
Anti-Angiogenic Activity of Drugs in Multiple Myeloma
 , ,
, , {kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
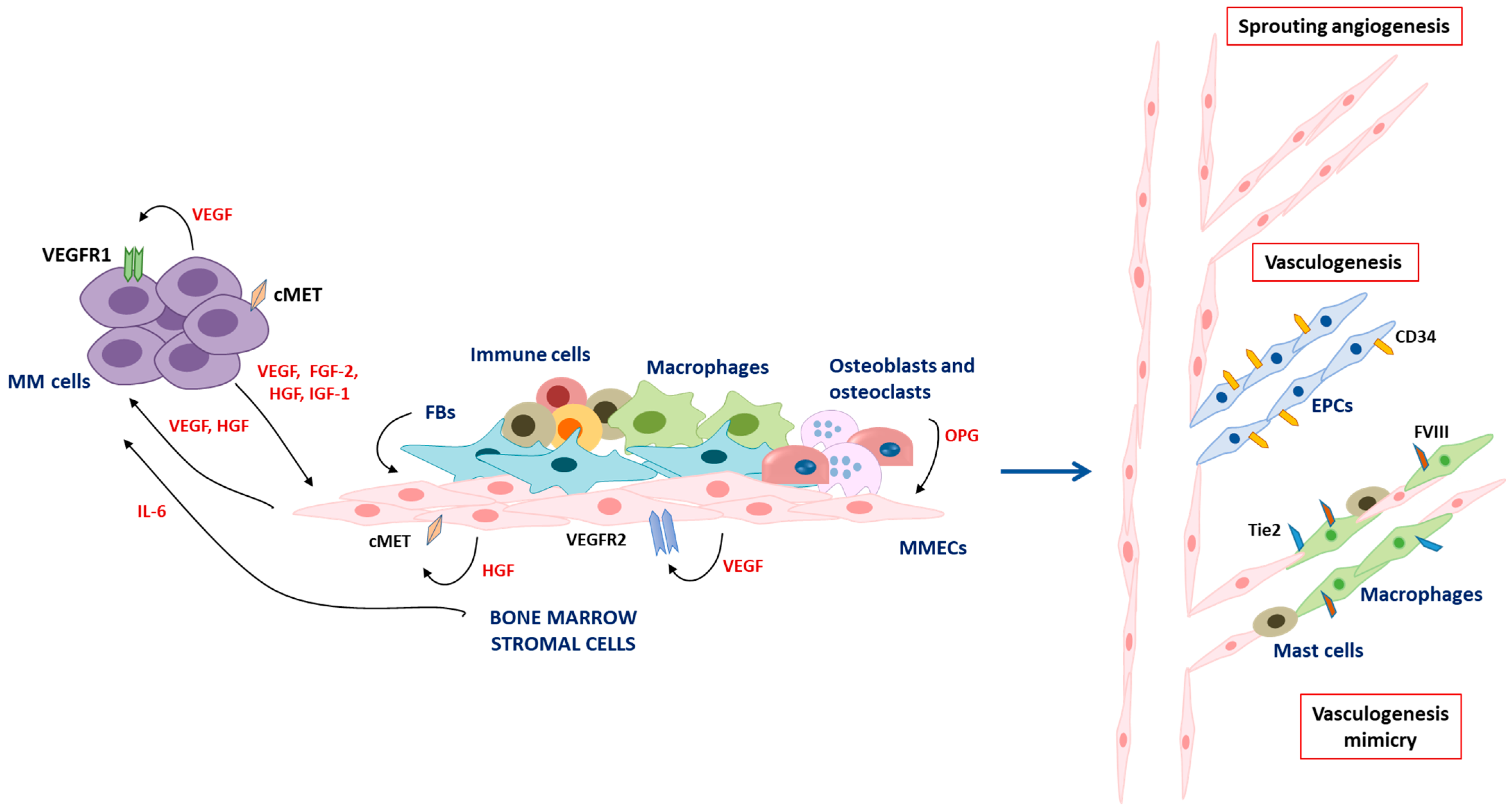
2. Angiogenesis and Vasculogenesis in Multiple Myeloma
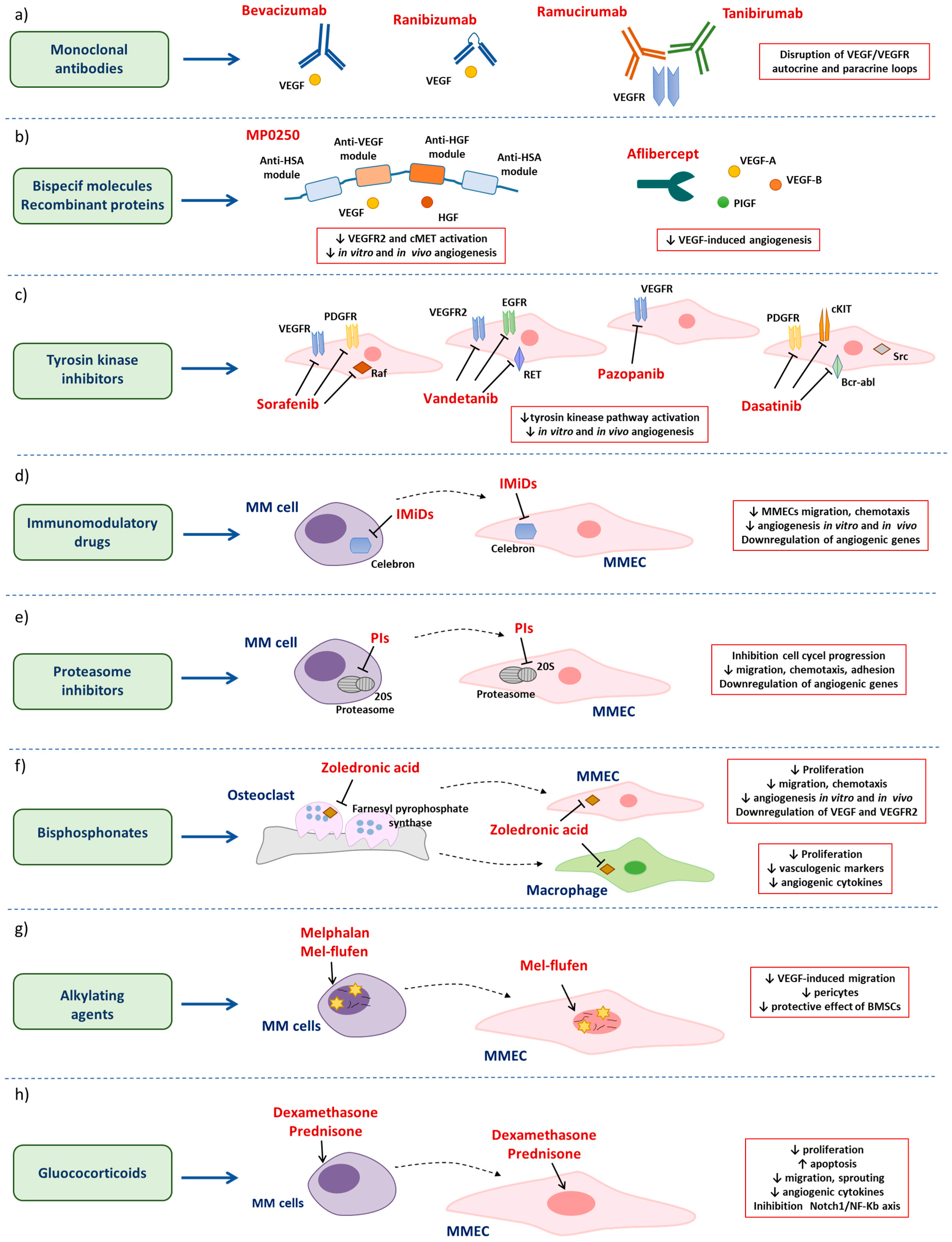
3. Anti-Angiogenic Drugs in MM
3.1. Anti-Angiogenic Drugs in Multiple Myeloma
3.1.1. Monoclonal Antibodies
3.1.2. Bispecific Molecules and Recombinant Molecules Targeting Angiogenic Cytokines
3.1.3. Tyrosine Kinase Inhibitors
3.2. Antimyeloma Drugs with Secondary Anti-Angiogenic Properties
3.2.1. Immunomodulatory Drugs
3.2.2. Proteasome Inhibitors
3.2.3. Bisphosphonates
3.2.4. Alkylating Agents
3.2.5. Glucocorticoids
4. Concluding Remarks
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kumar, S.K.; Rajkumar, V.; Kyle, R.A.; van Duin, M.; Sonneveld, P.; Mateos, M.V.; Gay, F.; Anderson, K.C. Multiple myeloma. Nat. Rev. Dis. Prim. 2017, 3, 17046. [Google Scholar] [CrossRef] [PubMed]
- Mateos, M.V.; Landgren, O. MGUS and Smoldering Multiple Myeloma: Diagnosis and Epidemiology. Cancer Treat. Res. 2016, 169, 3–12. [Google Scholar] [PubMed]
- Manier, S.; Sacco, A.; Leleu, X.; Ghobrial, I.M.; Roccaro, A.M. Bone marrow microenvironment in multiple myeloma progression. J. Biomed. Biotechnol. 2012, 2012, 157496. [Google Scholar] [CrossRef] [Green Version]
- Di Marzo, L.; Desantis, V.; Solimando, A.G.; Ruggieri, S.; Annese, T.; Nico, B.; Fumarulo, R.; Vacca, A.; Frassanito, M.A. Microenvironment drug resistance in multiple myeloma: Emerging new players. Oncotarget 2016, 7, 60698–60711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giuliani, N.; Colla, S.; Rizzoli, V. Angiogenic switch in multiple myeloma. Hematology 2004, 9, 377–381. [Google Scholar] [CrossRef] [Green Version]
- Vacca, A.; Ribatti, D.; Roncali, L.; Ranieri, G.; Serio, G.; Silvestris, F.; Dammacco, F. Bone marrow angiogenesis and progression in multiple myeloma. Br. J. Haematol. 1994, 87, 503–508. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Fonseca, R.; Dispenzieri, A.; Lacy, M.Q.; Lust, J.A.; Wellik, L.; Witzig, T.E.; Gertz, M.A.; Kyle, R.A.; Greipp, P.R.; et al. Prognostic value of angiogenesis in solitary bone plasmacytoma. Blood 2003, 10, 1715–1717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Gertz, M.A.; Dispenzieri, A.; Lacy, M.Q.; Wellik, L.A.; Fonseca, R.; Lust, J.A.; Witzig, T.E.; Kyle, R.A.; Greipp, P.R.; et al. Prognostic value of bone marrow angiogenesis in patients with multiple myeloma undergoing high-dose therapy. Bone Marrow Transplant. 2004, 34, 235–239. [Google Scholar] [CrossRef] [Green Version]
- Saltarella, I.; Morabito, F.; Giuliani, N.; Terragna, C.; Omedè, P.; Palumbo, A.; Bringhen, S.; De Paoli, L.; Martino, E.; Larocca, A.; et al. Prognostic or predictive value of circulating cytokines and angiogenic factors for initial treatment of multiple myeloma in the GIMEMA MM0305 randomized controlled trial. J. Hematol. Oncol. 2019, 12, 4. [Google Scholar] [CrossRef]
- Ribatti, D.; Vacca, A. New Insights in Anti-Angiogenesis in Multiple Myeloma. Int. J. Mol. Sci. 2018, 19, 2031. [Google Scholar] [CrossRef] [Green Version]
- Ria, R.; Melaccio, A.; Racanelli, V.; Vacca, A. Anti-VEGF Drugs in the Treatment of Multiple Myeloma Patients. J. Clin. Med. 2020, 9, 1765. [Google Scholar] [CrossRef] [PubMed]
- Reale, A.; Melaccio, A.; Lamanuzzi, A.; Saltarella, I.; Dammacco, F.; Vacca, A.; Ria, R. Functional and Biological Role of Endothelial Precursor Cells in Tumour Progression: A New Potential Therapeutic Target in Haematological Malignancies. Stem Cells Int. 2016, 2016, 7954580. [Google Scholar] [CrossRef] [Green Version]
- Fan, F.; Malvestiti, S.; Vallet, S.; Lind, J.; Garcia-Manteiga, J.M.; Morelli, E.; Jiang, Q.; Seckinger, A.; Hose, D.; Goldschmidt, H.; et al. JunB is a key regulator of multiple myeloma bone marrow angiogenesis. Leukemia 2021, 35, 3509–3525. [Google Scholar] [CrossRef] [PubMed]
- Hose, D.; Moreaux, J.; Meissner, T.; Seckinger, A.; Goldschmidt, H.; Benner, A.; Mahtouk, K.; Hillengass, J.; Rème, T.; De Vos, J.; et al. Induction of angiogenesis by normal and malignant plasma cells. Blood 2009, 114, 128–143. [Google Scholar] [CrossRef] [Green Version]
- Melaccio, A.; Reale, A.; Saltarella, I.; Desantis, V.; Lamanuzzi, A.; Cicco, S.; Frassanito, M.A.; Vacca, A.; Ria, R. Pathways of Angiogenic and Inflammatory Cytokines in Multiple Myeloma: Role in Plasma Cell Clonal Expansion and Drug Resistance. J. Clin. Med. 2022, 11, 6491. [Google Scholar] [CrossRef] [PubMed]
- Vacca, A.; Ria, R.; Ribatti, D.; Semeraro, F.; Djonov, V.; Di Raimondo, F.; Dammacco, F. A paracrine loop in the vascular endothelial growth factor pathway triggers tumor angiogenesis and growth in multiple myeloma. Haematologica 2003, 88, 176–185. [Google Scholar]
- Ria, R.; Vacca, A.; Russo, F.; Cirulli, T.; Massaia, M.; Tosi, P.; Cavo, M.; Guidolin, D.; Ribatti, D.; Dammacco, F. A VEGF-dependent autocrine loop mediates proliferation and capillarogenesis in bone marrow endothelial cells of patients with multiple myeloma. Thromb. Haemost. 2004, 92, 1438–1445. [Google Scholar] [CrossRef]
- Gupta, D.; Treon, S.P.; Shima, Y.; Hideshima, T.; Podar, K.; Tai, Y.T.; Lin, B.; Lentzsch, S.; Davies, F.E.; Chauhan, D.; et al. Adherence of multiple myeloma cells to bone marrow stromal cells upregulates vascular endothelial growth factor secretion: Therapeutic applications. Leukemia 2001, 15, 1950–1961. [Google Scholar] [CrossRef] [Green Version]
- Ferrucci, A.; Moschetta, M.; Frassanito, M.A.; Berardi, S.; Catacchio, I.; Ria, R.; Racanelli, V.; Caivano, A.; Solimando, A.G.; Vergara, D.; et al. A HGF/cMET autocrine loop is operative in multiple myeloma bone marrow endothelial cells and may represent a novel therapeutic target. Clin. Cancer Res. 2014, 20, 5796–5807. [Google Scholar] [CrossRef] [Green Version]
- Moschetta, M.; Basile, A.; Ferrucci, A.; Frassanito, M.A.; Rao, L.; Ria, R.; Solimando, A.G.; Giuliani, N.; Boccarelli, A.; Fumarola, F.; et al. Novel targeting of phospho-cMET overcomes drug resistance and induces antitumor activity in multiple myeloma. Clin. Cancer Res. 2013, 19, 4371–4382. [Google Scholar] [CrossRef] [Green Version]
- Rocci, A.; Gambella, M.; Aschero, S.; Baldi, I.; Trusolino, L.; Cavallo, F.; Gay, F.; Larocca, A.; Magarotto, V.; Omedè, P.; et al. MET dysregulation is a hallmark of aggressive disease in multiple myeloma patients. Br. J. Haematol. 2014, 164, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Hattori, Y.; Wenlin, D.; Yamada, T.; Kamata, T.; Kakimoto, T.; Okamoto, S.; Kawamura, C.; Kizaki, M.; Shimada, N.; et al. Elevated level of plasma basic fibroblast growth factor in multiple myeloma correlates with increased disease activity. Jpn. J. Cancer Res. 2002, 93, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Sezer, O.; Jakob, C.; Eucker, J.; Niemöller, K.; Gatz, F.; Wernecke, K.; Possinger, K. Serum levels of the angiogenic cytokines basic fibroblast growth factor (bFGF), vascular endothelial growth factor (VEGF) and hepatocyte growth factor (HGF) in multiple myeloma. Eur. J. Haematol. 2001, 66, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, N.; Colla, S.; Lazzaretti, M.; Sala, R.; Roti, G.; Mancini, C.; Bonomini, S.; Lunghi, P.; Hojden, M.; Genestreti, G.; et al. Proangiogenic properties of human myeloma cells: Production of angiopoietin-1 and its potential relationship to myeloma-induced angiogenesis. Blood 2003, 102, 638–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bieghs, L.; Johnsen, H.E.; Maes, K.; Menu, E.; Van Valckenborgh, E.; Overgaard, M.T.; Nyegaard, M.; Conover, C.A.; Vanderkerken, K.; De Bruyne, E. The insulin-like growth factor system in multiple myeloma: Diagnostic and therapeutic potential. Oncotarget 2016, 7, 48732–48752. [Google Scholar] [CrossRef]
- Vacca, A.; Ribatti, D.; Presta, M.; Minischetti, M.; Iurlaro, M.; Ria, R.; Albini, A.; Bussolino, F.; Dammacco, F. Bone marrow neovascularization, plasma cell angiogenic potential, and matrix metalloproteinase-2 secretion parallel progression of human multiple myeloma. Blood 1999, 93, 3064–3073. [Google Scholar] [CrossRef]
- Ria, R.; Reale, A.; De Luisi, A.; Ferrucci, A.; Moschetta, M.; Vacca, A. Bone marrow angiogenesis and progression in multiple myeloma. Am. J. Blood Res. 2011, 1, 76–89. [Google Scholar]
- Ria, R.; Catacchio, I.; Berardi, S.; De Luisi, A.; Caivano, A.; Piccoli, C.; Ruggieri, V.; Frassanito, M.A.; Ribatti, D.; Nico, B.; et al. HIF-1α of bone marrow endothelial cells implies relapse and drug resistance in patients with multiple myeloma and may act as a therapeutic target. Clin. Cancer Res. 2014, 20, 847–858. [Google Scholar] [CrossRef] [Green Version]
- Frassanito, M.A.; Rao, L.; Moschetta, M.; Ria, R.; Di Marzo, L.; De Luisi, A.; Racanelli, V.; Catacchio, I.; Berardi, S.; Basile, A.; et al. Bone marrow fibroblasts parallel multiple myeloma progression in patients and mice: In vitro and in vivo studies. Leukemia 2014, 28, 904–916. [Google Scholar] [CrossRef] [Green Version]
- Sfiridaki, K.; Pappa, C.A.; Tsirakis, G.; Kanellou, P.; Kaparou, M.; Stratinaki, M.; Sakellaris, G.; Kontakis, G.; Alexandrakis, M.G. Angiogenesis-related cytokines, RANKL, and osteoprotegerin in multiple myeloma patients in relation to clinical features and response to treatment. Mediat. Inflamm. 2011, 2011, 867576. [Google Scholar] [CrossRef] [Green Version]
- Ribatti, D.; Vacca, A. The role of inflammatory cells in angiogenesis in multiple myeloma. Adv. Exp. Med. Biol. 2014, 816, 361–376. [Google Scholar] [PubMed]
- Nico, B.; Mangieri, D.; Crivellato, E.; Vacca, A.; Ribatti, D. Mast cells contribute to vasculogenic mimicry in multiple myeloma. Stem Cells Dev. 2008, 17, 19–22. [Google Scholar] [CrossRef]
- Scavelli, C.; Nico, B.; Cirulli, T.; Ria, R.; Di Pietro, G.; Mangieri, D.; Bacigalupo, A.; Mangialardi, G.; Coluccia, A.M.; Caravita, T.; et al. Vasculogenic mimicry by bone marrow macrophages in patients with multiple myeloma. Oncogene 2008, 27, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Saltarella, I.; Apollonio, B.; Lamanuzzi, A.; Desantis, V.; Mariggiò, M.A.; Desaphy, J.F.; Vacca, A.; Frassanito, M.A. The Landscape of lncRNAs in Multiple Myeloma: Implications in the “Hallmarks of Cancer”, Clinical Perspectives and Therapeutic Opportunities. Cancers 2022, 14, 1963. [Google Scholar] [CrossRef] [PubMed]
- Umezu, T.; Tadokoro, H.; Azuma, K.; Yoshizawa, S.; Ohyashiki, K.; Ohyashiki, J.H. Exosomal miR-135b shed from hypoxic multiple myeloma cells enhances angiogenesis by targeting factor-inhibiting HIF-1. Blood 2014, 124, 3748–3757. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.Y.; She, X.M.; Qin, Y.; Chu, Z.B.; Chen, L.; Ai, L.S.; Zhang, L.; Hu, Y. miR-15a and miR-16 affect the angiogenesis of multiple myeloma by targeting VEGF. Carcinogenesis 2013, 34, 426–435. [Google Scholar] [CrossRef] [Green Version]
- Saltarella, I.; Lamanuzzi, A.; Apollonio, B.; Desantis, V.; Bartoli, G.; Vacca, A.; Frassanito, M.A. Role of Extracellular Vesicle-Based Cell-to-Cell Communication in Multiple Myeloma Progression. Cells 2021, 10, 3185. [Google Scholar] [CrossRef]
- Kazazi-Hyseni, F.; Beijnen, J.H.; Schellens, J.H. Bevacizumab. Oncologist 2010, 15, 819–825. [Google Scholar] [CrossRef]
- Braghiroli, M.I.; Sabbaga, J.; Hoff, P.M. Bevacizumab: Overview of the literature. Expert Rev. Anticancer Ther. 2012, 12, 567–580. [Google Scholar] [CrossRef]
- Attar-Schneider, O.; Drucker, L.; Zismanov, V.; Tartakover-Matalon, S.; Rashid, G.; Lishner, M. Bevacizumab attenuates major signaling cascades and eIF4E translation initiation factor in multiple myeloma cells. Lab. Investig. 2012, 92, 178–190. [Google Scholar] [CrossRef] [Green Version]
- Kong, D.H.; Kim, M.R.; Jang, J.H.; Na, H.J.; Lee, S. A Review of Anti-Angiogenic Targets for Monoclonal Antibody Cancer Therapy. Int. J. Mol. Sci. 2017, 18, 1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somlo, G.; Lashkari, A.; Bellamy, W.; Zimmerman, T.M.; Tuscano, J.M.; O’Donnell, M.R.; Mohrbacher, A.F.; Forman, S.J.; Frankel, P.; Chen, H.X.; et al. Phase II randomized trial of bevacizumab versus bevacizumab and thalidomide for relapsed/refractory multiple myeloma: A California Cancer Consortium trial. Br. J. Haematol. 2011, 154, 533–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, D.; Kassim, A.; Bhaskar, B.; Yi, J.; Wamstad, K.; Paton, V.E. Results from AMBER, a randomized phase 2 study of bevacizumab and bortezomib versus bortezomib in relapsed or refractory multiple myeloma. Cancer 2013, 119, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Lamanuzzi, A.; Saltarella, I.; Desantis, V.; Frassanito, M.A.; Leone, P.; Racanelli, V.; Nico, B.; Ribatti, D.; Ditonno, P.; Prete, M.; et al. Inhibition of mTOR complex 2 restrains tumor angiogenesis in multiple myeloma. Oncotarget 2018, 9, 20563–20577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saltarella, I.; Frassanito, M.A.; Lamanuzzi, A.; Brevi, A.; Leone, P.; Desantis, V.; Di Marzo, L.; Bellone, M.; Derudas, D.; Ribatti, D.; et al. Homotypic and Heterotypic Activation of the Notch Pathway in Multiple Myeloma-Enhanced Angiogenesis: A Novel Therapeutic Target? Neoplasia 2019, 21, 93–105. [Google Scholar] [CrossRef]
- Ferrara, N.; Damico, L.; Shams, N.; Lowman, H.; Kim, R. Development of ranibizumab, an anti-vascular endothelial growth factor antigen binding fragment, as therapy for neovascular age-related macular degeneration. Retina 2006, 26, 859–870. [Google Scholar] [CrossRef]
- Spratlin, J. Ramucirumab (IMC-1121B): Monoclonal antibody inhibition of vascular endothelial growth factor receptor-2. Curr. Oncol. Rep. 2011, 13, 97–102. [Google Scholar] [CrossRef]
- Kim, J.; Choi, S.H.; Ham, S.J.; Cho, Y.C.; Lee, S.I.; Kang, J.; Woo, D.C.; Lee, W.S.; Yoo, J.S.; Kim, K.W.; et al. Evaluation of drug mechanism and efficacy of a novel anti-angiogenic agent, TTAC-0001, using multi-modality bioimaging in a mouse breast cancer orthotopic model. PLoS ONE 2018, 13, e0187063. [Google Scholar] [CrossRef] [Green Version]
- Binz, H.K.; Bakker, T.R.; Phillips, D.J.; Cornelius, A.; Zitt, C.; Göttler, T.; Sigrist, G.; Fiedler, U.; Ekawardhani, S.; Dolado, I.; et al. Design and characterization of MP0250, a tri-specific anti-HGF/anti-VEGF DARPin® drug candidate. mAbs 2017, 9, 1262–1269. [Google Scholar] [CrossRef] [Green Version]
- Rao, L.; De Veirman, K.; Giannico, D.; Saltarella, I.; Desantis, V.; Frassanito, M.A.; Solimando, A.G.; Ribatti, D.; Prete, M.; Harstrick, A.; et al. Targeting angiogenesis in multiple myeloma by the VEGF and HGF blocking DARPin® protein MP0250: A preclinical study. Oncotarget 2018, 9, 13366–13381. [Google Scholar] [CrossRef] [Green Version]
- Knop, S.; Goldschmidt, H.; Raab, M.S.; Szarejko, M.; Jurczyszyn, A.; Dürig, J.; Bringhen, S.; Gamberi, B.; Vacca, A.; Acosta, J.C. MP0250 Combined with Bortezomib and Dexamethasone in Multiple Myeloma Patients Previoulsy Exposed to Proteasome Inhibitors and Immunomodulatory Drugs. Blood 2018, 132, 1980. [Google Scholar] [CrossRef]
- Al-Halafi, A.M. Vascular endothelial growth factor trap-eye and trap technology: Aflibercept from bench to bedside. Oman J. Ophthalmol. 2014, 7, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Ricci, V.; Ronzoni, M.; Fabozzi, T. Aflibercept a new target therapy in cancer treatment: A review. Crit. Rev. Oncol. Hematol. 2015, 96, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef] [Green Version]
- Strumberg, D.; Clark, J.W.; Awada, A.; Moore, M.J.; Richly, H.; Hendlisz, A.; Hirte, H.W.; Eder, J.P.; Lenz, H.J.; Schwartz, B. Safety, pharmacokinetics, and preliminary antitumor activity of sorafenib: A review of four phase I trials in patients with advanced refractory solid tumors. Oncol. 2007, 12, 426–437. [Google Scholar] [CrossRef] [Green Version]
- Kharaziha, P.; De Raeve, H.; Fristedt, C.; Li, Q.; Gruber, A.; Johnsson, P.; Kokaraki, G.; Panzar, M.; Laane, E.; Osterborg, A.; et al. Sorafenib has potent antitumor activity against multiple myeloma in vitro, ex vivo, and in vivo in the 5T33MM mouse model. Cancer Res. 2012, 72, 5348–5362. [Google Scholar] [CrossRef] [Green Version]
- Ramakrishnan, V.; Timm, M.; Haug, J.L.; Kimlinger, T.K.; Wellik, L.E.; Witzig, T.E.; Rajkumar, S.V.; Adjei, A.A.; Kumar, S. Sorafenib, a dual Raf kinase/vascular endothelial growth factor receptor inhibitor has significant anti-myeloma activity and synergizes with common anti-myeloma drugs. Oncogene 2010, 29, 1190–1202. [Google Scholar] [CrossRef] [Green Version]
- Yordanova, A.; Hose, D.; Neben, K.; Witzens-Harig, M.; Gütgemann, I.; Raab, M.S.; Moehler, T.; Goldschmidt, H.; Schmidt-Wolf, I.G. Sorafenib in patients with refractory or recurrent multiple myeloma. Hematol. Oncol. 2013, 31, 197–200. [Google Scholar] [CrossRef]
- Ryan, A.J.; Wedge, S.R. ZD6474--a novel inhibitor of VEGFR and EGFR tyrosine kinase activity. Br. J. Cancer. 2005, 92, S6–S13. [Google Scholar] [CrossRef] [Green Version]
- Wedge, S.R.; Ogilvie, D.J.; Dukes, M.; Kendrew, J.; Chester, R.; Jackson, J.A.; Boffey, S.J.; Valentine, P.J.; Curwen, J.O.; Musgrove, H.L.; et al. ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration. Cancer Res. 2002, 62, 4645–4655. [Google Scholar]
- Morabito, A.; Piccirillo, M.C.; Falasconi, F.; De Feo, G.; Del Giudice, A.; Bryce, J.; Di Maio, M.; De Maio, E.; Normanno, N.; Perrone, F. Vandetanib (ZD6474), a dual inhibitor of vascular endothelial growth factor receptor (VEGFR) and epidermal growth factor receptor (EGFR) tyrosine kinases: Current status and future directions. Oncologist 2009, 14, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, M.J.; Reece, D.E.; Marcellus, D.; Meyer, R.M.; Mathews, S.; Dong, R.P.; Eisenhauer, E. A phase II study of ZD6474, Zactima, a selective inhibitor of VEGFR and EGFR tyrosine kinase in patients with relapsed multiple myeloma—NCIC CTG IND.145. Investig. New Drugs 2006, 24, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Podar, K.; Tonon, G.; Sattler, M.; Tai, Y.T.; Legouill, S.; Yasui, H.; Ishitsuka, K.; Kumar, S.; Kumar, R.; Pandite, L.N.; et al. The small-molecule VEGF receptor inhibitor pazopanib (GW786034B) targets both tumor and endothelial cells in multiple myeloma. Proc. Natl. Acad. Sci. USA 2006, 103, 19478–19483. [Google Scholar] [CrossRef] [Green Version]
- Prince, H.M.; Hönemann, D.; Spencer, A.; Rizzieri, D.A.; Stadtmauer, E.A.; Roberts, A.W.; Bahlis, N.; Tricot, G.; Bell, B.; Demarini, D.J.; et al. Vascular endothelial growth factor inhibition is not an effective therapeutic strategy for relapsed or refractory multiple myeloma: A phase 2 study of pazopanib (GW786034). Blood 2009, 113, 4819–4820. [Google Scholar] [CrossRef] [PubMed]
- Santos, F.P.; Cortes, J. Dasatinib for the treatment of Philadelphia chromosome-positive leukemias. Expert Opin. Pharmacother. 2012, 13, 2381–2395. [Google Scholar] [CrossRef] [PubMed]
- Coluccia, A.M.; Cirulli, T.; Neri, P.; Mangieri, D.; Colanardi, M.C.; Gnoni, A.; Di Renzo, N.; Dammacco, F.; Tassone, P.; Ribatti, D.; et al. Validation of PDGFRbeta and c-Src tyrosine kinases as tumor/vessel targets in patients with multiple myeloma: Preclinical efficacy of the novel, orally available inhibitor dasatinib. Blood 2008, 112, 1346–1356. [Google Scholar] [CrossRef]
- Asatsuma-Okumura, T.; Ito, T.; Handa, H. Molecular Mechanisms of the Teratogenic Effects of Thalidomide. Pharmaceuticals 2020, 13, 95. [Google Scholar] [CrossRef]
- Latif, T.; Chauhan, N.; Khan, R.; Moran, A.; Usmani, S.Z. Thalidomide and its analogues in the treatment of Multiple Myeloma. Exp. Hematol. Oncol. 2012, 1, 27. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.X.; Kortuem, K.M.; Stewart, A.K. Molecular mechanism of action of immune-modulatory drugs thalidomide, lenalidomide and pomalidomide in multiple myeloma. Leuk. Lymphoma 2013, 54, 683–687. [Google Scholar] [CrossRef]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a primary target of thalidomide teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.X.; Braggio, E.; Shi, C.X.; Bruins, L.A.; Schmidt, J.E.; Van Wier, S.; Chang, X.B.; Bjorklund, C.C.; Fonseca, R.; Bergsagel, P.L.; et al. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood 2011, 118, 4771–4779. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Wang, L.; Guo, H.; Jia, J.; Gu, L.; Wang, X.; Yang, M.; Guan, H.; Yuan, W. Thalidomide Suppresses Angiogenesis Through the Signal Transducer and Activator of Transcription 3/SP4 Signaling Pathway in the Peritoneal Membrane. Front. Physiol. 2021, 12, 712147. [Google Scholar] [CrossRef] [PubMed]
- Yabu, T.; Tomimoto, H.; Taguchi, Y.; Yamaoka, S.; Igarashi, Y.; Okazaki, T. Thalidomide-induced antiangiogenic action is mediated by ceramide through depletion of VEGF receptors, and is antagonized by sphingosine-1-phosphate. Blood 2005, 106, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Fu, S.; Chen, H.; Feng, Q.; Gao, Y.; Xue, H.; Ge, Z.; Fang, J.; Xiao, S. Inhibition of endothelial Slit2/Robo1 signaling by thalidomide restrains angiogenesis by blocking the PI3K/Akt pathway. Dig. Dis. Sci. 2014, 59, 2958–2966. [Google Scholar] [CrossRef] [PubMed]
- Lepper, E.R.; Smith, N.F.; Cox, M.C.; Scripture, C.D.; Figg, W.D. Thalidomide metabolism and hydrolysis: Mechanisms and implications. Curr. Drug Metab. 2006, 7, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Holstein, S.A.; McCarthy, P.L. Immunomodulatory Drugs in Multiple Myeloma: Mechanisms of Action and Clinical Experience. Drugs 2017, 77, 505–520. [Google Scholar] [CrossRef]
- Price, D.K.; Ando, Y.; Kruger, E.A.; Weiss, M.; Figg, W.D. (5′-OH-thalidomide, a metabolite of thalidomide, inhibits angiogenesis. Ther. Drug Monit. 2002, 24, 104–110. [Google Scholar] [CrossRef]
- Gao, Y.; Ma, G.; Liu, S.; Teng, Y.; Wang, Y.; Su, Y. Thalidomide and multiple myeloma serum synergistically induce a hemostatic imbalance in endothelial cells in vitro. Thromb. Res. 2015, 135, 1154–1159. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Vacca, A. Novel therapeutic approaches targeting vascular endothelial growth factor and its receptors in haematological malignancies. Curr. Cancer Drug Targets 2005, 5, 573–578. [Google Scholar] [CrossRef]
- Vacca, A.; Scavelli, C.; Montefusco, V.; Di Pietro, G.; Neri, A.; Mattioli, M.; Bicciato, S.; Nico, B.; Ribatti, D.; Dammacco, F.; et al. Thalidomide downregulates angiogenic genes in bone marrow endothelial cells of patients with active multiple myeloma. J. Clin. Oncol. 2005, 23, 5334–5346. [Google Scholar] [CrossRef]
- De Luisi, A.; Ferrucci, A.; Coluccia, A.M.; Ria, R.; Moschetta, M.; de Luca, E.; Pieroni, L.; Maffia, M.; Urbani, A.; Di Pietro, G.; et al. Lenalidomide restrains motility and overangiogenic potential of bone marrow endothelial cells in patients with active multiple myeloma. Clin. Cancer Res. 2011, 17, 1935–1946. [Google Scholar] [CrossRef] [Green Version]
- Dredge, K.; Marriott, J.B.; Macdonald, C.D.; Man, H.W.; Chen, R.; Muller, G.W.; Stirling, D.; Dalgleish, A.G. Novel thalidomide analogues display anti-angiogenic activity independently of immunomodulatory effects. Br. J. Cancer 2010, 87, 1166–1172. [Google Scholar] [CrossRef]
- Kumar, S.; Witzig, T.E.; Dispenzieri, A.; Lacy, M.Q.; Wellik, L.E.; Fonseca, R.; Lust, J.A.; Gertz, M.A.; Kyle, R.A.; Greipp, P.R.; et al. Effect of thalidomide therapy on bone marrow angiogenesis in multiple myeloma. Leukemia 2004, 18, 624–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatjiharissi, E.; Terpos, E.; Papaioannou, M.; Hatjileontis, C.; Kaloutsi, V.; Galaktidou, G.; Gerotziafas, G.; Christakis, J.; Zervas, K. The combination of intermediate doses of thalidomide and dexamethasone reduces bone marrow micro-vessel density but not serum levels of angiogenic cytokines in patients with refractory/relapsed multiple myeloma. Hematol. Oncol. 2004, 22, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Cury, P.C.C.; Higashi, F.; Zacchi, F.F.S.; Palhares, R.B.; Quero, A.A.; Dias, A.L.M.S.; Crusoé, E.Q.; Hungria, V.T.M. Effect of thalidomide on bone marrow angiogenesis in multiple myeloma patients. Hematol. Transfus. Cell Ther. 2020, 42, 159–163. [Google Scholar] [CrossRef]
- Cibeira, M.T.; Rozman, M.; Segarra, M.; Lozano, E.; Rosiñol, L.; Cid, M.C.; Filella, X.; Bladé, J. Bone marrow angiogenesis and angiogenic factors in multiple myeloma treated with novel agents. Cytokine 2008, 41, 244–253. [Google Scholar] [CrossRef]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J.; Lee, K.P., Jr.; Boise, L.H. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, C.R.C.; Abdul-Majeed, S.; Cael, B.; Barta, S.K. Clinical Pharmacokinetics and Pharmacodynamics of Bortezomib. Clin. Pharmacokinet. 2019, 58, 157–168. [Google Scholar] [CrossRef]
- Ito, S. Proteasome Inhibitors for the Treatment of Multiple Myeloma. Cancers 2020, 12, 265. [Google Scholar] [CrossRef] [Green Version]
- Tamura, D.; Arao, T.; Tanaka, K.; Kaneda, H.; Matsumoto, K.; Kudo, K.; Aomatsu, K.; Fujita, Y.; Watanabe, T.; Saijo, N.; et al. Bortezomib potentially inhibits cellular growth of vascular endothelial cells through suppression of G2/M transition. Cancer Sci. 2010, 101, 1403–1408. [Google Scholar] [CrossRef]
- Belloni, D.; Veschini, L.; Foglieni, C.; Dell’Antonio, G.; Caligaris-Cappio, F.; Ferrarini, M.; Ferrero, E. Bortezomib induces autophagic death in proliferating human endothelial cells. Exp. Cell Res. 2010, 316, 1010–1018. [Google Scholar] [CrossRef] [PubMed]
- Di Lernia, G.; Leone, P.; Solimando, A.G.; Buonavoglia, A.; Saltarella, I.; Ria, R.; Ditonno, P.; Silvestris, N.; Crudele, L.; Vacca, A.; et al. Bortezomib Treatment Modulates Autophagy in Multiple Myeloma. J. Clin. Med. 2020, 9, 552. [Google Scholar] [CrossRef] [Green Version]
- Veschini, L.; Belloni, D.; Foglieni, C.; Cangi, M.G.; Ferrarini, M.; Caligaris-Cappio, F.; Ferrero, E. Hypoxia-inducible transcription factor-1 alpha determines sensitivity of endothelial cells to the proteosome inhibitor bortezomib. Blood 2007, 109, 2565–2570. [Google Scholar]
- Roccaro, A.M.; Hideshima, T.; Raje, N.; Kumar, S.; Ishitsuka, K.; Yasui, H.; Shiraishi, N.; Ribatti, D.; Nico, B.; Vacca, A.; et al. Bortezomib mediates antiangiogenesis in multiple myeloma via direct and indirect effects on endothelial cells. Cancer Res. 2006, 66, 184–191. [Google Scholar] [CrossRef] [Green Version]
- Politou, M.; Naresh, K.; Terpos, E.; Crawley, D.; Lampert, I.; Apperley, J.F.; Rahemtulla, A. Anti-angiogenic effect of bortezomib in patients with multiple myeloma. Blood 2005, 104, 4912. [Google Scholar] [CrossRef]
- Terpos, E.; Ntanasis-Stathopoulos, I.; Dimopoulos, M.A. Myeloma bone disease: From biology findings to treatment approaches. Blood 2019, 133, 1534–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drake, M.T.; Clarke, B.L.; Khosla, S. Bisphosphonates: Mechanism of action and role in clinical practice. Mayo Clin. Proc. 2008, 83, 1032–1045. [Google Scholar] [CrossRef] [Green Version]
- Salari, P.; Abdollahi, M. Long term bisphosphonate use in osteoporotic patients; a step forward, two steps back. J. Pharm. Pharm. Sci. 2012, 15, 305–317. [Google Scholar] [CrossRef] [Green Version]
- Tassone, P.; Forciniti, S.; Galea, E.; Morrone, G.; Turco, M.C.; Martinelli, V.; Tagliaferri, P.; Venuta, S. Growth inhibition and synergistic induction of apoptosis by zoledronate and dexamethasone in human myeloma cell lines. Leukemia 2000, 14, 841–844. [Google Scholar] [CrossRef] [Green Version]
- Ural, A.U.; Yilmaz, M.I.; Avcu, F.; Pekel, A.; Zerman, M.; Nevruz, O.; Sengul, A.; Yalcin, A. The bisphosphonate zoledronic acid induces cytotoxicity in human myeloma cell lines with enhancing effects of dexamethasone and thalidomide. Int. J. Hematol. 2003, 78, 443–449. [Google Scholar]
- Scavelli, C.; Di Pietro, G.; Cirulli, T.; Coluccia, M.; Boccarelli, A.; Giannini, T.; Mangialardi, G.; Bertieri, R.; Coluccia, A.M.; Ribatti, D.; et al. Zoledronic acid affects over-angiogenic phenotype of endothelial cells in patients with multiple myeloma. Mol. Cancer Ther. 2007, 6, 3256–3262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croucher, P.I.; De Hendrik, R.; Perry, M.J.; Hijzen, A.; Shipman, C.M.; Lippitt, J.; Green, J.; Van Marck, E.; Van Camp, B.; Vanderkerken, K. Zoledronic acid treatment of 5T2MM-bearing mice inhibits the development of myeloma bone disease: Evidence for decreased osteolysis, tumor burden and angiogenesis, and increased survival. J. Bone Miner. Res. 2003, 18, 482–492. [Google Scholar] [CrossRef] [PubMed]
- Moschetta, M.; Di Pietro, G.; Ria, R.; Gnoni, A.; Mangialardi, G.; Guarini, A.; Ditonno, P.; Musto, P.; D’Auria, F.; Ricciardi, M.R.; et al. Bortezomib and zoledronic acid on angiogenic and vasculogenic activities of bone marrow macrophages in patients with multiple myeloma. Eur. J. Cancer 2010, 46, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Falco, P.; Bringhen, S.; Avonto, I.; Gay, F.; Morabito, F.; Boccadoro, M.; Palumbo, A. Melphalan and its role in the management of patients with multiple myeloma. Expert Rev. Anticancer Ther. 2007, 7, 945–957. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Kumar, S.; Prasad, D.N.; Bhardwaj, T.R. Therapeutic journery of nitrogen mustard as alkylating anticancer agents: Historic to future perspectives. Eur. J. Med. Chem. 2018, 151, 401–433. [Google Scholar] [CrossRef]
- Poczta, A.; Rogalska, A.; Marczak, A. Treatment of Multiple Myeloma and the Role of Melphalan in the Era of Modern Therapies-Current Research and Clinical Approaches. J. Clin. Med. 2021, 10, 1841. [Google Scholar] [CrossRef]
- Begleiter, A.; Lam, H.Y.; Grover, J.; Froese, E.; Goldenberg, G.J. Evidence for active transport of melphalan by two amino acid carriers in L5178Y lymphoblasts in vitro. Cancer Res. 1979, 39, 353–359. [Google Scholar]
- Nath, C.E.; Shaw, P.J.; Trotman, J.; Zeng, L.; Duffull, S.B.; Hegarty, G.; McLachlan, A.J.; Gurney, H.; Kerridge, I.; Kwan, Y.L.; et al. Population pharmacokinetics of melphalan in patients with multiple myeloma undergoing high dose therapy. Br. J. Clin. Pharmacol. 2010, 69, 484–497. [Google Scholar] [CrossRef]
- Winter, U.; Mena, H.A.; Negrotto, S.; Arana, E.; Pascual-Pasto, G.; Laurent, V.; Suñol, M.; Chantada, G.L.; Carcaboso, A.M.; Schaiquevich, P. Schedule-Dependent Antiangiogenic and Cytotoxic Effects of Chemotherapy on Vascular Endothelial and Retinoblastoma Cells. PLoS ONE 2016, 11, e0160094. [Google Scholar] [CrossRef]
- Yang, Y.; Xing, Y.; Liang, C.; Hu, L.; Xu, F.; Mei, Q. In search of underlying mechanisms and potential drugs of melphalan-induced vascular toxicity through retinal endothelial cells using bioinformatics approach. Tumor Biol. 2016, 37, 6709–6718. [Google Scholar] [CrossRef]
- Ocio, E.M.; Nadeem, O.; Schjesvold, F.; Gay, F.; Touzeau, C.; Dimopoulos, M.A.; Richardson, P.G.; Mateos, M.V. Melflufen for the treatment of multiple myeloma. Expert Rev. Clin. Pharmacol. 2022, 15, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, J.J.; Kumari, R.; Traustadottir, G.A.; Huppunen, M.E.; Sergeev, P.; Majumder, M.M.; Schepsky, A.; Gudjonsson, T.; Lievonen, J.; Bazou, D.; et al. Aminopeptidase Expression in Multiple Myeloma Associates with Disease Progression and Sensitivity to Melflufen. Cancers 2021, 13, 1527. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Ray, A.; Viktorsson, K.; Spira, J.; Paba-Prada, C.; Munshi, N.; Richardson, P.; Lewensohn, R.; Anderson, K.C. In vitro and in vivo antitumor activity of a novel alkylating agent, melphalan-flufenamide, against multiple myeloma cells. Clin. Cancer Res. 2013, 19, 3019–3031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gebraad, A.; Ohlsbom, R.; Miettinen, J.J.; Emeh, P.; Pakarinen, T.K.; Manninen, M.; Eskelinen, A.; Kuismanen, K.; Slipicevic, A.; Lehmann, F.; et al. Growth Response and Differentiation of Bone Marrow-Derived Mesenchymal Stem/Stromal Cells in the Presence of Novel Multiple Myeloma Drug Melflufen. Cells 2022, 11, 1574. [Google Scholar] [CrossRef]
- Burwick, N.; Sharma, S. Glucocorticoids in multiple myeloma: Past, present, and future. Ann. Hematol. 2019, 98, 19–28. [Google Scholar] [CrossRef]
- Pufall, M.A. Glucocorticoids and Cancer. Adv. Exp. Med. Biol. 2015, 872, 315–333. [Google Scholar]
- King, T.; Faiman, B. Steroid-Associated Side Effects: A Symptom Management Update on Multiple Myeloma Treatment. Clin. J. Oncol. Nurs. 2017, 21, 240–249. [Google Scholar] [CrossRef]
- Chauhan, D.; Hideshima, T.; Rosen, S.; Reed, J.C.; Kharbanda, S.; Anderson, K.C. Apaf-1/cytochrome c-independent and Smac-dependent induction of apoptosis in multiple myeloma (MM) cells. J. Biol. Chem. 2001, 276, 24453–24456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauhan, D.; Auclair, D.; Robinson, E.K.; Hideshima, T.; Li, G.; Podar, K.; Gupta, D.; Richardson, P.; Schlossman, R.L.; Krett, N.; et al. Identification of genes regulated by dexamethasone in multiple myeloma cells using oligonucleotide arrays. Oncogene 2002, 21, 1346–1358. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Goodwin, J.E. The Effect of Glucocorticoids on Angiogenesis in the Treatment of Solid Tumors. J. Cell Signal 2020, 1, 42–49. [Google Scholar]
- Guo, L.P.; Niu, Y.F.; Shi, H.T.; Chen, H.M.; Zeng, T.M.; Tao, C.J.; Yuan, Z.G. Prednisolone and chlormethine inhibit multiple myeloma through inhibition of Notch/NF-kappa B-mediated angiogenesis. Int. J. Clin. Exp. Med. 2018, 11, 1699–1707. [Google Scholar]
- Morandi, F.; Horenstein, A.L.; Costa, F.; Giuliani, N.; Pistoia, V.; Malavasi, F. CD38: A Target for Immunotherapeutic Approaches in Multiple Myeloma. Front. Immunol. 2018, 9, 2722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonial, S.; Dimopoulos, M.; Palumbo, A.; White, D.; Grosicki, S.; Spicka, I.; Walter-Croneck, A.; Moreau, P.; Mateos, M.V.; Magen, H.; et al. Elotuzumab Therapy for Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2015, 373, 621–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terpos, E.; Katodritou, E.; Symeonidis, A.; Zagouri, F.; Gerofotis, A.; Christopoulou, G.; Gavriatopoulou, M.; Christoulas, D.; Ntanasis-Stathopoulos, I.; Kourakli, A.; et al. Effect of induction therapy with lenalidomide, doxorubicin and dexamethasone on bone remodeling and angiogenesis in newly diagnosed multiple myeloma. Int. J. Cancer 2019, 145, 559–568. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saltarella, I.; Altamura, C.; Campanale, C.; Laghetti, P.; Vacca, A.; Frassanito, M.A.; Desaphy, J.-F. Anti-Angiogenic Activity of Drugs in Multiple Myeloma. Cancers 2023, 15, 1990. https://doi.org/10.3390/cancers15071990
Saltarella I, Altamura C, Campanale C, Laghetti P, Vacca A, Frassanito MA, Desaphy J-F. Anti-Angiogenic Activity of Drugs in Multiple Myeloma. Cancers. 2023; 15(7):1990. https://doi.org/10.3390/cancers15071990
Chicago/Turabian StyleSaltarella, Ilaria, Concetta Altamura, Carmen Campanale, Paola Laghetti, Angelo Vacca, Maria Antonia Frassanito, and Jean-François Desaphy. 2023. "Anti-Angiogenic Activity of Drugs in Multiple Myeloma" Cancers 15, no. 7: 1990. https://doi.org/10.3390/cancers15071990