
Phase I Study of a Combination of Fluvastatin and Celecoxib in Children with Relapsing/Refractory Low-Grade or High-Grade Glioma (FLUVABREX)

 , , , , , , , and
, , , , , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
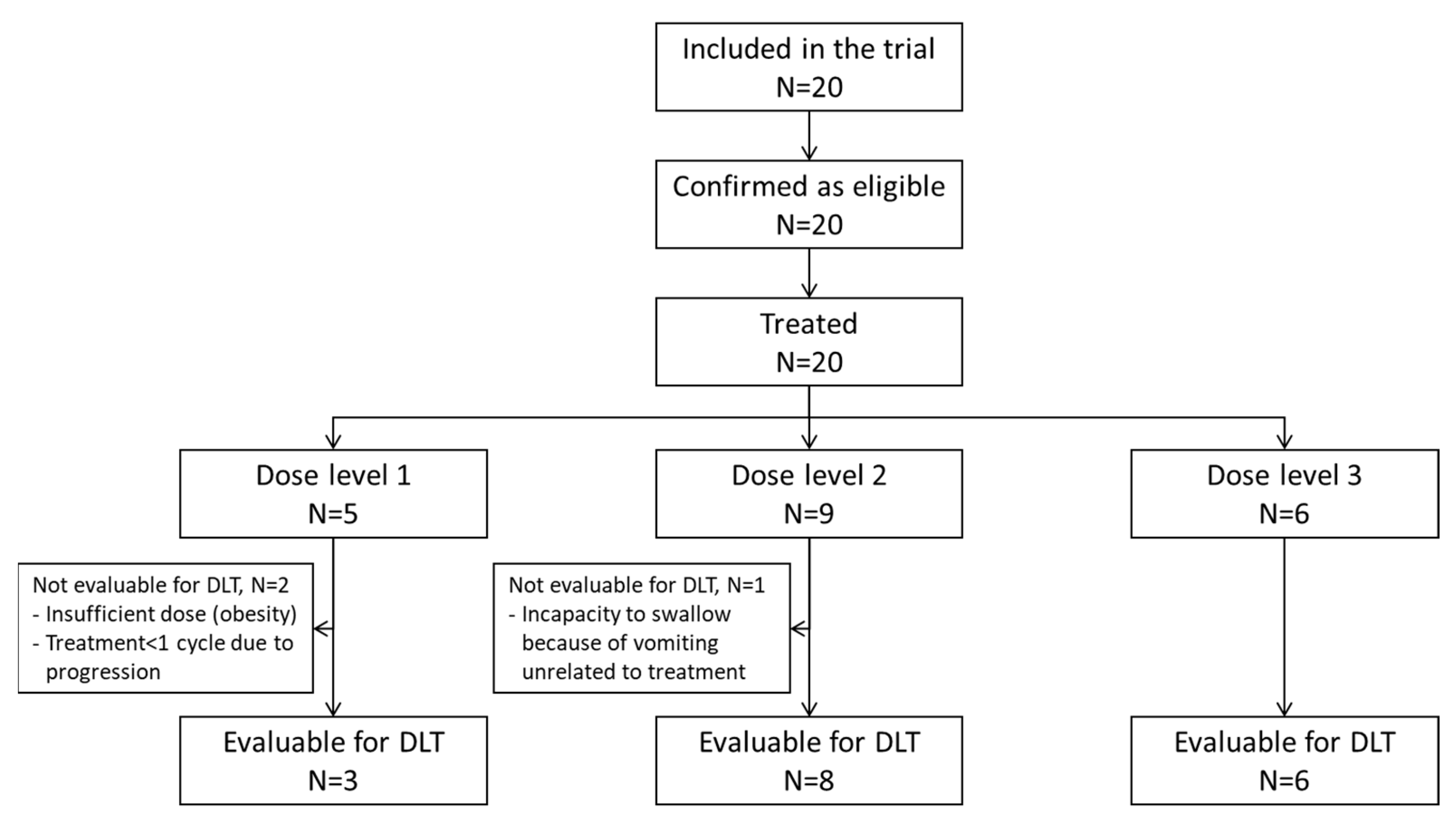
2.1. Study Population
2.2. Study Design and Treatment
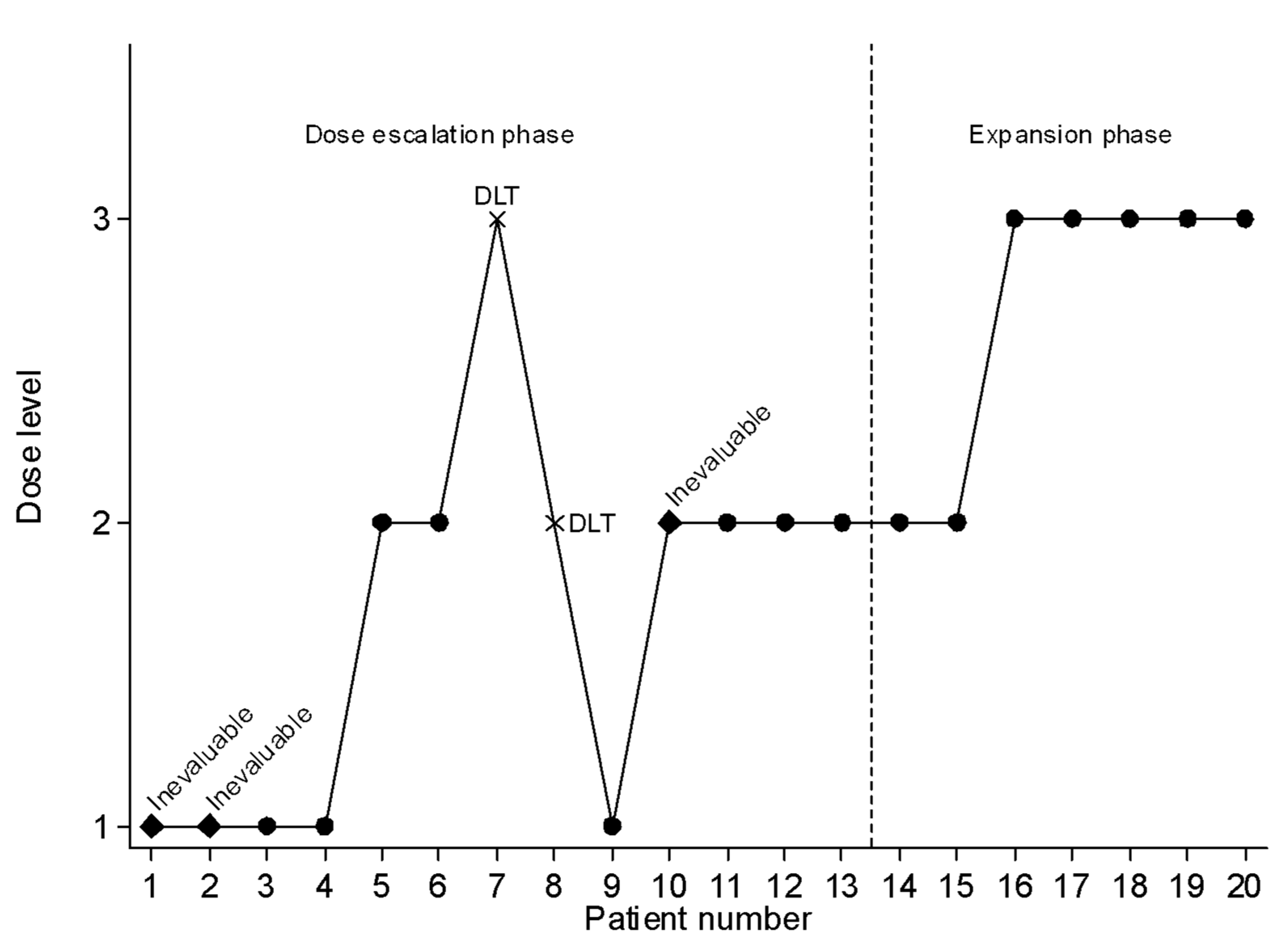
2.3. Dose-Escalation Phase
2.4. Expansion Phase
2.5. Safety Evaluation
2.6. Pharmacokinetic Assessments
2.7. Efficacy Evaluation
3. Results
3.1. Patient and Tumor Characteristics
3.2. Treatment Exposure
3.3. Toxicities
3.4. Pharmacokinetic Analysis
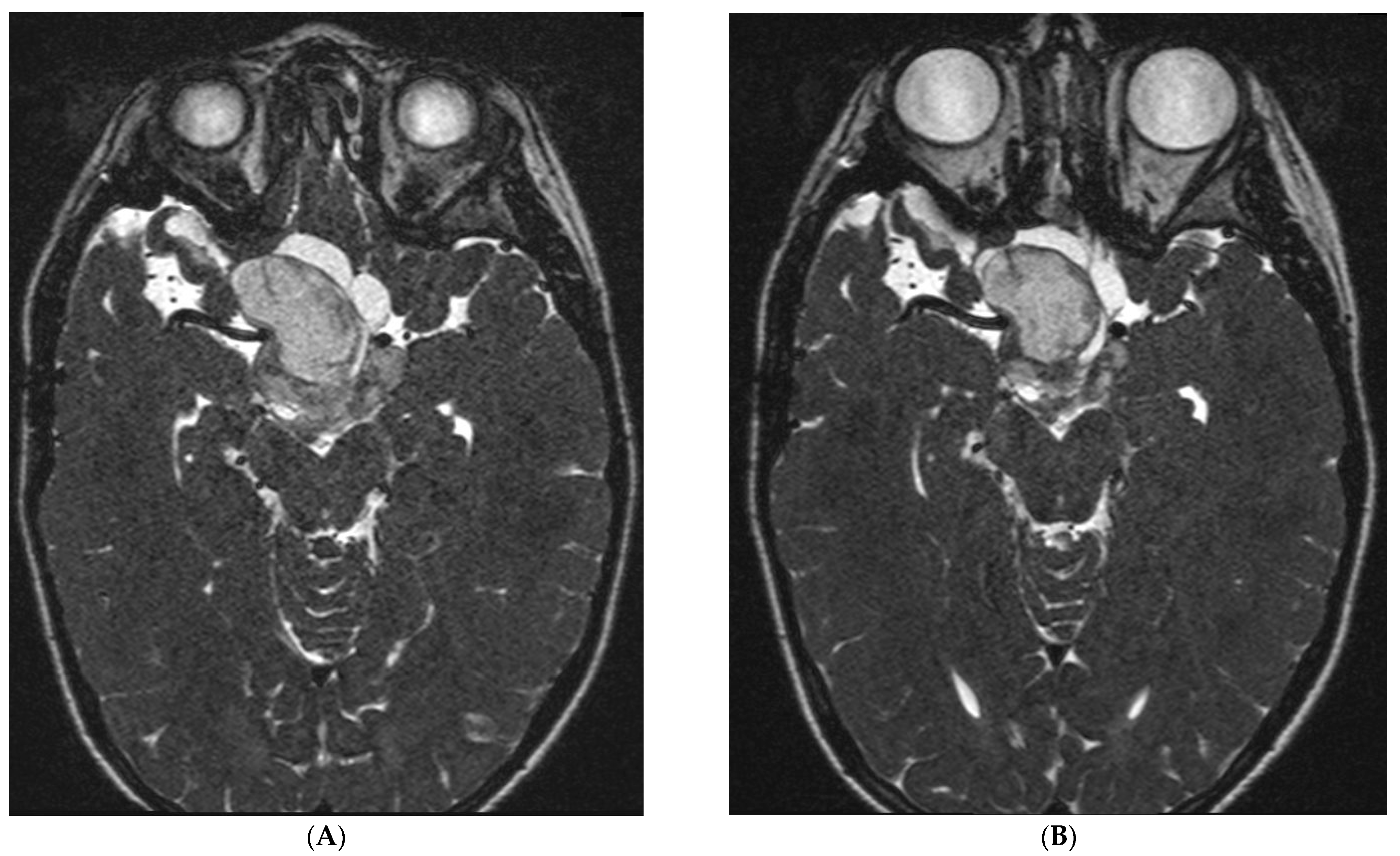
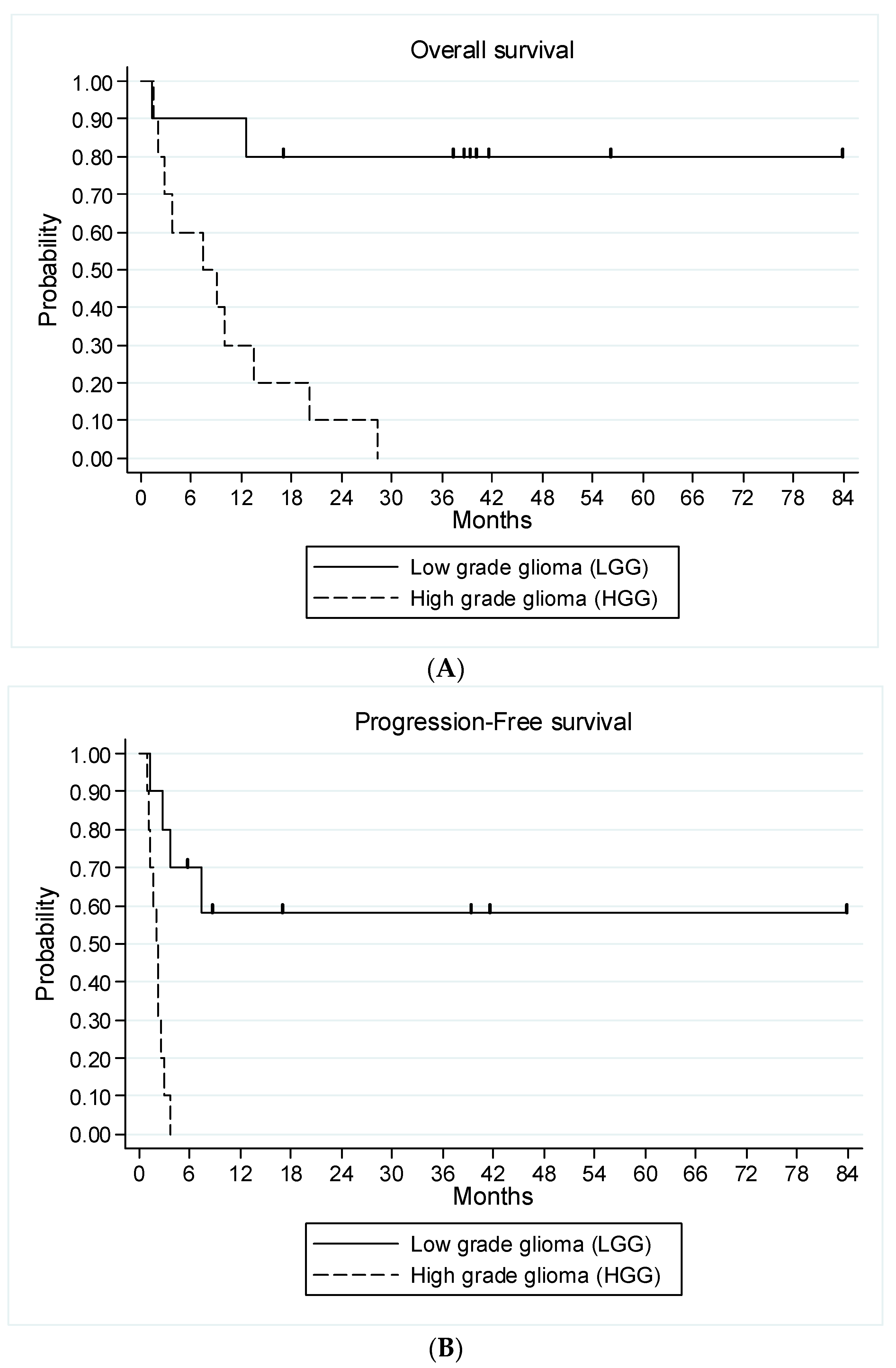
3.5. Efficacy
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Northcott, P.A.; Pfister, S.M.; Jones, D.T.W. Next-generation (epi)genetic drivers of childhood brain tumours and the outlook for targeted therapies. Lancet Oncol. 2015, 16, e293–e302. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; de Blank, P.M.; Kruchko, C.; Petersen, C.M.; Liao, P.; Finlay, J.L.; Stearns, D.S.; Wolff, J.E.; Wolinsky, Y.; Letterio, J.J.; et al. Alex’s Lemonade Stand Foundation Infant and Childhood Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2007–2011. Neuro Oncol. 2015, 16 (Suppl. 1), x1–x36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandopadhayay, P.; Bergthold, G.; London, W.B.; Goumnerova, L.C.; Morales La Madrid, A.; Marcus, K.J.; Guo, D.; Ullrich, N.J.; Robison, N.J.; Chi, S.N.; et al. Long-term outcome of 4,040 children diagnosed with pediatric low-grade gliomas: An analysis of the Surveillance Epidemiology and End Results (SEER) database. Pediatr. Blood Cancer 2014, 61, 1173–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnatry, R.; Zhukova, N.; Guerreiro Stucklin, A.S.; Pole, J.D.; Mistry, M.; Fried, I.; Ramaswamy, V.; Bartels, U.; Huang, A.; Laperriere, N.; et al. Clinical and treatment factors determining long-term outcomes for adult survivors of childhood low-grade glioma: A population-based study. Cancer 2016, 122, 1261–1269. [Google Scholar] [CrossRef]
- Colin, C.; Padovani, L.; Chappé, C.; Mercurio, S.; Scavarda, D.; Loundou, A.; Frassineti, F.; Andre, N.; Bouvier, C.; Korshunov, A.; et al. Outcome analysis of childhood pilocytic astrocytomas: A retrospective study of 148 cases at a single institution. Neuropathol. Appl. Neurobiol. 2013, 39, 693–705. [Google Scholar] [CrossRef]
- Jones, C.; Karajannis, M.A.; Jones, D.T.W.; Kieran, M.W.; Monje, M.; Baker, S.J.; Becher, O.J.; Cho, Y.J.; Gupta, N.; Hawkins, C.; et al. Pediatric high-grade glioma: Biologically and clinically in need of new thinking. Neuro Oncol. 2017, 19, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Fried, I.; Tabori, U.; Tihan, T.; Reginald, A.; Bouffet, E. Optic pathway gliomas: A review. CNS Oncol. 2013, 2, 143–159. [Google Scholar] [CrossRef]
- Ris, M.D.; Beebe, D.W.; Armstrong, F.D.; Fontanesi, J.; Holmes, E.; Sanford, R.A.; Wisoff, J.H.; Children’s Oncology Group. Cognitive and adaptive outcome in extracerebellar low-grade brain tumors in children: A report from the Children’s Oncology Group. J. Clin. Oncol. 2008, 26, 4765–4770. [Google Scholar] [CrossRef] [Green Version]
- Tacke, U.; Karger, D.; Spreer, J.; Berlis, A.; Nikkhah, G.; Korinthenberg, R. Incidence of vasculopathy in children with hypothalamic/chiasmatic gliomas treated with brachytherapy. Childs Nerv. Syst. 2011, 27, 961–966. [Google Scholar] [CrossRef]
- Grill, J.; Couanet, D.; Cappelli, C.; Habrand, J.L.; Rodriguez, D.; Sainte-Rose, C.; Kalifa, C. Radiation-induced cerebral vasculopathy in children with neurofibromatosis and optic pathway glioma. Ann. Neurol. 1999, 45, 393–396. [Google Scholar] [CrossRef]
- Perkins, S.M.; Fei, W.; Mitra, N.; Shinohara, E.T. Late causes of death in children treated for CNS malignancies. J. Neurooncol. 2013, 115, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Greenberger, B.A.; Pulsifer, M.B.; Ebb, D.H.; MacDonald, S.M.; Jones, R.M.; Butler, W.E.; Huang, M.S.; Marcus, K.J.; Oberg, J.A.; Tarbell, N.J.; et al. Clinical outcomes and late endocrine, neurocognitive, and visual profiles of proton radiation for pediatric low-grade gliomas. Int. J. Radiat. Oncol. Biol. Phys. 2014, 89, 1060–1068. [Google Scholar] [CrossRef] [PubMed]
- Gnekow, A.K.; Walker, D.A.; Kandels, D.; Picton, S.; Perilongo, G.; Grill, J.; Stokland, T.; Sandstrom, P.E.; Warmuth-Metz, M.; Pietsch, T.; et al. A European randomised controlled trial of the addition of etoposide to standard vincristine and carboplatin induction as part of an 18-month treatment programme for childhood (≤16 years) low grade glioma—A final report. Eur. J. Cancer 2017, 81, 206–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouffet, E.; Jakacki, R.; Goldman, S.; Hargrave, D.; Hawkins, C.; Shroff, M.; Hukin, J.; Bartels, U.; Foreman, N.; Kellie, S.; et al. Phase II study of weekly vinblastine in recurrent or refractory pediatric low-grade glioma. J. Clin. Oncol. 2012, 30, 1358–1363. [Google Scholar] [CrossRef]
- Ater, J.L.; Zhou, T.; Holmes, E.; Mazewski, C.M.; Booth, T.N.; Freyer, D.R.; Lazarus, K.H.; Packer, R.J.; Prados, M.; Sposto, R.; et al. Randomized study of two chemotherapy regimens for treatment of low-grade glioma in young children: A report from the Children’s Oncology Group. J. Clin. Oncol. 2012, 30, 2641–2647. [Google Scholar] [CrossRef] [Green Version]
- Lu, V.M.; Welby, J.P.; Nesvick, C.L.; Daniels, D.J. Efficacy and safety of bevacizumab in progressive pediatric low-grade glioma: A systematic review and meta-analysis of outcome rates. Neuro-Oncol. Pract. 2020, 7, 359–368. [Google Scholar] [CrossRef]
- Tchoghandjian, A.; Fernandez, C.; Colin, C.; El Ayachi, I.; Voutsinos-Porche, B.; Fina, F.; Scavarda, D.; Piercecchi-Marti, M.-D.; Intagliata, D.; Ouafik, L.; et al. Pilocytic astrocytoma of the optic pathway: A tumour deriving from radial glia cells with a specific gene signature. Brain 2009, 132, 1523–1535. [Google Scholar] [CrossRef] [Green Version]
- Mercurio, S.; Padovani, L.; Colin, C.; Carre, M.; Tchoghandjian, A.; Scavarda, D.; Lambert, S.; Baeza-Kallee, N.; Fernandez, C.; Chappe, C.; et al. Evidence for new targets and synergistic effect of metronomic celecoxib/fluvastatin combination in pilocytic astrocytoma. Acta Neuropathol. Commun. 2013, 1, 17. [Google Scholar] [CrossRef] [Green Version]
- López-Aguilar, E.; Sepúlveda-Vildósola, A.C.; Rivera-Márquez, H.; Cerecedo-Diaz, F.; Valdez-Sánchez, M.; Villasis-Keever, M.A. Security and maximal tolerated doses of fluvastatin in pediatric cancer patients. Arch. Med. Res. 1999, 30, 128–131. [Google Scholar] [CrossRef]
- López-Aguilar, E.; Sepúlveda-Vildósola, A.C.; Betanzos-Cabrera, Y.; Rocha-Moreno, Y.G.; Gascon-Lastiri, G.; Rivera-Marquez, H.; Wanzke-del-Angel, V.; Cerecedo-Diaz, F.; de la Cruz-Yanez, H. Phase II Study of Metronomic Chemotherapy with Thalidomide, Carboplatin-Vincristine-Fluvastatin in the Treatment of Brain Stem Tumors in Children. Arch. Med. Res. 2008, 39, 655–662. [Google Scholar] [CrossRef]
- Silverstein, R.L.; Febbraio, M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci. Signal. 2009, 2, re3. [Google Scholar] [CrossRef] [Green Version]
- Sławińska-Brych, A.; Zdzisińska, B.; Kandefer-Szerszeń, M. Fluvastatin inhibits growth and alters the malignant phenotype of the C6 glioma cell line. Pharmacol. Rep. 2014, 66, 121–129. [Google Scholar] [CrossRef]
- Chang, Y.-L.; Huang, L.-C.; Chen, Y.-C.; Wang, Y.-W.; Hueng, D.-Y.; Huang, S.-M. The synergistic effects of valproic acid and fluvastatin on apoptosis induction in glioblastoma multiforme cell lines. Int. J. Biochem. Cell Biol. 2017, 92, 155–163. [Google Scholar] [CrossRef]
- Tołoczko-Iwaniuk, N.; Dziemiańczyk-Pakieła, D.; Nowaszewska, B.K.; Celińska-Janowicz, K.; Miltyk, W. Celecoxib in Cancer Therapy and Prevention-Review. Curr. Drug Targets 2019, 20, 302–315. [Google Scholar] [CrossRef]
- Sato, A.; Mizobuchi, Y.; Nakajima, K.; Shono, K.; Fujihara, T.; Kageji, T.; Kitazato, K.; Matsuzaki, K.; Mure, H.; Kuwayama, K.; et al. Blocking COX-2 induces apoptosis and inhibits cell proliferation via the Akt/survivin- and Akt/ID3 pathway in low-grade-glioma. J. Neurooncol. 2017, 132, 231–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verschuur, A.; Heng-Maillard, M.-A.; Dory-Lautrec, P.; Truillet, R.; Jouve, E.; Chastagner, P.; Leblond, P.; Aerts, I.; Honore, S.; Entz-Werle, N.; et al. Metronomic Four-Drug Regimen Has Anti-tumor Activity in Pediatric Low-Grade Glioma; The Results of a Phase II Clinical Trial. Front. Pharmacol. 2018, 9, 00950. [Google Scholar] [CrossRef] [PubMed]
- van den Bent, M.J.; Wefel, J.S.; Schiff, D.; Taphoorn, M.J.B.; Jaeckle, K.; Armstrong, T.; Choucair, A.; Waldman, A.D.; Gorlia, T.; Chamberlain, M.; et al. Response assessment in neuro-oncology (a report of the RANO group): Assessment of outcome in trials of diffuse low-grade gliomas. Lancet Oncol. 2011, 12, 583–593. [Google Scholar] [CrossRef]
- Iasonos, A.; O’Quigley, J. Adaptive dose-finding studies: A review of model-guided phase I clinical trials. J. Clin. Oncol. 2014, 32, 2505–2511. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, A.; Jakacki, R.I.; Onar-Thomas, A.; Wu, S.; Nicolaides, T.; Poussaint, T.Y.; Fangusaro, J.; Phillips, J.; Perry, A.; Turner, D.; et al. A phase I trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low-grade glioma: A Pediatric Brain Tumor Consortium (PBTC) study. Neuro Oncol. 2017, 19, 1135–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fangusaro, J.; Onar-Thomas, A.; Young Poussaint, T.; Wu, S.; Ligon, A.H.; Lindeman, N.; Banerjee, A.; Packer, R.J.; Kilburn, L.B.; Glodman, S.; et al. Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: A multicentre, phase 2 trial. Lancet Oncol. 2019, 20, 1011–1022. [Google Scholar] [CrossRef]
- Bertolini, F.; Sukhatme, V.P.; Bouche, G. Drug repurposing in oncology--patient and health systems opportunities. Nat. Rev. Clin. Oncol. 2015, 12, 732–742. [Google Scholar] [CrossRef]
- Kline, C.; Felton, E.; Allen, I.E.; Tahir, P.; Mueller, S. Survival outcomes in pediatric recurrent high-grade glioma: Results of a 20-year systematic review and meta-analysis. J. Neurooncol. 2018, 137, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Fluvastatin-Summary of Product Characteristics. Available online: https://www.medicines.org.uk/emc/product/4465/smpc (accessed on 8 February 2023).
- Stempak, D.; Gammon, J.; Klein, J.; Koren, G.; Baruchel, S. Single-dose and steady-state pharmacokinetics of celecoxib in children. Clin. Pharmacol. Ther. 2002, 72, 490–497. [Google Scholar] [CrossRef] [PubMed]
- de Blank, P.; Bandopadhayay, P.; Haas-Kogan, D.; Fouladi, M.; Fangusaro, J. Management of pediatric low-grade glioma. Curr. Opin. Pediatr. 2019, 31, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Gururangan, S.; Fangusaro, J.; Poussaint, T.Y.; McLendon, R.E.; Onar-Thomas, A.; Wu, S.; Packer, R.J.; Banerjee, A.; Gilbertson, R.J.; Fahey, F.; et al. Efficacy of bevacizumab plus irinotecan in children with recurrent low-grade gliomas--a Pediatric Brain Tumor Consortium study. Neuro Oncol. 2014, 16, 310–317. [Google Scholar] [CrossRef] [Green Version]
- Hwang, E.I.; Jakacki, R.I.; Fisher, M.J.; Kilburn, L.B.; Horn, M.; Vezina, G.; Rood, B.R.; Packer, R.J. Long-term efficacy and toxicity of bevacizumab-based therapy in children with recurrent low-grade gliomas. Pediatr. Blood Cancer 2013, 60, 776–782. [Google Scholar] [CrossRef]
- Couec, M.-L.; André, N.; Thebaud, E.; Minckes, O.; Rialland, X.; Corradini, N.; Aerts, I.; Marec-Berard, P.; Bourdeaut, F.; Leblond, P.; et al. Bevacizumab and irinotecan in children with recurrent or refractory brain tumors: Toxicity and efficacy trends. Pediatr. Blood Cancer 2012, 59, 34–38. [Google Scholar] [CrossRef]
- Roux, C.; Revon-Rivière, G.; Gentet, J.C.; Verschuur, A.; Scavarda, D.; Saultier, P.; Appay, R.; Padovani, L.; Andre, N. Metronomic Maintenance with Weekly Vinblastine after Induction With Bevacizumab-Irinotecan in Children with Low-grade Glioma Prevents Early Relapse. J. Pediatr. Hematol. Oncol. 2021, 43, e630–e634. [Google Scholar] [CrossRef] [PubMed]
- Heng, M.A.; Padovani, L.; Dory-Lautrec, P.; Gentet, J.-C.; Verschuur, A.; Pasquier, E.; Figarella-Branger, D.; Scavarda, D.; Andre, N. Can metronomic maintenance with weekly vinblastine prevent early relapse/progression after bevacizumab-irinotecan in children with low-grade glioma? Cancer Med. 2016, 5, 1542–1545. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wu, G.; Miller, C.P.; Tatevossian, R.G.; Dalton, J.D.; Tang, B.; Orisme, W.; Punchihewa, C.; Parker, M.; Qaddoumi, I.; et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat. Genet. 2013, 45, 602–612. [Google Scholar]
- Kondyli, M.; Larouche, V.; Saint-Martin, C.; Ellezam, B.; Pouliot, L.; Sinnett, D.; Legault, G.; Crevier, L.; Weil, A.; Farmer, J.P.; et al. Trametinib for progressive pediatric low-grade gliomas. J. Neurooncol. 2018, 140, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.M.M.; Thompson, J.C.; Griesinger, A.M.; Amani, V.; Donson, A.D.; Birks, D.K.; Morgan, M.J.; Mirsky, D.M.; Handler, M.H.; Foreman, N.K.; et al. Autophagy Inhibition Improves Chemosensitivity in BRAFV600E Brain Tumors. Cancer Discov. 2014, 4, 773–780. [Google Scholar] [CrossRef] [Green Version]
- Mulcahy Levy, J.M.; Zahedi, S.; Griesinger, A.M.; Morin, A.; Davies, K.D.; Aisner, D.L.; Kleinschmidt-DeMasters, B.K.; Fitzwalter, B.E.; Goodall, M.L.; Thorburn, J.; et al. Autophagy inhibition overcomes multiple mechanisms of resistance to BRAF inhibition in brain tumors. Elife. 2017, 6, 358–367. [Google Scholar] [CrossRef]
- André, N.; Banavali, S.; Snihur, Y.; Pasquier, E. Has the time come for metronomics in low-income and middle-income countries? Lancet Oncol. 2013, 14, e239–e248. [Google Scholar] [CrossRef] [PubMed]
- Revon-Rivière, G.; Banavali, S.; Heississen, L.; Gomez Garcia, W.; Abdolkarimi, B.; Vaithilingum, M.; Li, C.-K.; Leung, P.C.; Malik, P.; Pasquier, E.; et al. Metronomic Chemotherapy for Children in Low- and Middle-Income Countries: Survey of Current Practices and Opinions of Pediatric Oncologists. J. Glob. Oncol. 2019, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Gyawali, B.; Bouche, G.; Crisp, N.; Andre, N. Challenges and opportunities for cancer clinical trials in low- and middle-income countries. Nat. Cancer 2020, 1, 142–145. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | N (%) |
|---|---|
| Sex | |
| Male | 13 (65%) |
| Female | 7 (35%) |
| Age in years | |
| Median (range) | 12.5 (5.9–19) |
| Neurofibromatosis type 1 | 3 (15%) |
| Metastatic at study entry | 2 (10%) |
| Grade | |
| Low-grade glioma | 10 (50%) |
| High-grade glioma | 10 (50%) |
| LGG (n = 10) | HGG (n = 10) | |||
|---|---|---|---|---|
| Histology | Pilocytic astrocytoma | 6 (30%) | Glioblastoma | 6 (30%) |
| Pilomyxoid astrocytoma | 1 (5%) | Anaplastic glioma | 2 (10%) | |
| Ganglioglioma (grade 2) | 1 (5%) | Anaplastic oligodendroglioma | 1 (5%) | |
| Diffuse astrocytoma (grade 2) | 1 (5%) | Anaplastic oligoastrocytoma | 1 (5%) | |
| No histology performed | 1 (5%) | |||
| BRAF rearrangement | Yes | 3 (30%) | ||
| No | 2 (20%) | |||
| Unknown | 5 (50%) | |||
| BRAF V600E mutation | Yes | 0 (0%) | ||
| No | 8 (80%) | |||
| Unknown | 2 (20%) | |||
| Prior treatment | Targeted or chemo-therapy | 10 (100%) | Chemotherapy | 10 (100%) |
| Radiation therapy | 2 (20%) | Radiation therapy | 10 (100%) | |
| Surgery | 7 (70%) | Surgery | 9 (90%) | |
| No. of prior lines | Median (range) | 4 (2–7) | Median (range) | 1.5 (1–4) |
| Previous therapy | Vinblastine | 10 (100%) | Temozolomide | 10 (100%) |
| Vincristine–Carboplatin | 9 (90%) | Bevacizumab | 5 (50%) | |
| Bevacizumab–Irinotecan | 3 (30%) | Lomustine | 4 (40%) | |
| Bevacizumab alone | 3 (30%) | Other regimens b | 7 (70%) | |
| Thioguanine–procarbazine–lomustine–vincristine (TPCV) | 3 (30%) | |||
| Other regimens a | 8 (80%) | |||
| Level (Dose) | Number of Patients | Evaluable for DLTs | DLT—Number (Details) |
|---|---|---|---|
| 1 (2 mg/kg/d) | 5 | 3 | / |
| 2 (4 mg/kg/d) | 9 | 8 | 1 (grade 3 maculopapular rash) |
| 3 (6 mg/kg/d) | 6 | 6 | 1 (grade 4 CPK increase) |
| 4 (8 mg/kg/d) | / | / | / |
| Adverse Event | Grade 1 | Grade 2 | Grade 3 | Grade 4 |
|---|---|---|---|---|
| Fatigue | 1 (DL1) | 1 (DL2) | ||
| Cough | 1 (DL1) | |||
| Nausea | 2 (DL1, DL3) | |||
| Constipation | 1 (DL3) | |||
| Diarrhea | 1 (DL2) | |||
| Abdominal pain | 1 (DL3) | 1 (DL2) | ||
| Vomiting | 2 (DL2, DL3) | |||
| Oral mucositis | 2 (DL2, DL3) | |||
| Myalgia | 1 (DL2) | 1 (DL3) | ||
| Maculopapular rash | 1 (DL2) | 1 (DL2) | ||
| Blood CPK increase | 1 (DL2) | 1 (DL3) | ||
| Hyperkaliemia | 1 (DL3) | |||
| Transaminases increase | 1 (DL3) | |||
| Bilirubin increase | 1 (DL3) |
| Fluvastatin | |||||||||
| Day | Dose | n | Tmax (h) median (range) | Cmax (ng/mL) | AUC0–24 h (h.ng/mL) | AUC0–∞ (h.ng/mL) | T1/2 (h) | CL/F (L/h/kg) | Vz/F (L/kg) |
| 1 (cycle 1) | 2 mg/kg | 5 | 2 (1–5) | 1238 ± 1030 | 2445 ± 1319 | 2459 ± 1324 | 4.6 ± 1.5 | 1.1 ± 0.7 | 6.5 ± 3.2 |
| 4 mg/kg | 7 | 2 (1–4) | 4540 ± 5401 | 10,420 ± 10,189 | 10,460 ± 10,198 | 4.1 ± 1.3 | 1 ± 1 | 5.5 ± 6.1 | |
| 6 mg/kg | 1 | 3 | 5336 | 14,515 | 14,535 | 3.7 | 0.4 | 2.2 | |
| 14 (cycle 1) | 2 mg/kg | 5 | 2 | 1206 ± 1502 | 3375 ± 1988 | - | 4.8 ± 1 | 0.8 ± 0.6 | 5.9 ± 4.4 |
| 4 mg/kg | 4 | 1.5 | 6220 ± 7769 | 17,367 ± 20,413 | - | 4.8 ± 2 | 0.7 ± 0.5 | 2.9 ± 3.2 | |
| 6 mg/kg | 1 | 5 | 4263 | 13,412 | - | 4.5 | 0.5 | 2.9 | |
| Celecoxib | |||||||||
| Day | Dose | n | Tmax (h) median (range) | Cmax (ng/mL) | AUC0–12 h (h.ng/mL) | AUC0–∞ (h.ng/mL) | T1/2 (h) | CL/F (L/h/kg) | Vz/F (L/kg) |
| 1 (cycle 2) | 200 mg | 6 | 3.5 (2–4) | 1475 ± 430 | 7577 ± 2032 | 11,065 ± 4222 | 6 ± 3 | 0.6 ± 0.5 | 4.2 ± 1.9 |
| 400 mg | 7 | 4 (3–8) | 1351 ± 317 | 7378 ± 2228 | 10,696 ± 3631 | 6 ± 3 | 0.6 ± 0.2 | 5.0 ± 2.4 | |
| 200 mg | 6 | 3.5 (2–4) | 1475 ± 430 | 7577 ± 2032 | 11,065 ± 4222 | 6 ± 3 | 0.6 ± 0.5 | 4.2 ± 1.9 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leblond, P.; Tresch-Bruneel, E.; Probst, A.; Néant, N.; Solas, C.; Sterin, A.; Boulanger, T.; Aerts, I.; Faure-Conter, C.; Bertozzi, A.-I.; et al. Phase I Study of a Combination of Fluvastatin and Celecoxib in Children with Relapsing/Refractory Low-Grade or High-Grade Glioma (FLUVABREX). Cancers 2023, 15, 2020. https://doi.org/10.3390/cancers15072020
Leblond P, Tresch-Bruneel E, Probst A, Néant N, Solas C, Sterin A, Boulanger T, Aerts I, Faure-Conter C, Bertozzi A-I, et al. Phase I Study of a Combination of Fluvastatin and Celecoxib in Children with Relapsing/Refractory Low-Grade or High-Grade Glioma (FLUVABREX). Cancers. 2023; 15(7):2020. https://doi.org/10.3390/cancers15072020
Chicago/Turabian StyleLeblond, Pierre, Emmanuelle Tresch-Bruneel, Alicia Probst, Nadège Néant, Caroline Solas, Arthur Sterin, Thomas Boulanger, Isabelle Aerts, Cécile Faure-Conter, Anne-Isabelle Bertozzi, and et al. 2023. "Phase I Study of a Combination of Fluvastatin and Celecoxib in Children with Relapsing/Refractory Low-Grade or High-Grade Glioma (FLUVABREX)" Cancers 15, no. 7: 2020. https://doi.org/10.3390/cancers15072020