

Mathematical Model of Clonal Evolution Proposes a Personalised Multi-Modal Therapy for High-Risk Neuroblastoma

 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
- 1
- The clonal competition between the treatment-sensitive, treatment-resistant, and non-malignant cell populations weakens the total cell population. For example, an evolution-guided application of paclitaxel was found to keep resistant cancer cells in check, thus prolonging the progression-free survival in preclinical breast cancer models [14].
- 2
- If the oncologist could hypothetically predict which mutations will be selected by the therapeutic agents, an evolutionary trap could theoretically be created (called a sucker’s gambit in the review [12]). In fact, the treatment-sensitive population could theoretically be maintained indefinitely by cycling between two complementary agents (evolutionary herding). It is known from experimental data that targetable mutations and alterations of oncogenic pathways in neuroblastoma are selected by chemotherapeutic agents and enriched at relapse [15]; examples are de novo mutations in ALK [16] and the genes encoding the RAS-MAPK pathway [17]. In fact, drugs targeting specific molecular aberrations in neuroblastoma are under active development and ALK inhibitors are the most notable examples because they are frontline treatment options [18,19]. Although neuroblastoma can develop resistance to ALK inhibitors too, the resistance mechanisms involved create other vulnerabilities, such as hypersensitivity to MEK inhibition [20].
- 3
- Chemotherapy will break the total cell population into smaller, fragmented (spatially distinct) [21], and genetically homogeneous (discussed above) cell populations. They are potentially vulnerable to even tiny stochastic perturbations induced by drugs or hypoxia. For instance, in a tumour, cells must cooperate to generate an angiogenic signal [22], so targeted therapies would kill them most effectively after the tumour breaks into clusters and before they can reconnect to build new blood vessels. To exploit the unique vulnerabilities of small populations, it is necessary to switch therapeutic agents when the vulnerabilities emerge.
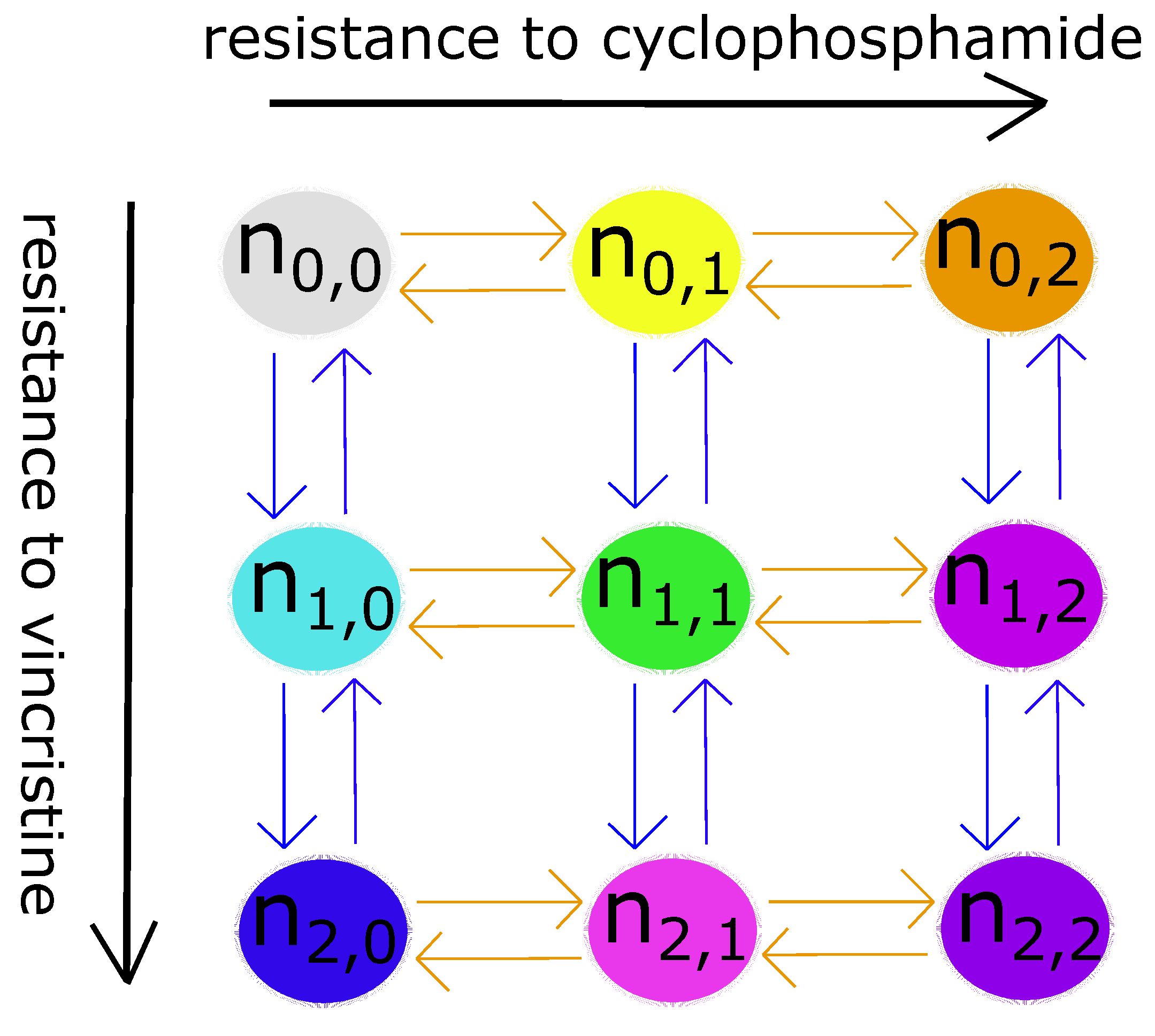
2. Quick Guide to Methodology
- 1
- A population of neuroblastoma cells (a clone) can undergo three processes only: growth (division minus natural death), mutation, and drug-induced mortality.
- 2
- Each clone follows logistic growth, limited by the other clones and the total carrying capacity (clonal competition).
- 3
- A neuroblastoma cell has three levels of genetic resistance to a drug: none, mild, and strong. It can only mutate and enter a clone whose resistance level is directly above or below its own. Mutation occurs randomly—uniformly in all directions—in the absence of drugs (selective pressures). Therefore, the mutation term is simply the growth term multiplied by the mutation rate.
- 4
- In addition to genetically conferred resistance, a neuroblastoma cell can phenotypically adapt to drugs after prolonged exposure to them. Adaptation costs energy, so both cell death and growth will decrease as a result. The extent of decrease depends linearly on the length of the exposure period.
- 5
- Drug delivery follows first-order pharmacokinetics.
- 6
- Spatial variations and stochastic effects are both assumed to be negligible.
3. Results
3.1. Mixtures of Fully Sensitive and VCR-Resistant Cells
3.2. Mixtures of Fully Sensitive and CPM-Resistant Cells

3.3. Mixtures of Fully Sensitive, VCR-Resistant, and CPM-Resistant Cells
4. Discussion
4.1. Therapeutic Strategies Based on General Evolutionary Principles
- 1
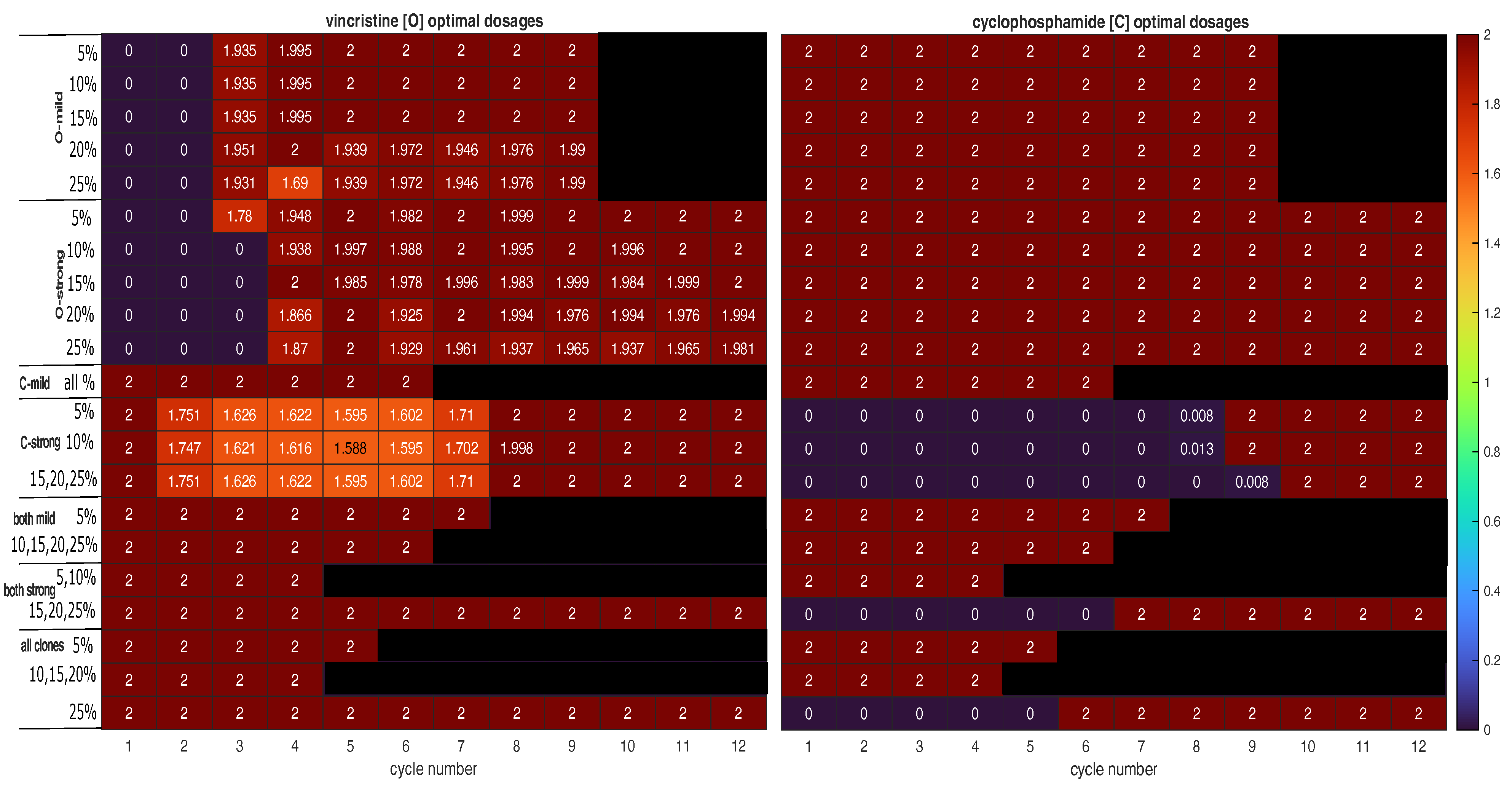
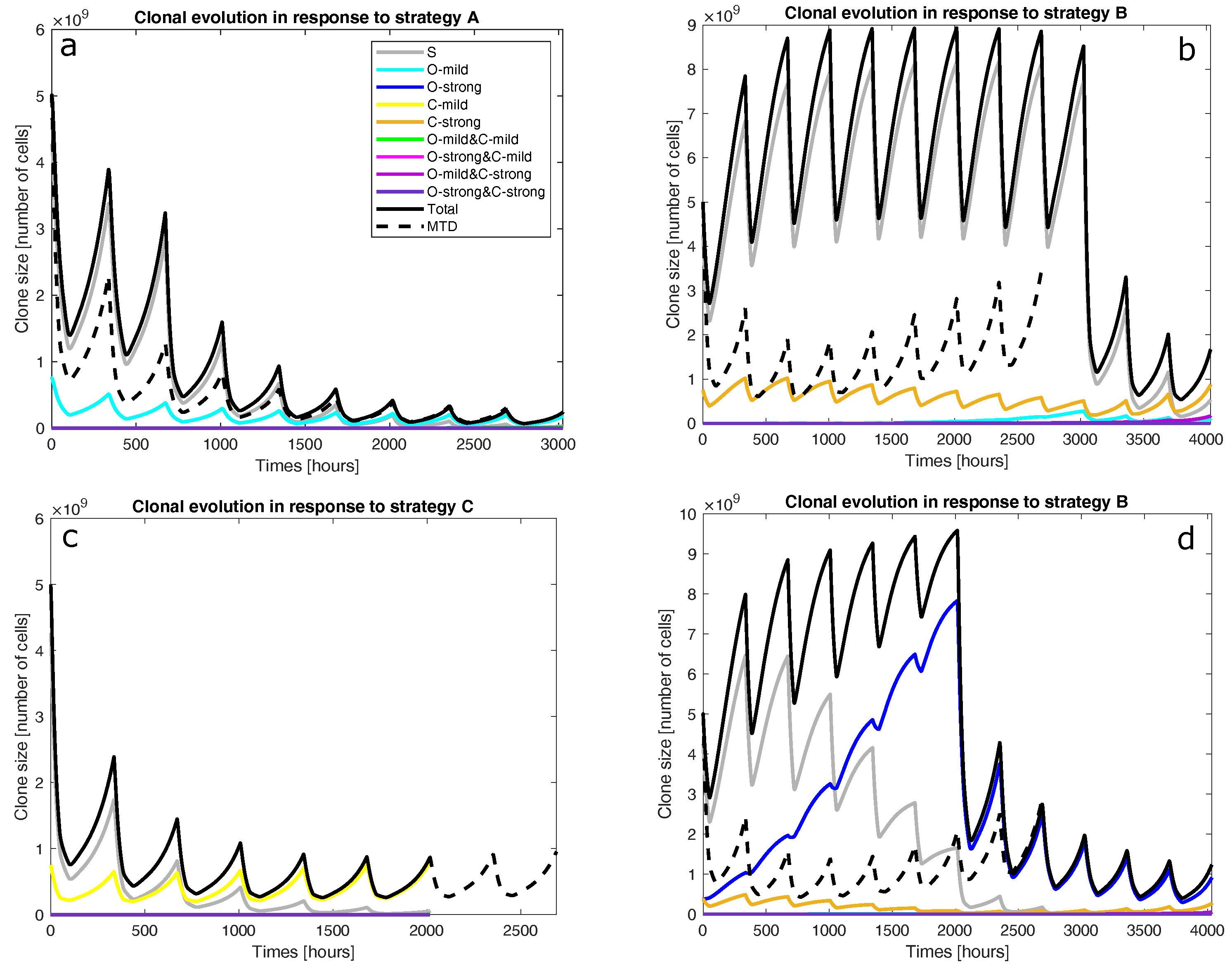
- Is the tumour to be treated already resistant to the less cytotoxic drug but not the other drug? If so, the optimal strategy is to apply the more effective drug at its MTD to exploit clonal evolution to kill the resistant clone effectively before adding the other drug to the regimen to shrink the tumour, which is mostly sensitive to the less effective drug at the end of the first stage. Finally, switch to a third drug (or another intervention) to exploit the final state of the tumour. This is strategy A. For instance, strategy A was found to be optimal for mixtures of fully sensitive and mildly VCR-resistant cancer cells (O-mild rows in Figure 2). Figure 3a presents the population dynamics of the nine clones induced by the optimal schedule corresponding to the third O-mild row (15%) in Figure 2.
- 2
- If the tumour is already resistant to the more cytotoxic drug only, is it mildly or strongly resistant to it? If it is strongly resistant, a similar two-stage strategy will work, but only the less effective drug is used in the first stage, while both drugs are used in the second stage. Furthermore, both stages should last longer than in strategy A to prolong clonal competition, thus maintaining a negative selection pressure on the resistant clone. This change is necessary because resistance to the more cytotoxic drug is harder to deal with. This is strategy B. For instance, strategy B was found to be optimal for mixtures of fully sensitive and strongly CPM-resistant cancer cells (C-strong rows in Figure 2). Figure 3b presents the population dynamics of the nine clones induced by the optimal schedule corresponding to the third C-strong row (15%) in Figure 2. During the dynamic simulation’s first stage, the strongly CPM-resistant clone (orange line) shrank even though the whole population (black line) and the fully sensitive clone (grey line) grew. If the tumour is mildly resistant to the more cytotoxic drug only, the optimal strategy is to use both drugs at their MTDs for a short duration to shrink the sensitive clone in the tumour and then switch to a third drug (or another intervention) targeting the presumably reduced and fragmented cell populations. This is strategy C. For instance, strategy C was found to be optimal for mixtures of fully sensitive and mildly CPM-resistant cancer cells (C-mild row in Figure 2). Figure 3c presents the population dynamics of the nine clones induced by the optimal schedule corresponding to the C-mild row in Figure 2; the resistant clone made up 15% of the initial population in this simulation.
- 3
- If the tumour is already resistant to both drugs, is it mildly or strongly resistant to the more cytotoxic drug, or are there both mildly and strongly resistant cells? If the tumour is only mildly resistant, strategy C is recommended.
- 4
- If the tumour is strongly resistant or contains both mildly and strongly resistant cells, what is the total fraction of cells that are resistant? A low fraction favours strategy C, while a high fraction favours strategy B. As a rule of thumb, based on the results presented in Figure 2, a fraction below 15% is considered low in a strongly resistant tumour and a fraction below 25% is considered low if the tumour contains both mildly and strongly resistant cells. For instance, strategy B was found to be optimal for the case where strongly VCR-resistant and strongly CPM-resistant cells constitute 15% of the initial population. Figure 3d shows the population dynamics of the nine clones induced by the optimal schedule corresponding to the second and last both strong rows in Figure 2; the resistant cells made up 15% of the initial population in this simulation. In the first stage, the CPM-resistant clone (orange line) was suppressed by the other clones in the presence of VCR only, but the VCR-resistant clone (blue line) expanded aggressively to dominate the population. In the second stage, the tumour dominated by the VCR-resistant clone responded to CPM effectively.
4.2. Clinical Translation
4.3. Design Choices
4.4. Validity and Future Work
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Matthay, K.; Maris, J.; Schleiermacher, G.; Nakagawara, A.; Mackall, C.; Diller, L.; Weiss, W. Neuroblastoma. Nat. Rev. Dis. Prim. 2016, 2, 16078. [Google Scholar] [CrossRef] [PubMed]
- Tomolonis, J.A.; Agarwal, S.; Shohet, J.M. Neuroblastoma pathogenesis: Deregulation of embryonic neural crest development. Cell Tissue Res. 2018, 372, 245–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kameneva, P.; Artemov, A.V.; Kastriti, M.E.; Faure, L.; Olsen, T.K.; Otte, J.; Erickson, A.; Semsch, B.; Andersson, E.R.; Ratz, M.; et al. Single-cell transcriptomics of human embryos identifies multiple sympathoblast lineages with potential implications for neuroblastoma origin. Nat. Genet. 2021, 53, 694–706. [Google Scholar] [CrossRef] [PubMed]
- Crane, J.F.; Trainor, P.A. Neural crest stem and progenitor cells. Annu. Rev. Cell Dev. Biol. 2006, 22, 267–286. [Google Scholar] [CrossRef]
- Schulte, J.; Lindner, S.; Bohrer, A.; Maurer, J.; De Preter, K.; Lefever, S.; Heukamp, L.; Schulte, S.; Molenaar, J.; Versteeg, R.; et al. MYCN and ALKF1174L are sufficient to drive neuroblastoma development from neural crest progenitor cells. Oncogene 2013, 32, 1059–1065. [Google Scholar] [CrossRef]
- Gautier, M.; Thirant, C.; Delattre, O.; Janoueix-Lerosey, I. Plasticity in neuroblastoma cell identity defines a noradrenergic-to-mesenchymal transition (NMT). Cancers 2021, 13, 2904. [Google Scholar] [CrossRef]
- Monclair, T.; Brodeur, G.M.; Ambros, P.F.; Brisse, H.J.; Cecchetto, G.; Holmes, K.; Kaneko, M.; London, W.B.; Matthay, K.K.; Nuchtern, J.G.; et al. The international neuroblastoma risk group (INRG) staging system: An INRG task force report. J. Clin. Oncol. 2009, 27, 298. [Google Scholar] [CrossRef] [Green Version]
- Morgenstern, D.A.; Baruchel, S.; Irwin, M.S. Current and future strategies for relapsed neuroblastoma: Challenges on the road to precision therapy. J. Pediatr. Hematol. 2013, 35, 337–347. [Google Scholar] [CrossRef]
- Garaventa, A.; Poetschger, U.; Valteau-Couanet, D.; Luksch, R.; Castel, V.; Elliott, M.; Ash, S.; Chan, G.C.F.; Laureys, G.; Beck-Popovic, M.; et al. Randomized Trial of Two Induction Therapy Regimens for High-Risk Neuroblastoma: HR-NBL1.5 International Society of Pediatric Oncology European Neuroblastoma Group Study, Journal of Clinical Oncology. J. Clin. Oncol. 2021, 39, 2552–2563. [Google Scholar] [CrossRef]
- Smith, V.; Foster, J. High-risk neuroblastoma treatment review. Children 2018, 5, 114. [Google Scholar] [CrossRef] [Green Version]
- Habib, E.E.; El-Kashef, A.T.; Fahmy, E.S. Management of neuroblastoma: A study of first- and second-line chemotherapy responses, a single institution experience. Oncol. Rev. 2012, 6, e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatenby, R.A.; Brown, J.S. Integrating evolutionary dynamics into cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 675–686. [Google Scholar] [CrossRef]
- K, S.; S, B.J.; S, D.W.; A, G.R. Optimizing Cancer Treatment Using Game Theory: A Review. JAMA Oncol. 2019, 5, 96–103. [Google Scholar]
- Enriquez-Navas, P.M.; Kam, Y.; Das, T.; Hassan, S.; Silva, A.; Foroutan, P.; Ruiz, E.; Martinez, G.; Minton, S.; Gillies, R.J.; et al. Exploiting evolutionary principles to prolong tumor control in preclinical models of breast cancer. Sci. Transl. Med. 2016, 8, 327ra24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schramm, A.; Köster, J.; Assenov, Y.; Althoff, K.; Peifer, M.; Mahlow, E.; Odersky, A.; Beisser, D.; Ernst, C.; Henssen, A.G.; et al. Mutational dynamics between primary and relapse neuroblastomas. Nat. Genet. 2015, 47, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Schleiermacher, G.; Javanmardi, N.; Bernard, V.; Leroy, Q.; Cappo, J.; Rio Frio, T.; Pierron, G.; Lapouble, E.; Combaret, V.; Speleman, F.; et al. Emergence of new ALK mutations at relapse of neuroblastoma. J. Clin. Oncol. 2014, 32, 2727–2734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eleveld, T.F.; Oldridge, D.A.; Bernard, V.; Koster, J.; Daage, L.C.; Diskin, S.J.; Schild, L.; Bentahar, N.B.; Bellini, A.; Chicard, M.; et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat. Genet. 2013, 47, 864–871. [Google Scholar] [CrossRef] [Green Version]
- Greengard, E.G. Molecularly targeted therapy for neuroblastoma. Children 2018, 5, 142. [Google Scholar] [CrossRef] [Green Version]
- Schulte, J.; Schulte, S.; Heukamp, L.; Astrahantseff, K.; Stephan, H.; Fischer, M.; Schramm, A.; Eggert, A. Targeted therapy for neuroblastoma: ALK inhibitors. Klin. PÄDiatrie 2013, 225, 303–308. [Google Scholar] [CrossRef]
- Berlak, M.; Tucker, E.; Dorel, M.; Winkler, A.; McGearey, A.; Rodriguez-Fos, E.; da Costa, B.M.; Barker, K.; Fyle, E.; Calton, E.; et al. Mutations in ALK signaling pathways conferring resistance to ALK inhibitor treatment lead to collateral vulnerabilities in neuroblastoma cells. Mol. Cancer 2022, 21, 126. [Google Scholar] [CrossRef]
- Armas, O.A.; Aprikian, A.G.; Melamed, J.; Cordon-Cardo, C.; Cohen, D.W.; Erlandson, R.; Fair, W.R.; Reuter, V.E. Clinical and pathobiological effects of neoadjuvant total androgen ablation therapy on clinically localized prostatic adenocarcinoma. Am. J. Surg. Pathol. 1994, 18, 979–991. [Google Scholar] [CrossRef] [PubMed]
- Gillies, R.J.; Brown, J.S.; Anderson, A.R.; Gatenby, R.A. Eco-evolutionary causes and consequences of temporal changes in intratumoural blood flow. Nat. Rev. Cancer 2018, 18, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, H.; Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist. 2019, 2, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habib, E.E.; El-Kashef, A.T.; Fahmy, E.S. The Role of Multidrug Resistance Efflux Pumps in Cancer: Revisiting a JNCI Publication Exploring Expression of the MDR1 (P-glycoprotein) Gene. J. Natl. Cancer Inst. 2015, 107, djv222. [Google Scholar]
- Feist, A.M.; Palsson, B.O. The biomass objective function. Curr. Opin. Microbiol. 2010, 13, 344–349. [Google Scholar] [CrossRef] [Green Version]
- Tumilowicz, J.J.; Nichols, W.W.; Cholon, J.J.; Greene, A.E. Definition of a continuous human cell line derived from neuroblastoma. Cancer Res. 1970, 30, 2110–2118. [Google Scholar]
- Jemaà, M.; Sime, W.; Abassi, Y.; Lasorsa, V.A.; Køhler, J.B.; Michaelis, M.; Cinatl, J.; Capasso, M.; Massoumi, R. Gene Expression Signature of Acquired Chemoresistance in Neuroblastoma Cells. Int. J. Mol. Sci. 2020, 21, 6811. [Google Scholar] [CrossRef]
- Kotchetkov, R.; Cinatl, J.; Blaheta, R.; Vogel, J.U.; Karaskova, J.; Squire, J.; Driever, P.H.; Klingebiel, T.; Cinatl Jr., J. Development of resistance to vincristine and doxorubicin in neuroblastoma alters malignant properties and induces additional karyotype changes: A preclinical model. Int. J. Cancer 2003, 104, 33–43. [Google Scholar] [CrossRef]
- Duesberg, P.; Stindl, R.; Hehlmann, R. Explaining the high mutation rates of cancer cells to drug and multidrug resistance by chromosome reassortments that are catalyzed by aneuploidy. Proc. Natl. Acad. Sci. USA 2000, 97, 14295–14300. [Google Scholar] [CrossRef] [Green Version]
- Zaizen, Y.; Nakagawara, A.; Ikeda, K. Patterns of Destruction of Mouse Neuroblastoma Cells by Extracellular Hydrogen Peroxide Formed by 6-Hydroxydopamine and Ascorbate. J. Cancer Res. Clin. Oncol. 1986, 111, 93–97. [Google Scholar] [CrossRef]
- Löschmann, N.; Michaelis, M.; Rothweiler, F.; Zehner, R.; Cinatl, J.; Voges, Y.; Sharifi, M.; Riecken, K.; Meyer, J.; von Deimling, A.; et al. Testing of SNS-032 in a Panel of Human Neuroblastoma Cell Lines with Acquired Resistance to a Broad Range of Drugs. Transl. Oncol. 2013, 6, 685–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chesler, L.; Goldenberg, D.D.; Collins, R.; Grimmer, M.; Kim, G.E.; Tihan, T.; Nguyen, K.; Yakovenko, S.; Matthay, K.K.; Weiss, W.A. Chemotherapy-Induced Apoptosis in a Transgenic Model of Neuroblastoma Proceeds Through p53 Induction. Neoplasia 2008, 10, 1268–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yogev, O.; Almeida, G.S.; Barker, K.T.; George, S.L.; Kwok, C.; Campbell, J.; Zarowiecki, M.; Kleftogiannis, D.; Smith, L.M.; Hallsworth, A.; et al. In vivo modeling of chemoresistant neuroblastoma provides new insights into chemorefractory disease and metastasis. Cancer Res. 2019, 79, 5382–5393. [Google Scholar] [CrossRef] [PubMed]
- Vincristine Dosing, Indications, Interactions, Adverse Effects, and More. Available online: https://reference.medscape.com/drug/oncovin-vincasar-pfs-vincristine-342097 (accessed on 5 December 2022).
- Cytoxan (cyclophosphamide) Dosing, Indications, Interactions, Adverse Effects, and More. Available online: https://reference.medscape.com/drug/cytoxan-cyclophosphamide-342214 (accessed on 5 December 2022).
- Groninger, E.; de Boar, T.M.; Koopmans, P.; Uges, D.; Sluiter, W.; Veerman, A.; Kamps, W.; de Graaf, S. Pharmacokinetics of Vincristine Monotherapy in Childhood Acute Lymphoblastic Leukemia. Pediatr. Res. 2002, 52, 113–118. [Google Scholar] [CrossRef] [Green Version]
- McCune, J.S.; Salinger, D.H.; Vicini, P.; Oglesby, C.; Blough, D.K.; Park, J.R. Population Pharmacokinetics of Cyclophosphamide and Metabolites in Children with Neuroblastoma: A Report from the Children’s Oncology Group. J. Clin. Pharmacol. 2009, 49, 88–102. [Google Scholar] [CrossRef] [Green Version]
- Feber, J.; Krásničanová, H. Measures of Body Surface Area in Children. In Handbook of Anthropometry: Physical Measures of Human Form in Health and Disease; Springer: Berlin/Heidelberg, Germany, 2012; pp. 1249–1256. [Google Scholar]
- Friis-Hansen, B.J.; Holiday, M.; Stapleton, T.; Wallace, W.M. Total body water in children. Pediatrics 1951, 7, 321–327. [Google Scholar] [CrossRef]
- Orr, K.E.; McHugh, K. The new international neuroblastoma response criteria. Pediatr. Radiol. 2019, 49, 1433–1440. [Google Scholar] [CrossRef] [PubMed]
- Logan, J.A.; Kelly, M.E.; Ayers, D.; Shipillis, N.; Baier, G.; Day, P.J. Systems biology and modeling in neuroblastoma: Practicalities and perspectives. Expert Rev. Mol. Diagn. 2010, 10, 131–145. [Google Scholar] [CrossRef]
- Hidalgo, M.R.; Amadoz, A.; Çubuk, C.; Carbonell-Caballero, J.; Dopazo, J. Models of cell signaling uncover molecular mechanisms of high-risk neuroblastoma and predict disease outcome. Biol. Direct 2018, 13, 16. [Google Scholar] [CrossRef]
- Fey, D.; Halasz, M.; Dreidax, D.; Kennedy, S.P.; Hastings, J.F.; Rauch, N.; Munoz, A.G.; Pilkington, R.; Fischer, M.; Westermann, F.; et al. Signaling pathway models as biomarkers: Patient-specific simulations of JNK activity predict the survival of neuroblastoma patients. Sci. Signal. 2015, 8, ra130. [Google Scholar] [CrossRef] [Green Version]
- Lombardo, S.D.; Presti, M.; Mangano, K.; Petralia, M.C.; Basile, M.S.; Libra, M.; Candido, S.; Fagone, P.; Mazzon, E.; Nicoletti, F.; et al. Prediction of PD-L1 expression in neuroblastoma via computational modeling. Brain Sci. 2019, 9, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlhaus, M.; Burkovski, A.; Hertwig, F.; Mussel, C.; Volland, R.; Fischer, M.; Debatin, K.M.; Kestler, H.A.; Beltinger, C. Boolean modeling identifies Greatwall/MASTL as an important regulator in the AURKA network of neuroblastoma. Cancer Lett. 2016, 371, 79–89. [Google Scholar] [CrossRef]
- He, Y.; Kodali, A.; Wallace, D.I. Predictive modeling of neuroblastoma growth dynamics in xenograft model after bevacizumab anti-VEGF therapy. Bull. Math. Biol. 2018, 80, 2026–2048. [Google Scholar] [CrossRef] [PubMed]
- Benzekry, S.; Sentis, C.; Coze, C.; Tessonnier, L.; André, N. Development and validation of a prediction model of overall survival in high-risk neuroblastoma using mechanistic modeling of metastasis. JCO Clin. Cancer Inform. 2021, 5, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Otero, J.G.; Alcamí, A.Á.A.; Belmonte-Beitia, J. Dynamics and analysis of a mathematical model of neuroblastoma treated with Celyvir. Appl. Math. Model. 2022, 110, 131–148. [Google Scholar] [CrossRef]
- Martí-Bonmatí, L.; Alberich-Bayarri, Á.; Ladenstein, R.; Blanquer, I.; Segrelles, J.D.; Cerdá-Alberich, L.; Gkontra, P.; Hero, B.; García-Aznar, J.; Keim, D.; et al. PRIMAGE project: Predictive in silico multiscale analytics to support childhood cancer personalised evaluation empowered by imaging biomarkers. Eur. Radiol. Exp. 2020, 4, 22. [Google Scholar] [CrossRef] [PubMed]
- Sainz-DeMena, D.; García-Aznar, J.M.; Pérez, M.Á.; Borau, C. Im2mesh: A Python Library to Reconstruct 3D Meshes from Scattered Data and 2D Segmentations, Application to Patient-Specific Neuroblastoma Tumour Image Sequences. Appl. Sci. 2022, 12, 11557. [Google Scholar] [CrossRef]
- de Melo Quintela, B.; Hervas-Raluy, S.; Garcia-Aznar, J.M.; Walker, D.; Wertheim, K.Y.; Viceconti, M. A theoretical analysis of the scale separation in a model to predict solid tumour growth. J. Theor. Biol. 2022, 547, 111173. [Google Scholar] [CrossRef]
- Veiga-Canuto, D.; Cerdà-Alberich, L.; Sangüesa Nebot, C.; Martínez de las Heras, B.; Pötschger, U.; Gabelloni, M.; Carot Sierra, J.M.; Taschner-Mandl, S.; Düster, V.; Cañete, A.; et al. Comparative Multicentric Evaluation of Inter-Observer Variability in Manual and Automatic Segmentation of Neuroblastic Tumors in Magnetic Resonance Images. Cancers 2022, 14, 3648. [Google Scholar] [CrossRef]
- Scapicchio, C.; Gabelloni, M.; Forte, S.M.; Alberich, L.C.; Faggioni, L.; Borgheresi, R.; Erba, P.; Paiar, F.; Marti-Bonmati, L.; Neri, E. DICOM-MIABIS integration model for biobanks: A use case of the EU PRIMAGE project. Eur. Radiol. Exp. 2021, 5, 20. [Google Scholar] [CrossRef]
- Collins, C.; Fister, K.R.; Key, B.; Williams, M. Blasting neuroblastoma using optimal control of chemotherapy. Math. Biosci. Eng. 2009, 6, 451. [Google Scholar]
- Panetta, J.C.; Schaiquevich, P.; Santana, V.M.; Stewart, C.F. Using pharmacokinetic and pharmacodynamic modeling and simulation to evaluate importance of schedule in topotecan therapy for pediatric neuroblastoma. Clin. Cancer Res. 2008, 14, 318–325. [Google Scholar] [CrossRef] [Green Version]
- Silva, A.S.; Kam, Y.; Khin, Z.P.; Minton, S.E.; Gillies, R.J.; Gatenby, R.A. Evolutionary Approaches to Prolong Progression-Free Survival in Breast Cancer. Cancer Res. 2012, 72, 6362–6370. [Google Scholar] [CrossRef] [Green Version]
- Schweizer, M.T.; Antonarakis, E.S.; Wang, H.; Ajiboye, A.S.; Spitz, A.; Cao, H.; Luo, J.; Haffner, M.C.; Yegnasubramanian, S.; Carducci, M.A.; et al. Effect of bipolar androgen therapy for asymptomatic men with castration-resistant prostate cancer: Results from a pilot clinical study. Sci. Transl. Med. 2015, 7, 269ra2. [Google Scholar] [CrossRef] [Green Version]
- Italia, M.; Dercole, F.; Lucchetti, R. Optimal chemotherapy counteracts cancer adaptive resistance in a cell-based, spatially-extended, evolutionary model. Phys. Biol. 2022, 19, 026004. [Google Scholar] [CrossRef]
- Bozic, I.; Reiter, J.G.; Allen, B.; Antal, T.; Chatterjee, K.; Shah, P.; Moon, Y.S.; Yaqubie, A.; Kelly, N.; Le, D.T.; et al. Evolutionary dynamics of cancer in response to targeted combination therapy. Elife 2013, 2, e00747. [Google Scholar] [CrossRef]
- Amirouchene-Angelozzi, N.; Swanton, C.; Bardelli, A. Tumor Evolution as a Therapeutic Target. Cancer Discov. 2017, 7, 805–817. [Google Scholar] [CrossRef] [Green Version]
- Weiss, T.; Taschner-Mandl, S.; Janker, L.; Bileck, A.; Rifatbegovic, F.; Kromp, F.; Sorger, H.; Kauer, M.O.; Frech, C.; Windhager, R.; et al. Schwann cell plasticity regulates neuroblastic tumor cell differentiation via epidermal growth factor-like protein 8. Nat. Commun. 2021, 12, 1624. [Google Scholar] [CrossRef]
- Blavier, L.; Yang, R.M.; DeClerck, Y.A. The tumor microenvironment in neuroblastoma: New players, new mechanisms of interaction and new perspectives. Cancers 2020, 12, 2912. [Google Scholar] [CrossRef]
- Kuo, F.Y.; Sloan, I.H. Lifting the curse of dimensionality. Not. AMS 2005, 52, 1320–1328. [Google Scholar]
- Strobl, M.A.; Gallaher, J.; West, J.; Robertson-Tessi, M.; Maini, P.K.; Anderson, A.R. Spatial structure impacts adaptive therapy by shaping intra-tumoral competition. Commun. Med. 2022, 2, 46. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Parameters | Values | Units | Meanings |
|---|---|---|---|
| h | VCR clearance rate | ||
| h | CPM clearance rate | ||
| h | Sensitive clone’s growth rate | ||
| h | VCR-10 clone’s growth rate | ||
| h | VCR-20 clone’s growth rate | ||
| h | CPM-20 clone’s growth rate | ||
| h | CPM-32 clone’s growth rate | ||
| h | VCR-10-CPM-20 clone’s growth rate | ||
| h | VCR-10-CPM-32 clone’s growth rate | ||
| h | VCR-20-CPM-20 clone’s growth rate | ||
| h | VCR-20-CPM-32 clone’s growth rate | ||
| K | cells | Carrying capacity of the tumour | |
| Dimensionless | Mutation rate | ||
| Dimensionless | Shape parameter in mortality function (VCR) | ||
| Dimensionless | Shape parameter in mortality function (VCR) | ||
| h | Sensitive clone’s maximum mortality rate due to VCR | ||
| h | VCR-10 clone’s maximum mortality rate due to VCR | ||
| 6 | h | VCR-20 clone’s maximum mortality rate due to VCR | |
| Dimensionless | Shape parameter in mortality function (CPM) | ||
| 1 | Dimensionless | Shape parameter in mortality function (CPM) | |
| h | Sensitive clone’s maximum mortality rate due to CPM | ||
| h | CPM-20 clone’s maximum mortality rate due to CPM | ||
| h | CPM-32 clone’s maximum mortality rate due to CPM | ||
| nuovo | 0 | days | nuovoMinimum memory period associated with phenotypic adaptation |
| 10 | days | nuovoMaximum memory period associated with phenotypic adaptation | |
| nuovo | 0 | Dimensionless | nuovoMinimum effect of phenotypic adaptation on growth |
| 1 | Dimensionless | Maximum effect of phenotypic adaptation on growth | |
| nuovo | 0 | Dimensionless | nuovoMinimum effect of phenotypic adaptation on drug-induced mortality |
| 2 | Dimensionless | Maximum effect of phenotypic adaptation on drug-induced mortality |
| VCR Size | Mild | Strong | |||||||
|---|---|---|---|---|---|---|---|---|---|
| N () | FS (%) | G (%) | N () | FS (%) | G (%) | ||||
| 5% | 1.17 | 2.34 | 16.09 | 0.53 | 1.06 | 50.48 | |||
| 10% | 1.80 | 3.6 | 21.18 | 0.85 | 1.7 | 50.29 | |||
| 15% | 2.38 | 4.76 | 21.40 | 1.11 | 2.22 | 51.35 | |||
| 20% | 2.91 | 5.82 | 21.07 | 1.36 | 2.7 | 51.00 | |||
| 25% | 3.42 | 6.84 | 19.46 | 1.60 | 3.2 | 50.25 | |||
| CPM Size | Mild | Strong | |||||||
| N () | FS (%) | G (%) | N () | FS (%) | G (%) | ||||
| 5% | 4.21 | 8.42 | 8.40 | 9.93 | 19.86 | 61.40 | |||
| 10% | 6.70 | 13.4 | 10.10 | 14.00 | 28 | 55.89 | |||
| 15% | 8.70 | 17.4 | 8.58 | 16.67 | 33.34 | 51.57 | |||
| 20% | 10.33 | 20.66 | 6.65 | 19.20 | 38.4 | 46.59 | |||
| 25% | 11.69 | 23.38 | 4.79 | 21.60 | 43.2 | 41.49 | |||
| Both Size | Mild | Strong | Mild and Strong | ||||||
| N () | FS (%) | G (%) | N () | FS (%) | G (%) | N () | FS (%) | G (%) | |
| 5% | 3.04 | 6.08 | 1.31 | 7.67 | 15.30 | 58.60 | 5.42 | 10.84 | 45.06 |
| 10% | 4.92 | 9.84 | 4.30 | 11.38 | 22.76 | 55.02 | 8.14 | 16.28 | 47.17 |
| 15% | 6.40 | 12.8 | 4.82 | 12.27 | 24.24 | 57.42 | 10.05 | 20.1 | 46.31 |
| 20% | 7.72 | 15.44 | 4.01 | 12.53 | 25.06 | 59.58 | 11.83 | 23.66 | 43.90 |
| 25% | 8.81 | 17.62 | 2.98 | 12.68 | 25.36 | 60.92 | 12.12 | 24.24 | 46.95 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Italia, M.; Wertheim, K.Y.; Taschner-Mandl, S.; Walker, D.; Dercole, F. Mathematical Model of Clonal Evolution Proposes a Personalised Multi-Modal Therapy for High-Risk Neuroblastoma. Cancers 2023, 15, 1986. https://doi.org/10.3390/cancers15071986
Italia M, Wertheim KY, Taschner-Mandl S, Walker D, Dercole F. Mathematical Model of Clonal Evolution Proposes a Personalised Multi-Modal Therapy for High-Risk Neuroblastoma. Cancers. 2023; 15(7):1986. https://doi.org/10.3390/cancers15071986
Chicago/Turabian StyleItalia, Matteo, Kenneth Y. Wertheim, Sabine Taschner-Mandl, Dawn Walker, and Fabio Dercole. 2023. "Mathematical Model of Clonal Evolution Proposes a Personalised Multi-Modal Therapy for High-Risk Neuroblastoma" Cancers 15, no. 7: 1986. https://doi.org/10.3390/cancers15071986