
Macrophages Promote Tumor Cell Extravasation across an Endothelial Barrier through Thin Membranous Connections
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Media
2.2. Antibodies and Reagents
2.3. Mice
2.4. Western Blot
2.5. Extravasation Trans-Endothelial Migration (eTEM) Assay
2.6. TMP Quantitation and Proximity Analysis
2.7. TMC Quantitation
2.8. Quantification of Lung Metastasis
2.9. Window for High Resolution Imaging of the Lung (WHRIL) and Intravital Imaging
3. Results
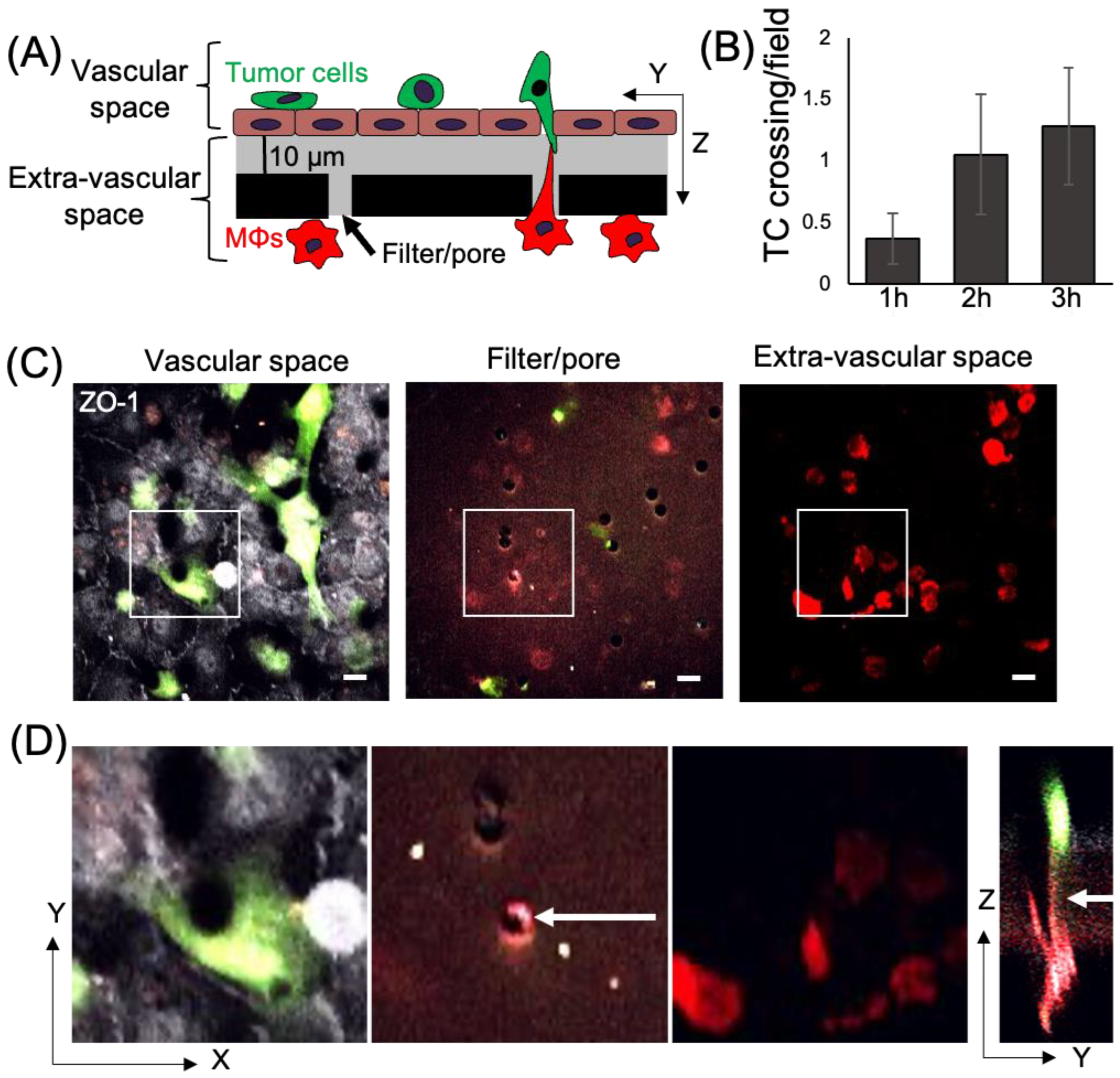
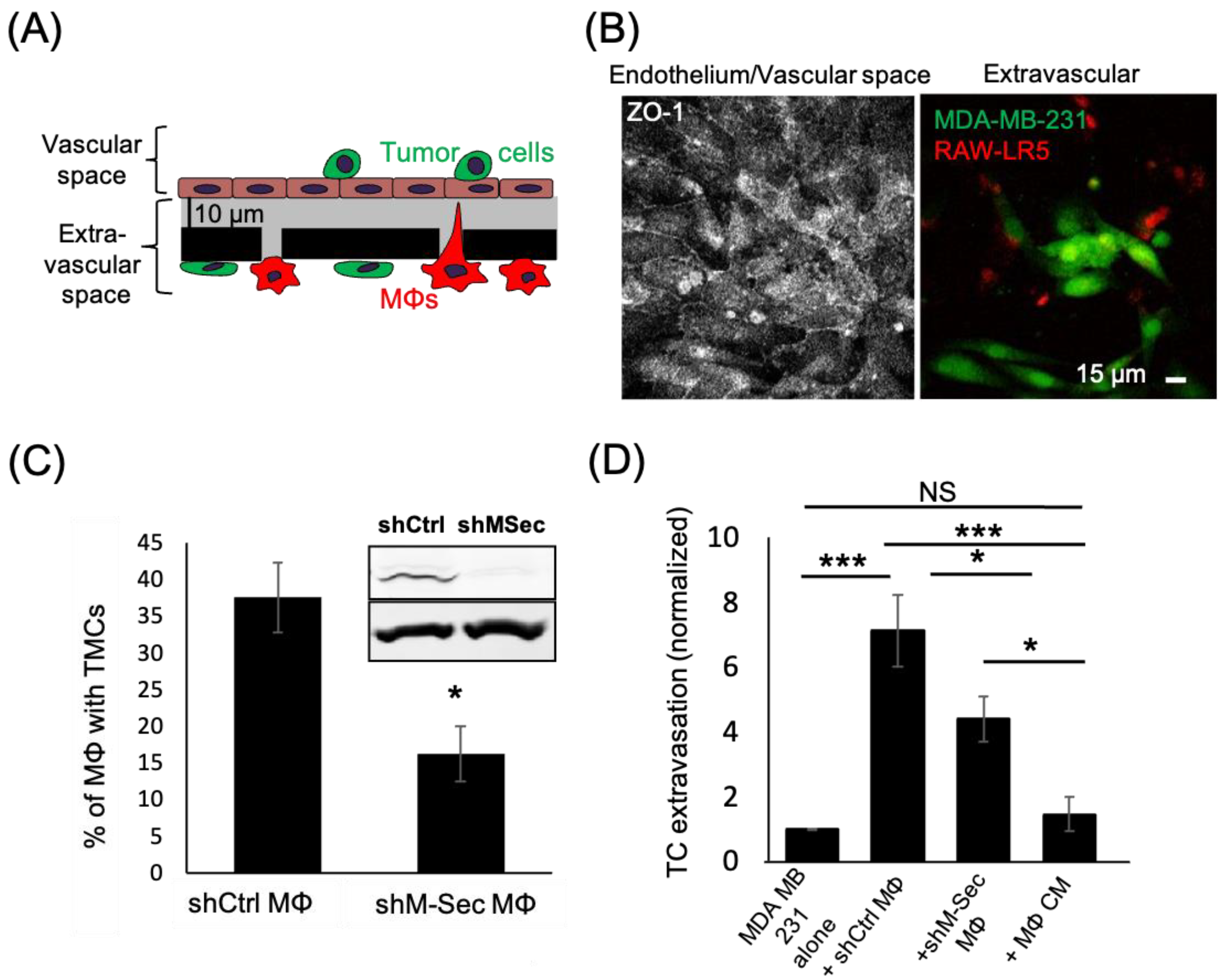
3.1. Macrophage TMCs Are Important in Promoting Tumor Cell Extravasation In Vitro
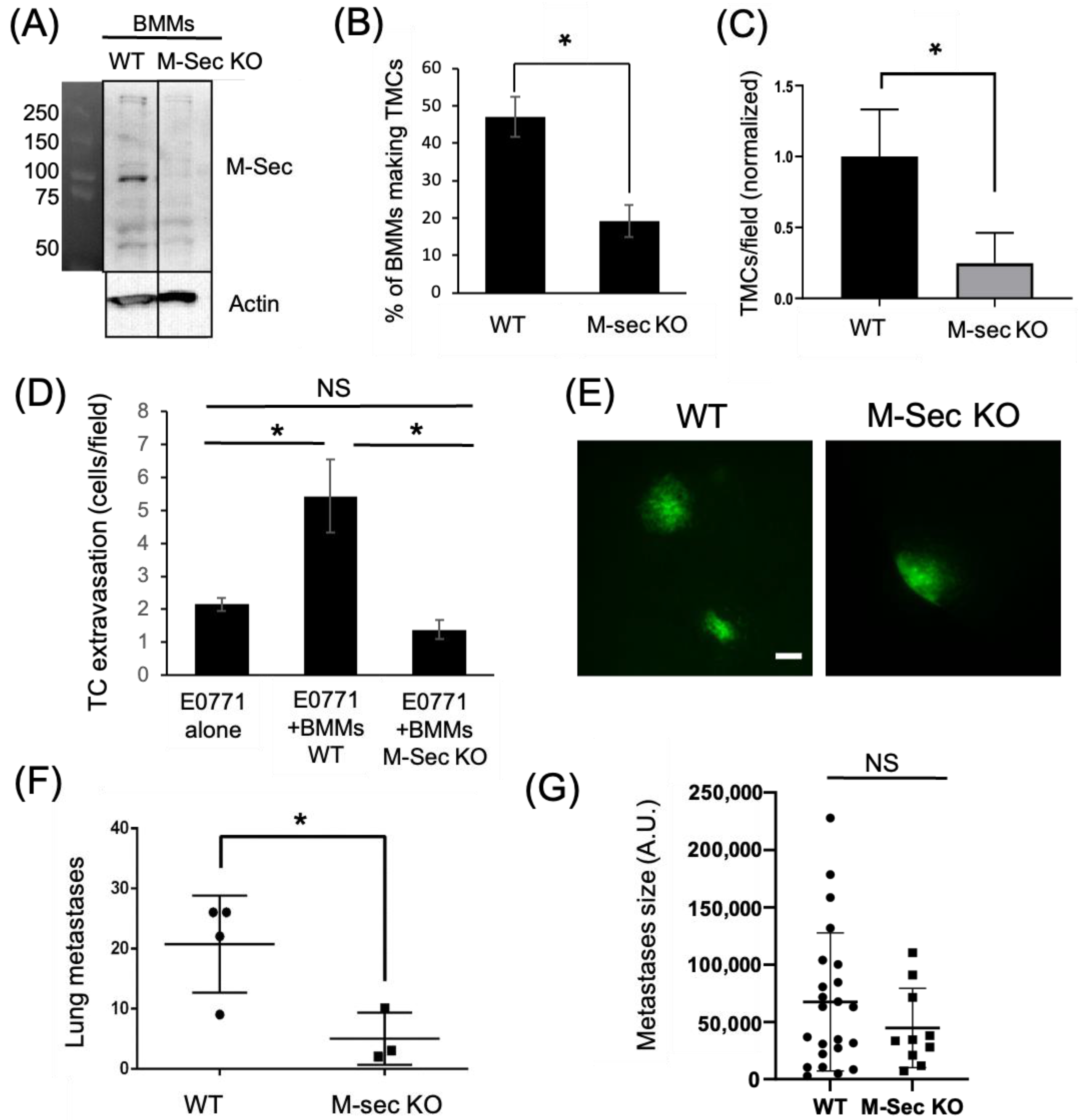
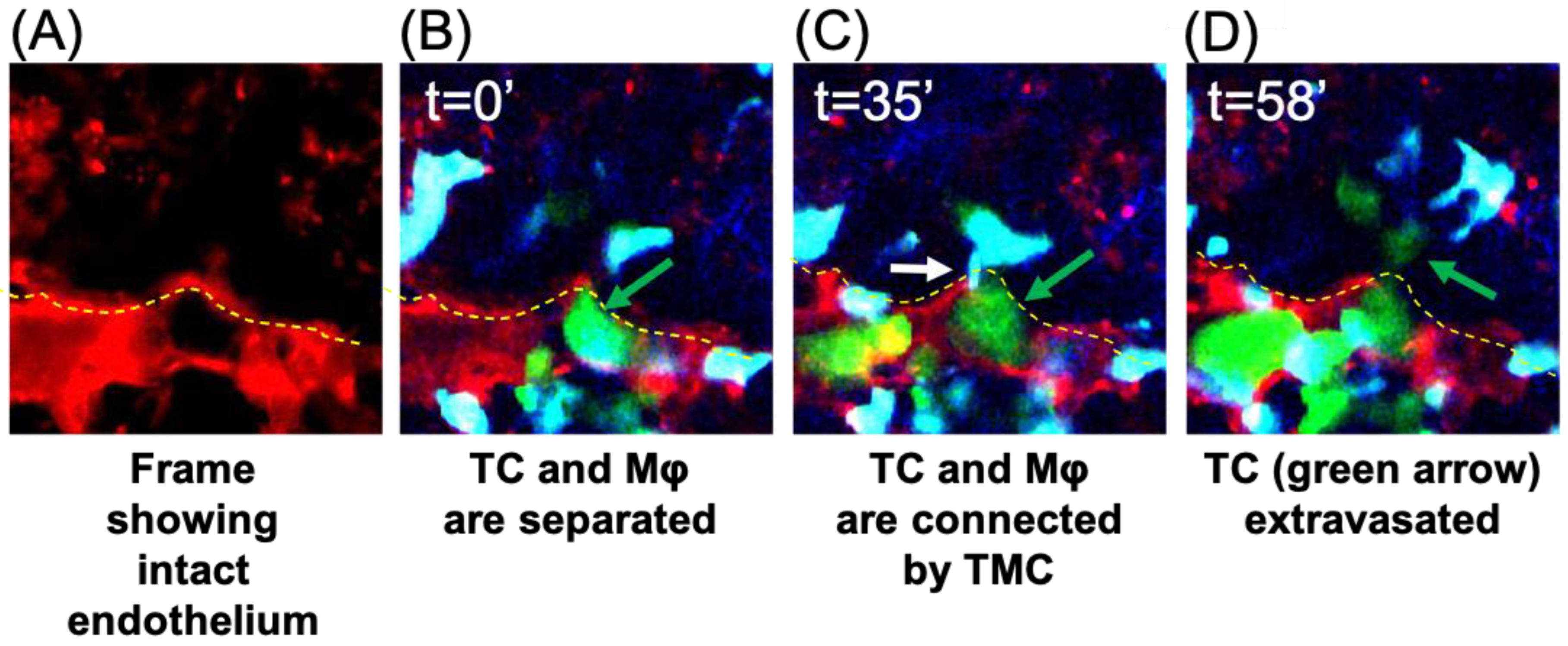
3.2. TMCs Promote Tumor Cell Extravasation In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Wyckoff, J.; Wang, W.; Lin, E.Y.; Wang, Y.; Pixley, F.; Stanley, E.R.; Graf, T.; Pollard, J.W.; Segall, J.; Condeelis, J. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 2004, 64, 7022–7029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Condeelis, J.; Pollard, J.W. Macrophages: Obligate partners for tumor cell migration, invasion, and metastasis. Cell 2006, 124, 263–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karagiannis, G.S.; Condeelis, J.S.; Oktay, M.H. Chemotherapy-Induced Metastasis: Molecular Mechanisms, Clinical Manifestations, Therapeutic Interventions. Cancer Res. 2019, 79, 4567–4576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, V.P.; Tang, B.; Wang, Y.; Duran, C.L.; Karagiannis, G.S.; Xue, E.A.; Entenberg, D.; Borriello, L.; Coste, A.; Eddy, R.J.; et al. Live tumor imaging shows macrophage induction and TMEM-mediated enrichment of cancer stem cells during metastatic dissemination. Nat. Commun. 2021, 12, 7300. [Google Scholar] [CrossRef]
- Borriello, L.; Karagiannis, G.S.; Duran, C.L.; Coste, A.; Oktay, M.H.; Entenberg, D.; Condeelis, J.S. The role of the tumor microenvironment in tumor cell intravasation and dissemination. Eur. J. Cell Biol. 2020, 99, 151098. [Google Scholar] [CrossRef]
- Bingle, L.; Brown, N.J.; Lewis, C.E. The role of tumour-associated macrophages in tumour progression: Implications for new anticancer therapies. J. Pathol. 2002, 196, 254–265. [Google Scholar] [CrossRef]
- Karagiannis, G.S.; Pastoriza, J.M.; Wang, Y.; Harney, A.S.; Entenberg, D.; Pignatelli, J.; Sharma, V.P.; Xue, E.A.; Cheng, E.; D’Alfonso, T.M.; et al. Neoadjuvant chemotherapy induces breast cancer metastasis through a TMEM-mediated mechanism. Sci. Transl. Med. 2017, 9, eaan0026. [Google Scholar] [CrossRef] [Green Version]
- Goswami, S.; Sahai, E.; Wyckoff, J.B.; Cammer, M.; Cox, D.; Pixley, F.J.; Stanley, E.R.; Segall, J.E.; Condeelis, J.S. Macrophages promote the invasion of breast carcinoma cells via a colony-stimulating factor-1/epidermal growth factor paracrine loop. Cancer Res. 2005, 65, 5278–5283. [Google Scholar] [CrossRef] [Green Version]
- Roussos, E.T.; Balsamo, M.; Alford, S.K.; Wyckoff, J.B.; Gligorijevic, B.; Wang, Y.; Pozzuto, M.; Stobezki, R.; Goswami, S.; Segall, J.E.; et al. Mena invasive (MenaINV) promotes multicellular streaming motility and transendothelial migration in a mouse model of breast cancer. J. Cell Sci. 2011, 124, 2120–2131. [Google Scholar] [CrossRef] [Green Version]
- Leung, E.; Xue, A.; Wang, Y.; Rougerie, P.; Sharma, V.P.; Eddy, R.; Cox, D.; Condeelis, J. Blood vessel endothelium-directed tumor cell streaming in breast tumors requires the HGF/C-Met signaling pathway. Oncogene 2017, 36, 2680–2692. [Google Scholar] [CrossRef] [Green Version]
- Harney, A.S.; Arwert, E.N.; Entenberg, D.; Wang, Y.; Guo, P.; Qian, B.Z.; Oktay, M.H.; Pollard, J.W.; Jones, J.G.; Condeelis, J.S. Real-Time Imaging Reveals Local, Transient Vascular Permeability, and Tumor Cell Intravasation Stimulated by TIE2hi Macrophage-Derived VEGFA. Cancer Discov. 2015, 5, 932–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borriello, L.; Coste, A.; Traub, B.; Sharma, V.P.; Karagiannis, G.S.; Lin, Y.; Wang, Y.; Ye, X.; Duran, C.L.; Chen, X.; et al. Primary tumor associated macrophages activate programs of invasion and dormancy in disseminating tumor cells. Nat. Commun. 2022, 13, 626. [Google Scholar] [CrossRef]
- Lin, E.Y.; Nguyen, A.V.; Russell, R.G.; Pollard, J.W. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J. Exp. Med. 2001, 193, 727–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, E.Y.; Jones, J.G.; Li, P.; Zhu, L.; Whitney, K.D.; Muller, W.J.; Pollard, J.W. Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am. J. Pathol. 2003, 163, 2113–2126. [Google Scholar] [CrossRef] [Green Version]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [Green Version]
- Qian, B.Z.; Pollard, J.W. New tricks for metastasis-associated macrophages. Breast Cancer Res. 2012, 14, 316. [Google Scholar] [CrossRef] [Green Version]
- Qian, B.; Deng, Y.; Im, J.H.; Muschel, R.J.; Zou, Y.; Li, J.; Lang, R.A.; Pollard, J.W. A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PLoS ONE 2009, 4, e6562. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, T.; Qian, B.Z.; Soong, D.; Cassetta, L.; Noy, R.; Sugano, G.; Kato, Y.; Li, J.; Pollard, J.W. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J. Exp. Med. 2015, 212, 1043–1059. [Google Scholar] [CrossRef]
- Rodriguez-Tirado, C.; Entenberg, D.; Li, J.; Qian, B.Z.; Condeelis, J.S.; Pollard, J.W. Interleukin 4 Controls the Pro-Tumoral Role of Macrophages in Mammary Cancer Pulmonary Metastasis in Mice. Cancers 2022, 14, 4336. [Google Scholar] [CrossRef] [PubMed]
- Entenberg, D.; Rodriguez-Tirado, C.; Kato, Y.; Kitamura, T.; Pollard, J.W.; Condeelis, J. In vivo subcellular resolution optical imaging in the lung reveals early metastatic proliferation and motility. Intravital 2015, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Entenberg, D.; Voiculescu, S.; Guo, P.; Borriello, L.; Wang, Y.; Karagiannis, G.S.; Jones, J.; Baccay, F.; Oktay, M.; Condeelis, J. A permanent window for the murine lung enables high-resolution imaging of cancer metastasis. Nat. Methods 2018, 15, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Chinnery, H.R.; Pearlman, E.; McMenamin, P.G. Cutting edge: Membrane nanotubes in vivo: A feature of MHC class II+ cells in the mouse cornea. J. Immunol. 2008, 180, 5779–5783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehberg, M.; Nekolla, K.; Sellner, S.; Praetner, M.; Mildner, K.; Zeuschner, D.; Krombach, F. Intercellular Transport of Nanomaterials is Mediated by Membrane Nanotubes In Vivo. Small 2016, 12, 1882–1890. [Google Scholar] [CrossRef] [PubMed]
- Alarcon-Martinez, L.; Villafranca-Baughman, D.; Quintero, H.; Kacerovsky, J.B.; Dotigny, F.; Murai, K.K.; Prat, A.; Drapeau, P.; Di Polo, A. Interpericyte tunnelling nanotubes regulate neurovascular coupling. Nature 2020, 585, 91–95. [Google Scholar] [CrossRef]
- Lou, E.; Fujisawa, S.; Barlas, A.; Romin, Y.; Manova-Todorova, K.; Moore, M.A.; Subramanian, S. Tunneling Nanotubes: A new paradigm for studying intercellular communication and therapeutics in cancer. Commun. Integr. Biol. 2012, 5, 399–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M.; et al. Brain tumour cells interconnect to a functional and resistant network. Nature 2015, 528, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Weil, S.; Osswald, M.; Solecki, G.; Grosch, J.; Jung, E.; Lemke, D.; Ratliff, M.; Hanggi, D.; Wick, W.; Winkler, F. Tumor microtubes convey resistance to surgical lesions and chemotherapy in gliomas. Neuro Oncol. 2017, 19, 1316–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abounit, S.; Zurzolo, C. Wiring through tunneling nanotubes--from electrical signals to organelle transfer. J. Cell Sci. 2012, 125, 1089–1098. [Google Scholar] [CrossRef] [Green Version]
- Lou, E.; Fujisawa, S.; Morozov, A.; Barlas, A.; Romin, Y.; Dogan, Y.; Gholami, S.; Moreira, A.L.; Manova-Todorova, K.; Moore, M.A. Tunneling nanotubes provide a unique conduit for intercellular transfer of cellular contents in human malignant pleural mesothelioma. PLoS ONE 2012, 7, e33093. [Google Scholar] [CrossRef] [Green Version]
- Jackson, M.V.; Morrison, T.J.; Doherty, D.F.; McAuley, D.F.; Matthay, M.A.; Kissenpfennig, A.; O’Kane, C.M.; Krasnodembskaya, A.D. Mitochondrial Transfer via Tunneling Nanotubes is an Important Mechanism by Which Mesenchymal Stem Cells Enhance Macrophage Phagocytosis in the In Vitro and In Vivo Models of ARDS. Stem Cells 2016, 34, 2210–2223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burt, R.; Dey, A.; Aref, S.; Aguiar, M.; Akarca, A.; Bailey, K.; Day, W.; Hooper, S.; Kirkwood, A.; Kirschner, K.; et al. Activated stromal cells transfer mitochondria to rescue acute lymphoblastic leukemia cells from oxidative stress. Blood 2019, 134, 1415–1429. [Google Scholar] [CrossRef] [PubMed]
- Hanna, S.J.; McCoy-Simandle, K.; Leung, E.; Genna, A.; Condeelis, J.; Cox, D. Tunneling nanotubes, a novel mode of tumor cell-macrophage communication in tumor cell invasion. J. Cell Sci. 2019, 132, jcs223321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCoy-Simandle, K.; Hanna, S.J.; Cox, D. Exosomes and nanotubes: Control of immune cell communication. Int. J. Biochem. Cell Biol. 2016, 71, 44–54. [Google Scholar] [CrossRef] [Green Version]
- Roehlecke, C.; Schmidt, M.H.H. Tunneling Nanotubes and Tumor Microtubes in Cancer. Cancers 2020, 12, 857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hase, K.; Kimura, S.; Takatsu, H.; Ohmae, M.; Kawano, S.; Kitamura, H.; Ito, M.; Watarai, H.; Hazelett, C.C.; Yeaman, C.; et al. M-Sec promotes membrane nanotube formation by interacting with Ral and the exocyst complex. Nat. Cell Biol. 2009, 11, 1427–1432. [Google Scholar] [CrossRef]
- Kimura, S.; Yamashita, M.; Yamakami-Kimura, M.; Sato, Y.; Yamagata, A.; Kobashigawa, Y.; Inagaki, F.; Amada, T.; Hase, K.; Iwanaga, T.; et al. Distinct Roles for the N- and C-terminal Regions of M-Sec in Plasma Membrane Deformation during Tunneling Nanotube Formation. Sci. Rep. 2016, 6, 33548. [Google Scholar] [CrossRef] [Green Version]
- Cox, D.; Chang, P.; Zhang, Q.; Reddy, P.G.; Bokoch, G.M.; Greenberg, S. Requirements for both Rac1 and Cdc42 in membrane ruffling and phagocytosis in leukocytes. J. Exp. Med. 1997, 186, 1487–1494. [Google Scholar] [CrossRef]
- Stanley, E.R. Murine bone marrow-derived macrophages. Methods Mol. Biol. 1997, 75, 301–304. [Google Scholar] [CrossRef]
- Barutta, F.; Kimura, S.; Hase, K.; Bellini, S.; Corbetta, B.; Corbelli, A.; Fiordaliso, F.; Barreca, A.; Papotti, M.G.; Ghiggeri, G.M.; et al. Protective Role of the M-Sec-Tunneling Nanotube System in Podocytes. J. Am. Soc. Nephrol. 2021, 32, 1114–1130. [Google Scholar] [CrossRef]
- Entenberg, D.; Wyckoff, J.; Gligorijevic, B.; Roussos, E.T.; Verkhusha, V.V.; Pollard, J.W.; Condeelis, J. Setup and use of a two-laser multiphoton microscope for multichannel intravital fluorescence imaging. Nat. Protoc. 2011, 6, 1500–1520. [Google Scholar] [CrossRef] [Green Version]
- Borriello, L.; Traub, B.; Coste, A.; Oktay, M.H.; Entenberg, D. A Permanent Window for Investigating Cancer Metastasis to the Lung. J. Vis. Exp. 2021, 173, e62761. [Google Scholar] [CrossRef]
- Hanna, S.J.; McCoy-Simandle, K.; Miskolci, V.; Guo, P.; Cammer, M.; Hodgson, L.; Cox, D. The Role of Rho-GTPases and actin polymerization during Macrophage Tunneling Nanotube Biogenesis. Sci. Rep. 2017, 7, 8547. [Google Scholar] [CrossRef] [PubMed]
- Dillekas, H.; Rogers, M.S.; Straume, O. Are 90% of deaths from cancer caused by metastases? Cancer Med. 2019, 8, 5574–5576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rescigno, M.; Rotta, G.; Valzasina, B.; Ricciardi-Castagnoli, P. Dendritic cells shuttle microbes across gut epithelial monolayers. Immunobiology 2001, 204, 572–581. [Google Scholar] [CrossRef]
- Naphade, S.; Sharma, J.; Gaide Chevronnay, H.P.; Shook, M.A.; Yeagy, B.A.; Rocca, C.J.; Ur, S.N.; Lau, A.J.; Courtoy, P.J.; Cherqui, S. Brief reports: Lysosomal cross-correction by hematopoietic stem cell-derived macrophages via tunneling nanotubes. Stem Cells 2015, 33, 301–309. [Google Scholar] [CrossRef] [Green Version]
- Carter, K.P.; Hanna, S.; Genna, A.; Lewis, D.; Segall, J.E.; Cox, D. Macrophages enhance 3D invasion in a breast cancer cell line by induction of tumor cell tunneling nanotubes. Cancer Rep. 2019, 2, e1213. [Google Scholar] [CrossRef]
- Qian, B.Z.; Zhang, H.; Li, J.; He, T.; Yeo, E.J.; Soong, D.Y.; Carragher, N.O.; Munro, A.; Chang, A.; Bresnick, A.R.; et al. FLT1 signaling in metastasis-associated macrophages activates an inflammatory signature that promotes breast cancer metastasis. J. Exp. Med. 2015, 212, 1433–1448. [Google Scholar] [CrossRef] [Green Version]
- Herrada-Manchon, H.; Celada, L.; Rodriguez-Gonzalez, D.; Alejandro Fernandez, M.; Aguilar, E.; Chiara, M.D. Three-dimensional bioprinted cancer models: A powerful platform for investigating tunneling nanotube-like cell structures in complex microenvironments. Mater. Sci. Eng. C Mater. Biol. Appl. 2021, 128, 112357. [Google Scholar] [CrossRef]
- Franchi, M.; Piperigkou, Z.; Riti, E.; Masola, V.; Onisto, M.; Karamanos, N.K. Long filopodia and tunneling nanotubes define new phenotypes of breast cancer cells in 3D cultures. Matrix Biol. Plus 2020, 6–7, 100026. [Google Scholar] [CrossRef]
- Jana, A.; Ladner, K.; Lou, E.; Nain, A.S. Tunneling Nanotubes between Cells Migrating in ECM Mimicking Fibrous Environments. Cancers 2022, 14, 1989. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Genna, A.; Duran, C.L.; Entenberg, D.; Condeelis, J.S.; Cox, D. Macrophages Promote Tumor Cell Extravasation across an Endothelial Barrier through Thin Membranous Connections. Cancers 2023, 15, 2092. https://doi.org/10.3390/cancers15072092
Genna A, Duran CL, Entenberg D, Condeelis JS, Cox D. Macrophages Promote Tumor Cell Extravasation across an Endothelial Barrier through Thin Membranous Connections. Cancers. 2023; 15(7):2092. https://doi.org/10.3390/cancers15072092
Chicago/Turabian StyleGenna, Alessandro, Camille L. Duran, David Entenberg, John S. Condeelis, and Dianne Cox. 2023. "Macrophages Promote Tumor Cell Extravasation across an Endothelial Barrier through Thin Membranous Connections" Cancers 15, no. 7: 2092. https://doi.org/10.3390/cancers15072092