
A CRISPR/Cas9-Based Assay for High-Throughput Studies of Cancer-Induced Innervation

Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
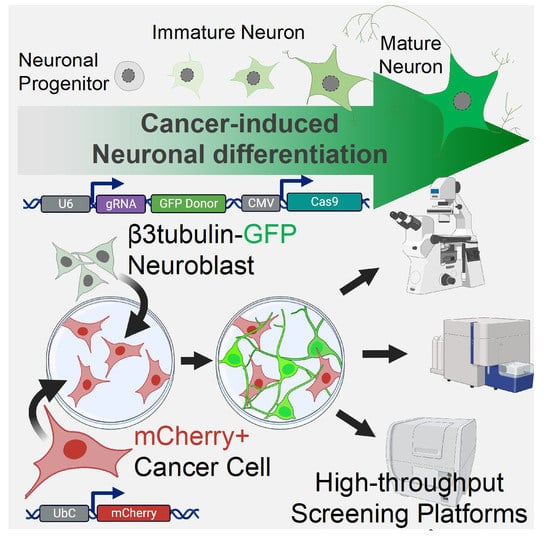
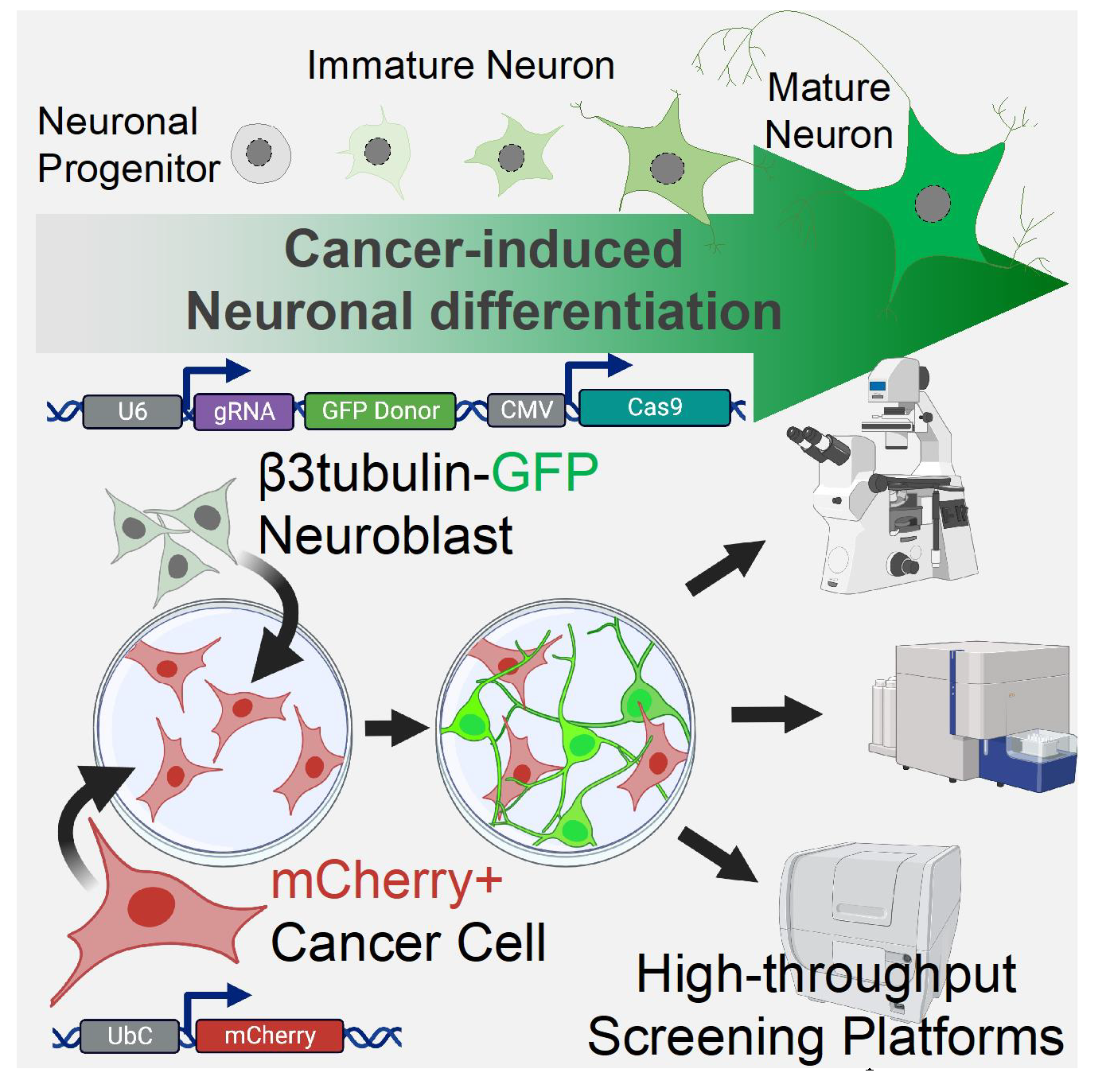
2.1. Cancer-Induced Neuronal Precursor Differentiation in Coculture In Vitro
2.2. Endogenous Labeling of β3-Tubulin Microtubule Protein in Neuronal Precursors
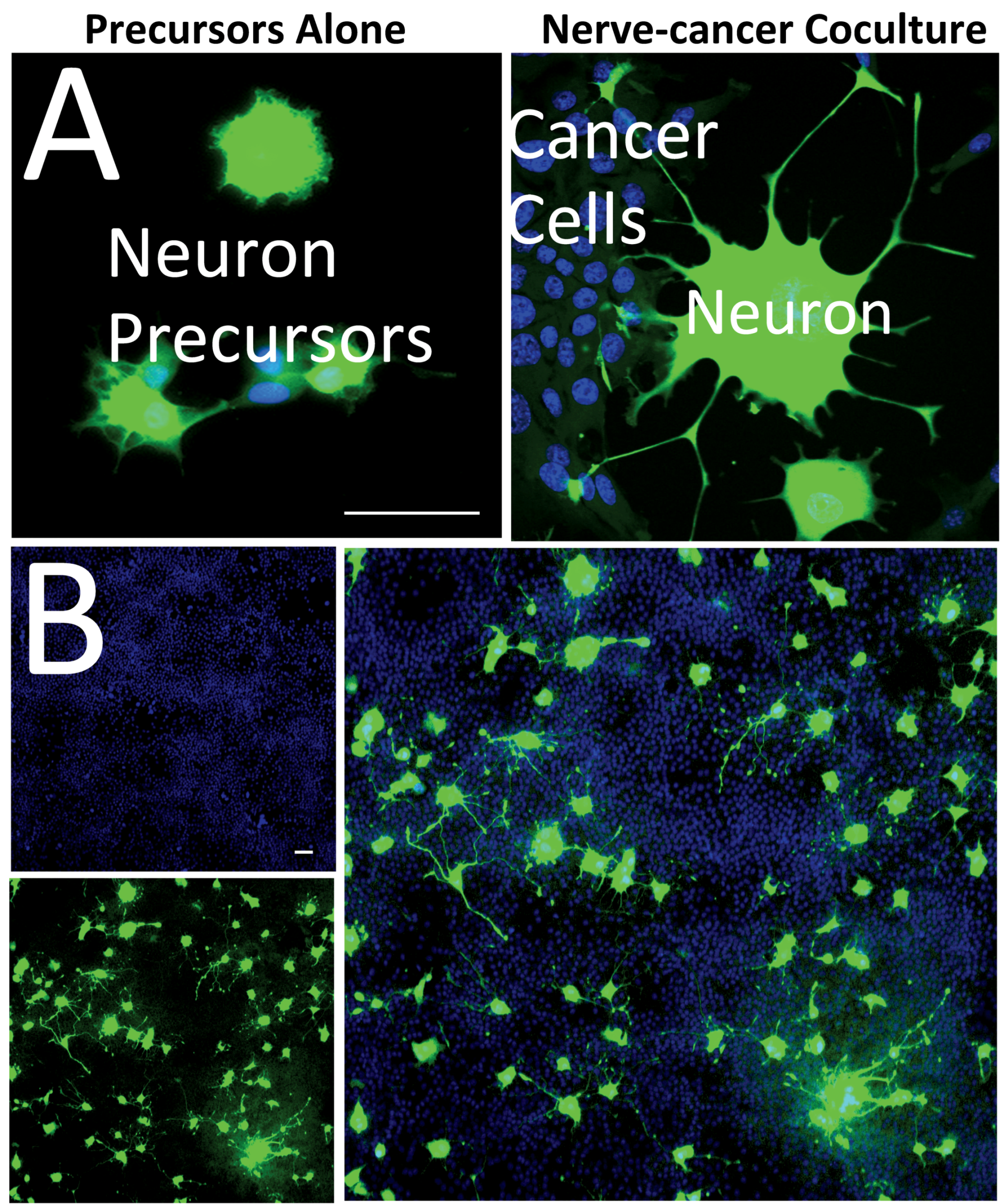
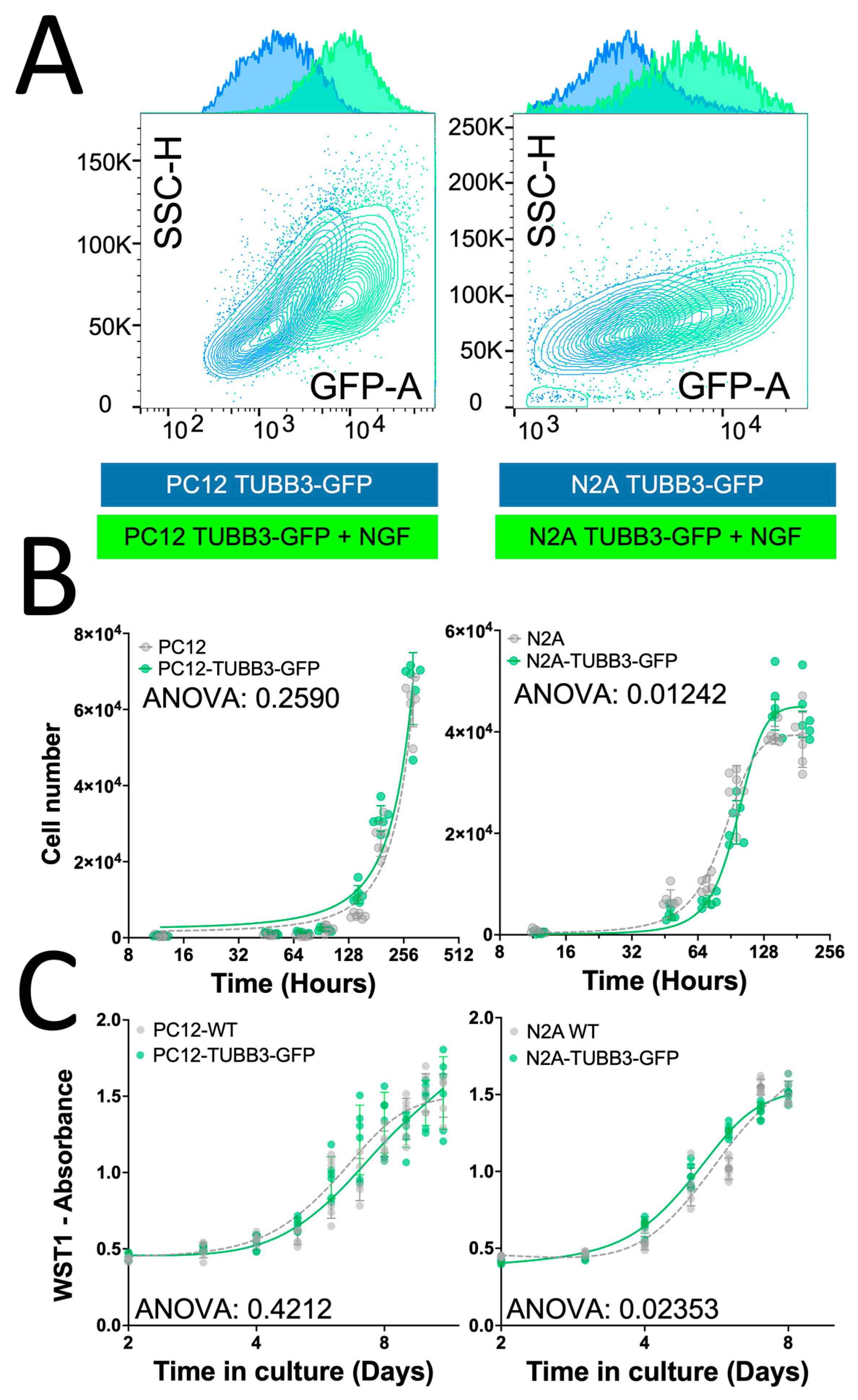
2.3. Characterization of the TUBB3-GFP Knock-in Approach
2.4. Validation of the Reporter Approach for High-Throughput Study of Cancer-Induced Neuronal Differentiation in Direct Coculture In Vitro
2.5. Breast Cancer Plasticity Controls Neuronal Precursors Differentiation
3. Discussion
4. Methods
4.1. Cell Culture and Reagents
4.2. Generation of PC12-GFP and PC12 mCherry
4.3. Generation of PC12-tubb3-GFP/ Neuro2A-tubb3-GFP Cell Lines
4.4. Flow Cytometry
4.5. Western Blot Analysis
4.6. Direct Counting Cell Proliferation Assay
4.7. WST1 Metabolic Activity Assay
4.8. Migration Assay
4.9. Fluorimeter and Microscopy Analysis
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, R.; Liu, S.; Zhang, S.; Min, L.; Zhu, S. Cellular and Extracellular Components in Tumor Microenvironment and Their Application in Early Diagnosis of Cancers. Anal. Cell. Pathol. 2020, 2020, 6283796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancino, M.; Ametller, E.; Gascón, P.; Almendro, V. The neuronal influence on tumor progression. Biochim. Biophys. Acta (BBA) Bioenerg. 2011, 1816, 105–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, S.R.; Jordan, M.; Nagarajan, P.; Amit, M. Nerve Density and Neuronal Biomarkers in Cancer. Cancers 2022, 14, 4817. [Google Scholar] [CrossRef]
- Marusyk, A.; Polyak, K. Tumor heterogeneity: Causes and consequences. Biochim. Biophys. Acta 2010, 1805, 105–117. [Google Scholar] [CrossRef] [Green Version]
- Monje, M.; Borniger, J.C.; D’Silva, N.J.; Deneen, B.; Dirks, P.B.; Fattahi, F.; Frenette, P.S.; Garzia, L.; Gutmann, D.H.; Hanahan, D.; et al. Roadmap for the Emerging Field of Cancer Neuroscience. Cell 2020, 181, 219–222. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Morishita, W.; Geraghty, A.C.; Silverbush, D.; Gillespie, S.M.; Arzt, M.; Tam, L.T.; Espenel, C.; Ponnuswami, A.; Ni, L.; et al. Electrical and synaptic integration of glioma into neural circuits. Nature 2019, 573, 539–545. [Google Scholar] [CrossRef]
- Magnon, C.; Hall, S.J.; Lin, J.; Xue, X.; Gerber, L.; Freedland, S.J.; Frenette, P.S. Autonomic Nerve Development Contributes to Prostate Cancer Progression. Science 2013, 341, 1236361. [Google Scholar] [CrossRef] [Green Version]
- Kaduri, M.; Sela, M.; Kagan, S.; Poley, M.; Abumanhal-Masarweh, H.; Mora-Raimundo, P.; Ouro, A.; Dahan, N.; Hershkovitz, D.; Shklover, J.; et al. Targeting neurons in the tumor microenvironment with bupivacaine nanoparticles reduces breast cancer progression and metastases. Sci. Adv. 2021, 7, eabj5435. [Google Scholar] [CrossRef]
- Zhao, C.-M.; Hayakawa, Y.; Kodama, Y.; Muthupalani, S.; Westphalen, C.B.; Andersen, G.T.; Flatberg, A.; Johannessen, H.; Friedman, R.A.; Renz, B.W.; et al. Denervation suppresses gastric tumorigenesis. Sci. Transl. Med. 2014, 6, 250ra115. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Na Hu, L.; Zheng, S.M.; La, T.; Wei, L.Y.; Zhang, X.J.; Zhang, Z.H.; Xing, J.; Wang, L.; Li, R.Q.; et al. High nerve density in breast cancer is associated with poor patient outcome. FASEB BioAdvances 2022, 4, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Ayala, G. Neuroepithelial Interactions in Cancer. Annu. Rev. Pathol. Mech. Dis. 2023, 18, 493–514. [Google Scholar] [CrossRef] [PubMed]
- Madeo, M.; Colbert, P.L.; Vermeer, D.W.; Lucido, C.T.; Cain, J.T.; Vichaya, E.G.; Grossberg, A.J.; Muirhead, D.; Rickel, A.P.; Hong, Z.; et al. Cancer exosomes induce tumor innervation. Nat. Commun. 2018, 9, 4284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferdoushi, A.; Griffin, N.; Marsland, M.; Xu, X.; Faulkner, S.; Gao, F.; Liu, H.; King, S.J.; Denham, J.W.; van Helden, D.F.; et al. Tumor innervation and clinical outcome in pancreatic cancer. Sci. Rep. 2021, 11, 7390. [Google Scholar] [CrossRef] [PubMed]
- Ayala, G.E.; Dai, H.; Powell, M.; Li, R.; Ding, Y.; Wheeler, T.M.; Shine, D.; Kadmon, D.; Thompson, T.; Miles, B.J.; et al. Cancer-Related Axonogenesis and Neurogenesis in Prostate Cancer. Clin. Cancer Res. 2008, 14, 7593–7603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schonkeren, S.; Thijssen, M.; Vaes, N.; Boesmans, W.; Melotte, V. The Emerging Role of Nerves and Glia in Colorectal Cancer. Cancers 2021, 13, 152. [Google Scholar] [CrossRef]
- Amit, M.; Takahashi, H.; Dragomir, M.P.; Lindemann, A.; Gleber-Netto, F.O.; Pickering, C.R.; Anfossi, S.; Osman, A.A.; Cai, Y.; Wang, R.; et al. Loss of p53 drives neuron reprogramming in head and neck cancer. Nature 2020, 578, 449–454. [Google Scholar] [CrossRef]
- Mauffrey, P.; Tchitchek, N.; Barroca, V.; Bemelmans, A.-P.; Firlej, V.; Allory, Y.; Roméo, P.-H.; Magnon, C. Progenitors from the central nervous system drive neurogenesis in cancer. Nature 2019, 569, 672–678. [Google Scholar] [CrossRef]
- Grelet, S.; Fréreux, C.; Obellianne, C.; Noguchi, K.; Howley, B.V.; Dalton, A.C.; Howe, P.H. TGFβ-induced expression of long noncoding lincRNA Platr18 controls breast cancer axonogenesis. Life Sci. Alliance 2021, 5, e202101261. [Google Scholar] [CrossRef]
- Han, H.; Yang, C.; Zhang, Y.; Han, C.; Zhang, G. Vascular Endothelial Growth Factor Mediates the Sprouted Axonogenesis of Breast Cancer in Rat. Am. J. Pathol. 2020, 191, 515–526. [Google Scholar] [CrossRef]
- Gysler, S.M.; Drapkin, R. Tumor innervation: Peripheral nerves take control of the tumor microenvironment. J. Clin. Investig. 2021, 131, e147276. [Google Scholar] [CrossRef]
- Zahalka, A.H.; Frenette, P.S. Nerves in cancer. Nat. Rev. Cancer 2020, 20, 143–157. [Google Scholar] [CrossRef]
- Hu, J.; Chen, W.; Shen, L.; Chen, Z.; Huang, J. Crosstalk between the peripheral nervous system and breast cancer influences tumor progression. Biochim. Biophys. Acta (BBA) Rev. Cancer 2022, 1877, 188828. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; He, D.; Florentin, D.; Frolov, A.; Hilsenbeck, S.; Ittmann, M.; Kadmon, D.; Miles, B.; Rowley, D.; Ayala, G. Semaphorin 4F as a Critical Regulator of Neuroepithelial Interactions and a Biomarker of Aggressive Prostate Cancer. Clin. Cancer Res. 2013, 19, 6101–6111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, C.C.; Marsland, M.; Wang, Y.; Dowdell, A.; Eden, E.; Gao, F.; Faulkner, S.; Jobling, P.; Li, X.; Liu, L.; et al. Tumor innervation is triggered by endoplasmic reticulum stress. Oncogene 2021, 41, 586–599. [Google Scholar] [CrossRef] [PubMed]
- Le, T.T.; Payne, S.L.; Buckwald, M.N.; Hayes, L.A.; Parker, S.R.; Burge, C.B.; Oudin, M.J. Sensory nerves enhance triple-negative breast cancer invasion and metastasis via the axon guidance molecule PlexinB3. NPJ Breast Cancer 2022, 8, 1–11. [Google Scholar] [CrossRef]
- Faulkner, S.; Jobling, P.; March, B.; Jiang, C.C.; Hondermarck, H. Tumor Neurobiology and the War of Nerves in Cancer. Cancer Discov. 2019, 9, 702–710. [Google Scholar] [CrossRef] [Green Version]
- Pletto, D.; Capra, S.; Finardi, A.; Colciaghi, F.; Nobili, P.; Battaglia, G.S.; Locatelli, D.; Cagnoli, C. Axon outgrowth and neuronal differentiation defects after a-SMN and FL-SMN silencing in primary hippocampal cultures. PLoS ONE 2018, 13, e0199105. [Google Scholar] [CrossRef]
- Greene, L.A.; Tischler, A. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc. Natl. Acad. Sci. USA 1976, 73, 2424–2428. [Google Scholar] [CrossRef] [Green Version]
- Hu, R.; Cao, Q.; Sun, Z.; Chen, J.; Zheng, Q.; Xiao, F. A novel method of neural differentiation of PC12 cells by using Opti-MEM as a basic induction medium. Int. J. Mol. Med. 2017, 41, 195–201. [Google Scholar] [CrossRef] [Green Version]
- Sanz, C.G.; Barsan, M.M.; Enache, T.A. PC-12 Cell Line as a Neuronal Cell Model for Biosensing Applications Daniela Oprea. Biosensors 2022, 12, 500. [Google Scholar]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Gaj, T.; Sirk, S.J.; Shui, S.-L.; Liu, J. Genome-Editing Technologies: Principles and Applications. Cold Spring Harb. Perspect. Biol. 2016, 8, a023754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, K.; Tsunekawa, Y.; Hernandez-Benitez, R.; Wu, J.; Zhu, J.; Kim, E.J.; Hatanaka, F.; Yamamoto, M.; Araoka, T.; Li, Z.; et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 2016, 540, 144–149. [Google Scholar] [CrossRef] [Green Version]
- Katsetos, C.D.; Legido, A.; Perentes, E.; Mörk, S.J. Class III β-Tubulin Isotype: A Key Cytoskeletal Protein at the Crossroads of Developmental Neurobiology and Tumor Neuropathology. J. Child Neurol. 2003, 18, 851–866. [Google Scholar] [CrossRef]
- Tischfield, M.A.; Baris, H.N.; Wu, C.; Rudolph, G.; Van Maldergem, L.; He, W.; Chan, W.-M.; Andrews, C.; Demer, J.L.; Robertson, R.L.; et al. Human TUBB3 Mutations Perturb Microtubule Dynamics, Kinesin Interactions, and Axon Guidance. Cell 2010, 140, 74–87. [Google Scholar] [CrossRef] [Green Version]
- Sferra, A.; Nicita, F.; Bertini, E. Microtubule Dysfunction: A Common Feature of Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 7354. [Google Scholar] [CrossRef]
- Willems, J.; de Jong, A.P.H.; Scheefhals, N.; Mertens, E.; Catsburg, L.A.E.; Poorthuis, R.B.; de Winter, F.; Verhaagen, J.; Meye, F.J.; MacGillavry, H.D. ORANGE: A CRISPR/Cas9-based genome editing toolbox for epitope tagging of endogenous proteins in neurons. PLOS Biol. 2020, 18, e3000665. [Google Scholar] [CrossRef] [Green Version]
- Greene, L.A. Nerve growth factor prevents the death and stimulates the neuronal differentiation of clonal PC12 pheochromocytoma cells in serum-free medium. J. Cell Biol. 1978, 78, 747–755. [Google Scholar] [CrossRef] [Green Version]
- Tremblay, R.G.; Sikorska, M.; Sandhu, J.K.; Lanthier, P.; Ribecco-Lutkiewicz, M.; Bani-Yaghoub, M. Differentiation of mouse Neuro 2A cells into dopamine neurons. J. Neurosci. Methods 2010, 186, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Limoli, C.L.; Rola, R.; Giedzinski, E.; Mantha, S.; Huang, T.-T.; Fike, J.R. Cell-density-dependent regulation of neural precursor cell function. Proc. Natl. Acad. Sci. USA 2004, 101, 16052–16057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelopoulos, I.; Gakis, G.; Birmpas, K.; Kyrousi, C.; Habeos, E.E.; Kaplani, K.; Lygerou, Z.; Habeos, I.; Taraviras, S. Metabolic regulation of the neural stem cell fate: Unraveling new connections, establishing new concepts. Front. Neurosci. 2022, 16, 10091250. [Google Scholar] [CrossRef]
- Grelet, S.; Link, L.A.; Howley, B.; Obellianne, C.; Palanisamy, V.; Gangaraju, V.K.; Diehl, J.A.; Howe, P.H. A regulated PNUTS mRNA to lncRNA splice switch mediates EMT and tumour progression. Nature 2017, 19, 1105–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, M.; Johnson, K.R.; Wheelock, M.J. Cadherin switching: Essential for behavioral but not morphological changes during an epithelium-to-mesenchyme transition. J. Cell Sci. 2005, 118, 873–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viloria-Petit, A.M.; David, L.; Jia, J.Y.; Erdemir, T.; Bane, A.L.; Pinnaduwage, D.; Roncari, L.; Narimatsu, M.; Bose, R.; Moffat, J.; et al. A role for the TGFβ-Par6 polarity pathway in breast cancer progression. Proc. Natl. Acad. Sci. USA 2009, 106, 14028–14033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jolly, M.K.; Celià-Terrassa, T. Dynamics of Phenotypic Heterogeneity Associated with EMT and Stemness during Cancer Progression. J. Clin. Med. 2019, 8, 1542. [Google Scholar] [CrossRef] [Green Version]
- Jonkman, J.E.N.; Cathcart, J.A.; Xu, F.; Bartolini, M.E.; Amon, J.E.; Stevens, K.M.; Colarusso, P. An introduction to the wound healing assay using live-cell microscopy. Cell Adhes. Migr. 2014, 8, 440–451. [Google Scholar] [CrossRef] [Green Version]
- Barrallo-Gimeno, A.; Nieto, M.A. The Snail genes as inducers of cell movement and survival: Implications in development and cancer. Development 2005, 132, 3151–3161. [Google Scholar] [CrossRef] [Green Version]
- Mousa, A.; Bakhiet, M. Role of Cytokine Signaling during Nervous System Development. Int. J. Mol. Sci. 2013, 14, 13931–13957. [Google Scholar] [CrossRef] [Green Version]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homem, C.; Repic, M.; Knoblich, J.A. Proliferation control in neural stem and progenitor cells. Nat. Rev. Neurosci. 2015, 16, 647–659. [Google Scholar] [CrossRef] [PubMed]
- Grelet, S.; Geslain, R.; Howe, P.H. EMT does not work regular shifts. Cell Cycle 2018, 17, 141–142. [Google Scholar] [CrossRef] [Green Version]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galappaththi, S.L.; Katz, B.; Howze, P.H., IV; Hoover, G.; Grelet, S. A CRISPR/Cas9-Based Assay for High-Throughput Studies of Cancer-Induced Innervation. Cancers 2023, 15, 2026. https://doi.org/10.3390/cancers15072026
Galappaththi SL, Katz B, Howze PH IV, Hoover G, Grelet S. A CRISPR/Cas9-Based Assay for High-Throughput Studies of Cancer-Induced Innervation. Cancers. 2023; 15(7):2026. https://doi.org/10.3390/cancers15072026
Chicago/Turabian StyleGalappaththi, Sapthala Loku, Brenna Katz, Patrick H. Howze, IV, Gregory Hoover, and Simon Grelet. 2023. "A CRISPR/Cas9-Based Assay for High-Throughput Studies of Cancer-Induced Innervation" Cancers 15, no. 7: 2026. https://doi.org/10.3390/cancers15072026