
Microparticle Phosphatidylserine Mediates Coagulation: Involvement in Tumor Progression and Metastasis
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. MPs Involved in Tumor Progression
2.1. The Effect of MPs on Tumor Progression
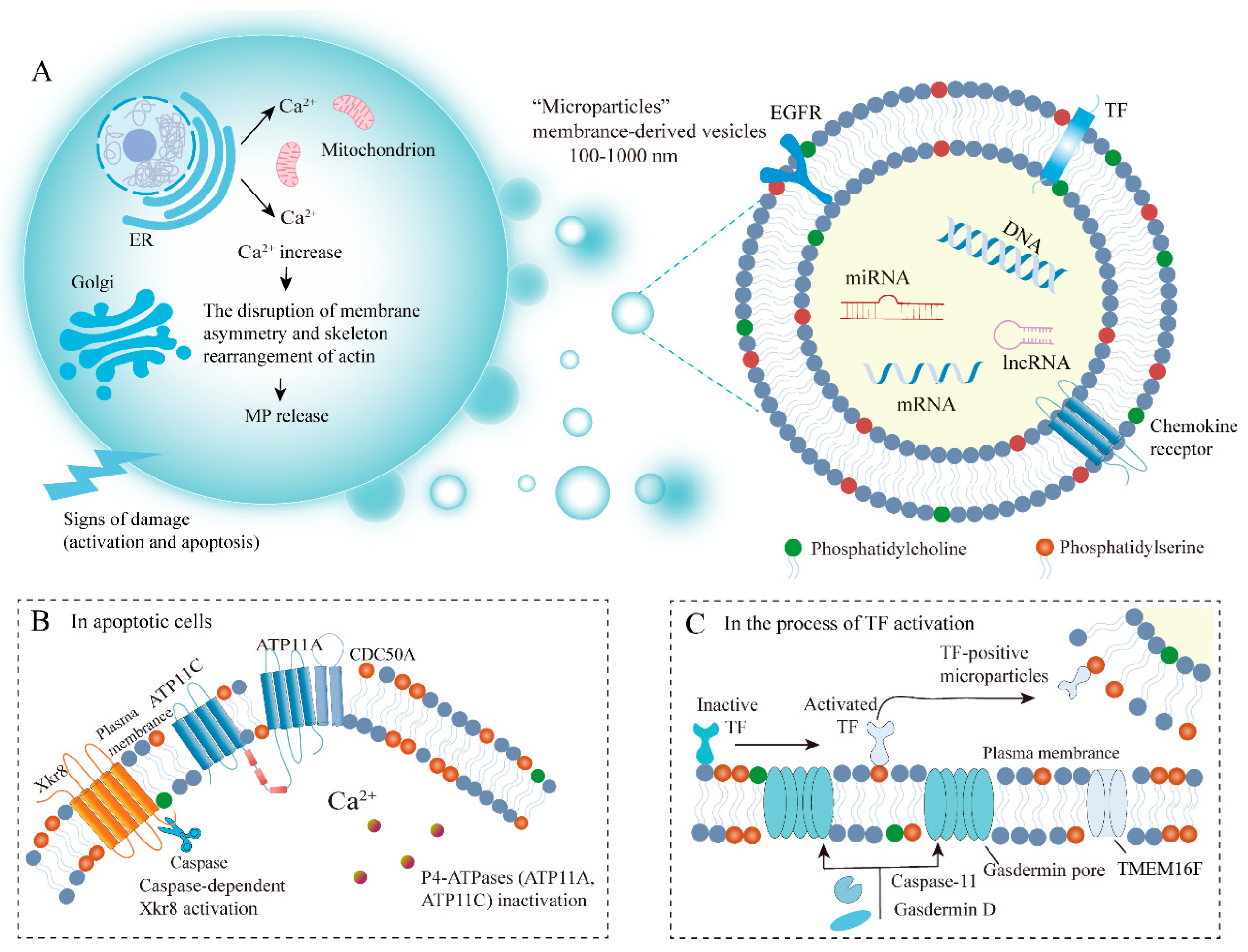
2.2. Biogenesis of MPs
2.3. Factors and Mechanisms Influencing MP Biogenesis
3. PS Externalization on MPs Involved in Tumor Coagulation
3.1. The Mechanisms of PS Externalization
3.2. PS Involved in Tumor Coagulation
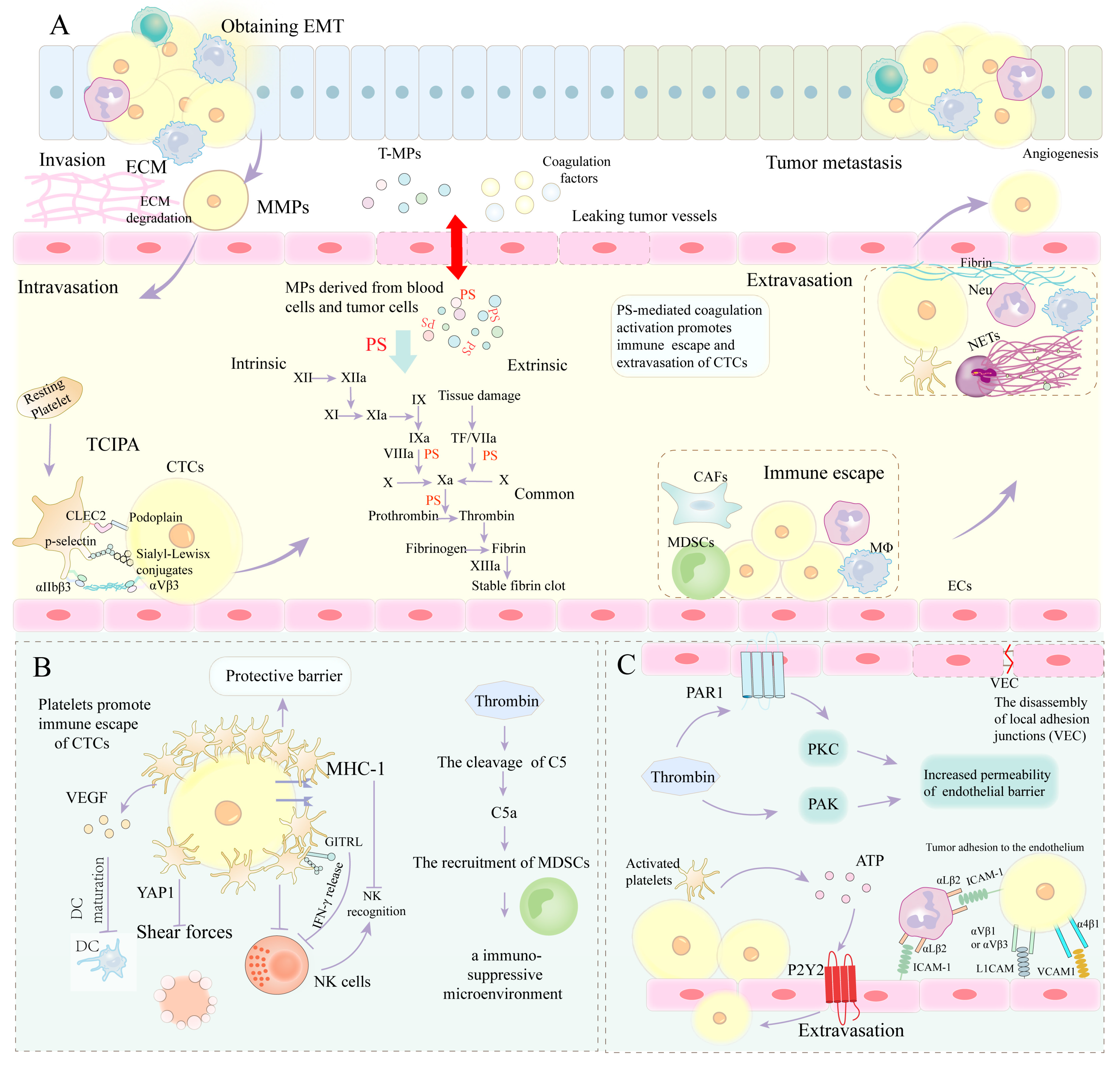
4. PS Involvement in Tumor Metastasis by Mediating Coagulation
4.1. PS-Mediated Coagulation Cascade Involvement in EMT
HIF Involvement in EMT
4.2. PS-Mediated Coagulation Cascade Involved in ECM Remodeling
Plasminogen Activator/Plasmin System
4.3. PS-Mediated Coagulation Cascade Involved in Immune Escape
4.4. PS-Mediated Coagulation Cascade Involved in Extravasation of CTCs
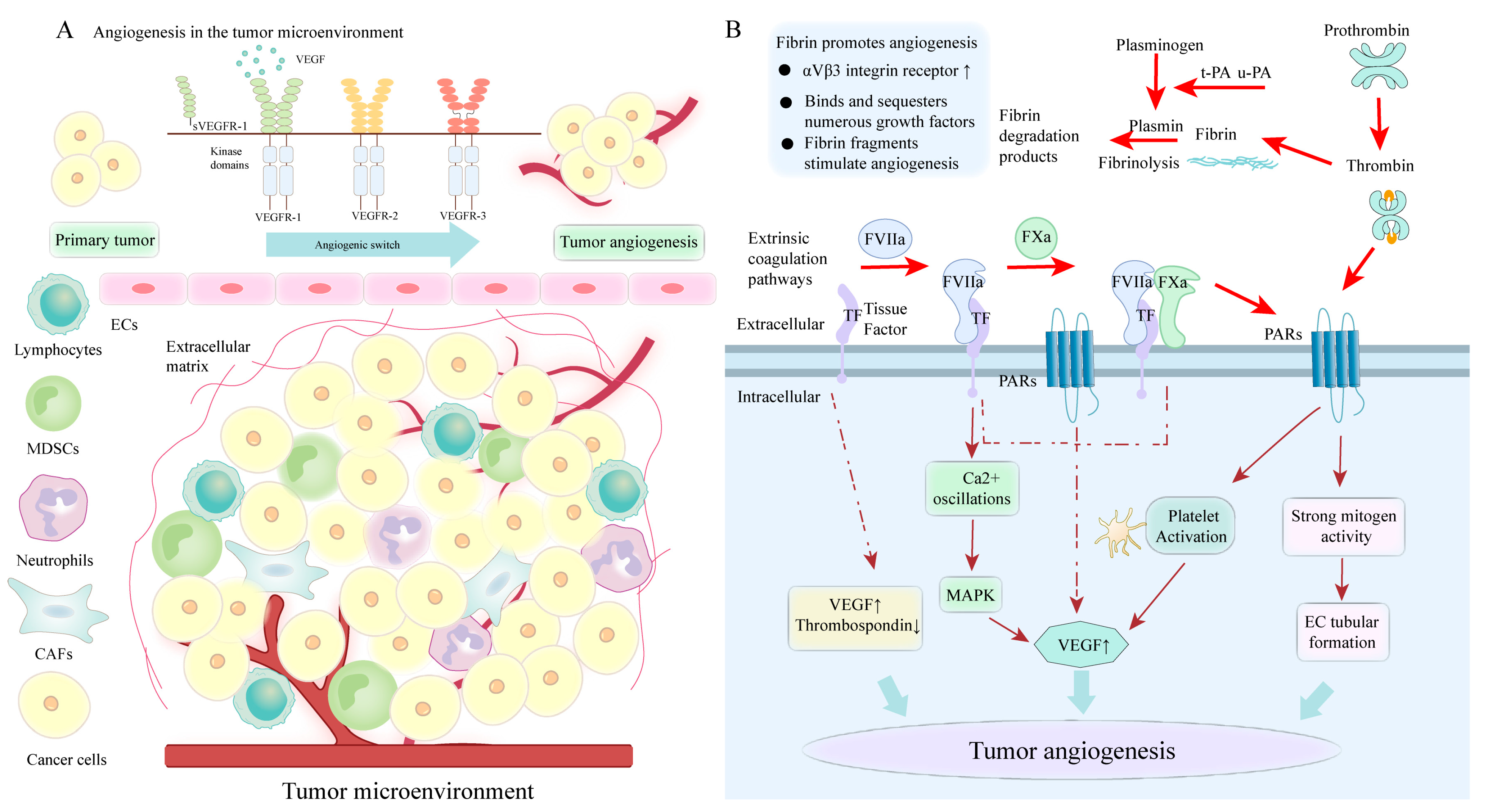
4.5. PS-Mediated Coagulation Cascade Involved in Angiogenesis
4.5.1. The Role of TF in Tumor Angiogenesis
4.5.2. The Role of Thrombin in Tumor Angiogenesis
4.5.3. The Role of Fibrin and FXIII in Tumor Angiogenesis
5. Treatment Strategy
5.1. Blocking PS on Tumor Cells and Their MPs
5.2. Cancer Treatment Strategies Targeting Thrombosis
5.3. Other Treatments
5.3.1. Targeting HIF Therapy
5.3.2. Targeting VEGF Therapy
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kalluri, R. The biology and function of exosomes in cancer. J. Clin. Investig. 2016, 126, 1208–1215. [Google Scholar] [CrossRef]
- Willms, E.; Cabañas, C.; Mäger, I.; Wood, M.J.A.; Vader, P. Extracellular vesicle heterogeneity: Subpopulations, isolation techniques, and diverse functions in cancer progression. Front. Immunol. 2018, 9, 738. [Google Scholar] [CrossRef] [Green Version]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Tricarico, C.; Clancy, J.; D’Souza-Schorey, C. Biology and biogenesis of shed microvesicles. Small GTPases 2017, 2, 220–232. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Qiu, S.; Jin, K.; Zheng, X.; Zhou, X.; Jin, D.; Xu, B.; Jin, X. Tumor-derived microparticles promote the progression of triple-negative breast cancer via PD-L1-associated immune suppression. Cancer Lett. 2021, 523, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Rak, J. Microparticles in cancer. Semin. Thromb. Hemost. 2010, 36, 888–906. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Luo, M.; Shao, B.; Yang, J.Y.; Tong, A.; Wang, R.B.; Liu, Y.T.; Jun, R.; Liu, T.; Yi, T.; et al. Phosphatidylserine released from apoptotic cells in tumor induces M2-like macrophage polarization through the PSR-STAT3-JMJD3 axis. Cancer Commun. 2022, 42, 205–222. [Google Scholar] [CrossRef]
- Lal, I.; Dittus, K.; Holmes, C.E. Platelets, coagulation and fibrinolysis in breast cancer progression. Breast Cancer Res. 2013, 15, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanda, M.; Tanaka, C.; Kobayashi, D.; Mizuno, A.; Tanaka, Y.; Takami, H.; Iwata, N.; Hayashi, M.; Niwa, Y.; Yamada, S.; et al. Proposal of the coagulation score as a predictor for short-term and long-term outcomes of patients with resectable gastric cancer. Ann. Surg. Oncol. 2017, 24, 502–509. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, Z.; Zhou, P.; Yu, M.; Li, B.; Liu, Y.; Jin, J.; Liu, W.; Jing, H.; Du, J.; et al. Phosphatidylserine-exposing tumor-derived microparticles exacerbate coagulation and cancer cell transendothelial migration in triple-negative breast cancer. Theranostics 2021, 11, 6445–6460. [Google Scholar] [CrossRef]
- Rajeev Krishnan, S.; De Rubis, G.; Suen, H.; Joshua, D.; Lam Kwan, Y.; Bebawy, M. A liquid biopsy to detect multidrug resistance and disease burden in multiple myeloma. Blood Cancer J. 2020, 10, 37. [Google Scholar] [CrossRef] [Green Version]
- Cao, M.; Li, T.; He, Z.; Wang, L.; Yang, X.; Kou, Y.; Zou, L.; Dong, X.; Novakovic, V.A.; Bi, Y.; et al. Promyelocytic extracellular chromatin exacerbates coagulation and fibrinolysis in acute promyelocytic leukemia. Blood 2017, 129, 1855–1864. [Google Scholar] [CrossRef]
- Mezouar, S.; Mege, D.; Darbousset, R.; Farge, D.; Debourdeau, P.; Dignat-George, F.; Panicot-Dubois, L.; Dubois, C. Involvement of platelet-derived microparticles in tumor progression and thrombosis. Semin. Oncol. 2014, 41, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Berckmans, R.J.; Nieuwland, R.; Böing, A.N.; Romijn, F.P.; Hack, C.E.; Sturk, A. Cell-derived microparticles circulate in healthy humans and support low grade thrombin generation. Thromb. Haemost. 2001, 85, 639–646. [Google Scholar] [PubMed]
- VanWijk, M.J.; Boer, K.; Berckmans, R.J.; Meijers, J.C.; van der Post, J.A.; Sturk, A.; Van Bavel, E.; Nieuwland, R. Enhanced coagulation activation in preeclampsia: The role of APC resistance, microparticles and other plasma constituents. Thromb. Haemost. 2002, 88, 415–420. [Google Scholar] [PubMed]
- Cauwenberghs, S.; Feijge, M.A.; Harper, A.G.; Sage, S.O.; Curvers, J.; Heemskerk, J.W. Shedding of procoagulant microparticles from unstimulated platelets by integrin-mediated destabilization of actin cytoskeleton. FEBS Lett. 2006, 580, 5313–5320. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, N.; Rubenich, D.S.; Zaręba, Ł.; Siewiera, J.; Pieper, J.; Braganhol, E.; Reichert, T.E.; Szczepański, M.J. Potential roles of tumor cell- and stroma cell-derived small extracellular vesicles in promoting a pro-angiogenic tumor microenvironment. Cancers 2020, 12, 3599. [Google Scholar] [CrossRef]
- Li, D.; Jia, H.; Zhang, H.; Lv, M.; Liu, J.; Zhang, Y.; Huang, T.; Huang, B. TLR4 signaling induces the release of microparticles by tumor cells that regulate inflammatory cytokine IL-6 of macrophages via microRNA let-7b. Oncoimmunology. 2012, 1, 687–693. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Sun, W.; Zhang, H.; Ma, J.; Xu, P.; Yu, Y.; Fang, H.; Zhou, L.; Lv, J.; Xie, J.; et al. Macrophages reprogrammed by lung cancer microparticles promote tumor development via release of IL-1β. Cell Mol. Immunol. 2020, 17, 1233–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Zhang, H.; Tang, K.; Huang, B. Tumor-derived microparticles in tumor immunology and immunotherapy. Eur. J. Immunol. 2020, 50, 1653–1662. [Google Scholar] [CrossRef]
- Huang, B.; Zhao, J.; Li, H.; He, K.L.; Chen, Y.; Chen, S.H.; Mayer, L.; Unkeless, J.C.; Xiong, H. Toll-like receptors on tumor cells facilitate evasion of immune surveillance. Cancer Res. 2005, 65, 5009–5014. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.G.; Zhang, W.; Liu, B.; Man, Q.W.; Xiong, X.P.; Li, C.; Zhu, J.Y.; Wang, W.M.; Jia, J.; Sun, Z.J.; et al. Clinical significance and roles in angiogenesis of circulating microparticles in oral cancer. J. Dent. Res. 2016, 95, 860–867. [Google Scholar] [CrossRef]
- Ren, J.G.; Man, Q.W.; Zhang, W.; Li, C.; Xiong, X.P.; Zhu, J.Y.; Wang, W.M.; Sun, Z.J.; Jia, J.; Zhang, W.F.; et al. Elevated level of circulating platelet-derived microparticles in oral cancer. J. Dent. Res. 2016, 95, 87–93. [Google Scholar] [CrossRef] [Green Version]
- Ma, R.; Ji, T.; Chen, D.; Dong, W.; Zhang, H.; Yin, X.; Ma, J.; Liang, X.; Zhang, Y.; Shen, G.; et al. Tumor cell-derived microparticles polarize M2 tumor-associated macrophages for tumor progression. Oncoimmunology 2016, 5, e1118599. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wang, S.; Xia, H.; Tan, X.; Song, S.; Zhang, S.; Meng, D.; Chen, Q.; Jin, Y. The potential applications of microparticles in the diagnosis, treatment, and prognosis of lung cancer. J. Transl. Med. 2022, 20, 404. [Google Scholar] [CrossRef]
- Sheu, J.J.; Lee, F.Y.; Wallace, C.G.; Tsai, T.H.; Leu, S.; Chen, Y.L.; Chai, H.T.; Lu, H.I.; Sun, C.K.; Yip, H.K. Administered circulating microparticles derived from lung cancer patients markedly improved angiogenesis, blood flow and ischemic recovery in rat critical limb ischemia. J. Transl. Med. 2015, 13, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesselaar, M.E.; Romijn, F.P.; Van Der Linden, I.K.; Prins, F.A.; Bertina, R.M.; Osanto, S. Microparticle-associated tissue factor activity: A link between cancer and thrombosis? J. Thromb. Haemost. 2007, 5, 520–527. [Google Scholar] [CrossRef]
- Mege, D.; Panicot-Dubois, L.; Ouaissi, M.; Robert, S.; Sielezneff, I.; Sastre, B.; Dignat-George, F.; Dubois, C. The origin and concentration of circulating microparticles differ according to cancer type and evolution: A prospective single-center study. Int. J. Cancer 2016, 138, 939–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bian, X.; Xiao, Y.T.; Wu, T.; Yao, M.; Du, L.; Ren, S.; Wang, J. Microvesicles and chemokines in tumor microenvironment: Mediators of intercellular communications in tumor progression. Mol. Cancer 2019, 18, 50. [Google Scholar] [CrossRef] [PubMed]
- Ko, S.F.; Yip, H.K.; Zhen, Y.Y.; Lee, C.C.; Li, J.H.; Lee, C.C.; Leu, S.; Huang, C.C.; Ng, S.H.; Lin, J.W. Cancer patient-derived circulating microparticles enhance lung metastasis in a rat model: Dual-source CT, cellular, and molecular studies. Mol. Imaging Biol. 2016, 18, 490–499. [Google Scholar] [CrossRef]
- Haemmerle, M.; Stone, R.L.; Menter, D.G.; Afshar-Kharghan, V.; Sood, A.K. The Platelet lifeline to cancer: Challenges and opportunities. Cancer Cell 2018, 33, 965–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.K.; Song, K.S.; Park, Y.S.; Kang, Y.H.; Lee, Y.J.; Lee, K.R.; Kim, H.K.; Ryu, K.W.; Bae, J.M.; Kim, S. Elevated levels of circulating platelet microparticles, VEGF, IL-6 and RANTES in patients with gastric cancer: Possible role of a metastasis predictor. Eur. J. Cancer 2003, 39, 184–191. [Google Scholar] [CrossRef]
- Liu, T.; Wang, J.; Liu, Y.; Wu, J.; Yuan, Y.; Wang, C.; Fang, X.; Li, H. Prediction of the therapeutic effects of pembrolizumab and nivolumab in advanced non-small cell lung cancer by platelet-derived microparticles in circulating blood. Technol. Cancer Res. Treat 2021, 20, 1533033821997817. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan-Chari, V.; Clancy, J.W.; Sedgwick, A.; D’Souza-Schorey, C. Microvesicles: Mediators of extracellular communication during cancer progression. J. Cell Sci. 2010, 123 Pt 10, 1603–1611. [Google Scholar] [CrossRef] [Green Version]
- Piccin, A.; Murphy, W.G.; Smith, O.P. Circulating microparticles: Pathophysiology and clinical implications. Blood Rev. 2007, 21, 157–171. [Google Scholar] [CrossRef]
- Jurj, A.; Zanoaga, O.; Braicu, C.; Lazar, V.; Tomuleasa, C.; Irimie, A.; Berindan-Neagoe, I. A Comprehensive Picture of Extracellular Vesicles and Their Contents. Molecular Transfer to Cancer Cells. Cancers 2020, 12, 298. [Google Scholar] [CrossRef] [Green Version]
- Del Conde, I.; Shrimpton, C.N.; Thiagarajan, P.; López, J.A. Tissue-factor-bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood 2005, 106, 1604–1611. [Google Scholar] [CrossRef] [PubMed]
- Pollet, H.; Conrard, L.; Cloos, A.S.; Tyteca, D. Plasma membrane lipid domains as platforms for vesicle biogenesis and shedding? Biomolecules 2018, 8, 94. [Google Scholar] [CrossRef] [Green Version]
- Muralidharan-Chari, V.; Clancy, J.; Plou, C.; Romao, M.; Chavrier, P.; Raposo, G.; D’Souza-Schorey, C. ARF6-regulated shedding of tumor cell-derived plasma membrane microvesicles. Curr. Biol. 2009, 19, 1875–1885. [Google Scholar] [CrossRef] [Green Version]
- McConnell, R.E.; Higginbotham, J.N.; Shifrin, D.A., Jr.; Tabb, D.L.; Coffey, R.J.; Tyska, M.J. The enterocyte microvillus is a vesicle-generating organelle. J. Cell Biol. 2009, 185, 1285–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McConnell, R.E.; Tyska, M.J. Myosin-1a powers the sliding of apical membrane along microvillar actin bundles. J. Cell Biol. 2007, 177, 671–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seetharaman, S.; Etienne-Manneville, S. Cytoskeletal crosstalk in cell migration. Trends Cell Biol. 2020, 30, 720–735. [Google Scholar] [CrossRef] [PubMed]
- Barger, S.R.; Gauthier, N.C.; Krendel, M. Squeezing in a Meal: Myosin Functions in Phagocytosis. Trends Cell Biol. 2020, 30, 157–167. [Google Scholar] [CrossRef]
- Schlienger, S.; Campbell, S.; Claing, A. ARF1 regulates the Rho/MLC pathway to control EGF-dependent breast cancer cell invasion. Mol. Biol. Cell 2014, 25, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Antonyak, M.A.; Zhang, J.; Cerione, R.A. RhoA triggers a specific signaling pathway that generates transforming microvesicles in cancer cells. Oncogene 2012, 31, 4740–4749. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Liu, C.; Bi, Z.Y.; Zhou, Q.; Zhang, H.; Li, L.L.; Zhang, J.; Zhu, W.; Song, Y.Y.; Zhang, F.; et al. Comprehensive landscape of extracellular vesicle-derived RNAs in cancer initiation, progression, metastasis and cancer immunology. Mol. Cancer 2020, 19, 102. [Google Scholar] [CrossRef]
- Zhu, S.; Li, S.; Yi, M.; Li, N.; Wu, K. Roles of microvesicles in tumor progression and clinical applications. Int. J. Nanomed. 2021, 16, 7071–7090. [Google Scholar] [CrossRef] [PubMed]
- Kholia, S.; Jorfi, S.; Thompson, P.R.; Causey, C.P.; Nicholas, A.P.; Inal, J.M.; Lange, S. A novel role for peptidylarginine deiminases in microvesicle release reveals therapeutic potential of PAD inhibition in sensitizing prostate cancer cells to chemotherapy. J. Extracell. Vesicles 2015, 4, 26192. [Google Scholar] [CrossRef] [Green Version]
- Liao, C.F.; Lin, S.H.; Chen, H.C.; Tai, C.J.; Chang, C.C.; Li, L.T.; Yeh, C.M.; Yeh, K.T.; Chen, Y.C.; Hsu, T.H.; et al. CSE1L, a novel microvesicle membrane protein, mediates Ras-triggered microvesicle generation and metastasis of tumor cells. Mol. Med. 2012, 18, 1269–1280. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, Y.; Wang, L.; Zhao, R.; Qiao, Y.; Han, D.; Sun, Q.; Dong, N.; Liu, Y.; Wu, D.; et al. miR-200a targets Gelsolin: A novel mechanism regulating secretion of microvesicles in hepatocellular carcinoma cells. Oncol. Rep. 2017, 37, 2711–2719. [Google Scholar] [CrossRef] [Green Version]
- Kalita-de Croft, P.; Sharma, S.; Sobrevia, L.; Salomon, C. Extracellular vesicle interactions with the external and internal exposome in mediating carcinogenesis. Mol. Aspects Med. 2022, 87, 101039. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.; Fa, H.; Xiao, D.; Wang, J. Targeting phosphatidylserine for cancer therapy: Prospects and challenges. Theranostics 2020, 10, 9214–9229. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, S.; Minamisawa, T.; Suga, K.; Kishita, H.; Akagi, T.; Ichiki, T.; Ichikawa, Y.; Shiba, K. Subtypes of tumour cell-derived small extracellular vesicles having differently externalized phosphatidylserine. J. Extracell. Vesicles 2019, 8, 1579541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallabhapurapu, S.D.; Blanco, V.M.; Sulaiman, M.K.; Vallabhapurapu, S.L.; Chu, Z.; Franco, R.S.; Qi, X. Variation in human cancer cell external phosphatidylserine is regulated by flippase activity and intracellular calcium. Oncotarget 2015, 6, 34375–34388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, X.; Liu, C.; Wang, H.; Wang, L.; Xiao, F.; Guo, Z.; Zhang, H. Surface phosphatidylserine is responsible for the internalization on microvesicles derived from hypoxia-induced human bone marrow mesenchymal stem cells into human endothelial cells. PLoS ONE 2016, 11, e0147360. [Google Scholar] [CrossRef] [Green Version]
- Birge, R.B.; Boeltz, S.; Kumar, S.; Carlson, J.; Wanderley, J.; Calianese, D.; Barcinski, M.; Brekken, R.A.; Huang, X.; Hutchins, J.T.; et al. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ. 2016, 23, 962–978. [Google Scholar] [CrossRef] [Green Version]
- Sharma, R.; Huang, X.; Brekken, R.A.; Schroit, A.J. Detection of phosphatidylserine-positive exosomes for the diagnosis of early-stage malignancies. Br. J. Cancer 2017, 117, 545–552. [Google Scholar] [CrossRef] [Green Version]
- Takatsu, H.; Takayama, M.; Naito, T.; Takada, N.; Tsumagari, K.; Ishihama, Y.; Nakayama, K.; Shin, H.W. Phospholipid flippase ATP11C is endocytosed and downregulated following Ca2+-mediated protein kinase C activation. Nat. Commun. 2017, 8, 1423. [Google Scholar] [CrossRef] [Green Version]
- Shin, H.W.; Takatsu, H. Phosphatidylserine exposure in living cells. Crit. Rev. Biochem. Mol. Biol. 2020, 55, 166–178. [Google Scholar] [CrossRef]
- Shin, H.W.; Takatsu, H. Substrates of P4-ATPases: Beyond aminophospholipids (phosphatidylserine and phosphatidylethanolamine). FASEB J. 2019, 33, 3087–3096. [Google Scholar] [CrossRef]
- Coleman, J.A.; Molday, R.S. Critical role of the beta-subunit CDC50A in the stable expression, assembly, subcellular localization, and lipid transport activity of the P4-ATPase ATP8A2. J. Biol. Chem. 2011, 286, 17205–17216. [Google Scholar] [CrossRef] [Green Version]
- Whitlock, J.M.; Hartzell, H.C. Anoctamins/TMEM16 proteins: Chloride channels flirting with lipids and extracellular vesicles. Annu. Rev. Physiol. 2017, 79, 119–143. [Google Scholar] [CrossRef] [Green Version]
- Sakuragi, T.; Kosako, H.; Nagata, S. Phosphorylation-mediated activation of mouse Xkr8 scramblase for phosphatidylserine exposure. Proc. Natl. Acad. Sci. USA 2019, 116, 2907–2912. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Cheng, X.; Tang, Y.; Qiu, X.; Wang, Y.; Kang, H.; Wu, J.; Wang, Z.; Liu, Y.; Chen, F. Bacterial endotoxin activates the coagulation cascade through Gasdermin D-dependent phosphatidylserine exposure. Immunity 2019, 51, 983–996.e6. [Google Scholar] [CrossRef] [PubMed]
- Falanga, A.; Russo, L.; Milesi, V.; Vignoli, A. Mechanisms and risk factors of thrombosis in cancer. Crit. Rev. Oncol. Hematol. 2017, 118, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Campello, E.; Ilich, A.; Simioni, P.; Key, N.S. The relationship between pancreatic cancer and hypercoagulability: A comprehensive review on epidemiological and biological issues. Br. J. Cancer 2019, 121, 359–371. [Google Scholar] [CrossRef]
- Fang, L.; Xu, Q.; Qian, J.; Zhou, J.Y. Aberrant factors of fibrinolysis and coagulation in pancreatic cancer. Onco. Targets Ther. 2021, 14, 53–65. [Google Scholar] [CrossRef]
- Date, K.; Ettelaie, C.; Maraveyas, A. Tissue factor-bearing microparticles and inflammation: A potential mechanism for the development of venous thromboembolism in cancer. J. Thromb. Haemost. 2017, 15, 2289–2299. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Guo, B.; Li, Y.; Zhu, B. Tissue factor in tumor microenvironment: A systematic review. J. Hematol. Oncol. 2014, 7, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nickel, K.F.; Ronquist, G.; Langer, F.; Labberton, L.; Fuchs, T.A.; Bokemeyer, C.; Sauter, G.; Graefen, M.; Mackman, N.; Stavrou, E.X.; et al. The polyphosphate-factor XII pathway drives coagulation in prostate cancer-associated thrombosis. Blood 2015, 126, 1379–1389. [Google Scholar] [CrossRef] [Green Version]
- Obermann, W.M.J.; Brockhaus, K.; Eble, J.A. Platelets, constant and cooperative companions of sessile and disseminating tumor cells, crucially contribute to the tumor microenvironment. Front. Cell Dev. Biol. 2021, 9, 674553. [Google Scholar] [CrossRef]
- Malvezzi, M.; Chalat, M.; Janjusevic, R.; Picollo, A.; Terashima, H.; Menon, A.K.; Accardi, A. Ca2+-dependent phospholipid scrambling by a reconstituted TMEM16 ion channel. Nat. Commun. 2013, 4, 2367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connolly, G.C.; Francis, C.W. Cancer-associated thrombosis. Hematol. Am. Soc. Hematol. Educ. Program 2013, 684–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sang, Y.; Roest, M.; de Laat, B.; de Groot, P.G.; Huskens, D. Interplay between platelets and coagulation. Blood Rev. 2021, 46, 100733. [Google Scholar] [CrossRef]
- Yu, L.X.; Yan, L.; Yang, W.; Wu, F.Q.; Ling, Y.; Chen, S.Z.; Tang, L.; Tan, Y.X.; Cao, D.; Wu, M.C.; et al. Platelets promote tumour metastasis via interaction between TLR4 and tumour cell-released high-mobility group box1 protein. Nat. Commun. 2014, 5, 5256. [Google Scholar] [CrossRef] [Green Version]
- Lova, P.; Canobbio, I.; Guidetti, G.F.; Balduini, C.; Torti, M. Thrombin induces platelet activation in the absence of functional protease activated receptors 1 and 4 and glycoprotein Ib-IX-V. Cell Signal 2010, 22, 1681–1687. [Google Scholar] [CrossRef]
- Stefanini, L.; Paul, D.S.; Robledo, R.F.; Chan, E.R.; Getz, T.M.; Campbell, R.A.; Kechele, D.O.; Casari, C.; Piatt, R.; Caron, K.M.; et al. RASA3 is a critical inhibitor of RAP1-dependent platelet activation. J. Clin. Investig. 2015, 125, 1419–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, W.; Tao, L.; Wang, Y.; Zhang, F.; Li, M.; Huang, S.; Wang, A.; Chen, W.; Yue, Z.; Chen, L.; et al. Downregulation of integrins in cancer cells and anti-platelet properties are involved in holothurian glycosaminoglycan-mediated disruption of the interaction of cancer cells and platelets in hematogenous metastasis. J. Vasc. Res. 2015, 52, 197–209. [Google Scholar] [CrossRef]
- Mittal, V. Epithelial mesenchymal transition in tumor metastasis. Annu. Rev. Pathol. 2018, 13, 395–412. [Google Scholar] [CrossRef]
- Popper, H. Primary tumor and metastasis-sectioning the different steps of the metastatic cascade. Transl. Lung Cancer Res. 2020, 9, 2277–2300. [Google Scholar] [CrossRef]
- Hugo, H.; Ackland, M.L.; Blick, T.; Lawrence, M.G.; Clements, J.A.; Williams, E.D.; Thompson, E.W. Epithelial--mesenchymal and mesenchymal--epithelial transitions in carcinoma progression. J. Cell Physiol. 2007, 213, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Keleg, S.; Büchler, P.; Ludwig, R.; Büchler, M.W.; Friess, H. Invasion and metastasis in pancreatic cancer. Mol. Cancer 2003, 2, 14. [Google Scholar] [CrossRef] [Green Version]
- Kuczynski, E.A.; Vermeulen, P.B.; Pezzella, F.; Kerbel, R.S.; Reynolds, A.R. Vessel co-option in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 469–493. [Google Scholar] [CrossRef] [PubMed]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in signaling and disease: Beyond discovery and development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef]
- Haen, P.; Mege, D.; Crescence, L.; Dignat-George, F.; Dubois, C.; Panicot-Dubois, L. Thrombosis risk associated with head and neck cancer: A review. Int. J. Mol. Sci. 2019, 20, 2838. [Google Scholar] [CrossRef] [Green Version]
- Rickles, F.R.; Patierno, S.; Fernandez, P.M. Tissue factor, thrombin, and cancer. Chest 2003, 124 (Suppl. 3), 58S–68S. [Google Scholar] [CrossRef]
- Palumbo, J.S.; Talmage, K.E.; Massari, J.V.; La Jeunesse, C.M.; Flick, M.J.; Kombrinck, K.W.; Jirousková, M.; Degen, J.L. Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells. Blood 2005, 105, 178–185. [Google Scholar] [CrossRef] [Green Version]
- Jolly, M.K.; Boareto, M.; Huang, B.; Jia, D.; Lu, M.; Ben-Jacob, E.; Onuchic, J.N.; Levine, H. Implications of the hybrid epithelial/mesenchymal phenotype in metastasis. Front. Oncol. 2015, 5, 155. [Google Scholar] [CrossRef] [Green Version]
- Lecharpentier, A.; Vielh, P.; Perez-Moreno, P.; Planchard, D.; Soria, J.C.; Farace, F. Detection of circulating tumour cells with a hybrid (epithelial/mesenchymal) phenotype in patients with metastatic non-small cell lung cancer. Br. J. Cancer 2011, 105, 1338–1341. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, A.J.; Marengo, M.S.; Oltean, S.; Kemeny, G.; Bitting, R.L.; Turnbull, J.D.; Herold, C.I.; Marcom, P.K.; George, D.J.; Garcia-Blanco, M.A. Circulating tumor cells from patients with advanced prostate and breast cancer display both epithelial and mesenchymal markers. Mol. Cancer Res. 2011, 9, 997–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Said, N.A.; Williams, E.D. Growth factors in induction of epithelial-mesenchymal transition and metastasis. Cells Tissues Organs 2011, 193, 85–97. [Google Scholar] [CrossRef]
- Liu, Y.; Jiang, P.; Capkova, K.; Xue, D.; Ye, L.; Sinha, S.C.; Mackman, N.; Janda, K.D.; Liu, C. Tissue factor-activated coagulation cascade in the tumor microenvironment is critical for tumor progression and an effective target for therapy. Cancer Res. 2011, 71, 6492–6502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milsom, C.C.; Yu, J.L.; Mackman, N.; Micallef, J.; Anderson, G.M.; Guha, A.; Rak, J.W. Tissue factor regulation by epidermal growth factor receptor and epithelial-to-mesenchymal transitions: Effect on tumor initiation and angiogenesis. Cancer Res. 2008, 68, 10068–10076. [Google Scholar] [CrossRef] [Green Version]
- Otsuki, T.; Fujimoto, D.; Hirono, Y.; Goi, T.; Yamaguchi, A. Thrombin conducts epithelial-mesenchymal transition via protease-activated receptor-1 in human gastric cancer. Int. J. Oncol. 2014, 45, 2287–2294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar-Shavit, R.; Turm, H.; Salah, Z.; Maoz, M.; Cohen, I.; Weiss, E.; Uziely, B.; Grisaru-Granovsky, S. PAR1 plays a role in epithelial malignancies: Transcriptional regulation and novel signaling pathway. IUBMB Life 2011, 63, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Booden, M.A.; Eckert, L.B.; Der, C.J.; Trejo, J. Persistent signaling by dysregulated thrombin receptor trafficking promotes breast carcinoma cell invasion. Mol. Cell Biol. 2004, 24, 1990–1999. [Google Scholar] [CrossRef] [Green Version]
- Labelle, M.; Begum, S.; Hynes, R.O. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 2011, 20, 576–590. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Wu, M.; Hu, Z.; Ma, Y.; Qi, W.; Zhang, Y.; Li, Y.; Yu, M.; Wang, H.; Mo, W. Thrombin is a therapeutic target for non-small-cell lung cancer to inhibit vasculogenic mimicry formation. Signal Transduct Target Ther. 2020, 5, 117. [Google Scholar] [CrossRef]
- Wu, Q.; You, L.; Nepovimova, E.; Heger, Z.; Wu, W.; Kuca, K.; Adam, V. Hypoxia-inducible factors: Master regulators of hypoxic tumor immune escape. J. Hematol. Oncol. 2022, 15, 77. [Google Scholar] [CrossRef]
- Bian, Y.; Yin, G.; Wang, G.; Liu, T.; Liang, L.; Yang, X.; Zhang, W.; Tang, D. Degradation of HIF-1α induced by curcumol blocks glutaminolysis and inhibits epithelial-mesenchymal transition and invasion in colorectal cancer cells. Cell Biol. Toxicol. 2022, 2022, 1–22. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, T.; Liu, X.; Li, Z.; Zhou, D.; Xu, W. Melatonin suppresses epithelial-to-mesenchymal transition in the MG-63 cell line. Mol. Med. Rep. 2020, 21, 1356–1364. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Lan, L.; Jiang, Y.G.; Zhao, J.H.; Li, M.C.; Wei, N.B.; Lin, Y.H. Epithelial-mesenchymal transition and migration of prostate cancer stem cells is driven by cancer-associated fibroblasts in an HIF-1α/β-catenin-dependent pathway. Mol. Cells 2013, 36, 138–144. [Google Scholar] [CrossRef]
- Shin, D.H.; Jo, J.Y.; Kim, S.H.; Choi, M.; Han, C.; Choi, B.K.; Kim, S.S. Midkine Is a Potential Therapeutic Target of Tumorigenesis, Angiogenesis, and Metastasis in Non-Small Cell Lung Cancer. Cancers 2020, 12, 2402. [Google Scholar] [CrossRef]
- Islam, S.U.; Ahmed, M.B.; Sonn, J.K.; Jin, E.J.; Lee, Y.S. PRP4 induces epithelial-mesenchymal transition and drug resistance in colon cancer cells via activation of p53. Int. J. Mol. Sci. 2022, 23, 3092. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.X.; Wang, D.W.; Liu, T.; Liu, W.X.; Xia, W.B.; Xu, J.; Zhang, Y.H.; Qu, Y.K.; Guo, L.Q.; Ding, L.; et al. Effects of the HIF-1α and NF-κB loop on epithelial-mesenchymal transition and chemoresistance induced by hypoxia in pancreatic cancer cells. Oncol. Rep. 2014, 31, 1891–1898. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.D.; Hu, B.; Cen, G.; Yang, Y.H.; Chen, W.W.; Guo, Z.Y.; Wang, X.F.; Zhao, Q.; Qiu, Z.J. MiR-301a transcriptionally activated by HIF-2α promotes hypoxia-induced epithelial-mesenchymal transition by targeting TP63 in pancreatic cancer. World J. Gastroenterol. 2020, 26, 2349–2373. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Liu, F.; Han, L.; Zhao, L.; Chen, J.; Olopade, O.I.; He, M.; Wei, M. HIF-2α promotes conversion to a stem cell phenotype and induces chemoresistance in breast cancer cells by activating Wnt and Notch pathways. J. Exp. Clin. Cancer Res. 2018, 37, 256. [Google Scholar] [CrossRef] [Green Version]
- Shaker, H.; Bundred, N.J.; Landberg, G.; Pritchard, S.A.; Albadry, H.; Nicholson, S.L.; Harries, L.J.; Heah, J.Y.E.; Castle, J.; Kirwan, C.C. Breast cancer stromal clotting activation (Tissue Factor and thrombin): A pre-invasive phenomena that is prognostic in invasion. Cancer Med. 2020, 9, 1768–1778. [Google Scholar] [CrossRef] [Green Version]
- Ruf, W.; Disse, J.; Carneiro-Lobo, T.C.; Yokota, N.; Schaffner, F. Tissue factor and cell signalling in cancer progression and thrombosis. J. Thromb. Haemost. 2011, 9 (Suppl. 1), 306–315. [Google Scholar] [CrossRef] [Green Version]
- Das, K.; Prasad, R.; Ansari, S.A.; Roy, A.; Mukherjee, A.; Sen, P. Matrix metalloproteinase-2: A key regulator in coagulation proteases mediated human breast cancer progression through autocrine signaling. Biomed. Pharmacother. 2018, 105, 395–406. [Google Scholar] [CrossRef]
- Wang, X.; Wang, E.; Kavanagh, J.J.; Freedman, R.S. Ovarian cancer, the coagulation pathway, and inflammation. J. Transl. Med. 2005, 3, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Q.; Luo, J.; Wang, T.; Ren, J.; Hu, K.; Wu, G. The activation of protease-activated receptor 1 mediates proliferation and invasion of nasopharyngeal carcinoma cells. Oncol. Rep. 2012, 28, 255–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso-Escolano, D.; Strongin, A.Y.; Chung, A.W.; Deryugina, E.I.; Radomski, M.W. Membrane type-1 matrix metalloproteinase stimulates tumour cell-induced platelet aggregation: Role of receptor glycoproteins. Br. J. Pharmacol. 2004, 141, 241–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danø, K.; Andreasen, P.A.; Grøndahl-Hansen, J.; Kristensen, P.; Nielsen, L.S.; Skriver, L. Plasminogen activators, tissue degradation, and cancer. Adv. Cancer Res. 1985, 44, 139–266. [Google Scholar] [PubMed]
- Bramhall, S.R.; Neoptolemos, J.P.; Stamp, G.W.; Lemoine, N.R. Imbalance of expression of matrix metalloproteinases (MMPs) and tissue inhibitors of the matrix metalloproteinases (TIMPs) in human pancreatic carcinoma. J. Pathol. 1997, 182, 347–355. [Google Scholar] [CrossRef]
- Cantero, D.; Friess, H.; Deflorin, J.; Zimmermann, A.; Bründler, M.A.; Riesle, E.; Korc, M.; Büchler, M.W. Enhanced expression of urokinase plasminogen activator and its receptor in pancreatic carcinoma. Br. J. Cancer 1997, 75, 388–395. [Google Scholar] [CrossRef] [Green Version]
- Del Rosso, M.; Fibbi, G.; Pucci, M.; D’Alessio, S.; Del Rosso, A.; Magnelli, L.; Chiarugi, V. Multiple pathways of cell invasion are regulated by multiple families of serine proteases. Clin. Exp. Metastasis 2002, 19, 193–207. [Google Scholar] [CrossRef]
- Yoshida, E.; Verrusio, E.N.; Mihara, H.; Oh, D.; Kwaan, H.C. Enhancement of the expression of urokinase-type plasminogen activator from PC-3 human prostate cancer cells by thrombin. Cancer Res. 1994, 54, 3300–3304. [Google Scholar]
- Rosenthal, R.; Cadieux, E.L.; Salgado, R.; Bakir, M.A.; Moore, D.A.; Hiley, C.T.; Lund, T.; Tanić, M.; Reading, J.L.; Joshi, K.; et al. Neoantigen-directed immune escape in lung cancer evolution. Nature 2019, 567, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Menter, T.; Tzankov, A. Mechanisms of immune evasion and immune modulation by lymphoma cells. Front. Oncol. 2018, 8, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, Z.; Wu, S.; Zhang, Z.; Ding, S. Mechanism of tumor cells escaping from immune surveillance of NK cells. Immunopharmacol. Immunotoxicol. 2020, 42, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer. 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Kim, T.H.; Fouladdel, S.; Zhang, Z.; Soni, P.; Qin, A.; Zhao, L.; Azizi, E.; Lawrence, T.S.; Ramnath, N.; et al. PD-L1 expression in circulating tumor cells increases during radio(chemo)therapy and indicates poor prognosis in non-small cell lung cancer. Sci. Rep. 2019, 9, 566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Wang, J.; Zhang, X.; Liu, P.; Zhang, X.; Wang, J.; Zheng, X.; Wei, L.; Peng, Q.; Liu, C.; et al. Proinflammatory S100A8 induces pd-l1 expression in macrophages, mediating tumor immune escape. J. Immunol. 2020, 204, 2589–2599. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Navas, C.; de Miguel-Perez, D.; Exposito-Hernandez, J.; Bayarri, C.; Amezcua, V.; Ortigosa, A.; Valdivia, J.; Guerrero, R.; Garcia Puche, J.L.; Lorente, J.A.; et al. Cooperative and escaping mechanisms between circulating tumor cells and blood constituents. Cells 2019, 8, 1382. [Google Scholar] [CrossRef] [Green Version]
- Kumagai, Y.; Ohzawa, H.; Miyato, H.; Horie, H.; Hosoya, Y.; Lefor, A.K.; Sata, N.; Kitayama, J. Surgical stress increases circulating low-density neutrophils which may promote tumor recurrence. J. Surg. Res. 2020, 246, 52–61. [Google Scholar] [CrossRef] [Green Version]
- Coffelt, S.B.; Kersten, K.; Doornebal, C.W.; Weiden, J.; Vrijland, K.; Hau, C.S.; Verstegen, N.J.M.; Ciampricotti, M.; Hawinkels, L.J.A.C.; Jonkers, J.; et al. IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature 2015, 522, 345–348. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Stang, A.; Schweickert, P.G.; Lanman, N.A.; Paul, E.N.; Monia, B.P.; Revenko, A.S.; Palumbo, J.S.; Mullins, E.S.; Elzey, B.D.; et al. Thrombin signaling promotes pancreatic adenocarcinoma through par-1-dependent immune evasion. Cancer Res. 2019, 79, 3417–3430. [Google Scholar] [CrossRef]
- Markiewski, M.M.; DeAngelis, R.A.; Benencia, F.; Ricklin-Lichtsteiner, S.K.; Koutoulaki, A.; Gerard, C.; Coukos, G.; Lambris, J.D. Modulation of the antitumor immune response by complement. Nat. Immunol. 2008, 9, 1225–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palumbo, J.S.; Barney, K.A.; Blevins, E.A.; Shaw, M.A.; Mishra, A.; Flick, M.J.; Kombrinck, K.W.; Talmage, K.E.; Souri, M.; Ichinose, A.; et al. Factor XIII transglutaminase supports hematogenous tumor cell metastasis through a mechanism dependent on natural killer cell function. J. Thromb. Haemost. 2008, 6, 812–819. [Google Scholar] [CrossRef]
- Haemmerle, M.; Taylor, M.L.; Gutschner, T.; Pradeep, S.; Cho, M.S.; Sheng, J.; Lyons, Y.M.; Nagaraja, A.S.; Dood, R.L.; Wen, Y.; et al. Platelets reduce anoikis and promote metastasis by activating YAP1 signaling. Nat. Commun. 2017, 8, 310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Placke, T.; Örgel, M.; Schaller, M.; Jung, G.; Rammensee, H.G.; Kopp, H.G.; Salih, H.R. Platelet-derived MHC class I confers a pseudonormal phenotype to cancer cells that subverts the antitumor reactivity of natural killer immune cells. Cancer Res. 2012, 72, 440–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Placke, T.; Salih, H.R.; Kopp, H.G. GITR ligand provided by thrombopoietic cells inhibits NK cell antitumor activity. J. Immunol. 2012, 189, 154–160. [Google Scholar] [CrossRef] [Green Version]
- Kopp, H.G.; Placke, T.; Salih, H.R. Platelet-derived transforming growth factor-beta down-regulates NKG2D thereby inhibiting natural killer cell antitumor reactivity. Cancer Res. 2009, 69, 7775–7783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sierko, E.; Wojtukiewicz, M.Z. Inhibition of platelet function: Does it offer a chance of better cancer progression control? Semin. Thromb. Hemost. 2007, 33, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Schnoor, M.; Alcaide, P.; Voisin, M.B.; van Buul, J.D. Crossing the vascular wall: Common and unique mechanisms exploited by different leukocyte subsets during extravasation. Mediators Inflamm. 2015, 2015, 946509. [Google Scholar] [CrossRef] [Green Version]
- Sökeland, G.; Schumacher, U. The functional role of integrins during intra- and extravasation within the metastatic cascade. Mol. Cancer 2019, 18, 12. [Google Scholar] [CrossRef]
- Labelle, M.; Begum, S.; Hynes, R.O. Platelets guide the formation of early metastatic niches. Proc. Natl. Acad. Sci. USA 2014, 111, E3053–E3061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cools-Lartigue, J.; Spicer, J.; Najmeh, S.; Ferri, L. Neutrophil extracellular traps in cancer progression. Cell Mol. Life Sci. 2014, 71, 4179–4194. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [Green Version]
- Häuselmann, I.; Roblek, M.; Protsyuk, D.; Huck, V.; Knopfova, L.; Grässle, S.; Bauer, A.T.; Schneider, S.W.; Borsig, L. Monocyte induction of E-Selectin-mediated endothelial activation releases VE-Cadherin junctions to promote tumor cell extravasation in the metastasis cascade. Cancer Res. 2016, 76, 5302–5312. [Google Scholar] [CrossRef] [Green Version]
- Qian, B.; Deng, Y.; Im, J.H.; Muschel, R.J.; Zou, Y.; Li, J.; Lang, R.A.; Pollard, J.W. A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PLoS ONE 2009, 4, e6562. [Google Scholar] [CrossRef] [Green Version]
- Konstantoulaki, M.; Kouklis, P.; Malik, A.B. Protein kinase C modifications of VE-cadherin, p120, and beta-catenin contribute to endothelial barrier dysregulation induced by thrombin. Am. J. Physiol. Lung Cell Mol. Physiol. 2003, 285, L434–L442. [Google Scholar] [CrossRef]
- Yokoyama, K.; Erickson, H.P.; Ikeda, Y.; Takada, Y. Identification of amino acid sequences in fibrinogen gamma -chain and tenascin C C-terminal domains critical for binding to integrin alpha vbeta 3. J. Biol. Chem. 2000, 275, 16891–16898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Ozdemir, T.; Chung, C.Y.; Robertson, G.P.; Dong, C. Sequential binding of αVβ3 and ICAM-1 determines fibrin-mediated melanoma capture and stable adhesion to CD11b/CD18 on neutrophils. J. Immunol. 2011, 186, 242–254. [Google Scholar] [CrossRef] [Green Version]
- Sandoval, R.; Malik, A.B.; Minshall, R.D.; Kouklis, P.; Ellis, C.A.; Tiruppathi, C. Ca(2+) signalling and PKCalpha activate increased endothelial permeability by disassembly of VE-cadherin junctions. J. Physiol. 2001, 533 Pt 2, 433–445. [Google Scholar] [CrossRef]
- Stockton, R.A.; Schaefer, E.; Schwartz, M.A. p21-activated kinase regulates endothelial permeability through modulation of contractility. J. Biol. Chem. 2004, 279, 46621–46630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dejana, E.; Orsenigo, F.; Lampugnani, M.G. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J. Cell Sci. 2008, 121 Pt 13, 2115–2122. [Google Scholar] [CrossRef] [Green Version]
- Dudek, S.M.; Garcia, J.G. Cytoskeletal regulation of pulmonary vascular permeability. J. Appl. Physiol. (1985) 2001, 91, 1487–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonsdorf, A.S.; Krämer, B.F.; Fahrleitner, M.; Schönberger, T.; Gnerlich, S.; Ring, S.; Gehring, S.; Schneider, S.W.; Kruhlak, M.J.; Meuth, S.G.; et al. Engagement of αIIbβ3 (GPIIb/IIIa) with ανβ3 integrin mediates interaction of melanoma cells with platelets: A connection to hematogenous metastasis. J. Biol. Chem. 2012, 287, 2168–2178. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, D.; Strilic, B.; Sivaraj, K.K.; Wettschureck, N.; Offermanns, S. Platelet-derived nucleotides promote tumor-cell transendothelial migration and metastasis via P2Y2 receptor. Cancer Cell 2013, 24, 130–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mammadova-Bach, E.; Zigrino, P.; Brucker, C.; Bourdon, C.; Freund, M.; De Arcangelis, A.; Abrams, S.I.; Orend, G.; Gachet, C.; Mangin, P.H. Platelet integrin α6β1 controls lung metastasis through direct binding to cancer cell-derived ADAM9. JCI Insight 2016, 1, e88245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.R.; Yousef, G.M.; Ni, H. Cancer and platelet crosstalk: Opportunities and challenges for aspirin and other antiplatelet agents. Blood 2018, 131, 1777–1789. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, Y.; Friess, H.; Buchler, M.; Beger, H.G.; Uchida, E.; Onda, M.; Kobrin, M.S.; Korc, M. Overexpression of acidic and basic fibroblast growth factors in human pancreatic cancer correlates with advanced tumor stage. Cancer Res. 1993, 53, 5289–5296. [Google Scholar]
- Welsh, J.; Smith, J.D.; Yates, K.R.; Greenman, J.; Maraveyas, A.; Madden, L.A. Tissue factor expression determines tumour cell coagulation kinetics. Int. J. Lab. Hematol. 2012, 34, 396–402. [Google Scholar] [CrossRef]
- Hong, H.; Zhang, Y.; Nayak, T.R.; Engle, J.W.; Wong, H.C.; Liu, B.; Barnhart, T.E.; Cai, W. Immuno-PET of tissue factor in pancreatic cancer. J. Nucl. Med. 2012, 53, 1748–1754. [Google Scholar] [CrossRef] [Green Version]
- Cao, R.; Björndahl, M.A.; Religa, P.; Clasper, S.; Garvin, S.; Galter, D.; Meister, B.; Ikomi, F.; Tritsaris, K.; Dissing, S.; et al. PDGF-BB induces intratumoral lymphangiogenesis and promotes lymphatic metastasis. Cancer Cell 2004, 6, 333–345. [Google Scholar] [CrossRef] [Green Version]
- van den Berg, Y.W.; Osanto, S.; Reitsma, P.H.; Versteeg, H.H. The relationship between tissue factor and cancer progression: Insights from bench and bedside. Blood 2012, 119, 924–932. [Google Scholar] [CrossRef] [Green Version]
- Uusitalo-Jarvinen, H.; Kurokawa, T.; Mueller, B.M.; Andrade-Gordon, P.; Friedlander, M.; Ruf, W. Role of protease activated receptor 1 and 2 signaling in hypoxia-induced angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1456–1462. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, J.E.; Zakarija, A.; Cundiff, D.L.; Doll, J.A.; Hymen, E.; Cornwell, M.; Crawford, S.E.; Liu, N.; Signaevsky, M.; Soff, G.A. Alternatively spliced human tissue factor promotes tumor growth and angiogenesis in a pancreatic cancer tumor model. Thromb. Res. 2007, 120 (Suppl. 2), S13–S21. [Google Scholar] [CrossRef]
- van den Berg, Y.W.; van den Hengel, L.G.; Myers, H.R.; Ayachi, O.; Jordanova, E.; Ruf, W.; Spek, C.A.; Reitsma, P.H.; Bogdanov, V.Y.; Versteeg, H.H. Alternatively spliced tissue factor induces angiogenesis through integrin ligation. Proc. Natl. Acad. Sci. USA 2009, 106, 19497–19502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.Q.; Li, J.J.; Hu, L.; Lee, M.; Karpatkin, S. Thrombin induces increased expression and secretion of VEGF from human FS4 fibroblasts, DU145 prostate cells and CHRF megakaryocytes. Thromb. Haemost. 2001, 86, 1094–1098. [Google Scholar]
- Xu, Y.; Gu, Y.; Keep, R.F.; Heth, J.; Muraszko, K.M.; Xi, G.; Hua, Y. Thrombin up-regulates vascular endothelial growth factor in experimental gliomas. Neurol. Res. 2009, 31, 759–765. [Google Scholar] [CrossRef]
- Tsopanoglou, N.E.; Maragoudakis, M.E. On the mechanism of thrombin-induced angiogenesis. Potentiation of vascular endothelial growth factor activity on endothelial cells by up-regulation of its receptors. J. Biol. Chem. 1999, 274, 23969–23976. [Google Scholar] [CrossRef] [Green Version]
- Hughes, P.E.; Pfaff, M. Integrin affinity modulation. Trends Cell Biol. 1998, 8, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Battinelli, E.M.; Markens, B.A.; Kulenthirarajan, R.A.; Machlus, K.R.; Flaumenhaft, R.; Italiano, J.E., Jr. Anticoagulation inhibits tumor cell-mediated release of platelet angiogenic proteins and diminishes platelet angiogenic response. Blood 2014, 123, 101–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnamoorthy, S.; Jin, R.; Cai, Y.; Maddipati, K.R.; Nie, D.; Pagès, G.; Tucker, S.C.; Honn, K.V. 12-Lipoxygenase and the regulation of hypoxia-inducible factor in prostate cancer cells. Exp. Cell Res. 2010, 316, 1706–1715. [Google Scholar] [CrossRef] [Green Version]
- Timar, J.; Bazaz, R.; Tang, D.G.; Kimler, V.; Taylor, J.D.; Honn, K.V. Post-translational regulation of surface integrin expression in tumor cells by 12(S)-HETE. Adv. Exp. Med. Biol. 1997, 400B, 757–763. [Google Scholar]
- Beausoleil, M.S.; Schulze, E.B.; Goodale, D.; Postenka, C.O.; Allan, A.L. Deletion of the thrombin cleavage domain of osteopontin mediates breast cancer cell adhesion, proteolytic activity, tumorgenicity, and metastasis. BMC Cancer 2011, 11, 25. [Google Scholar] [CrossRef] [Green Version]
- Karpatkin, S. Growth regulated oncogene is pivotal in thrombin-induced angiogenesis. Thromb. Res. 2007, 120 (Suppl. 2), S71–S74. [Google Scholar] [CrossRef] [PubMed]
- Maragoudakis, M.E.; Kraniti, N.; Giannopoulou, E.; Alexopoulos, K.; Matsoukas, J. Modulation of angiogenesis and progelatinase a by thrombin receptor mimetics and antagonists. Endothelium 2001, 8, 195–205. [Google Scholar] [CrossRef]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar]
- Falanga, A.; Panova-Noeva, M.; Russo, L. Procoagulant mechanisms in tumour cells. Best Pract. Res. Clin. Haematol. 2009, 22, 49–60. [Google Scholar] [CrossRef]
- Dasgupta, S.K.; Guchhait, P.; Thiagarajan, P. Lactadherin binding and phosphatidylserine expression on cell surface-comparison with annexin A5. Transl. Res. 2006, 148, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Digumarti, R.; Bapsy, P.P.; Suresh, A.V.; Bhattacharyya, G.S.; Dasappa, L.; Shan, J.S.; Gerber, D.E. Bavituximab plus paclitaxel and carboplatin for the treatment of advanced non-small-cell lung cancer. Lung Cancer 2014, 86, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Gerber, D.E.; Stopeck, A.T.; Wong, L.; Rosen, L.S.; Thorpe, P.E.; Shan, J.S.; Ibrahim, N.K. Phase I safety and pharmacokinetic study of bavituximab, a chimeric phosphatidylserine-targeting monoclonal antibody, in patients with advanced solid tumors. Clin. Cancer Res. 2011, 17, 6888–6896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, M.D.; Chen, W.Y.; Li, L.; Hertzmark, E.; Spiegelman, D.; Hankinson, S.E. Aspirin intake and survival after breast cancer. J. Clin. Oncol. 2010, 28, 1467–1472. [Google Scholar] [CrossRef] [PubMed]
- Rothwell, P.M.; Wilson, M.; Price, J.F.; Belch, J.F.; Meade, T.W.; Mehta, Z. Effect of daily aspirin on risk of cancer metastasis: A study of incident cancers during randomised controlled trials. Lancet 2012, 379, 1591–1601. [Google Scholar] [CrossRef]
- Amirkhosravi, A.; Mousa, S.A.; Amaya, M.; Blaydes, S.; Desai, H.; Meyer, T.; Francis, J.L. Inhibition of tumor cell-induced platelet aggregation and lung metastasis by the oral GpIIb/IIIa antagonist XV454. Thromb. Haemost. 2003, 90, 549–554. [Google Scholar]
- Rhee, J.S.; Black, M.; Schubert, U.; Fischer, S.; Morgenstern, E.; Hammes, H.P.; Preissner, K.T. The functional role of blood platelet components in angiogenesis. Thromb. Haemost. 2004, 92, 394–402. [Google Scholar] [PubMed]
- Harbi, M.H.; Smith, C.W.; Nicolson, P.L.R.; Watson, S.P.; Thomas, M.R. Novel antiplatelet strategies targeting GPVI, CLEC-2 and tyrosine kinases. Platelets 2021, 32, 29–41. [Google Scholar] [CrossRef]
- Schlesinger, M. Role of platelets and platelet receptors in cancer metastasis. J. Hematol. Oncol. 2018, 11, 125. [Google Scholar] [CrossRef] [Green Version]
- Versteeg, H.H. Tissue factor: Old and new links with cancer biology. Semin. Thromb. Hemost. 2015, 41, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Versteeg, H.H.; Schaffner, F.; Kerver, M.; Petersen, H.H.; Ahamed, J.; Felding-Habermann, B.; Takada, Y.; Mueller, B.M.; Ruf, W. Inhibition of tissue factor signaling suppresses tumor growth. Blood 2008, 111, 190–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojtukiewicz, M.Z.; Hempel, D.; Sierko, E.; Tucker, S.C.; Honn, K.V. Thrombin-unique coagulation system protein with multifaceted impacts on cancer and metastasis. Cancer Metastasis Rev. 2016, 35, 213–233. [Google Scholar] [CrossRef]
- Shi, K.; Damhofer, H.; Daalhuisen, J.; Ten Brink, M.; Richel, D.J.; Spek, C.A. Dabigatran potentiates gemcitabine-induced growth inhibition of pancreatic cancer in mice. Mol. Med. 2017, 23, 13–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asanuma, K.; Wakabayashi, H.; Hayashi, T.; Okuyama, N.; Seto, M.; Matsumine, A.; Kusuzaki, K.; Suzuki, K.; Uchida, A. Thrombin inhibitor, argatroban, prevents tumor cell migration and bone metastasis. Oncology 2004, 67, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Nierodzik, M.L.; Karpatkin, S. Thrombin induces tumor growth, metastasis, and angiogenesis: Evidence for a thrombin-regulated dormant tumor phenotype. Cancer Cell 2006, 10, 355–362. [Google Scholar] [CrossRef] [Green Version]
- Ward, M.P.; Kane, L.E.; Norris, L.A.; Mohamed, B.M.; Kelly, T.; Bates, M.; Clarke, A.; Brady, N.; Martin, C.M.; Brooks, R.D.; et al. Platelets, immune cells and the coagulation cascade; friend or foe of the circulating tumour cell? Mol. Cancer 2021, 20, 59. [Google Scholar] [CrossRef] [PubMed]
- Farge, D.; Frere, C.; Connors, J.M.; Khorana, A.A.; Kakkar, A.; Ay, C.; Muñoz, A.; Brenner, B.; Prata, P.H.; Brilhante, D.; et al. 2022 international clinical practice guidelines for the treatment and prophylaxis of venous thromboembolism in patients with cancer, including patients with COVID-19. Lancet Oncol. 2022, 23, e334–e347. [Google Scholar] [CrossRef] [PubMed]
- Ventura, J.S.; Faria, F.; Batista, I.F.; Simons, S.M.; Oliveira, D.G.; Morais, K.L.; Chudzinski-Tavassi, A.M. A Kunitz-type FXa inhibitor affects tumor progression, hypercoagulable state and triggers apoptosis. Biomed. Pharmacother. 2013, 67, 192–196. [Google Scholar] [CrossRef]
- Lazo-Langner, A.; Goss, G.D.; Spaans, J.N.; Rodger, M.A. The effect of low-molecular-weight heparin on cancer survival. A systematic review and meta-analysis of randomized trials. J. Thromb. Haemost. 2007, 5, 729–737. [Google Scholar] [CrossRef]
- Sørensen, H.T.; Mellemkjaer, L.; Olsen, J.H.; Baron, J.A. Prognosis of cancers associated with venous thromboembolism. N. Engl. J. Med. 2000, 343, 1846–1850. [Google Scholar] [CrossRef] [PubMed]
- Piccioli, A.; Falanga, A.; Prandoni, P. Anticoagulants and cancer survival. Semin. Thromb. Hemost. 2006, 32, 810–813. [Google Scholar] [CrossRef]
- Huang, R.; Rofstad, E.K. Integrins as therapeutic targets in the organ-specific metastasis of human malignant melanoma. J. Exp. Clin. Cancer Res. 2018, 37, 92. [Google Scholar] [CrossRef]
- Trikha, M.; Zhou, Z.; Nemeth, J.A.; Chen, Q.; Sharp, C.; Emmell, E.; Giles-Komar, J.; Nakada, M.T. CNTO 95, a fully human monoclonal antibody that inhibits alphav integrins, has antitumor and antiangiogenic activity in vivo. Int. J. Cancer 2004, 110, 326–335. [Google Scholar] [CrossRef]
- Jiang, Y.; Dai, J.; Yao, Z.; Shelley, G.; Keller, E.T. Abituzumab targeting of αV-class integrins inhibits prostate cancer progression. Mol. Cancer Res. 2017, 15, 875–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenthal, M.A.; Davidson, P.; Rolland, F.; Campone, M.; Xue, L.; Han, T.H.; Mehta, A.; Berd, Y.; He, W.; Lombardi, A. Evaluation of the safety, pharmacokinetics and treatment effects of an alpha(nu) beta (3) integrin inhibitor on bone turnover and disease activity in men with hormone-refractory prostate cancer and bone metastases. Asia Pac. J. Clin. Oncol. 2010, 6, 42–48. [Google Scholar] [CrossRef]
- Lin, T.H.; Tan, T.W.; Tsai, T.H.; Chen, C.C.; Hsieh, T.F.; Lee, S.S.; Liu, H.H.; Chen, W.C.; Tang, C.H. D-pinitol inhibits prostate cancer metastasis through inhibition of αVβ3 integrin by modulating FAK, c-Src and NF-κB pathways. Int. J. Mol. Sci. 2013, 14, 9790–9802. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, J.; Huang, H. Recent agents targeting HIF-1α for cancer therapy. J. Cell Biochem. 2013, 114, 498–509. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Tang, B.; Sun, X. Development of inhibitors targeting hypoxia-inducible factor 1 and 2 for cancer therapy. Yonsei Med. J. 2017, 58, 489–496. [Google Scholar] [CrossRef]
- Murugesan, T.; Rajajeyabalachandran, G.; Kumar, S.; Nagaraju, S.; Jegatheesan, S.K. Targeting HIF-2α as therapy for advanced cancers. Drug Discov. Today 2018, 23, 1444–1451. [Google Scholar] [CrossRef]
- Motto, I.; Bordogna, A.; Soshilov, A.A.; Denison, M.S.; Bonati, L. New aryl hydrocarbon receptor homology model targeted to improve docking reliability. J. Chem. Inf. Model. 2011, 51, 2868–2881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrara, N. Vascular endothelial growth factor as a target for anticancer therapy. Oncologist 2004, 9 (Suppl. 1), 2–10. [Google Scholar] [CrossRef] [PubMed]
- Wedam, S.B.; Low, J.A.; Yang, S.X.; Chow, C.K.; Choyke, P.; Danforth, D.; Hewitt, S.M.; Berman, A.; Steinberg, S.M.; Liewehr, D.J.; et al. Antiangiogenic and antitumor effects of bevacizumab in patients with inflammatory and locally advanced breast cancer. J. Clin. Oncol. 2006, 24, 769–777. [Google Scholar] [CrossRef]
- Chekhonin, V.P.; Shein, S.A.; Korchagina, A.A.; Gurina, O.I. VEGF in tumor progression and targeted therapy. Curr. Cancer Drug Targets 2013, 13, 423–443. [Google Scholar] [CrossRef]
- Wachsberger, P.R.; Burd, R.; Cardi, C.; Thakur, M.; Daskalakis, C.; Holash, J.; Yancopoulos, G.D.; Dicker, A.P. VEGF trap in combination with radiotherapy improves tumor control in u87 glioblastoma. Int. J. Radiat. Oncol. Biol. Phys. 2007, 67, 1526–1537. [Google Scholar] [CrossRef] [Green Version]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jing, H.; Wu, X.; Xiang, M.; Wang, C.; Novakovic, V.A.; Shi, J. Microparticle Phosphatidylserine Mediates Coagulation: Involvement in Tumor Progression and Metastasis. Cancers 2023, 15, 1957. https://doi.org/10.3390/cancers15071957
Jing H, Wu X, Xiang M, Wang C, Novakovic VA, Shi J. Microparticle Phosphatidylserine Mediates Coagulation: Involvement in Tumor Progression and Metastasis. Cancers. 2023; 15(7):1957. https://doi.org/10.3390/cancers15071957
Chicago/Turabian StyleJing, Haijiao, Xiaoming Wu, Mengqi Xiang, Chengyue Wang, Valerie A. Novakovic, and Jialan Shi. 2023. "Microparticle Phosphatidylserine Mediates Coagulation: Involvement in Tumor Progression and Metastasis" Cancers 15, no. 7: 1957. https://doi.org/10.3390/cancers15071957