawson, T. A conserved amino-terminal Shc domain binds to phosphotyrosine motifs in activated receptors and phosphopeptides. Curr. Biol. 1995, 5, 404–412. [Google Scholar] [CrossRef]
- Chardin, P.; Camonis, J.H.; Gale, N.W.; van Aelst, L.; Schlessinger, J.; Wigler, M.H.; Bar-Sagi, D. Human Sos1: A guanine nucleotide exchange factor for Ras that binds to GRB2. Science 1993, 260, 1338–1343. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Batzer, A.; Daly, R.; Yajnik, V.; Skolnik, E.; Chardin, P.; Bar-Sagi, D.; Margolis, B.; Schlessinger, J. Guanine-nucleotide-releasing factor hSos1 binds to Grb2 and links receptor tyrosine kinases to Ras signalling. Nature 1993, 363, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Egan, S.E.; Giddings, B.W.; Brooks, M.W.; Buday, L.; Sizeland, A.M.; Weinberg, R.A. Association of Sos Ras exchange protein with Grb2 is implicated in tyrosine kinase signal transduction and transformation. Nature 1993, 363, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Rozakis-Adcock, M.; Fernley, R.; Wade, J.; Pawson, T.; Bowtell, D. The SH2 and SH3 domains of mammalian Grb2 couple the EGF receptor to the Ras activator mSos1. Nature 1993, 363, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.A.; Schreiber, S.L. Grb2 SH3 binding to peptides from Sos: Evaluation of a general model for SH3-ligand interactions. Chem. Biol. 1995, 2, 53–60. [Google Scholar] [CrossRef]
- Boriack-Sjodin, P.A.; Margarit, S.M.; Bar-Sagi, D.; Kuriyan, J. The structural basis of the activation of Ras by Sos. Nature 1998, 394, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Brtva, T.R.; Drugan, J.K.; Ghosh, S.; Terrell, R.S.; Campbell-Burk, S.; Bell, R.M.; Der, C.J. Two distinct Raf domains mediate interaction with Ras. J. Biol. Chem. 1995, 270, 9809–9812. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Zang, M.; Waelde, C.A.; Wen, R.; Luo, Z. Phosphorylation of 338SSYY341 regulates specific interaction between Raf-1 and MEK1. J. Biol. Chem. 2002, 277, 44996–45003. [Google Scholar] [CrossRef] [PubMed]
- Zang, M.; Gong, J.; Luo, L.; Zhou, J.; Xiang, X.; Huang, W.; Luo, X.; Olbrot, M.; Peng, Y.; Chen, C.; et al. Characterization of Ser338 phosphorylation for Raf-1 activation. J. Biol. Chem. 2008, 283, 31429–31437. [Google Scholar] [CrossRef] [PubMed]
- Diaz, B.; Barnard, D.; Filson, A. Phosphorylation of Raf-1 serine 338-serine 339 is an essential regulatory event for Ras-dependent activation and biological signaling. Mol. Cell. Biol. 1997, 17, 4509–4516. [Google Scholar] [CrossRef] [PubMed]
- Fabian, J.R.; Daar, I.O.; Morrison, D.K. Critical tyrosine residues regulate the enzymatic and biological activity of Raf-1 kinase. Mol. Cell. Biol. 1993, 13, 7170–7179. [Google Scholar] [CrossRef] [PubMed]
- Bondzi, C.; Grant, S.; Krystal, G.W. A novel assay for the measurement of Raf-1 kinase activity. Oncogene 2000, 19, 5030–5033. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, N.; Premkumar Reddy, E. Signaling by dual specificity kinases. Oncogene 1998, 17, 1447–1455. [Google Scholar] [CrossRef] [PubMed]
- Sasaoka, T.; Langlois, W.J.; Leitner, J.W.; Draznin, B.; Olefsky, J.M. The Signaling Pathway Coupling Epidermal Growth Factor Receptors to Activation of p21ras. J. Biol. Chem. 1994, 269, 32621–32625. [Google Scholar] [PubMed]
- Lanzerstorfer, P.; Borgmann, D.; Schütz, G.; Winkler, S.M.; Höglinger, O.; Weghuber, J. Quantification and kinetic analysis of Grb2-EGFR interaction on micro-patterned surfaces for the characterization of EGFR-modulating substances. PLoS ONE 2014, 9, e92151. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Kim, I.S.; Park, J.B.; Lee, M.N.; Lee, H.Y.; Suh, P.-G.; Ryu, S.H. The phox homology domain of phospholipase D activates dynamin GTPase activity and accelerates EGFR endocytosis. Nat. Cell Biol. 2006, 8, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Du, G.; Skowronek, K.; Frohman, M.A.; Bar-Sagi, D. Phospholipase D2-generated phosphatidic acid couples EGFR stimulation to Ras activation by Sos. Nat. Cell Biol. 2007, 9, 706–712. [Google Scholar] [CrossRef] [PubMed]
- Soubeyran, P.; Kowanetz, K.; Szymkiewicz, I.; Langdon, W.Y.; Dikic, I. Cbl-CIN85-endophilin complex mediates ligand-induced downregulation of EGF receptors. Nature 2002, 416, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Waterman, H.; Katz, M.; Rubin, C.; Shtiegman, K.; Lavi, S.; Elson, A.; Jovin, T.; Yarden, Y. A mutant EGF-receptor defective in ubiquitylation and endocytosis unveils a role for Grb2 in negative signaling. EMBO J. 2002, 21, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Fukazawa, T.; Miyake, S.; Band, V.; Band, H. Tyrosine phosphorylation of Cbl upon epidermal growth factor (EGF) stimulation and its association with EGF receptor and downstream signaling proteins. J. Biol. Chem. 1996, 271, 14554–14559. [Google Scholar] [CrossRef] [PubMed]
- Sigismund, S.; Algisi, V.; Nappo, G.; Conte, A.; Pascolutti, R.; Cuomo, A.; Bonaldi, T.; Argenzio, E.; Verhoef, L.G.G.C.; Maspero, E.; et al. Threshold-controlled ubiquitination of the EGFR directs receptor fate. EMBO J. 2013, 32, 2140–2157. [Google Scholar] [CrossRef] [PubMed]
- Gureasko, J.; Galush, W.J.; Boykevisch, S.; Sondermann, H.; Bar-Sagi, D.; Groves, J.T.; Kuriyan, J. Membrane-dependent signal integration by the Ras activator Son of sevenless. Nat. Struct. Mol. Biol. 2008, 15, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Chang, J.S.; Park, S.K.; Hwang, J.I.; Ryu, S.H.; Suh, P.G. Direct interaction of SOS1 Ras exchange protein with the SH3 domain of phospholipase C-γ1. Biochemistry 2000, 39, 8674–8682. [Google Scholar] [CrossRef] [PubMed]
- Maertens, O.; Cichowski, K. An expanding role for RAS GTPase activating proteins (RAS GAPs) in cancer. Adv. Biol. Regul. 2014, 55, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Bunda, S.; Heir, P.; Srikumar, T.; Cook, J.D.; Burrell, K.; Kano, Y.; Lee, J.E.; Zadeh, G.; Raught, B.; Ohh, M. Src promotes GTPase activity of Ras via tyrosine 32 phosphorylation. Proc. Natl. Acad. Sci. USA 2014, 111, E3785–E3794. [Google Scholar] [CrossRef] [PubMed]
- Bunda, S.; Burrell, K.; Heir, P.; Zeng, L.; Alamsahebpour, A.; Kano, Y.; Raught, B.; Zhang, Z.-Y.; Zadeh, G.; Ohh, M. Inhibition of SHP2-mediated dephosphorylation of Ras suppresses oncogenesis. Nat. Commun. 2015, 6, 8859. [Google Scholar] [CrossRef] [PubMed]
- Kano, Y.; Cook, J.D.; Lee, J.E.; Ohh, M. New structural and functional insight into the regulation of Ras. Semin. Cell Dev. Biol. 2016, 58, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Findlay, G.M.; Pawson, T. How is SOS activated? Let us count the ways. Nat. Struct. Mol. Biol. 2008, 15, 538–540. [Google Scholar] [CrossRef] [PubMed]
- Hofer, F.; Fields, S.; Schneider, C.; Martin, G.S. Activated Ras interacts with the Ral guanine nucleotide dissociation stimulator. Proc. Natl. Acad. Sci. USA 1994, 91, 11089–11093. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Viciana, P.; Warne, P.H.; Dhand, R.; Vanhaesebroeck, B.; Gout, I.; Fry, M.J.; Waterfield, M.D.; Downward, J. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature 1994, 370, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Medarde, A.; Santos, E. Ras in cancer and developmental diseases. Genes Cancer 2011, 2, 344–358. [Google Scholar] [CrossRef] [PubMed]
- Hallberg, B.; Rayter, S.I.; Downward, J. Interaction of Ras and Raf in intact mammalian cells upon extracellular stimulation. J. Biol. Chem. 1994, 269, 3913–3916. [Google Scholar] [PubMed]
- O’Neill, A.K.; Niederst, M.J.; Newton, A.C. Suppression of survival signalling pathways by the phosphatase PHLPP. FEBS J. 2013, 280, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Rendueles, M.E.R.; Ricarte-Filho, J.C.; Untch, B.R.; Landa, I.; Knauf, J.A.; Voza, F.; Smith, V.E.; Ganly, I.; Taylor, B.S.; Persaud, Y.; et al. NF2 loss promotes oncogenic RAS-induced thyroid cancers via YAP-dependent transactivation of RAS proteins and sensitizes them to MEK inhibition. Cancer Discov. 2015, 5, 1178–1193. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.D.; Kariya, K.I.; Tamada, M.; Akasaka, K.; Shirouzu, M.; Yokoyama, S.; Kataoka, T. Cysteine-rich region of Raf-1 interacts with activator domain of post-translationally modified Ha-Ras. J. Biol. Chem. 1995, 270, 30274–30277. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.D.; Kariya, K.I.; Kotani, G.; Shirouzu, M.; Yokoyama, S.; Kataoka, T. Coassociation of Rap1A and Ha-Ras with Raf-1 N-terminal region interferes with Ras-dependent activation of Raf-1. J. Biol. Chem. 1997, 272, 11702–11705. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Diaz, B.; Marshall, M.S.; Avruch, J. An intact Raf zinc finger is required for optimal binding to processed Ras and for ras-dependent Raf activation in situ. Mol. Cell. Biol. 1997, 17, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Lane, A.; Yan, J.; McPherson, R.; Hancock, J.F. Activity of plasma membrane-recruited Raf-1 is regulated by Ras via the Raf zinc finger. J. Biol. Chem. 1997, 272, 20139–20145. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, A.S.; Kolch, W. Untying the regulation of the Raf-1 kinase. Arch. Biochem. Biophys. 2002, 404, 3–9. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Futreal, P.A. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Garnett, M.J.; Marais, R. Guilty as charged: B-RAF is a human oncogene. Cancer Cell 2004, 6, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Marais, R.; Marshall, C.J. Control of the ERK MAP kinase cascade by Ras and Raf. Cancer Surv. 1996, 27, 101–125. [Google Scholar] [PubMed]
- Mason, C.S.; Springer, C.J.; Cooper, R.G.; Superti-Furga, G.; Marshall, C.J.; Marais, R. Serine and tyrosine phosphorylations cooperate in Raf-1, but not B-Raf activation. EMBO J. 1999, 18, 2137–2148. [Google Scholar] [CrossRef] [PubMed]
- King, A.J.; Sun, H.; Diaz, B.; Barnard, D.; Miao, W.; Bagrodia, S.; Marshall, M.S. The protein kinase Pak3 positively regulates Raf-1 activity through phosphorylation of serine 338. Nature 1998, 396, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Chiloeches, A.; Mason, C.S.; Marais, R. S338 phosphorylation of Raf-1 is independent of phosphatidylinositol 3-kinase and Pak3. Mol. Cell. Biol. 2001, 21, 2423–2434. [Google Scholar] [CrossRef] [PubMed]
- Frost, J.A.; Steen, H.; Shapiro, P.; Lewis, T.; Ahn, N.; Shaw, P.E.; Cobb, M.H. Cross-cascade activation of ERKs and ternary complex factors by Rho family proteins. EMBO J. 1997, 16, 6426–6438. [Google Scholar] [CrossRef] [PubMed]
- Slack-Davis, J.K.; Eblen, S.T.; Zecevic, M.; Boerner, S.A.; Tarcsafalvi, A.; Diaz, H.B.; Marshall, M.S.; Weber, M.J.; Parsons, J.T.; Catling, A.D. PAK1 phosphorylation of MEK1 regulates fibronectin-stimulated MAPK activation. J. Cell Biol. 2003, 162, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, A.; King, W.G.; Mattaliano, M.D.; Frost, J.A.; Diaz, B.; Morrison, D.K.; Cobb, M.H.; Marshall, M.S.; Brugge, J.S. Phosphatidylinositol 3-kinase regulates Raf1 through Pak phosphorylation of serine 338. Curr. Biol. 2000, 10, 551–554. [Google Scholar] [CrossRef]
- Zhu, J.; Balan, V.; Bronisz, A.; Balan, K.; Sun, H.; Leicht, D.T.; Luo, Z.; Qin, J.; Avruch, J.; Tzivion, G. Identification of Raf-1 S471 as a novel phosphorylation site critical for Raf-1 and B-Raf kinase activities and for MEK binding. Mol. Biol Cell 2005, 16, 4733–4744. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.; Lee, J.; Guan, K.L. Positive and negative regulation of Raf kinase activity and function by phosphorylation. EMBO J. 2001, 20, 3716–3727. [Google Scholar] [CrossRef] [PubMed]
- Michaud, N.R.; Fabian, J.R.; Mathes, K.D.; Morrison, D.K. 14-3-3 is not essential for Raf-1 function: Identification of Raf-1 proteins that are biologically activated in a 14-3-3- and Ras-independent manner. Mol. Cell. Biol. 1995, 15, 3390–3397. [Google Scholar] [CrossRef] [PubMed]
- Muslin, A.J.; Tanner, J.W.; Allen, P.M.; Shaw, A.S. Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell 1996, 84, 889–897. [Google Scholar] [CrossRef]
- Zimmermann, S.; Moelling, K. Phosphorylation and regulation of Raf by Akt (protein kinase B). Science 1999, 286, 1741–1744. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; McPherson, R.A.; Apolloni, A.; Yan, J.; Lane, A.; Clyde-Smith, J. 14-3-3 Facilitates Ras-Dependent Raf-1 Activation In Vitro and In Vivo 14-3-3 Facilitates Ras-Dependent Raf-1 Activation In Vitro and In Vivo. Mol. Cell. Biol. 1998, 18, 3947–3955. [Google Scholar] [CrossRef] [PubMed]
- Rommel, C.; Radziwill, G.; Lovrić, J.; Noeldeke, J.; Heinicke, T.; Jones, D.; Aitken, A.; Moelling, K. Activated Ras displaces 14-3-3 protein from the amino terminus of c-Raf-1. Oncogene 1996, 12, 609–619. [Google Scholar] [PubMed]
- Kubicek, M.; Pacher, M.; Abraham, D.; Podar, K.; Eulitz, M.; Baccarini, M. Dephosphorylation of Ser-259 regulates Raf-1 membrane association. J. Biol. Chem. 2002, 277, 7913–7919. [Google Scholar] [CrossRef] [PubMed]
- Abraham, D.; Podar, K.; Pacher, M.; Kubicek, M.; Welzel, N.; Hemmings, B.A.; Dilworth, S.M.; Mischak, H.; Kolch, W.; Baccarini, M. Raf-1-associated protein phosphatase 2A as a positive regulator of kinase activation. J. Biol. Chem. 2000, 275, 22300–22304. [Google Scholar] [CrossRef] [PubMed]
- Tzivion, G.; Luo, Z.; Avruch, J. A dimeric 14-3-3 protein is an essential cofactor for Raf kinase activity. Nature 1998, 394, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Mischak, H.; Seitz, T.; Janosch, P.; Eulitz, M.; Steen, H.; Schellerer, M.; Philipp, A.; Kolch, W. Negative regulation of Raf-1 by phosphorylation of serine 621. Mol. Cell. Biol. 1996, 16, 5409–5418. [Google Scholar] [CrossRef] [PubMed]
- Thorson, J.A.; Yu, L.W.; Hsu, A.L.; Shih, N.Y.; Graves, P.R.; Tanner, J.W.; Allen, P.M.; Piwnica-Worms, H.; Shaw, A.S. 14-3-3 proteins are required for maintenance of Raf-1 phosphorylation and kinase activity. Mol. Cell. Biol. 1998, 18, 5229–5238. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, M.K.; Müller, J.; Ritt, D.A.; Zhou, M.; Zhou, X.Z.; Copeland, T.D.; Conrads, T.P.; Veenstra, T.D.; Lu, K.P.; Morrison, D.K. Regulation of Raf-1 by direct feedback phosphorylation. Mol. Cell 2005, 17, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Rushworth, L.K.; Hindley, A.D.; Neill, E.O.; Kolch, W. Regulation and Role of Raf-1 / B-Raf Heterodimerization. Mol. Cell. Biol. 2006, 26, 2262–2272. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. ERK1/2 MAP kinases: Structure, function, and regulation. Pharmacol. Res. 2012, 66, 105–143. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, A.; Terai, K.; Itoh, R.E.; Aoki, K.; Nakamura, T.; Kuroda, S.; Nishida, E.; Matsuda, M. Dynamics of the Ras/ERK MAPK cascade as monitored by fluorescent probes. J. Biol. Chem. 2006, 281, 8917–8926. [Google Scholar] [CrossRef] [PubMed]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Pagès, G.; Pouysségur, J. Constitutively active mutants of MAP kinase kinase (MEK1) induce growth factor-relaxation and oncogenicity when expressed in fibroblasts. Oncogene 1994, 9, 3379–3387. [Google Scholar] [PubMed]
- Cowley, S.; Paterson, H.; Kemp, P.; Marshall, C.J. Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell 1994, 77, 841–852. [Google Scholar] [CrossRef]
- Lefloch, R.; Pouysségur, J.; Lenormand, P. Total ERK1/2 activity regulates cell proliferation. Cell Cycle 2009, 8, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Seger, R. The extracellular signal-regulated kinase: Multiple substrates regulate diverse cellular functions. Growth Factors 2006, 24, 21–44. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.A.; Dreisbach, V.C.; Murphy, L.O.; Blenis, J. Characterization of regulatory events associated with membrane targeting of p90 ribosomal S6 kinase 1. Mol. Cell. Biol. 2001, 21, 7470–7480. [Google Scholar] [CrossRef] [PubMed]
- Dalby, K.N.; Morrice, N.; Caudwell, F.B.; Avruch, J.; Cohen, P. Identification of regulatory phosphorylation sites in mitogen-activated protein kinase (MAPK)-activated protein kinase-1a/p90(rsk) that are inducible by MAPK. J. Biol. Chem. 1998, 273, 1496–1505. [Google Scholar] [CrossRef] [PubMed]
- Jensen, C.J.; Buch, M.B.; Krag, T.O.; Hemmings, B.A.; Gammeltoft, S.; Frödin, M. 90-kDa ribosomal S6 kinase is phosphorylated and activated by 3- phosphoinositide-dependent protein kinase-1. J. Biol. Chem. 1999, 274, 27168–27176. [Google Scholar] [CrossRef] [PubMed]
- Anjum, R.; Blenis, J. The RSK family of kinases: Emerging roles in cellular signalling. Nat. Rev. Mol. Cell. Biol. 2008, 9, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [PubMed]
- Murphy, L.O.; Blenis, J. MAPK signal specificity: The right place at the right time. Trends Biochem Sci 2006, 31, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Stacey, D.W.; Watson, T.; Kung, H.F.; Curran, T. Microinjection of transforming ras protein induces c-fos expression. Mol. Cell. Biol. 1987, 7, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Quantin, B.; Breathnach, R. Epidermal growth factor stimulates transcription of the c-jun proto-oncogene in rat fibroblasts. Nature 1988, 334, 538–539. [Google Scholar] [CrossRef] [PubMed]
- Hollenhorst, P.C.; McIntosh, L.P.; Graves, B.J. Genomic and biochemical insights into the specificity of ETS transcription factors. Annu. Rev. Biochem. 2011, 80, 437–471. [Google Scholar] [CrossRef] [PubMed]
- Buchwalter, G.; Gross, C.; Wasylyk, B. Ets ternary complex transcription factors. Gene 2004, 324, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Herber, B.; Truss, M.; Beato, M.; Muller, R. Inducible regulatory elements in the human Cyclin D1 promoter. Oncogene 1994, 9, 1295–1304. [Google Scholar] [PubMed]
- Albanese, C.; Johnson, J.; Watanabe, G.; Eklund, N.; Vu, D.; Arnold, A.; Pestell, R.G. Transforming p21(ras) mutants and c-Ets-2 activate the Cyclin D1 promoter through distinguishable regions. J. Biol. Chem. 1995, 270, 23589–23597. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, K.; Sagata, N. The Mos/MAP kinase pathway stabilizes c-fos by phosphorylation and augments its transforming activity in NIH 3T3 cells. EMBO J. 1995, 14, 5048–5059. [Google Scholar] [PubMed]
- Morton, S.; Davis, R.J.; McLaren, A.; Cohen, P. A reinvestigation of the multisite phosphorylation of the transcription factor c-Jun. EMBO J. 2003, 22, 3876–3886. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Han, B.; Chen, J.; Wiedemeyer, R.; Orsulic, S.; Bose, S.; Zhang, X.; Karlan, B.Y.; Giuliano, A.E.; Cui, Y.; et al. FOXC1 is a critical mediator of EGFR function in human basal-like breast cancer. Ann. Surg. Oncol. 2014, 21, S758–S766. [Google Scholar] [CrossRef] [PubMed]
- Berry, F.B.; Mirzayans, F.; Walter, M.A. Regulation of FOXC1 stability and transcriptional activity by an epidermal growth factor-activated mitogen-activated protein kinase signaling cascade. J. Biol. Chem. 2006, 281, 10098–10104. [Google Scholar] [CrossRef] [PubMed]
- Chambard, J.C.; Lefloch, R.; Pouysségur, J.; Lenormand, P. ERK implication in cell cycle regulation. Biochim. Biophys. Acta Mol. Cell Res 2007, 1773, 1299–1310. [Google Scholar] [CrossRef] [PubMed]
- Dibble, C.C.; Cantley, L.C. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015, 25, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Whitman, M.; Downes, C.P.; Keeler, M.; Keller, T.; Cantley, L. Type I phosphatidylinositol kinase makes a novel inositol phospholipid, phosphatidylinositol-3-phosphate. Nature 1988, 332, 644–666. [Google Scholar] [CrossRef] [PubMed]
- Bjorge, J.D.; Chan, T.O.; Antczak, M.; Kung, H.J.; Fujita, D.J. Activated type I phosphatidylinositol kinase is associated with the epidermal growth factor (EGF) receptor following EGF stimulation. Proc. Natl. Acad. Sci. USA 1990, 87, 3816–3820. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, L.M.; Yuzugullu, H.; Zhao, J.J. PI3K in cancer: Divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer 2015, 15, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Cantley, L.C. Phosphoinositide 3-kinase in immunological systems. Semin. Immunol. 2002, 14, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Auger, K.R.; Serunian, L.A.; Soltoff, S.P.; Libby, P.; Cantley, L.C. PDGF-dependent tyrosine phosphorylation stimulates production of novel polyphosphoinositides in intact cells. Cell 1989, 57, 167–175. [Google Scholar] [CrossRef]
- Carpenter, C.L.; Duckworth, B.C.; Auger, K.R.; Cohen, B.; Schaffhausen, B.S.; Cantley, L.C. Purification and characterization of phosphoinositide 3-kinase from rat liver. J. Biol. Chem. 1990, 265, 19704–19711. [Google Scholar] [PubMed]
- Vogt, P.K.; Hart, J.R.; Gymnopoulos, M.; Jiang, H.; Kang, S.; Bader, A.G.; Zhao, L.; Denley, A. Phosphatidylinositol 3-kinase: The oncoprotein. Curr. Top. Microbiol. Immunol. 2010, 347, 79–104. [Google Scholar] [CrossRef] [PubMed]
- Salmena, L.; Carracedo, A.; Pandolfi, P.P. Tenets of PTEN tumor suppression. Cell 2008, 133, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Stephens, L.; Hawkins, P. PI3K signalling: The path to discovery and understanding. Nat. Rev. Mol. Cell. Biol. 2012, 13, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Soltoff, S.P.; Carraway, K.L.; Prigent, S.A.; Gullick, W.G.; Cantley, L.C. ErbB3 is involved in activation of phosphatidylinositol 3-kinase by epidermal growth factor. Mol. Cell. Biol. 1994, 14, 3550–3558. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.H.; Sierke, S.L.; Koland, J.G. Epidermal growth factor-dependent association of phosphatidylinositol 3-kinase with the erbB3 gene product. J. Biol. Chem. 1994, 269, 24747–24755. [Google Scholar] [PubMed]
- Soltoff, S.P.; Cantley, L.C. p120cbl is a cytosolic adapter protein that associates with phosphoinositide 3-kinase in response to epidermal growth factor in PC12 and other cells. J. Biol. Chem. 1996, 271, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Mattoon, D.R.; Lamothe, B.; Lax, I.; Schlessinger, J. The docking protein Gab1 is the primary mediator of EGF-stimulated activation of the PI-3K/Akt cell survival pathway. BMC Biol. 2004, 2, 24. [Google Scholar] [CrossRef] [PubMed]
- Lock, L.S.; Royal, I.; Naujokas, M.A.; Park, M. Identification of an atypical Grb2 carboxyl-terminal SH3 domain binding site in Gab docking proteins reveals Grb2-dependent and -independent recruitment of Gab1 to receptor tyorosine kinases. J. Biol. Chem. 2000, 275, 31536–31545. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Rohrschneider, L.R. The gift of Gab. FEBS Lett. 2002, 515, 1–7. [Google Scholar] [CrossRef]
- Maroun, C.R.; Holgado-Madruga, M.; Royal, I.; Naujokas, M.A.; Fournier, T.M.; Wong, A.J.; Park, M. The Gab1 PH domain is required for localization of Gab1 at sites of cell-cell contact and epithelial morphogenesis downstream from the met receptor tyrosine kinase. Mol. Cell. Biol. 1999, 19, 1784–1799. [Google Scholar] [CrossRef] [PubMed]
- Sjölander, A.; Yamamoto, K.; Huber, B.E.; Lapetina, E.G. Association of p21ras with phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. USA 1991, 88, 7908–7912. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Viciana, P.; Warne, P.H.; Vanhaesebroeck, B.; Waterfield1l, M.D.; Downward, J. Activation of phosphoinositide 3-kinase by interaction with Ras and by point mutation. EMBO J. 1996, 15, 2442–2451. [Google Scholar] [PubMed]
- Cuevas, B.D.; Lu, Y.; Mao, M.; Zhang, J.; LaPushin, R.; Siminovitch, K.; Mills, G.B. Tyrosine Phosphorylation of p85 Relieves Its Inhibitory Activity on Phosphatidylinositol 3-Kinase. J. Biol. Chem. 2001, 276, 27455–27461. [Google Scholar] [CrossRef] [PubMed]
- Franke, T.F.; Kaplan, D.R.; Cantley, L.C.; Toker, A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science 1997, 275, 665–668. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.H.; Li, C.F.; Yang, W.L.; Gao, Y.; Lee, S.W.; Feng, Z.; Huang, H.Y.; Tsai, K.K.C.; Flores, L.G.; Shao, Y. The Skp2-SCF E3 ligase regulates akt ubiquitination, glycolysis, herceptin sensitivity, and tumorigenesis. Cell 2012, 149, 1098–1111. [Google Scholar] [CrossRef] [PubMed]
- Toker, A.; Newton, A.C. Akt/protein kinase B is regulated by autophosphorylation at the hypothetical PDK-2 site. J. Biol. Chem. 2000, 275, 8271–8274. [Google Scholar] [CrossRef] [PubMed]
- Pearce, L.R.; Komander, D.; Alessi, D.R. The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell. Biol. 2010, 11, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Stephens, L.; Anderson, K.; Stokoe, D.; Erdjument-Bromage, H.; Painter, G.F.; Holmes, A.B.; Gaffney, P.R.; Reese, C.B.; McCormick, F.; Tempst, P.; et al. Protein kinase B kinases that mediate phosphatidylinositol 3,4,5- trisphosphate-dependent activation of protein kinase B. Science 1998, 279, 710–714. [Google Scholar] [CrossRef] [PubMed]
- Casamayor, A.; Morrice, N.A.; Alessi, D.R. Phosphorylation of Ser-241 is essential for the activity of 3-phosphoinositide-dependent protein kinase-1: Identification of five sites of phosphorylation in vivo. Biochem. J. 1999, 342, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Furnari, F.; Newton, A.C. PHLPP: A phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol. Cell 2005, 18, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Brognard, J.; Sierecki, E.; Gao, T.; Newton, A.C. PHLPP and a Second Isoform, PHLPP2, Differentially Attenuate the Amplitude of Akt Signaling by Regulating Distinct Akt Isoforms. Mol. Cell 2007, 25, 917–931. [Google Scholar] [CrossRef] [PubMed]
- Andjelković, M.; Jakubowicz, T.; Cron, P.; Ming, X.F.; Han, J.W.; Hemmings, B.A. Activation and phosphorylation of a pleckstrin homology domain containing protein kinase (RAC-PK/PKB) promoted by serum and protein phosphatase inhibitors. Proc. Natl. Acad. Sci. USA 1996, 93, 5699–5704. [Google Scholar] [CrossRef] [PubMed]
- Santi, S.A.; Lee, H. The Akt isoforms are present at distinct subcellular locations. Am. J. Physiol. Cell Physiol. 2010, 298, C580–C591. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Tschopp, O. Protein kinase Bα/Akt1 regulates placental development and fetal growth. J. Biol. Chem. 2003, 278, 32124–32131. [Google Scholar] [CrossRef] [PubMed]
- Zinda, M.J.; Johnson, M.A.; Paul, J.D.; Horn, C.; Konicek, B.W.; Zhao, H.L.; Sandusky, G.; Thomas, J.E.; Neubauer, B.L.; Lai, M.T.; et al. AKT-1, -2, and -3 are expressed in both normal and tumor tissues of the lung, breast, prostate, and colon. Clin. Cancer Res. 2001, 7, 2475–2479. [Google Scholar] [CrossRef] [PubMed]
- Cariaga-Martinez, A.E.; López-Ruiz, P.; Nombela-Blanco, M.P.; Motiño, O.; González-Corpas, A.; Rodriguez-Ubreva, J.; Lobo, M.V.T.; Cortés, M.A.; Colás, B. Distinct and specific roles of AKT1 and AKT2 in androgen-sensitive and androgen-independent prostate cancer cells. Cell Signal. 2013, 25, 1586–1597. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Thorvaldsen, J.L.; Chu, Q.; Feng, F.; Birnbaum, M.J. Akt1/PKBalpha Is Required for Normal Growth but Dispensable for Maintenance of Glucose Homeostasis in Mice. J. Biol. Chem. 2001, 276, 38349–38352. [Google Scholar] [CrossRef] [PubMed]
- Dummler, B.; Tschopp, O.; Hynx, D.; Yang, Z.-Z.; Dirnhofer, S.; Hemmings, B.A. Life with a single isoform of Akt: Mice lacking Akt2 and Akt3 are viable but display impaired glucose homeostasis and growth deficiencies. Mol. Cell. Biol. 2006, 26, 8042–8051. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, R.S.; Orena, S.J.; Rafidi, K.; Torchia, A.J.; Stock, J.L.; Hildebrandt, A.L.; Coskran, T.; Black, S.C.; Brees, D.J.; Wicks, J.R.; et al. Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKB?? J. Clin. Investig. 2003, 112, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Arboleda, M.J.; Lyons, J.F.; Kabbinavar, F.F.; Bray, M.R.; Snow, B.E.; Ayala, R.; Danino, M.; Karlan, B.Y.; Slamon, D.J. Overexpression of AKT2/protein kinase B beta leads to up- regulation of beta 1 integrins, increased invasion, and metastasis of human breast and ovarian cancer cells. Cancer Res. 2003, 63, 196–206. [Google Scholar] [PubMed]
- Irie, H.; Pearline, R.; Grueneberg, D. Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial–mesenchymal transition. J. Cell Boil. 2005, 17, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Ju, X.; Katiyar, S.; Wang, C.; Liu, M.; Jiao, X.; Li, S.; Zhou, J.; Turner, J.; Lisanti, M.P.; Russell, R.G.; et al. Akt1 governs breast cancer progression in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 7438–7443. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.W.; Kim, D.S.; Lee, J.H.; Lee, B.S.; Lee, S.H.; Jung, H.L.; Sung, K.W.; Kim, H.T.; Yoo, K.H.; Koo, H.H. Roles of AKT1 and AKT2 in non-small cell lung cancer cell survival, growth, and migration. Cancer Sci. 2011, 102, 1822–1828. [Google Scholar] [CrossRef] [PubMed]
- Héron-Milhavet, L.; Franckhauser, C.; Rana, V.; Berthenet, C.; Fisher, D.; Hemmings, B.A.; Fernandez, A.; Lamb, N.J.C. Only Akt1 is required for proliferation, while Akt2 promotes cell cycle exit through p21 binding. Mol. Cell. Biol. 2006, 26, 8267–8280. [Google Scholar] [CrossRef] [PubMed]
- Linnerth-Petrik, N.M.; Santry, L.A.; Petrik, J.J.; Wootton, S.K. Opposing functions of Akt isoforms in lung tumor initiation and progression. PLoS ONE 2014, 9, e94595. [Google Scholar] [CrossRef] [PubMed]
- Stål, O.; Pérez-Tenorio, G.; Akerberg, L.; Olsson, B.; Nordenskjöld, B.; Skoog, L.; Rutqvist, L.E. Akt kinases in breast cancer and the results of adjuvant therapy. Breast Cancer Res. 2003, 5, R37–R44. [Google Scholar] [CrossRef] [PubMed]
- Staal, S.P. Molecular cloning of the akt oncogene and its human homologues AKT1 and AKT2: Amplification of AKT1 in a primary human gastric adenocarcinoma. Proc. Natl. Acad. Sci. USA 1987, 84, 5034–5037. [Google Scholar] [CrossRef] [PubMed]
- Carpten, J.D.; Faber, A.L.; Horn, C.; Donoho, G.P.; Briggs, S.L.; Robbins, C.M.; Hostetter, G.; Boguslawski, S.; Moses, T.Y.; Savage, S.; et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 2007, 448, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.L.; Xu, P.Z.; Peng, X.D.; Chen, W.S.; Guzman, G.; Yang, X.; Di Cristofano, A.; Pandolfi, P.P.; Hay, N. The deficiency of Akt1 is sufficient to suppress tumor development in Pten+/− mice. Genes Dev. 2006, 20, 1569–1574. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.Q.; Ruggeri, B.; Klein, W.M.; Sonoda, G.; Altomare, D.A.; Watson, D.K.; Testa, J.R. Amplification of AKT2 in human pancreatic cells and inhibition of AKT2 expression and tumorigenicity by antisense RNA. Proc. Natl. Acad. Sci. USA 1996, 93, 3636–3641. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.Q.; Godwin, A.K.; Bellacosa, A.; Taguchi, T.; Franke, T.F.; Hamilton, T.C.; Tsichlis, P.N.; Testa, J.R. AKT2, a putative oncogene encoding a member of a subfamily of protein-serine/threonine kinases, is amplified in human ovarian carcinomas. Proc. Natl. Acad. Sci. USA 1992, 89, 9267–9271. [Google Scholar] [CrossRef] [PubMed]
- Roy, H.K.; Olusola, B.F.; Clemens, D.L.; Karolski, W.J.; Ratashak, A.; Lynch, H.T.; Smyrk, T.C. AKT proto-oncogene overexpression is an early event during sporadic colon carcinogenesis. Carcinogenesis 2002, 23, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Sakon, M.; Nagano, H.; Hiraoka, N.; Yamamoto, H.; Hayashi, N.; Dono, K.; Nakamori, S.; Umeshita, K.; Ito, Y.; et al. Akt2 expression correlates with prognosis of human hepatocellular carcinoma. Oncol. Rep. 2004, 11, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Altomare, D.A.; Testa, J.R. Perturbations of the AKT signaling pathway in human cancer. Oncogene 2005, 24, 7455–7464. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Wang, T.-L.; Samuels, Y.; Bardelli, A.; Cummins, J.M.; DeLong, L.; Silliman, N.; Ptak, J.; Szabo, S.; Willson, J.K.V.; et al. Colorectal cancer: Mutations in a signalling pathway. Nature 2005, 436, 792. [Google Scholar] [CrossRef] [PubMed]
- Okano, J.; Gaslightwala, I.; Birnbaum, M.J.; Rustgi, A.K.; Nakagawa, H. Akt/protein kinase B isoforms are differentially regulated by epidermal growth factor stimulation. J. Biol. Chem. 2000, 275, 30934–30942. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.P.; Hu, M.C.; Miller, S.A.; Yu, Z.; Xia, W.; Lin, S.Y.; Hung, M.C. HER-2/neu blocks tumor necrosis factor-induced apoptosis via the Akt/NF-kappaB pathway. J. Biol. Chem. 2000, 275, 8027–8031. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.R.; Dudek, H.; Xu, T.; Masters, S.; Haian, F.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell- intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef]
- Cardone, M.H. Regulation of Cell Death Protease Caspase-9 by Phosphorylation. Science 1998, 282, 1318–1321. [Google Scholar] [CrossRef] [PubMed]
- Tang, E.D.; Nuñez, G.; Barr, F.G.; Guan, K.L. Negative regulation of the forkhead transcription factor FKHR by Akt. J. Biol. Chem. 1999, 274, 16741–16746. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- Ogawara, Y.; Kishishita, S.; Obata, T.; Isazawa, Y.; Suzuki, T.; Tanaka, K.; Masuyama, N.; Gotoh, Y. Akt enhances Mdm2-mediated ubiquitination and degradation of p53. J. Biol. Chem. 2002, 277, 21843–21850. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.P.; Liao, Y.; Xia, W.; Zou, Y.; Spohn, B.; Hung, M.C. HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat. Cell Biol. 2001, 3, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Tamaskovic, R.; Yang, Z.; Brazil, D.P.; Merlo, A.; Hess, D.; Hemmings, B.A. Stabilization of Mdm2 via decreased ubiquitination is mediated by protein kinase B/Akt-dependent phosphorylation. J. Biol. Chem. 2004, 279, 35510–35517. [Google Scholar] [CrossRef] [PubMed]
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. DEPTOR Is an mTOR Inhibitor Frequently Overexpressed in Multiple Myeloma Cells and Required for Their Survival. Cell 2009, 137, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J.; Cantley, L.C. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006, 441, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Manning, B.D. The TSC1-TSC2 complex: A molecular switchboard controlling cell growth. Biochem. J. 2008, 412, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.-L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Tee, A.R.; Logsdon, M.N.; Blenis, J.; Cantley, L.C. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/Akt pathway. Mol. Cell 2002, 10, 151–162. [Google Scholar] [CrossRef]
- Ma, L.; Chen, Z.; Erdjument-Bromage, H.; Tempst, P.; Pandolfi, P.P. Phosphorylation and functional inactivation of TSC2 by Erk: Implications for tuberous sclerosis and cancer pathogenesis. Cell 2005, 121, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Tee, A.R.; Fingar, D.C.; Manning, B.D.; Kwiatkowski, D.J.; Cantley, L.C.; Blenis, J. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc. Natl. Acad. Sci. USA 2002, 99, 13571–13576. [Google Scholar] [CrossRef] [PubMed]
- Scott, P.H.; Brunn, G.J.; Kohn, A.D.; Roth, R.A.; Lawrence, J.C., Jr. Evidence of insulin-stimulated phosphorylation and activation of the mammalian target of rapamycin mediated by a protein kinase B signaling pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 7772–7777. [Google Scholar] [CrossRef] [PubMed]
- Dan, H.C.; Ebbs, A.; Pasparakis, M.; Van Dyke, T.; Basseres, D.S.; Baldwin, A.S. Akt-dependent activation of mTORC1 complex involves phosphorylation of mTOR (mammalian target of rapamycin) by IκB kinaseα (IKKα). J. Biol. Chem. 2014, 289, 25227–25240. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Jaquez, H.A.; Keller, J.A.; Foster, K.G.; Ekim, B.; Soliman, G.A.; Feener, E.P.; Ballif, B.A.; Fingar, D.C. Site-specific mTOR phosphorylation promotes mTORC1-mediated signaling and cell growth. Mol. Cell. Biol. 2009, 29, 4308–4324. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R.T. mTOR as a positive regulator of tumor cell responses to hypoxia. Curr. Top. Microbiol. Immunol. 2004, 279, 299–319. [Google Scholar] [PubMed]
- Grewe, M.; Gansauge, F.; Schmid, R.M.; Adler, G.; Seufferlein, T. Regulation of cell growth and Cyclin D1 expression by the constitutively active FRAP-p70(s6K) pathway in human pancreatic cancer cells. Cancer Res. 1999, 59, 3581–3587. [Google Scholar] [PubMed]
- Averous, J.; Fonseca, B.D.; Proud, C.G. Regulation of cyclin D1 expression by mTORC1 signaling requires eukaryotic initiation factor 4E-binding protein 1. Oncogene 2008, 27, 1106–1113. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Elstrom, R.L.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.M.; et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004, 64, 3892–3899. [Google Scholar] [CrossRef] [PubMed]
- Courtnay, R.; Ngo, D.C.; Malik, N.; Ververis, K.; Tortorella, S.M.; Karagiannis, T.C. Cancer metabolism and the Warburg effect: The role of HIF-1 and PI3K. Mol. Biol. Rep. 2015, 42, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Lien, E.C.; Lyssiotis, C.A.; Cantley, L.C. Metabolic Reprogramming by the PI3K-Akt-mTOR Pathway in Cancer. Recent Res. Cancer Res. 2016, 207, 39–72. [Google Scholar] [CrossRef]
- Nakamura, Y.; Fukami, K. Roles of phospholipase C isozymes in organogenesis and embryonic development. Physiology 2009, 24, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Ji, Q.S.; Winnier, G.E.; Niswender, K.D.; Horstman, D.; Wisdom, R.; Magnuson, M.A.; Carpenter, G. Essential role of the tyrosine kinase substrate phospholipase C-γ1 in mammalian growth and development. Proc. Natl. Acad. Sci. USA 1997, 94, 2999–3003. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.J.; Kume, T.; McKay, C.; Xu, M.J.; Ihle, J.N.; Carpenter, G. Absence of erythrogenesis and vasculogenesis in Plcg1-deficient mice. J. Biol. Chem. 2002, 277, 9335–9341. [Google Scholar] [CrossRef] [PubMed]
- Sala, G.; Dituri, F.; Raimondi, C.; Previdi, S.; Maffucci, T.; Mazzoletti, M.; Rossi, C.; Iezzi, M.; Lattanzio, R.; Piantelli, M.; et al. Phospholipase Cgamma1 is required for metastasis development and progression. Cancer Res. 2008, 68, 10187–10196. [Google Scholar] [CrossRef] [PubMed]
- Lattanzio, R.; Piantelli, M.; Falasca, M. Role of phospholipase C in cell invasion and metastasis. Adv. Biol. Regul. 2013, 53, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Rotin, D.; Margolis, B.; Mohammadi, M.; Daly, R.J.; Daum, G.; Li, N.; Fischer, E.H.; Burgess, W.H.; Ullrich, A.; Schlessinger, J. SH2 domains prevent tyrosine dephosphorylation of the EGF receptor: Identification of Tyr992 as the high-affinity binding site for SH2 domains of phospholipase C gamma. EMBO J. 1992, 11, 559–567. [Google Scholar] [PubMed]
- Anderson, D.; Koch, C.A.; Grey, L.; Ellis, C.; Moran, M.F.; Pawson, T. Binding of SH2 domains of phospholipase C gamma 1, GAP, and Src to activated growth factor receptors. Science 1990, 250, 979–982. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, A.; Vecchi, M.; Ji, Q.S.; Mernaugh, R.; Carpenter, G. The role of individual SH2 domains in mediating association of phospholipase C-γ1 with the activated EGF receptor. J. Biol. Chem. 1999, 274, 26091–26097. [Google Scholar] [CrossRef] [PubMed]
- Falasca, M.; Logan, S.K.; Lehto, V.P.; Baccante, G.; Lemmon, M.A.; Schlessinger, J. Activation of phospholipase C gamma by PI 3-kinase-induced PH domain- mediated membrane targeting. EMBO J. 1998, 17, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Nishibe, S.; Wahl, M.I.; Hernández-Sotomayor, S.M.; Tonks, N.K.; Rhee, S.G.; Carpenter, G. Increase of the catalytic activity of phospholipase C-gamma 1 by tyrosine phosphorylation. Science 1990, 250, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Serrano, C.J.; Graham, L.; DeBell, K.; Rawat, R.; Veri, M.C.; Bonvini, E.; Rellahan, B.L.; Reischl, I.G. A New Tyrosine Phosphorylation Site in PLCγ1: The Role of Tyrosine 775 in Immune Receptor Signaling. J. Immunol. 2005, 174, 6233–6237. [Google Scholar] [CrossRef] [PubMed]
- Gresset, A.; Hicks, S.N.; Harden, T.K.; Sondek, J. Mechanism of phosphorylation-induced activation of phospholipase C-gamma isozymes. J. Biol. Chem. 2010, 285, 35836–35847. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.H.; Toita, R.; Kim, C.W.; Katayama, Y. Protein kinase C (PKC) isozyme-specific substrates and their design. Biotechnol. Adv. 2012, 30, 1662–1672. [Google Scholar] [CrossRef] [PubMed]
- Cazaubon, S.; Bornancin, F.; Parker, P.J. Threonine-497 is a critical site for permissive activation of protein kinase C alpha. Biochem. J. 1994, 301 (Pt 2), 443–448. [Google Scholar] [CrossRef] [PubMed]
- Cazaubon, S.M.; Parker, P.J. Identification of the phosphorylated region responsible for the permissive activation of protein kinase C. J. Biol. Chem. 1993, 268, 17559–17563. [Google Scholar] [PubMed]
- Lund, K.A.; Lazar, C.S.; Chen, W.S.; Walsh, B.J.; Welsh, J.B.; Herbst, J.J.; Walton, G.M.; Rosenfeld, M.G.; Gill, G.N.; Wiley, H.S. Phosphorylation of the epidermal growth factor receptor at threonine 654 inhibits ligand-induced internalization and down-regulation. J. Biol. Chem. 1990, 265, 20517–20523. [Google Scholar] [PubMed]
- Lee, C.S.; Kim, K.L.; Jang, J.H.; Choi, Y.S.; Suh, P.-G.; Ryu, S.H. The roles of phospholipase D in EGFR signaling. Biochim. Biophys. Acta 2009, 1791, 862–868. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Han, J.M.; Park, J.B.; Lee, S.D.; Oh, Y.S.; Chung, C.; Lee, T.G.; Kim, J.H.; Park, S.K.; Yoo, J.S.; et al. Phosphorylation and activation of phospholipase D1 by protein kinase C in vivo: Determination of multiple phosphorylation sites. Biochemistry 1999, 38, 10344–10351. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Jiang, Y.; Foster, D. Epidermal growth factor induces the production of biologically distinguishable diglyceride species from phosphatidylinositol and phosphatidylcholine via the independent activation of type C and type D phospholipases. Cell Growth Differ. 1994, 5, 79–85. [Google Scholar] [PubMed]
- Lu, Z.; Hornia, A.; Joseph, T.; Sukezane, T.; Frankel, P.; Zhong, M.; Bychenok, S.; Xu, L.; Feig, L.A.; Foster, D.A. Phospholipase D and RalA cooperate with the epidermal growth factor receptor to transform 3Y1 rat fibroblasts. Mol. Cell. Biol. 2000, 20, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Joseph, T.; Wooden, R.; Bryant, A.; Zhong, M.; Lu, Z.; Foster, D.A. Transformation of cells overexpressing a tyrosine kinase by phospholipase D1 and D2. Biochem. Biophys. Res. Commun. 2001, 289, 1019–1024. [Google Scholar] [CrossRef] [PubMed]
- Park, J.B.; Lee, C.S.; Jang, J.-H.; Ghim, J.; Kim, Y.-J.; You, S.; Hwang, D.; Suh, P.-G.; Ryu, S.H. Phospholipase signalling networks in cancer. Nat. Rev. Cancer 2012, 12, 782–792. [Google Scholar] [CrossRef] [PubMed]
- Rous, P. A Sarcoma of the Fowl Transmissible By an Agent Separable From the Tumor Cells. J. Exp. Med. 1911, 13, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Harrison, S.C.; Eck, M.J. Three-dimensional structure of the tyrosine kinase c-Src. Nature 1997, 385, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.C.; Weijland, A.; Gonfloni, S.; Thompson, A.; Courtneidge, S.A.; Superti-Furga, G.; Wierenga, R.K. The 2.35 A crystal structure of the inactivated form of chicken Src: A dynamic molecule with multiple regulatory interactions. J. Mol. Biol. 1997, 274, 757–775. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.A.; Gould, K.L.; Cartwright, C.A.; Hunter, T. Tyr527 is phosphorylated in pp60c-src: Implications for regulation. Science 1986, 231, 1431–1434. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Nada, S.; Yamanashi, Y.; Yamamoto, T.; Nakagawa, H. CSK: A protein-tyrosine kinase involved in regulation of src family kinases. J. Biol. Chem. 1991, 266, 24249–24252. [Google Scholar] [PubMed]
- Okada, M. Regulation of the Src family kinases by Csk. Int. J. Biol. Sci. 2012, 8, 1385–1397. [Google Scholar] [CrossRef] [PubMed]
- Somani, A.K.; Bignon, J.S.; Mills, G.B.; Siminovitch, K.A.; Branch, D.R. Src kinase activity is regulated by the SHP-1 protein-tyrosine phosphatase. J. Biol. Chem. 1997, 272, 21113–21119. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.Y.; Cartwright, C.A. Regulation of the Src tyrosine kinase and Syp tyrosine phosphatase by their cellular association. Oncogene 1995, 11, 1955–1962. [Google Scholar] [PubMed]
- Fang, K.S.; Sabe, H.; Saito, H.; Hanafusa, H. Comparative study of three protein-tyrosine phosphatases. Chicken protein-tyrosine phosphatase lambda dephosphorylates c- Src tyrosine 527. J. Biol. Chem. 1994, 269, 20194–20200. [Google Scholar] [PubMed]
- Charbonneau, H.; Tonks, N.K.; Kumar, S.; Diltz, C.D.; Harrylock, M.; Cool, D.E.; Krebs, E.G.; Fischer, E.H.; Walsh, K.A. Human placenta protein-tyrosine-phosphatase: Amino acid sequence and relationship to a family of receptor-like proteins. Proc. Natl. Acad. Sci. USA 1989, 86, 5252–5256. [Google Scholar] [CrossRef] [PubMed]
- Biscardi, J.S.; Maa, M.-C.; Tice, D.A.; Leu, T.-H.; Parsons, S.J. c-Src-mediated Phosphorylation of the Epidermal Growth Factor Receptor on Tyr845 and Tyr1101 Is Associated with Modulation of Receptor Function. J. Biol. Chem. 1999, 274, 8335–8343. [Google Scholar] [CrossRef] [PubMed]
- Hunter, T.; Sefton, B.M. Transforming gene product of Rous sarcoma virus phosphorylates tyrosine. Proc. Natl. Acad. Sci. USA 1980, 77, 1311–1315. [Google Scholar] [CrossRef] [PubMed]
- Collett, M.S.; Purchio, A.F.; Erikson, R.L. Avian sarcoma virus-transforming protein, pp60_src_ shows protein kinase activity specific for tyrosine. Nature 1980, 285, 167–169. [Google Scholar] [CrossRef] [PubMed]
- Levinson, A.D.; Oppermann, H.; Varmus, H.E.; Bishop, J.M. The purified product of the transforming gene of avian sarcoma virus phosphorylates tyrosine. J. Biol. Chem. 1980, 255, 11973–11980. [Google Scholar] [PubMed]
- Sen, B.; Johnson, F.M. Regulation of Src Family Kinases in Human Cancers. J. Signal Transduct. 2011, 2011, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.M.; Brugge, J.S. Cellular functions regulated by Src family kinases. Annu. Rev. Cell Dev. Biol. 1997, 13, 513–609. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Gur, G.; Yarden, Y. Src promotes destruction of c-Cbl: Implications for oncogenic synergy between Src and growth factor receptors. Proc. Natl. Acad. Sci. USA 2003, 100, 2438–2443. [Google Scholar] [CrossRef] [PubMed]
- Tice, D.A.; Biscardi, J.S.; Nickles, A.l.; Parsons, S.J. Mechanism of biological synergy between cellular Src and epidermal growth factor receptor. Proc. Natl. Acad. Sci. USA 1999, 96, 1415–1420. [Google Scholar] [CrossRef] [PubMed]
- Goi, T.; Shipitsin, M.; Lu, Z.; Foster, D.A.; Klinz, S.G.; Feig, L.A. An EGF receptor/Ral-GTPase signaling cascade regulates c-Src activity and substrate specificity. EMBO J. 2000, 19, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Maa, M.C.; Leu, T.H.; McCarley, D.J.; Schatzman, R.C.; Parsons, S.J. Potentiation of epidermal growth factor receptor-mediated oncogenesis by c-Src: Implications for the etiology of multiple human cancers. Proc. Natl. Acad. Sci. USA 1995, 92, 6981–6985. [Google Scholar] [CrossRef] [PubMed]
- Poole, E.M.; Curtin, K.; Hsu, L.; Kulmacz, R.J.; Duggan, D.J.; Makar, K.W.; Xiao, L.; Carlson, C.S.; Slattery, M.L.; Caan, B.J.; et al. Genetic variability in EGFR, Src and HER2 and risk of colorectal adenoma and cancer. Int. J. Mol. Epidemiol. Genet. 2011, 2, 300–315. [Google Scholar] [PubMed]
- Olayioye, M.A.; Beuvink, I.; Horsch, K.; Daly, J.M.; Hynes, N.E. ErbB receptor-induced activation of Stat transcription factors is mediated by Src tyrosine kinases. J. Biol. Chem. 1999, 274, 17209–17218. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Sato, A.; Aoto, M.; Fukami, Y. c-Src phosphorylates epidermal growth factor receptor on tyrosine 845. Biochem. Biophys. Res. Commun. 1995, 215, 1078–1087. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.; Eastwood, M.P.; Zhang, X.; Kim, E.T.; Arkhipov, A.; Dror, R.O.; Jumper, J.; Kuriyan, J.; Shaw, D.E. Oncogenic mutations counteract intrinsic disorder in the EGFR kinase and promote receptor dimerization. Cell 2012, 149, 860–870. [Google Scholar] [CrossRef] [PubMed]
- Endoh, H.; Ishibashi, Y.; Yamaki, E.; Yoshida, T.; Yajima, T.; Kimura, H.; Kosaka, T.; Onozato, R.; Tanaka, S.; Mitsudomi, T.; et al. Immunohistochemical analysis of phosphorylated epidermal growth factor receptor might provide a surrogate marker of EGFR mutation. Lung Cancer 2009, 63, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Chung, B.M.; Dimri, M.; George, M.; Reddi, A.L.; Chen, G.; Band, V.; Band, H. The role of cooperativity with Src in oncogenic transformation mediated by non-small cell lung cancer-associated EGF receptor mutants. Oncogene 2009, 28, 1821–1832. [Google Scholar] [CrossRef] [PubMed]
- Mattila, E.; Pellinen, T.; Nevo, J.; Vuoriluoto, K.; Arjonen, A.; Ivaska, J. Negative regulation of EGFR signalling through integrin-alpha1beta1-mediated activation of protein tyrosine phosphatase TCPTP. Nat. Cell Biol. 2005, 7, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Baumdick, M.; Brüggemann, Y.; Schmick, M.; Xouri, G.; Sabet, O.; Davis, L.; Chin, J.W.; Bastiaens, P.I.H. EGF-dependent re-routing of vesicular recycling switches spontaneous phosphorylation suppression to EGFR signaling. Elife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Koppikar, P.; Choi, S.-H.; Egloff, A.M.; Cai, Q.; Suzuki, S.; Freilino, M.; Nozawa, H.; Thomas, S.M.; Gooding, W.E.; Siegfried, J.M.; et al. Combined inhibition of c-Src and epidermal growth factor receptor abrogates growth and invasion of head and neck squamous cell carcinoma. Clin. Cancer Res. 2008, 14, 4284–4291. [Google Scholar] [CrossRef] [PubMed]
- Gargalionis, A.N.; Karamouzis, M.V.; Papavassiliou, A.G. The molecular rationale of Src inhibition in colorectal carcinomas. Int. J. Cancer 2014, 134, 2019–2029. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.Y.; Makino, K.; Xia, W.; Matin, a.; Wen, Y.; Kwong, K.Y.; Bourguignon, L.; Hung, M.C. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat. Cell Biol. 2001, 3, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Lei, Z.M.; Bian, L.; Rao, C.V. Functional nuclear epidermal growth factor receptors in human choriocarcinoma JEG-3 cells and normal human placenta. Endocrinology 1995, 136, 3163–3172. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.-W.; Hung, M.-C. Nuclear EGFR signalling network in cancers: Linking EGFR pathway to cell cycle progression, nitric oxide pathway and patient survival. Br. J. Cancer 2006, 94, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-N.; Yamaguchi, H.; Hsu, J.-M.; Hung, M.-C. Nuclear trafficking of the epidermal growth factor receptor family membrane proteins. Oncogene 2010, 29, 3997–4006. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.W.; Hsu, S.C.; Ali-Seyed, M.; Gunduz, M.; Xia, W.; Wei, Y.; Bartholomeusz, G.; Shih, J.Y.; Hung, M.C. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer Cell 2005, 7, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-J.; Lan, L.; Peng, G.; Chang, W.-C.; Hsu, M.-C.; Wang, Y.-N.; Cheng, C.-C.; Wei, L.; Nakajima, S.; Chang, S.-S.; et al. Tyrosine 370 phosphorylation of ATM positively regulates DNA damage response. Cell Res. 2015, 25, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Kamio, T.; Shigematsu, K.; Sou, H.; Kawai, K.; Tsuchiyama, H. Immunohistochemical expression of epidermal growth factor receptors in human adrenocortical carcinoma. Hum. Pathol. 1990, 21, 277–282. [Google Scholar] [CrossRef]
- Lo, H.W.; Xia, W.; Wei, Y.; Ali-Seyed, M.; Huang, S.F.; Hung, M.C. Novel prognostic value of nuclear epidermal growth factor receptor in breast cancer. Cancer Res. 2005, 65, 338–348. [Google Scholar] [PubMed]
- Marti, U.; Ruchti, C.; Kämpf, J.; Thomas, G.A.; Williams, E.D.; Peter, H.J.; Gerber, H.; Bürgi, U. Nuclear localization of epidermal growth factor and epidermal growth factor receptors in human thyroid tissues. Thyroid 2001, 11, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Psyrri, A.; Yu, Z.; Weinberger, P.M.; Sasaki, C.; Haffty, B.; Camp, R.; Rimm, D.; Burtness, B.A. Quantitative determination of nuclear and cytoplasmic epidermal growth factor receptor expression in oropharyngeal squamous cell cancer by using automated quantitative analysis. Clin. Cancer Res. 2005, 11, 5856–5862. [Google Scholar] [CrossRef] [PubMed]
- Dittmann, K.; Mayer, C.; Fehrenbacher, B.; Schaller, M.; Kehlbach, R.; Rodemann, H.P. Nuclear EGFR shuttling induced by ionizing radiation is regulated by phosphorylation at residue Thr654. FEBS Lett. 2010, 584, 3878–3884. [Google Scholar] [CrossRef] [PubMed]
- Liccardi, G.; Hartley, J.A.; Hochhauser, D. EGFR nuclear translocation modulates DNA repair following cisplatin and ionizing radiation treatment. Cancer Res. 2011, 71, 1103–1114. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.C.; Miller, S.A.; Wang, Y.; Hung, M.C. Nuclear EGFR is required for cisplatin resistance and DNA repair. Am. J. Transl. Res. 2009, 1, 249–258. [Google Scholar] [PubMed]
- De Angelis Campos, A.C.; Rodrigues, M.A.; de Andrade, C.; de Goes, A.M.; Nathanson, M.H.; Gomes, D.A. Epidermal growth factor receptors destined for the nucleus are internalized via a clathrin-dependent pathway. Biochem. Biophys. Res. Commun. 2011, 412, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.W.; Ali-Seyed, M.; Wu, Y.; Bartholomeusz, G.; Hsu, S.C.; Hung, M.C. Nuclear-cytoplasmic transport of EGFR involves receptor endocytosis, importin beta1 and CRM1. J. Cell. Biochem. 2006, 98, 1570–1583. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.N.; Wang, H.; Yamaguchi, H.; Lee, H.J.; Lee, H.H.; Hung, M.C. COPI-mediated retrograde trafficking from the Golgi to the ER regulates EGFR nuclear transport. Biochem. Biophys. Res. Commun. 2010, 399, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Jaganathan, S.; Yue, P.; Paladino, D.C.; Bogdanovic, J.; Huo, Q.; Turkson, J. A functional nuclear epidermal growth factor receptor, Src and Stat3 heteromeric complex in pancreatic cancer cells. PLoS ONE 2011, 6, e19605. [Google Scholar] [CrossRef] [PubMed]
- Huo, L.; Wang, Y.-N.; Xia, W.; Hsu, S.-C.; Lai, C.-C.; Li, L.-Y.; Chang, W.-C.; Wang, Y.; Hsu, M.-C.; Yu, Y.-L. RNA helicase A is a DNA-binding partner for EGFR-mediated transcriptional activation in the nucleus. Proc. Natl. Acad. Sci. USA 2010, 107, 16125–16130. [Google Scholar] [CrossRef] [PubMed]
- Bitler, B.; Goverdhan, A.; Schroeder, J. MUC1 regulates nuclear localization and function of the EGFR. J. Cell Sci. 2010, 123, 1716–1723. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, E.; De Palma, R.; Orditura, M.; De Vita, F.; Ciardiello, F. Anti-epidermal growth factor receptor monoclonal antibodies in cancer therapy. Clin. Exp. Immunol. 2009, 158, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, F.; Tortora, G.; Magrassi, S.F.; Lanzara, A. EGFR Antagonists in Cancer Treatment. N. Engl. J. Med. 2008, 358, 1160–1174. [Google Scholar] [CrossRef] [PubMed]
- Modjtahedi, H.; Essapen, S. Epidermal growth factor receptor inhibitors in cancer treatment: Advances, challenges and opportunities. Anticancer Drugs 2009, 20, 851–855. [Google Scholar] [CrossRef] [PubMed]
- Shukuya, T.; Takahashi, T.; Kaira, R.; Ono, A.; Nakamura, Y.; Tsuya, A.; Kenmotsu, H.; Naito, T.; Kaira, K.; Murakami, H. Efficacy of gefitinib for non-adenocarcinoma non-small-cell lung cancer patients harboring epidermal growth factor receptor mutations: A pooled analysis of published reports. Cancer Sci. 2011, 102, 1032–1037. [Google Scholar] [CrossRef] [PubMed]
- Harandi, A.; Zaidi, A.S.; Stocker, A.M.; Laber, D.A. Clinical efficacy and toxicity of anti-EGFR therapy in common cancers. J. Oncol. 2009. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating Mutations in the Epidermal Growth Factor Receptor Underlying Responsiveness of Non–Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Bertotti, A.; Papp, E.; Jones, S.; Adleff, V.; Anagnostou, V.; Lupo, B.; Sausen, M.; Phallen, J.; Hruban, C.A.; Tokheim, C.; et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature 2015, 526, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Krasinskas, A.M. EGFR Signaling in Colorectal Carcinoma. Pathol. Res. Int. 2011, 2011, 932932. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.Y.; Shia, J.; Kemeny, N.E.; Shah, M.; Schwartz, G.K.; Tse, A.; Hamilton, A.; Pan, D.; Schrag, D.; Schwartz, L.; et al. Cetuximab shows activity in colorectal cancer patients with tumors that do not express the epidermal growth factor receptor by immunohistochemistry. J. Clin. Oncol. 2005, 23, 1803–1810. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, D.; Humblet, Y.; Siena, S.; Khayat, D.; Bleiberg, H.; Santoro, A.; Bets, D.; Mueser, M.; Harstrick, A.; Verslype, C.; et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N. Engl. J. Med. 2004, 351, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Laurent-Puig, P.; Cayre, A.; Manceau, G.; Buc, E.; Bachet, J.B.; Lecomte, T.; Rougier, P.; Lievre, A.; Landi, B.; Boige, V.; et al. Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. J. Clin. Oncol. 2009, 27, 5924–5930. [Google Scholar] [CrossRef] [PubMed]
- Cleary, J.M.; Reardon, D.A.; Azad, N.; Gandhi, L.; Shapiro, G.I.; Chaves, J.; Pedersen, M.; Ansell, P.; Ames, W.; Xiong, H.; et al. A phase 1 study of ABT-806 in subjects with advanced solid tumors. Investig. New Drugs 2015, 33, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Benvenuti, S.; Sartore-bianchi, A.; Di Nicolantonio, F.; Zanon, C.; Moroni, M.; Veronese, S.; Siena, S.; Bardelli, A. Oncogenic Activation of the RAS / RAF Signaling Pathway Impairs the Response of Metastatic Colorectal Cancers to Anti—Epidermal Growth Factor Receptor Antibody Therapies. Cancer Res. 2007, 67, 2643–2648. [Google Scholar] [CrossRef] [PubMed]
- Di Fiore, F.; Blanchard, F.; Charbonnier, F.; Le Pessot, F.; Lamy, A.; Galais, M.P.; Bastit, L.; Killian, A.; Sesboue, R.; Tuech, J.J.; et al. Clinical relevance of KRAS mutation detection in metastatic colorectal cancer treated by Cetuximab plus chemotherapy. Br. J. Cancer 2007, 96, 1166–1169. [Google Scholar] [CrossRef] [PubMed]
- Lievre, A.; Bachet, J.-B.; Boige, V.; Cayre, A.; Le Corre, D.; Buc, E.; Ychou, M.; Bouche, O.; Landi, B.; Louvet, C.; et al. KRAS Mutations As an Independent Prognostic Factor in Patients With Advanced Colorectal Cancer Treated With Cetuximab. J. Clin. Oncol. 2008, 26, 374–379. [Google Scholar] [CrossRef] [PubMed]
- De Roock, W.; Piessevaux, H.; De Schutter, J.; Janssens, M.; De Hertogh, G.; Personeni, N.; Biesmans, B.; Van Laethem, J.L.; Peeters, M.; Humblet, Y.; et al. KRAS wild-type state predicts survival and is associated to early radiological response in metastatic colorectal cancer treated with cetuximab. Ann. Oncol. 2008, 19, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Cripps, C.; Gill, S.; Ahmed, S.; Colwell, B.; Dowden, S.; Kennecke, H.; Maroun, J.; Samson, B.; Thirlwell, M.; Wong, R. Consensus recommendations for the use of anti-egfr therapies in metastatic colorectal cancer. Curr. Oncol. 2010, 17, 39–45. [Google Scholar] [PubMed]
- Sorich, M.J.; Wiese, M.D.; Rowland, A.; Kichenadasse, G.; McKinnon, R.A.; Karapetis, C.S. Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: A meta-analysis of randomized controlled trials. Ann. Oncol. 2015, 26, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Lovly, C.L.; Horn, G.; Oxnard, W.P. EGFR c.2369C>T (T790M) Mutation in Non-Small Cell Lung Cancer. My Cancer Genome. Available online: Https//www.mycancergenome.org/content/disease/lung-cancer/egfr/4/ (accessed on 15 October 2015).
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; LaFramboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 2012, 44, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Yauch, R.L.; Januario, T.; Eberhard, D.A.; Cavet, G.; Zhu, W.; Fu, L.; Pham, T.Q.; Soriano, R.; Stinson, J.; Seshagiri, S.; et al. Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin. Cancer Res. 2005, 11, 8686–8698. [Google Scholar] [CrossRef] [PubMed]
- Uramoto, H.; Iwata, T.; Onitsuka, T.; Shimokawa, H.; Hanagiri, T.; Oyama, T. Epithelial-mesenchymal transition in EGFR-TKI acquired resistant lung adenocarcinoma. Anticancer Res. 2010, 30, 2513–2517. [Google Scholar]
- Suda, K.; Tomizawa, K.; Fujii, M.; Murakami, H.; Osada, H.; Maehara, Y.; Yatabe, Y.; Sekido, Y.M.T. Epithelial to mesenchymal transition in an epidermal growth factor receptor-mutant lung cancer cell line with acquired resistance to erlotinib. J. Thorac. Oncol. 2011, 7, 1152–1161. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, K.R.; Demuth, C.; Sorensen, B.S.; Nielsen, A.L. The role of epithelial to mesenchymal transition in resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Transl. Lung Cancer Res. 2016, 5, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Peeters, M.; Price, T.J.; Cervantes, A.; Sobrero, A.F.; Ducreux, M.; Hotko, Y.; André, T.; Chan, E.; Lordick, F.; Punt, C.J.A.; et al. Randomized phase III study of panitumumab with fluorouracil, leucovorin, and irinotecan (FOLFIRI) compared with FOLFIRI alone as second-line treatment in patients with metastatic colorectal cancer. J. Clin. Oncol. 2010, 28, 4706–4713. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, E.P.; Lacouture, M.; Shearer, H.; Iannotti, N.; Piperdi, B.; Pillai, M.; Xu, F.; Yassine, M. Final STEPP results of prophylacatic versus reactive skin toxicity (ST) treatment (tx) for panitumumab (pmab)-related ST in patients (pts) with metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2009, 27, CRA4027. [Google Scholar] [CrossRef]
- Park, K.; Tan, E.H.; O’Byrne, K.; Zhang, L.; Boyer, M.; Mok, T.; Hirsh, V.; Yang, J.C.H.; Lee, K.H.; Lu, S.; et al. Afatinib versus gefitinib as first-line treatment of patients with EGFR mutation-positive non-small-cell lung cancer (LUX-Lung 7): A phase 2B, open-label, randomised controlled trial. Lancet Oncol. 2016, 17, 577–589. [Google Scholar] [CrossRef]
- Huillard, O.; Bakalian, S.; Levy, C.; Desjardins, L.; Lumbroso-Le Rouic, L.; Pop, S.; Sablin, M.P.; Le Tourneau, C. Ocular adverse events of molecularly targeted agents approved in solid tumours: A systematic review. Eur. J. Cancer 2014, 50, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.S.; Levin, F.; Chu, D.S. Persistent corneal epithelial defect associated with erlotinib treatment. Cornea 2009, 28, 706–707. [Google Scholar] [CrossRef] [PubMed]
- Carrasco-García, E.; Saceda, M.; Grasso, S.; Rocamora-Reverte, L.; Conde, M.; Gómez-Martínez, Á.; García-Morales, P.; Ferragut, J.A.; Martínez-Lacaci, I. Small tyrosine kinase inhibitors interrupt EGFR signaling by interacting with erbB3 and erbB4 in glioblastoma cell lines. Exp. Cell Res. 2011, 317, 1476–1489. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.F.; Liu, Z.C.; Xie, B.F.; Li, Z.M.; Feng, G.K.; Yang, D.; Zeng, Y.X. EGFR tyrosine kinase inhibitor AG1478 inhibits cell proliferation and arrests cell cycle in nasopharyngeal carcinoma cells. Cancer Lett. 2001, 169, 27–32. [Google Scholar] [CrossRef]
- Giocanti, N.; Hennequin, C.; Rouillard, D.; Defrance, R.; Favaudon, V. Additive interaction of gefitinib (‘Iressa’, ZD1839) and ionising radiation in human tumour cells in vitro. Br. J. Cancer 2004, 91, 2026–2033. [Google Scholar] [CrossRef] [PubMed]
- Busse, D.; Doughty, R.S.; Ramsey, T.T.; Russell, W.E.; Price, J.O.; Flanagan, W.M.; Shawver, L.K.; Arteaga, C.L. Reversible G1 arrest induced by inhibition of the epidermal growth factor receptor tyrosine kinase requires up-regulation of p27(KIP1) independent of MAPK activity. J. Biol. Chem. 2000, 275, 6987–6995. [Google Scholar] [CrossRef] [PubMed]
- Kiyota, A.; Shintani, S.; Mihara, M.; Nakahara, Y.; Ueyama, Y.; Matsumura, T.; Tachikawa, T.; Wong, D.T. Anti-epidermal growth factor receptor monoclonal antibody 225 upregulates p27(KIP1) and p15(INK4B) and induces G1 arrest in oral squamous carcinoma cell lines. Oncology 2002, 63, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, A.J.; Fry, D.W. G1 cell cycle arrest due to the inhibition of erbB family receptor tyrosine kinases does not require the retinoblastoma protein. Exp. Cell Res. 2005, 303, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Huether, A.; Höpfner, M.; Sutter, A.P.; Schuppan, D.; Scherübl, H. Erlotinib induces cell cycle arrest and apoptosis in hepatocellular cancer cells and enhances chemosensitivity towards cytostatics. J. Hepatol. 2005, 43, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Markaverich, B.M.; Vijjeswarapu, M.; Shoulars, K.; Rodriguez, M. Luteolin and gefitinib regulation of EGF signaling pathway and cell cycle pathway genes in PC-3 human prostate cancer cells. J. Steroid Biochem. Mol. Biol. 2010, 122, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.H.; Jeong, E.H.; Lee, T.G.; Kim, S.Y.; Kim, H.R.; Kim, C.H. Gefitinib induces cytoplasmic translocation of the CDK inhibitor p27 and its binding to a cleaved intermediate of caspase 8 in non-small cell lung cancer cells. Cell. Oncol. 2014, 37, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Yan, H.; Zhou, Q. AG1478 inhibits the migration and invasion of cisplatin-resistant human lung adenocarcinoma cells via the cell cycle regulation by matrix metalloproteinase-9. Oncol. Lett. 2014, 8, 921–927. [Google Scholar] [CrossRef]
- Haraldsdottir, S.; Bekaii-Saab, T. Integrating anti-EGFR therapies in metastatic colorectal cancer. J. Gastrointest. Oncol. 2013, 4, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhou, S.; Li, X.; Wang, D.; Wang, Y.; Zhou, C. Schmid-Bindert, G. Gefitinib-resistance is related to BIM expression in non-small cell lung cancer cell lines. Cancer Biother. Radiopharm. 2013, 28, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Fu, L.-W. Redundant kinase activation and resistance of EGFR-tyrosine kinase inhibitors. Am. J. Cancer Res. 2014, 4, 608–628. [Google Scholar] [PubMed]
- Misale, S.; Arena, S.; Lamba, S.; Siravegna, G.; Lallo, A.; Hobor, S.; Russo, M.; Buscarino, M.; Lazzari, L.; Sartore-Bianchi, A.; et al. Blockade of EGFR and MEK intercepts heterogeneous mechanisms of acquired resistance to anti-EGFR therapies in colorectal cancer. Sci. Transl. Med. 2014, 6, 224ra26. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. Improved Survival with MEK Inhibition in BRAF-Mutated Melanoma. N. Engl. J. Med. 2012, 367, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.H.; Lee, J.H.; Chang, Y.J.; Tsai, H.H.; Lin, Y.L.; Lin, A.M.Y.; Yang, J.C.H. MEK inhibitors reverse resistance in epidermal growth factor receptor mutation lung cancer cells with acquired resistance to gefitinib. Mol. Oncol. 2013, 7, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Turke, A.B.; Song, Y.; Costa, C.; Cook, R.; Arteaga, C.L.; Asara, J.M.; Engelman, J.A. MEK inhibition leads to PI3K/AKT activation by relieving a negative feedback on ERBB receptors. Cancer Res. 2012, 72, 3228–3237. [Google Scholar] [CrossRef] [PubMed]
- Rexer, B.N.; Ghosh, R.; Arteaga, C.L. Inhibition of PI3K and MEK: It is all about combinations and biomarkers. Clin. Cancer Res. 2009, 15, 4518–4520. [Google Scholar] [CrossRef] [PubMed]
- Posch, C.; Moslehi, H.; Feeney, L.; Green, G.A.; Ebaee, A.; Feichtenschlager, V.; Chong, K.; Peng, L.; Dimon, M.T.; Phillips, T.; et al. Combined targeting of MEK and PI3K/mTOR effector pathways is necessary to effectively inhibit NRAS mutant melanoma in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2013, 110, 4015–4020. [Google Scholar] [CrossRef] [PubMed]
- Saini, K.S.; Loi, S.; de Azambuja, E.; Metzger-Filho, O.; Saini, M.L.; Ignatiadis, M.; Dancey, J.E.; Piccart-Gebhart, M.J. Targeting the PI3K/AKT/mTOR and Raf/MEK/ERK pathways in the treatment of breast cancer. Cancer Treat. Rev. 2013, 39, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef] [PubMed]
- Harrington, L.S.; Findlay, G.M.; Gray, A.; Tolkacheva, T.; Wigfield, S.; Rebholz, H.; Barnett, J.; Leslie, N.R.; Cheng, S.; Shepherd, P.R.; et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J. Cell Biol. 2004, 166, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Chandarlapaty, S.; Sawai, A.; Scaltriti, M.; Rodrik-Outmezguine, V.; Grbovic-Huezo, O.; Serra, V.; Serra, V.; Majumder, P.K.; Baselga, J.; Rosen, N. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell 2011, 19, 58–71. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, A.; Sanchez, V.; Kuba, M.G.; Rinehart, C.; Arteaga, C.L. Feedback upregulation of HER3 (ErbB3) expression and activity attenuates antitumor effect of PI3K inhibitors. Proc. Natl. Acad. Sci. USA 2012, 109, 2718–2723. [Google Scholar] [CrossRef] [PubMed]
- Serra, V.; Scaltriti, M.; Prudkin, L.; Eichhorn, P.J.; Ibrahim, Y.H.; Chandarlapaty, S.; Markman, B.; Rodriguez, O.; Guzman, M.; Rodriguez, S.; et al. PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-overexpressing breast cancer. Oncogene 2011, 30, 2547–2557. [Google Scholar] [CrossRef] [PubMed]
- Rodrik-Outmezguine, V.S.; Chandarlapaty, S.; Pagano, N.C.; Poulikakos, P.I.; Scaltriti, M.; Moskatel, E.; Moskatel, E.; Baselga, J.; Guichard, S.; Rosen, N. mTOR kinase inhibition causes feedback-dependent biphasic regulation of AKT signaling. Cancer Discov. 2011, 1, 248–259. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, K.E.; Rojo, F.; She, Q.-B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Amin, D.N.; Sergina, N.; Lim, L.; Goga, A.; Moasser, M.M. HER3 signalling is regulated through a multitude of redundant mechanisms in HER2-driven tumour cells. Biochem. J. 2012, 447, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.J.; Castel, P.; Radosevic-Robin, N.; Elkabets, M.; Auricchio, N.; Aceto, N.; Weitsman, G.; Barber, P.; Vojnovic, B.; Ellis, H.; et al. Antagonism of EGFR and HER3 enhances the response to inhibitors of the PI3K-Akt pathway in triple-negative breast cancer. Sci. Signal. 2014, 7, ra29. [Google Scholar] [CrossRef] [PubMed]
- Garrett, J.T.; Sutton, C.R.; Kurupi, R.; Bialucha, C.U.; Ettenberg, S.A.; Collins, S.D.; Sheng, Q.; Wallweber, J.; Defazio-Eli, L.; Arteaga, C.L. Combination of antibody that inhibits ligand-independent HER3 dimerization and a p110?? inhibitor potently blocks pi3k signaling and growth of HER2+ breast cancers. Cancer Res. 2013, 73, 6013–6023. [Google Scholar] [CrossRef] [PubMed]
- Guertin, D.A.; Sabatini, D.M. Defining the Role of mTOR in Cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Rehman, S.K.; Zhang, W.; Wen, A.; Yao, L.; Zhang, J. Autophagy is a therapeutic target in anticancer drug resistance. Biochim. Biophys. Acta 2010, 1806, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wu, Z.; Wong, G.; Pectasides, E.; Nagaraja, A.; Stachler, M.; Zhang, H.; Chen, T.; Liu, D. CDK4/6 or MAPK blockade enhances efficacy of EGFR inhibition in oesophageal squamous cell carcinoma. Nat. Commun. 2017, 8, 13897. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.M.; Kazlauskas, A. Growth-factor-dependent mitogenesis requires two distinct phases of signalling. Nat. Cell Biol. 2001, 3, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Pennock, S.; Wang, Z.X. Stimulation of cell proliferation by endosomal epidermal growth factor receptor as revealed through two distinct phases of signaling. Mol. Cell. Biol. 2003, 23, 5803–5815. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V.; Pardee, A.B. The restriction point of the cell cycle. Cell Cycle 2002, 1, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Shimamura, T.; Monti, S.; Steidl, U.; Hetherington, C.J.; Lowell, A.M.; Golub, T.; Meyerson, M.; Tenen, D.G.; Shapiro, G.L.; et al. Transcriptional profiling identifies Cyclin D1 as a critical downstream effector of mutant epidermal growth factor receptor signaling. Cancer Res. 2006, 66, 11389–11398. [Google Scholar] [CrossRef] [PubMed]
- Reissmann, P.T.; Koga, H.; Figlin, R.A.; Holmes, E.C.; Slamon, D.J. Amplification and overexpression of the Cyclin D1 and epidermal growth factor receptor genes in non-small-cell lung cancer. Lung Cancer Study Group. J. Cancer Res. Clin. Oncol. 1999, 125, 61–70. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, G.G.; Anderson, J.J.; Milton, I.; Steward, M.; Parr, A.H.; Thomas, M.D.; Henry, J.A.; Angus, B.; Lennard, T.W.; Horne, C.H. Determination of the prognostic value of Cyclin D1 overexpression in breast cancer. Oncogene 1995, 11, 885–891. [Google Scholar] [PubMed]
- Perry, J.E.; Grossmann, M.E.; Tindall, D.J. Epidermal growth factor induces Cyclin D1 in a human prostate cancer cell line. Prostate 1998, 35, 117–124. [Google Scholar] [CrossRef]
- Narayanan, R.; Kim, H.N.; Narayanan, N.K.; Nargi, D.; Narayanan, B.A. Epidermal growth factor-stimulated human cervical cancer cell growth is associated with EGFR and Cyclin D1 activation, independent of COX-2 expression levels. Int. J. Oncol. 2012, 40, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.J.; Luan, J.; Zhang, H.S.; Ruan, C.P.; Xu, X.Y.; Li, Q.Q.; Wang, N.H. EGFR-mediated G1/S transition contributes to the multidrug resistance in breast cancer cells. Mol. Biol. Rep. 2012, 39, 5465–5471. [Google Scholar] [CrossRef] [PubMed]
- Cobrinik, D. Pocket proteins and cell cycle control. Oncogene 2005, 24, 2796–2809. [Google Scholar] [CrossRef] [PubMed]
- Rubin, S.M. Deciphering the retinoblastoma protein phosphorylation code. Trends Biochem. Sci. 2013, 38, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, M.; Brill, J.A.; Fink, G.R.; Weinberg, R.A. Collaboration of G1 cyclins in the functional inactivation of the retinoblastoma protein. Genes Dev. 1994, 8, 1759–1771. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Ebisuya, M.; Ashida, F.; Okamoto, K.; Yonehara, S.; Nishida, E. Continuous ERK Activation Downregulates Antiproliferative Genes throughout G1 Phase to Allow Cell-Cycle Progression. Curr. Biol. 2006, 16, 1171–1182. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Klippel, A.; Muslin, A.J.; Fantl, W.J.; Williams, L.T.; Anthony, J.; Wendy, J.; Lewis, T.; Hu, Q.; Klippel, A.; et al. Ras-dependent induction of cellular responses by constitutively active phosphatidylinositol-3 kinase. Science 1995, 268, 100–102. [Google Scholar] [CrossRef] [PubMed]
- Ghosh Choudhury, G.; Kim, Y.S.; Simon, M.; Wozney, J.; Harris, S.; Ghosh-Choudhury, N.; Abboud, H.E.; Ghosh Choundhury, G.; Ghosh-Choundhury, N. Bone morphogenetic protein 2 inhibits platelet-derived growth factor-induced c-fos gene transcription and DNA synthesis in mesangial cells. Involvement of mitogen-activated protein kinase. J. Biol. Chem. 1999, 274, 10897–10902. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Fernandez de Mattos, S.; van der Horst, A.; Klompmaker, R.; Kops, G.J.P.L.; Lam, E.W.-F.; Burgering, B.M.T.; Medema, R.H. Cell cycle inhibition by FoxO forkhead transcription factors involves downregulation of cyclin D. Mol. Cell. Biol. 2002, 22, 7842–7852. [Google Scholar] [CrossRef] [PubMed]
- Torii, S.; Yamamoto, T.; Tsuchiya, Y.; Nishida, E. ERK MAP kinase in G1 cell cycle progression and cancer. Cancer Sci. 2006, 97, 697–702. [Google Scholar] [CrossRef] [PubMed]
- Diehl, J.A.; Cheng, M.; Roussel, M.F.; Sherr, C.J. Glycogen synthase kinase-3beta regulates Cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998, 12, 3499–3511. [Google Scholar] [CrossRef] [PubMed]
- Welcker, M.; Singer, J.; Loeb, K.R.; Grim, J.; Bloecher, A.; Gurien-West, M.; Clurman, B.E.; Roberts, J.M. Multisite phosphorylation by Cdk2 and GSK3 controls cyclin E degradation. Mol. Cell 2003, 12, 381–392. [Google Scholar] [CrossRef]
- Tullai, J.W.; Graham, J.R.; Cooper, G.M. A GSK-3-mediated transcriptional network maintains repression of immediate early genes in quiescent cells. Cell Cycle 2011, 10, 3072–3077. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, C.; Leighton, I.A.; Cohen, P. Inactivation of glycogen synthase kinase-3fl by phosphorylation: New kinase connections in insulin and growth-factor signalling. Biochem. J. 1993, 296, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Stambolic, V.; Woodgett, J.R. Mitogen inactivation of glycogen synthase kinase-3 beta in intact cells via serine 9 phosphorylation. Biochem. J. 1994, 303 (Pt 3), 701–704. [Google Scholar] [CrossRef] [PubMed]
- Waskiewicz, A.J.; Flynn, A.; Proud, C.G.; Cooper, J.A. Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J. 1997, 16, 1909–1920. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Flynn, A.; Waskiewicz, A.J.; Webb, B.L.J.; Vries, R.G.; Baines, I.A.; Cooper, J.A.; Proud, C.G. The phosphorylation of eukaryotic initiation factor eIF4E in response to phorbol esters, cell stresses, and cytokines is mediated by distinct MAP kinase pathways. J. Biol. Chem. 1998, 273, 9373–9377. [Google Scholar] [CrossRef] [PubMed]
- Fry, D.W.; Harvey, P.J.; Keller, P.R.; Elliott, W.L.; Meade, M.; Trachet, E.; Albassam, M.; Zheng, X.; Leopold, W.R.; Pryer, N.K.; et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol. Cancer Ther. 2004, 3, 1427–1438. [Google Scholar] [PubMed]
- Toogood, P.L.; Harvey, P.J.; Repine, J.T.; Sheehan, D.J.; VanderWel, S.N.; Zhou, H.; Keller, P.R.; McNamara, D.J.; Sherry, D.; Zhu, T.; et al. Discovery of a potent and selective inhibitor of cyclin-dependent kinase 4/6. J. Med. Chem. 2005, 48, 2388–2406. [Google Scholar] [CrossRef] [PubMed]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [PubMed]
- An, H.X.; Beckmann, M.W.; Reifenberger, G.; Bender, H.G.; Niederacher, D. Gene amplification and overexpression of CDK4 in sporadic breast carcinomas is associated with high tumor cell proliferation. Am. J. Pathol. 1999, 154, 113–118. [Google Scholar] [CrossRef]
- Mayer, E.L. Targeting Breast Cancer with CDK Inhibitors. Curr. Oncol. Rep. 2015, 17, 443. [Google Scholar] [CrossRef] [PubMed]
- Toyoshima, H.; Hunter, T. p27, A novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell 1994, 78, 67–74. [Google Scholar] [CrossRef]
- Cheng, M.; Sexl, V.; Sherr, C.J.; Roussel, M.F. Assembly of cyclin D-dependent kinase and titration of p27KIP1 regulated by mitogen-activated protein kinase kinase (MEK1). Proc. Natl. Acad. Sci. USA 1998, 95, 1091–1096. [Google Scholar] [CrossRef] [PubMed]
- Katayose, Y.; Kim, M.; Rakkar, A.N.S.; Li, Z.; Cowan, K.H.; Seth, P. Promoting apoptosis: A novel activity associated with the cyclin- dependent kinase inhibitor p27. Cancer Res. 1997, 57, 5441–5445. [Google Scholar] [PubMed]
- Nickeleit, I.; Zender, S.; Kossatz, U.; Malek, N.P. P27KIP1: A Target for Tumor Therapies? Cell Div. 2007, 2, 13. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Meng, Y.; Li, J.; Zhang, D.; Yu, Y.; Lepor, H.; Wu, X.R.; Huang, H. p27KIP1 down-regulates egfr expression via inhibition of JNK/C-JUN transactivation: A novel function of p27KIP1 in tumor suppression deficient in metastatic bladder cancer cells. J. Urol. 2013, 189, e463. [Google Scholar] [CrossRef]
- Fang, Y.; Wang, Y.; Wang, Y.; Meng, Y.; Zhu, J.; Jin, H.; Li, J.; Zhang, D.; Yu, Y.; Wu, X.-R.; et al. A new tumour suppression mechanism by p27Kip1: EGFR down-regulation mediated by JNK/c-Jun pathway inhibition. Biochem. J. 2014, 463, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Rivard, N.; Boucher, M.-J.; Asselin, C.; L’Allemain, G. MAP kinase cascade is required for p27 downregulation and S phase entry in fibroblasts and epithelial cells. Am. J. Physiol. 1999, 277, C652–C664. [Google Scholar] [PubMed]
- Delmas, C.; Manenti, S.; Boudjelal, A.; Peyssonnaux, C.; Eychène, A.; Darbon, J.M. The p42/p44 mitogen-activated protein kinase activation triggers p27KIP1 degradation independently of CDK2/cyclin E in NIH 3T3 cells. J. Biol. Chem. 2001, 276, 34958–34965. [Google Scholar] [CrossRef] [PubMed]
- Kortylewski, M.; Heinrich, P.C.; Kauffmann, M.E.; Bohm, M.; Mackiewicz, A.; Behrmann, I. Mitogen-activated protein kinases control p27/Kip1 expression and growth of human melanoma cells. Biochem. J. 2001, 357, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Kawada, M.; Yamagoe, S.; Murakami, Y.; Suzuki, K.; Mizuno, S.; Uehara, Y. Induction of p27KIP1 degradation and anchorage independence by Ras through the MAP kinase signaling pathway. Oncogene 1997, 15, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Cetinkaya, C.; Munoz-Alonso, M.J.; von der Lehr, N.; Bahram, F.; Beuger, V.; Eilers, M.; Leon, J.; Larsson, L.-G. Myc represses differentiation-induced p21CIP1 expression via Miz-1-dependent interaction with the p21 core promoter. Oncogene 2003, 22, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Sahin, O.; Fröhlich, H.; Löbke, C.; Korf, U.; Burmester, S.; Majety, M.; Mattern, J.; Schupp, I.; Chaouiya, C.; Thieffry, D.; et al. Modeling ERBB receptor-regulated G1/S transition to find novel targets for de novo trastuzumab resistance. BMC Syst. Biol. 2009, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Zubovitz, J.; Petrocelli, T.; Kotchetkov, R.; Connor, M.K.; Han, K.; Lee, J.-H.; Ciarallo, S.; Catzavelos, C.; Beniston, R.; et al. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat. Med. 2002, 8, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Katayama, K.; Fujita, N.; Tsuruo, T. Akt/protein kinase B-dependent phosphorylation and inactivation of WEE1Hu promote cell cycle progression at G2/M transition. Mol. Cell. Biol. 2005, 25, 5725–5737. [Google Scholar] [CrossRef] [PubMed]
- Medema, R.H.; Kops, G.J.; Bos, J.L.; Burgering, B.M. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature 2000, 404, 782–787. [Google Scholar] [CrossRef] [PubMed]
- Dijkers, P.F.; Medema, R.H.; Pals, C.; Banerji, L.; Thomas, N.S.; Lam, E.W.; Burgering, B.M.; Raaijmakers, J.A.; Lammers, J.W.; Koenderman, L.; et al. Forkhead transcription factor FKHR-L1 modulates cytokine-dependent transcriptional regulation of p27(KIP1). Mol. Cell. Biol. 2000, 20, 9138–9148. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhou, Y.; Wang, X.; Evers, B.M. p27KIP1 nuclear localization and cyclin-dependent kinase inhibitory activity are regulated by glycogen synthase kinase-3 in human colon cancer cells. Cell Death Differ. 2008, 15, 908–919. [Google Scholar] [CrossRef] [PubMed]
- Goh, L.K.; Huang, F.; Kim, W.; Gygi, S.; Sorkin, A. Multiple mechanisms collectively regulate clathrin-mediated endocytosis of the epidermal growth factor receptor. J. Cell Biol. 2010, 189, 871–883. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chen, X.; Wang, Z. Dimerization drives EGF receptor endocytosis through two sets of compatible endocytic codes. J. Cell Sci. 2015, 128, 935–950. [Google Scholar] [CrossRef] [PubMed]
- Levkowitz, G.; Waterman, H.; Zamir, E.; Kam, Z.; Oved, S.; Langdon, W.Y.; Beguinot, L.; Geiger, B.; Yarden, Y. c-Cb1/Sli-1 regulates endocytic sorting and ubiquitination of the epidermal growth factor receptor. Genes Dev. 1998, 12, 3663–3674. [Google Scholar] [CrossRef] [PubMed]
- Sousa, L.P.; Lax, I.; Shen, H.; Ferguson, S.M.; De Camilli, P.; Schlessinger, J. Suppression of EGFR endocytosis by dynamin depletion reveals that EGFR signaling occurs primarily at the plasma membrane. Proc. Natl. Acad. Sci. USA 2012, 109, 4419–4424. [Google Scholar] [CrossRef] [PubMed]
- Noh, T.; Kook, Y.H.; Park, C.; Youn, H.; Kim, H.; Oh, E.T.; Beguinot, L.; Geiger, B.; Yarden, Y. Block copolymer micelles conjugated with anti-EGFR antibody for targeted delivery of anticancer drug. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 7321–7331. [Google Scholar] [CrossRef]
- Maya, S.; Sarmento, B.; Lakshmanan, V.K.; Menon, D.; Jayakumar, R. Actively targeted cetuximab conjugated gamma-poly(glutamic acid)-docetaxel nanomedicines for epidermal growth factor receptor over expressing colon cancer cells. J. Biomed. Nanotechnol. 2014, 10, 1416–1428. [Google Scholar] [CrossRef] [PubMed]
- Wee, P.; Shi, H.; Jiang, J.; Wang, Y.; Wang, Z. EGF stimulates the activation of EGF receptors and the selective activation of major signaling pathways during mitosis. Cell Signal. 2015, 27, 638–651. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Shi, H.; Chen, X.; Wang, Z. Regulation of EGF-stimulated EGF receptor endocytosis during M phase. Traffic 2011, 12, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Dangi, S.; Shapiro, P. Cdc2-mediated inhibition of epidermal growth factor activation of the extracellular signal-regulated kinase pathway during mitosis. J. Biol. Chem. 2005, 280, 24524–24531. [Google Scholar] [CrossRef] [PubMed]
- Kiyokawa, N. Mitosis-specific Negative Regulation of Epidermal Growth Factor Receptor, Triggered by a Decrease in Ligand Binding and Dimerization, Can Be Overcome by Overexpression of Receptor. J. Biol. Chem. 1997, 272, 18656–18665. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. https://doi.org/10.3390/cancers9050052
Wee P, Wang Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers. 2017; 9(5):52. https://doi.org/10.3390/cancers9050052
Chicago/Turabian StyleWee, Ping, and Zhixiang Wang. 2017. "Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways" Cancers 9, no. 5: 52. https://doi.org/10.3390/cancers9050052