
DNA Repair and Therapeutic Strategies in Cancer Stem Cells


Abstract
:Simple Summary
Abstract
1. Introduction
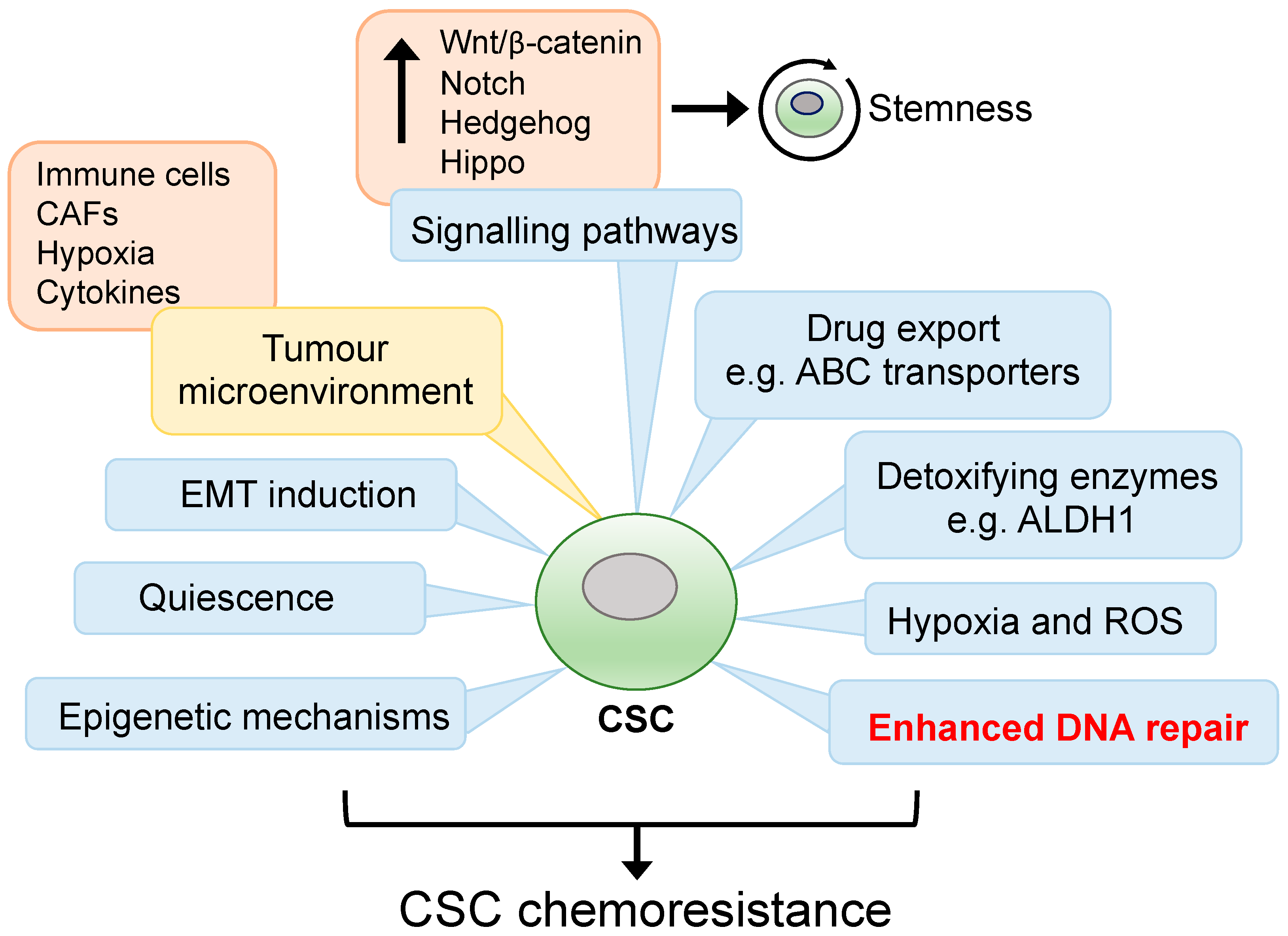
2. Therapy Resistance in CSCs
3. The DNA Damage Response (DDR)
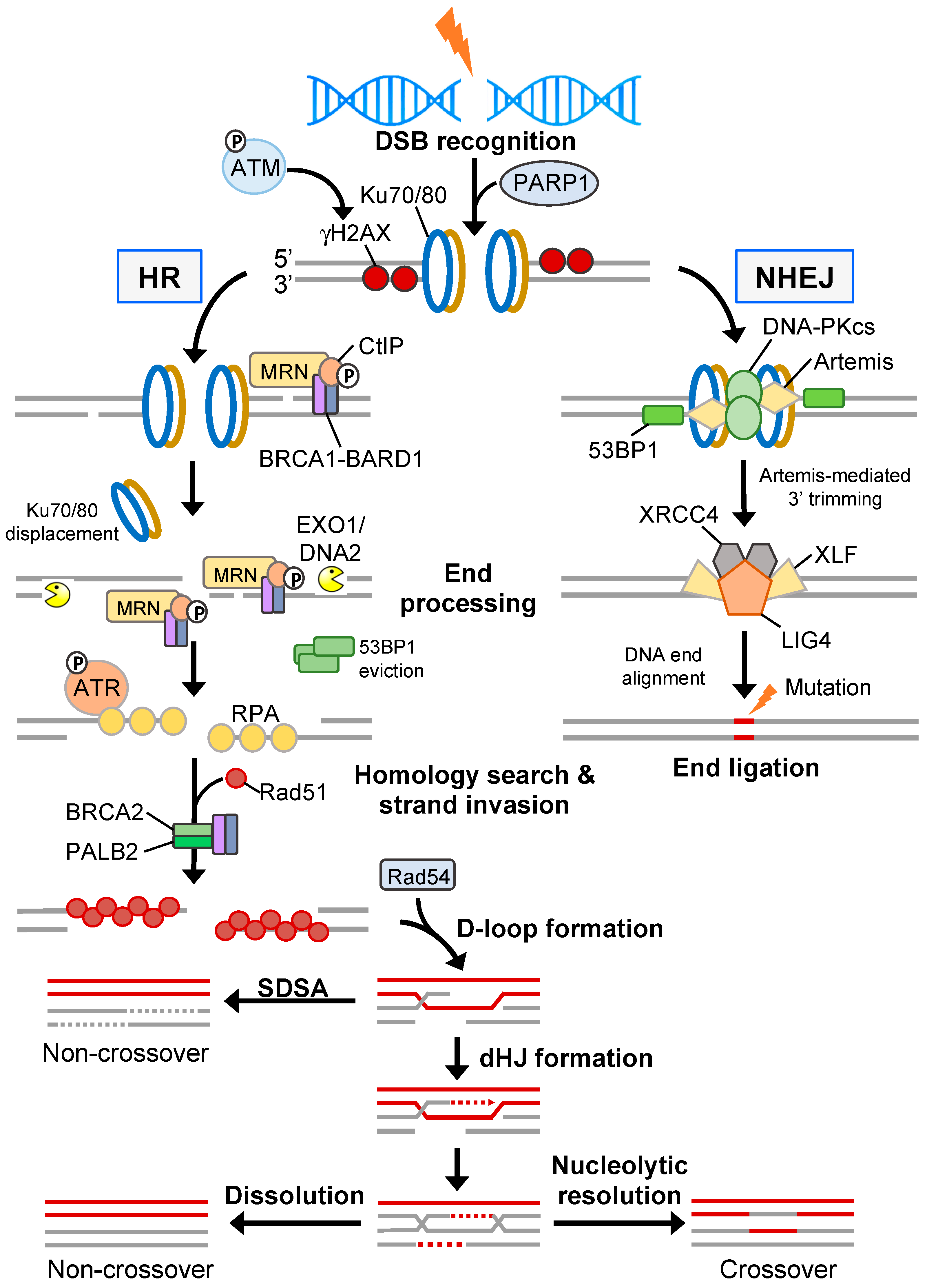
3.1. Double-Strand Break Repair
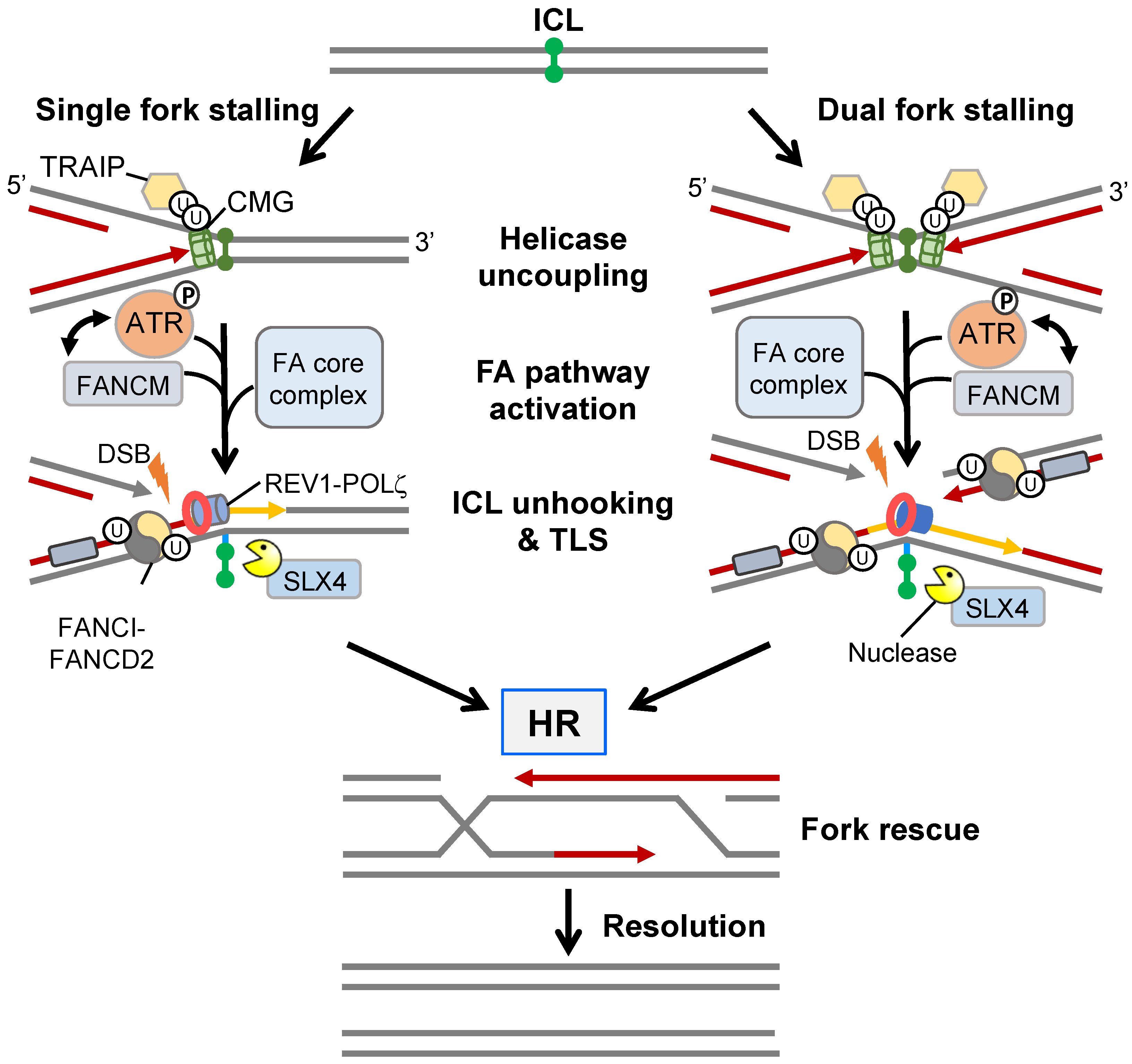
3.2. Interstrand Crosslink Repair by the Fanconi Anaemia Pathway
3.3. Single-Strand Break Repair Pathways
4. DNA Damage Repair in Non-Transformed Stem Cells
5. DNA Damage Repair in CSCs and Therapeutic Intervention
5.1. Drug Targeting the ATR-CHK1-WEE1 Checkpoint Pathway in CSCs
5.2. ATM Inhibitors in CSCs
5.3. PARP Inhibitors in CSCs
5.4. Drug Targeting HR Factors in CSCs
5.5. Drug Targeting c-NHEJ Factors in CSCs
5.6. DNA Repair Gene Splicing as a Mechanism of CSC Chemoresistance
6. Challenges and Opportunities for the Future
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bonnet, D.; Dick, J.E. Human Acute Myeloid Leukemia Is Organized as a Hierarchy That Originates from a Primitive Hematopoietic Cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A Cell Initiating Human Acute Myeloid Leukaemia after Transplantation into SCID Mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective Identification of Tumorigenic Breast Cancer Cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective Identification of Tumorigenic Prostate Cancer Stem Cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a Cancer Stem Cell in Human Brain Tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct Populations of Cancer Stem Cells Determine Tumor Growth and Metastatic Activity in Human Pancreatic Cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef] [Green Version]
- Eramo, A.; Lotti, F.; Sette, G.; Pilozzi, E.; Biffoni, M.; Virgilio, A.D.; Conticello, C.; Ruco, L.; Peschle, C.; Maria, R.D. Identification and Expansion of the Tumorigenic Lung Cancer Stem Cell Population. Cell Death Differ. 2008, 15, 504–514. [Google Scholar] [CrossRef]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A Human Colon Cancer Cell Capable of Initiating Tumour Growth in Immunodeficient Mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef]
- Prager, B.C.; Xie, Q.; Bao, S.; Rich, J.N. Cancer Stem Cells: The Architects of the Tumor Ecosystem. Cell Stem Cell 2019, 24, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Trumpp, A.; Haas, S. Cancer Stem Cells: The Adventurous Journey from Hematopoietic to Leukemic Stem Cells. Cell 2022, 185, 1266–1270. [Google Scholar] [CrossRef]
- Shackleton, M.; Quintana, E.; Fearon, E.R.; Morrison, S.J. Heterogeneity in Cancer: Cancer Stem Cells versus Clonal Evolution. Cell 2009, 138, 822–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barker, N.; Ridgway, R.A.; van Es, J.H.; van de Wetering, M.; Begthel, H.; van den Born, M.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt Stem Cells as the Cells-of-Origin of Intestinal Cancer. Nature 2009, 457, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Molyneux, G.; Geyer, F.C.; Magnay, F.-A.; McCarthy, A.; Kendrick, H.; Natrajan, R.; Mackay, A.; Grigoriadis, A.; Tutt, A.; Ashworth, A.; et al. BRCA1 Basal-like Breast Cancers Originate from Luminal Epithelial Progenitors and Not from Basal Stem Cells. Cell Stem Cell 2010, 7, 403–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blokzijl, F.; de Ligt, J.; Jager, M.; Sasselli, V.; Roerink, S.; Sasaki, N.; Huch, M.; Boymans, S.; Kuijk, E.; Prins, P.; et al. Tissue-Specific Mutation Accumulation in Human Adult Stem Cells during Life. Nature 2016, 538, 260–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, X.; Jörg, D.J.; Cavalli, F.M.G.; Richards, L.M.; Nguyen, L.V.; Vanner, R.J.; Guilhamon, P.; Lee, L.; Kushida, M.M.; Pellacani, D.; et al. Fate Mapping of Human Glioblastoma Reveals an Invariant Stem Cell Hierarchy. Nature 2017, 549, 227–232. [Google Scholar] [CrossRef] [Green Version]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [Green Version]
- Hall, A.E.; Pohl, S.Ö.-G.; Cammareri, P.; Aitken, S.; Younger, N.T.; Raponi, M.; Billard, C.V.; Carrancio, A.B.; Bastem, A.; Freile, P.; et al. RNA Splicing Is a Key Mediator of Tumour Cell Plasticity and a Therapeutic Vulnerability in Colorectal Cancer. Nat. Commun. 2022, 13, 2791. [Google Scholar] [CrossRef]
- Schwitalla, S.; Fingerle, A.A.; Cammareri, P.; Nebelsiek, T.; Göktuna, S.I.; Ziegler, P.K.; Canli, O.; Heijmans, J.; Huels, D.J.; Moreaux, G.; et al. Intestinal Tumorigenesis Initiated by Dedifferentiation and Acquisition of Stem-Cell-like Properties. Cell 2013, 152, 25–38. [Google Scholar] [CrossRef] [Green Version]
- Mohan, A.; Rajan, R.R.; Mohan, G.; Puthenveettil, P.K.; Maliekal, T.T. Markers and Reporters to Reveal the Hierarchy in Heterogeneous Cancer Stem Cells. Front. Cell Dev. Biol. 2021, 9, 668851. [Google Scholar] [CrossRef]
- Zhang, M.; Behbod, F.; Atkinson, R.L.; Landis, M.D.; Kittrell, F.; Edwards, D.; Medina, D.; Tsimelzon, A.; Hilsenbeck, S.; Green, J.E.; et al. Identification of Tumor-Initiating Cells in a P53-Null Mouse Model of Breast Cancer. Cancer Res. 2008, 68, 4674–4682. [Google Scholar] [CrossRef] [Green Version]
- Lim, E.; Vaillant, F.; Wu, D.; Forrest, N.C.; Pal, B.; Hart, A.H.; Asselin-Labat, M.-L.; Gyorki, D.E.; Ward, T.; Partanen, A.; et al. Aberrant Luminal Progenitors as the Candidate Target Population for Basal Tumor Development in BRCA1 Mutation Carriers. Nat. Med. 2009, 15, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Wright, M.H.; Calcagno, A.M.; Salcido, C.D.; Carlson, M.D.; Ambudkar, S.V.; Varticovski, L. Brca1 Breast Tumors Contain Distinct CD44+/CD24− and CD133+cells with Cancer Stem Cell Characteristics. Breast Cancer Res. 2008, 10, R10. [Google Scholar] [CrossRef] [Green Version]
- Britton, K.M.; Eyre, R.; Harvey, I.J.; Stemke-Hale, K.; Browell, D.; Lennard, T.W.J.; Meeson, A.P. Breast Cancer, Side Population Cells and ABCG2 Expression. Cancer Lett. 2012, 323, 97–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginestier, C.; Hur, M.-H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 Is a Marker of Normal and Malignant Human Mammary Stem Cells and a Predictor of Poor Clinical Outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricardo, S.; Vieira, A.F.; Gerhard, R.; Leitão, D.; Pinto, R.; Cameselle-Teijeiro, J.F.; Milanezi, F.; Schmitt, F.; Paredes, J. Breast Cancer Stem Cell Markers CD44, CD24 and ALDH1: Expression Distribution within Intrinsic Molecular Subtype. J. Clin. Pathol. 2011, 64, 937. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cong, Y.; Wang, D.; Sun, Y.; Deng, L.; Liu, Y.; Martin-Trevino, R.; Shang, L.; McDermott, S.P.; Landis, M.D.; et al. Breast Cancer Stem Cells Transition between Epithelial and Mesenchymal States Reflective of Their Normal Counterparts. Stem Cell Rep. 2014, 2, 78–91. [Google Scholar] [CrossRef]
- Akrap, N.; Andersson, D.; Bom, E.; Gregersson, P.; Ståhlberg, A.; Landberg, G. Identification of Distinct Breast Cancer Stem Cell Populations Based on Single-Cell Analyses of Functionally Enriched Stem and Progenitor Pools. Stem Cell Rep 2016, 6, 121–136. [Google Scholar] [CrossRef] [Green Version]
- Harrison, H.; Farnie, G.; Howell, S.J.; Rock, R.E.; Stylianou, S.; Brennan, K.R.; Bundred, N.J.; Clarke, R.B. Regulation of Breast Cancer Stem Cell Activity by Signaling through the Notch4 Receptor. Cancer Res. 2010, 70, 709–718. [Google Scholar] [CrossRef] [Green Version]
- Harrison, H.; Rogerson, L.; Gregson, H.J.; Brennan, K.R.; Clarke, R.B.; Landberg, G. Contrasting Hypoxic Effects on Breast Cancer Stem Cell Hierarchy Is Dependent on ER-α Status. Cancer Res. 2013, 73, 1420–1433. [Google Scholar] [CrossRef] [Green Version]
- Richichi, C.; Brescia, P.; Alberizzi, V.; Fornasari, L.; Pelicci, G. Marker-Independent Method for Isolating Slow-Dividing Cancer Stem Cells in Human Glioblastoma. Neoplasia 2013, 15, 840-IN39. [Google Scholar] [CrossRef] [Green Version]
- Topatana, W.; Juengpanich, S.; Li, S.; Cao, J.; Hu, J.; Lee, J.; Suliyanto, K.; Ma, D.; Zhang, B.; Chen, M.; et al. Advances in Synthetic Lethality for Cancer Therapy: Cellular Mechanism and Clinical Translation. J. Hematol. Oncol. 2020, 13, 118. [Google Scholar] [CrossRef] [PubMed]
- Rezayatmand, H.; Razmkhah, M.; Razeghian-Jahromi, I. Drug Resistance in Cancer Therapy: The Pandora’s Box of Cancer Stem Cells. Stem Cell Res. 2022, 13, 181. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Vila, M.; Takahashi, R.-U.; Usuba, W.; Kohama, I.; Ochiya, T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int. J. Mol. Sci. 2017, 18, 2574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.-G.; Lee, S.-H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer Stem Cells (CSCs) in Drug Resistance and Their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018, 2018, 5416923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Wang, Z.; Ajani, J.A.; Song, S. Drug Resistance and Cancer Stem Cells. Cell Commun. Signal. 2021, 19, 19. [Google Scholar] [CrossRef] [PubMed]
- Gaggianesi, M.; Franco, S.D.; Pantina, V.D.; Porcelli, G.; D’Accardo, C.; Verona, F.; Veschi, V.; Colarossi, L.; Faldetta, N.; Pistone, G.; et al. Messing Up the Cancer Stem Cell Chemoresistance Mechanisms Supported by Tumor Microenvironment. Front. Oncol. 2021, 11, 702642. [Google Scholar] [CrossRef]
- Wu, A.; Wei, J.; Kong, L.-Y.; Wang, Y.; Priebe, W.; Qiao, W.; Sawaya, R.; Heimberger, A.B. Glioma Cancer Stem Cells Induce Immunosuppressive Macrophages/Microglia. Neuro-Oncology 2010, 12, 1113–1125. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Li, S.; Zhang, J.; Wang, L.; Wu, X.; Wang, J.; Huang, Q.; Lai, M. Cancer Stemness, Immune Cells, and Epithelial–Mesenchymal Transition Cooperatively Predict Prognosis in Colorectal Carcinoma. Clin. Color. Canc. 2018, 17, e579–e592. [Google Scholar] [CrossRef]
- Sang, X.; Wu, F.; Wu, D.; Lin, S.; Li, J.; Zhao, N.; Chen, X.; Xu, A. Human Hepatic Cancer Stem Cells (HCSCs) Markers Correlated With Immune Infiltrates Reveal Prognostic Significance of Hepatocellular Carcinoma. Front. Genet. 2020, 11, 112. [Google Scholar] [CrossRef]
- Lee, Y.; Shin, J.H.; Longmire, M.; Wang, H.; Kohrt, H.E.; Chang, H.Y.; Sunwoo, J.B. CD44+ Cells in Head and Neck Squamous Cell Carcinoma Suppress T-Cell–Mediated Immunity by Selective Constitutive and Inducible Expression of PD-L1. Clin. Cancer Res. 2016, 22, 3571–3581. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Fillmore, C.M.; Koyama, S.; Wu, H.; Zhao, Y.; Chen, Z.; Herter-Sprie, G.S.; Akbay, E.A.; Tchaicha, J.H.; Altabef, A.; et al. Loss of Lkb1 and Pten Leads to Lung Squamous Cell Carcinoma with Elevated PD-L1 Expression. Cancer Cell 2014, 25, 590–604. [Google Scholar] [CrossRef] [Green Version]
- Hsu, J.-M.; Xia, W.; Hsu, Y.-H.; Chan, L.-C.; Yu, W.-H.; Cha, J.-H.; Chen, C.-T.; Liao, H.-W.; Kuo, C.-W.; Khoo, K.-H.; et al. STT3-Dependent PD-L1 Accumulation on Cancer Stem Cells Promotes Immune Evasion. Nat. Commun. 2018, 9, 1908. [Google Scholar] [CrossRef] [Green Version]
- Galassi, C.; Musella, M.; Manduca, N.; Maccafeo, E.; Sistigu, A. The Immune Privilege of Cancer Stem Cells: A Key to Understanding Tumor Immune Escape and Therapy Failure. Cells 2021, 10, 2361. [Google Scholar] [CrossRef] [PubMed]
- Magni, M.; Shammah, S.; Schiro, R.; Mellado, W.; Dalla-Favera, R.; Gianni, A. Induction of Cyclophosphamide-Resistance by Aldehyde-Dehydrogenase Gene Transfer. Blood 1996, 87, 1097–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.; Schuetz, J.D.; Bunting, K.D.; Colapietro, A.-M.; Sampath, J.; Morris, J.J.; Lagutina, I.; Grosveld, G.C.; Osawa, M.; Nakauchi, H.; et al. The ABC Transporter Bcrp1/ABCG2 Is Expressed in a Wide Variety of Stem Cells and Is a Molecular Determinant of the Side-Population Phenotype. Nat. Med. 2001, 7, 1028–1034. [Google Scholar] [CrossRef] [PubMed]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the Role of ABC Transporters in Multidrug-Resistant Cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef]
- Kim, H.; Lin, Q.; Glazer, P.M.; Yun, Z. The Hypoxic Tumor Microenvironment in vivo Selects the Cancer Stem Cell Fate of Breast Cancer Cells. Breast Cancer Res. 2018, 20, 16. [Google Scholar] [CrossRef] [Green Version]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem Cell-Associated Heterogeneity in Glioblastoma Results from Intrinsic Tumor Plasticity Shaped by the Microenvironment. Nat. Commun. 2019, 10, 1787. [Google Scholar] [CrossRef] [Green Version]
- DeNardo, D.G.; Brennan, D.J.; Rexhepaj, E.; Ruffell, B.; Shiao, S.L.; Madden, S.F.; Gallagher, W.M.; Wadhwani, N.; Keil, S.D.; Junaid, S.A.; et al. Leukocyte Complexity Predicts Breast Cancer Survival and Functionally Regulates Response to Chemotherapy. Cancer Discov. 2011, 1, 54–67. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Wang, W.; Wang, X.; Xu, C.; Zhang, N.; Di, W. Cisplatin-Stimulated Macrophages Promote Ovarian Cancer Migration via the CCL20-CCR6 Axis. Cancer Lett. 2020, 472, 59–69. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Marjanovic, N.D.; Lee, T.; Bell, G.; Kleer, C.G.; Reinhardt, F.; D’Alessio, A.C.; Young, R.A.; Weinberg, R.A. Poised Chromatin at the ZEB1 Promoter Enables Breast Cancer Cell Plasticity and Enhances Tumorigenicity. Cell 2013, 154, 61–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, P.P.; Dupre, T.V.; Siskind, L.J.; Beverly, L.J. Common Cytotoxic Chemotherapeutics Induce Epithelial-Mesenchymal Transition (EMT) Downstream of ER Stress. Oncotarget 2017, 8, 22625–22639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Lewis, M.T.; Huang, J.; Gutierrez, C.; Osborne, C.K.; Wu, M.-F.; Hilsenbeck, S.G.; Pavlick, A.; Zhang, X.; Chamness, G.C.; et al. Intrinsic Resistance of Tumorigenic Breast Cancer Cells to Chemotherapy. Jnci. J. Natl. Cancer Inst. 2008, 100, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-P.; Yang, C.-J.; Huang, M.-S.; Yeh, C.-T.; Wu, A.T.H.; Lee, Y.-C.; Lai, T.-C.; Lee, C.-H.; Hsiao, Y.-W.; Lu, J.; et al. Cisplatin Selects for Multidrug-Resistant CD133+ Cells in Lung Adenocarcinoma by Activating Notch Signaling. Cancer Res. 2013, 73, 406–416. [Google Scholar] [CrossRef] [Green Version]
- Ooki, A.; Dinalankara, W.; Marchionni, L.; Tsay, J.-C.J.; Goparaju, C.; Maleki, Z.; Rom, W.N.; Pass, H.I.; Hoque, M.O. Epigenetically Regulated PAX6 Drives Cancer Cells toward a Stem-like State via GLI-SOX2 Signaling Axis in Lung Adenocarcinoma. Oncogene 2018, 37, 5967–5981. [Google Scholar] [CrossRef]
- Bellio, C.; DiGloria, C.; Foster, R.; James, K.; Konstantinopoulos, P.A.; Growdon, W.B.; Rueda, B.R. PARP Inhibition Induces Enrichment of DNA Repair Proficient CD133 and CD117 Positive Ovarian Cancer Stem Cells. Mol. Cancer Res. 2018, 17, 431–445. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Li, Y.; Yu, T.-S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A Restricted Cell Population Propagates Glioblastoma Growth after Chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [Green Version]
- Cheung-Ong, K.; Giaever, G.; Nislow, C. DNA-Damaging Agents in Cancer Chemotherapy: Serendipity and Chemical Biology. Chem. Biol. 2013, 20, 648–659. [Google Scholar] [CrossRef] [Green Version]
- WANG, W.; SHENG, W.; YU, C.; CAO, J.; ZHOU, J.; WU, J.; ZHANG, H.; ZHANG, S. REV3L Modulates Cisplatin Sensitivity of Non-Small Cell Lung Cancer H1299 Cells. Oncol. Rep. 2015, 34, 1460–1468. [Google Scholar] [CrossRef]
- Oliver, T.G.; Mercer, K.L.; Sayles, L.C.; Burke, J.R.; Mendus, D.; Lovejoy, K.S.; Cheng, M.-H.; Subramanian, A.; Mu, D.; Powers, S.; et al. Chronic Cisplatin Treatment Promotes Enhanced Damage Repair and Tumor Progression in a Mouse Model of Lung Cancer. Gene Dev. 2010, 24, 837–852. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.K.; Matulonis, U.A. PARP Inhibitor Resistance Mechanisms and Implications for Post-Progression Combination Therapies. Cancers 2020, 12, 2054. [Google Scholar] [CrossRef] [PubMed]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA Double-Strand Break Repair-Pathway Choice in Somatic Mammalian Cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escribano-Díaz, C.; Orthwein, A.; Fradet-Turcotte, A.; Xing, M.; Young, J.T.F.; Tkáč, J.; Cook, M.A.; Rosebrock, A.P.; Munro, M.; Canny, M.D.; et al. A Cell Cycle-Dependent Regulatory Circuit Composed of 53BP1-RIF1 and BRCA1-CtIP Controls DNA Repair Pathway Choice. Mol. Cell 2013, 49, 872–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodarzi, A.A.; Yu, Y.; Riballo, E.; Douglas, P.; Walker, S.A.; Ye, R.; Härer, C.; Marchetti, C.; Morrice, N.; Jeggo, P.A.; et al. DNA-PK Autophosphorylation Facilitates Artemis Endonuclease Activity. Embo J. 2006, 25, 3880–3889. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-Homologous DNA End Joining and Alternative Pathways to Double-Strand Break Repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Mohiuddin, I.S.; Kang, M.H. DNA-PK as an Emerging Therapeutic Target in Cancer. Front. Oncol. 2019, 9, 635. [Google Scholar] [CrossRef] [Green Version]
- Schrempf, A.; Slyskova, J.; Loizou, J.I. Targeting the DNA Repair Enzyme Polymerase θ in Cancer Therapy. Trends Cancer 2021, 7, 98–111. [Google Scholar] [CrossRef]
- Mateos-Gomez, P.A.; Gong, F.; Nair, N.; Miller, K.M.; Lazzerini-Denchi, E.; Sfeir, A. Mammalian Polymerase θ Promotes Alternative NHEJ and Suppresses Recombination. Nature 2015, 518, 254–257. [Google Scholar] [CrossRef] [Green Version]
- Della-Maria, J.; Zhou, Y.; Tsai, M.-S.; Kuhnlein, J.; Carney, J.P.; Paull, T.T.; Tomkinson, A.E. Human Mre11/Human Rad50/Nbs1 and DNA Ligase IIIα/XRCC1 Protein Complexes Act Together in an Alternative Nonhomologous End Joining Pathway*. J. Biol. Chem. 2011, 286, 33845–33853. [Google Scholar] [CrossRef] [Green Version]
- Ceccaldi, R.; Liu, J.C.; Amunugama, R.; Hajdu, I.; Primack, B.; Petalcorin, M.I.R.; O’Connor, K.W.; Konstantinopoulos, P.A.; Elledge, S.J.; Boulton, S.J.; et al. Homologous-Recombination-Deficient Tumours Are Dependent on Polθ-Mediated Repair. Nature 2015, 518, 258–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyatt, D.W.; Feng, W.; Conlin, M.P.; Yousefzadeh, M.J.; Roberts, S.A.; Mieczkowski, P.; Wood, R.D.; Gupta, G.P.; Ramsden, D.A. Essential Roles for Polymerase θ-Mediated End Joining in the Repair of Chromosome Breaks. Mol. Cell 2016, 63, 662–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myler, L.R.; Gallardo, I.F.; Soniat, M.M.; Deshpande, R.A.; Gonzalez, X.B.; Kim, Y.; Paull, T.T.; Finkelstein, I.J. Single-Molecule Imaging Reveals How Mre11-Rad50-Nbs1 Initiates DNA Break Repair. Mol. Cell 2017, 67, 891–898.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-H.; Paull, T.T. ATM Activation by DNA Double-Strand Breaks through the Mre11-Rad50-Nbs1 Complex. Science 2005, 308, 551–554. [Google Scholar] [CrossRef]
- Stucki, M.; Clapperton, J.A.; Mohammad, D.; Yaffe, M.B.; Smerdon, S.J.; Jackson, S.P. MDC1 Directly Binds Phosphorylated Histone H2AX to Regulate Cellular Responses to DNA Double-Strand Breaks. Cell 2005, 123, 1213–1226. [Google Scholar] [CrossRef] [Green Version]
- Stewart, G.S.; Wang, B.; Bignell, C.R.; Taylor, A.M.R.; Elledge, S.J. MDC1 Is a Mediator of the Mammalian DNA Damage Checkpoint. Nature 2003, 421, 961–966. [Google Scholar] [CrossRef]
- Doil, C.; Mailand, N.; Bekker-Jensen, S.; Menard, P.; Larsen, D.H.; Pepperkok, R.; Ellenberg, J.; Panier, S.; Durocher, D.; Bartek, J.; et al. RNF168 Binds and Amplifies Ubiquitin Conjugates on Damaged Chromosomes to Allow Accumulation of Repair Proteins. Cell 2009, 136, 435–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huen, M.S.Y.; Grant, R.; Manke, I.; Minn, K.; Yu, X.; Yaffe, M.B.; Chen, J. RNF8 Transduces the DNA-Damage Signal via Histone Ubiquitylation and Checkpoint Protein Assembly. Cell 2007, 131, 901–914. [Google Scholar] [CrossRef] [Green Version]
- Kolas, N.K.; Chapman, J.R.; Nakada, S.; Ylanko, J.; Chahwan, R.; Sweeney, F.D.; Panier, S.; Mendez, M.; Wildenhain, J.; Thomson, T.M.; et al. Orchestration of the DNA-Damage Response by the RNF8 Ubiquitin Ligase. Science 2007, 318, 1637–1640. [Google Scholar] [CrossRef] [Green Version]
- Mailand, N.; Bekker-Jensen, S.; Faustrup, H.; Melander, F.; Bartek, J.; Lukas, C.; Lukas, J. RNF8 Ubiquitylates Histones at DNA Double-Strand Breaks and Promotes Assembly of Repair Proteins. Cell 2007, 131, 887–900. [Google Scholar] [CrossRef] [Green Version]
- Stewart, G.S.; Panier, S.; Townsend, K.; Al-Hakim, A.K.; Kolas, N.K.; Miller, E.S.; Nakada, S.; Ylanko, J.; Olivarius, S.; Mendez, M.; et al. The RIDDLE Syndrome Protein Mediates a Ubiquitin-Dependent Signaling Cascade at Sites of DNA Damage. Cell 2009, 136, 420–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panier, S.; Boulton, S.J. Double-Strand Break Repair: 53BP1 Comes into Focus. Nat. Rev. Mol. Cell Biol. 2014, 15, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Cho, N.W.; Cui, G.; Manion, E.M.; Shanbhag, N.M.; Botuyan, M.V.; Mer, G.; Greenberg, R.A. Acetylation Limits 53BP1 Association with Damaged Chromatin to Promote Homologous Recombination. Nat. Struct. Mol. Biol. 2013, 20, 317–325. [Google Scholar] [CrossRef]
- Becker, J.R.; Clifford, G.; Bonnet, C.; Groth, A.; Wilson, M.D.; Chapman, J.R. BARD1 Reads H2A Lysine 15 Ubiquitination to Direct Homologous Recombination. Nature 2021, 596, 433–437. [Google Scholar] [CrossRef]
- Nakamura, K.; Saredi, G.; Becker, J.R.; Foster, B.M.; Nguyen, N.V.; Beyer, T.E.; Cesa, L.C.; Faull, P.A.; Lukauskas, S.; Frimurer, T.; et al. H4K20me0 Recognition by BRCA1–BARD1 Directs Homologous Recombination to Sister Chromatids. Nat. Cell Biol. 2019, 21, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Densham, R.M.; Garvin, A.J.; Stone, H.R.; Strachan, J.; Baldock, R.A.; Daza-Martin, M.; Fletcher, A.; Blair-Reid, S.; Beesley, J.; Johal, B.; et al. Human BRCA1-BARD1 Ubiquitin Ligase Activity Counteracts Chromatin Barriers to DNA Resection. Nat. Struct. Mol. Biol. 2016, 23, 647–655. [Google Scholar] [CrossRef] [Green Version]
- Shibata, A.; Moiani, D.; Arvai, A.S.; Perry, J.; Harding, S.M.; Genois, M.-M.; Maity, R.; van Rossum-Fikkert, S.; Kertokalio, A.; Romoli, F.; et al. DNA Double-Strand Break Repair Pathway Choice Is Directed by Distinct MRE11 Nuclease Activities. Mol. Cell 2014, 53, 7–18. [Google Scholar] [CrossRef] [Green Version]
- Nimonkar, A.V.; Genschel, J.; Kinoshita, E.; Polaczek, P.; Campbell, J.L.; Wyman, C.; Modrich, P.; Kowalczykowski, S.C. BLM–DNA2–RPA–MRN and EXO1–BLM–RPA–MRN Constitute Two DNA End Resection Machineries for Human DNA Break Repair. Gene Dev. 2011, 25, 350–362. [Google Scholar] [CrossRef] [Green Version]
- Delacroix, S.; Wagner, J.M.; Kobayashi, M.; Yamamoto, K.; Karnitz, L.M. The Rad9-Hus1-Rad1 (9-1-1) Clamp Activates Checkpoint Signaling via TopBP1. Genes Dev. 2007, 21, 1472–1477. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Steinfeld, J.B.; Liang, F.; Chen, X.; Maranon, D.G.; Ma, C.J.; Kwon, Y.; Rao, T.; Wang, W.; Sheng, C.; et al. BRCA1–BARD1 Promotes RAD51-Mediated Homologous DNA Pairing. Nature 2017, 550, 360–365. [Google Scholar] [CrossRef] [Green Version]
- Semlow, D.R.; Walter, J.C. Mechanisms of Vertebrate DNA Interstrand Cross-Link Repair. Annu. Rev. Biochem. 2021, 90, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Rennie, M.L.; Lemonidis, K.; Arkinson, C.; Chaugule, V.K.; Clarke, M.; Streetley, J.; Spagnolo, L.; Walden, H. Differential Functions of FANCI and FANCD2 Ubiquitination Stabilize ID2 Complex on DNA. Embo Rep. 2020, 21, e50133. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; van Twest, S.; Leis, A.; Bythell-Douglas, R.; Murphy, V.J.; Sharp, M.; Parker, M.W.; Crismani, W.; Deans, A.J. Monoubiquitination by the Human Fanconi Anemia Core Complex Clamps FANCI:FANCD2 on DNA in Filamentous Arrays. Elife 2020, 9, e54128. [Google Scholar] [CrossRef] [PubMed]
- Stoepker, C.; Hain, K.; Schuster, B.; Hilhorst-Hofstee, Y.; Rooimans, M.A.; Steltenpool, J.; Oostra, A.B.; Eirich, K.; Korthof, E.T.; Nieuwint, A.W.M.; et al. SLX4, a Coordinator of Structure-Specific Endonucleases, Is Mutated in a New Fanconi Anemia Subtype. Nat. Genet. 2011, 43, 138–141. [Google Scholar] [CrossRef]
- Ciccia, A.; McDonald, N.; West, S.C. Structural and Functional Relationships of the XPF/MUS81 Family of Proteins. Annu. Rev. Biochem. 2008, 77, 259–287. [Google Scholar] [CrossRef]
- Budzowska, M.; Graham, T.G.; Sobeck, A.; Waga, S.; Walter, J.C. Regulation of the Rev1–Pol ζ Complex during Bypass of a DNA Interstrand Cross-link. Embo J. 2015, 34, 1971–1985. [Google Scholar] [CrossRef] [Green Version]
- van Twest, S.; Murphy, V.J.; Hodson, C.; Tan, W.; Swuec, P.; O’Rourke, J.J.; Heierhorst, J.; Crismani, W.; Deans, A.J. Mechanism of Ubiquitination and Deubiquitination in the Fanconi Anemia Pathway. Mol. Cell 2017, 65, 247–259. [Google Scholar] [CrossRef] [Green Version]
- García-Rubio, M.L.; Pérez-Calero, C.; Barroso, S.I.; Tumini, E.; Herrera-Moyano, E.; Rosado, I.V.; Aguilera, A. The Fanconi Anemia Pathway Protects Genome Integrity from R-Loops. PLoS Genet. 2015, 11, e1005674. [Google Scholar] [CrossRef] [Green Version]
- Sirbu, B.M.; McDonald, W.H.; Dungrawala, H.; Badu-Nkansah, A.; Kavanaugh, G.M.; Chen, Y.; Tabb, D.L.; Cortez, D. Identification of Proteins at Active, Stalled, and Collapsed Replication Forks Using Isolation of Proteins on Nascent DNA (IPOND) Coupled with Mass Spectrometry. J. Biol. Chem. 2013, 288, 31458–31467. [Google Scholar] [CrossRef] [Green Version]
- Brosh, R.M.; Wu, Y. An Emerging Picture of FANCJ’s Role in G4 Resolution to Facilitate DNA Replication. NAR Cancer 2021, 3, zcab034. [Google Scholar] [CrossRef]
- Michl, J.; Zimmer, J.; Tarsounas, M. Interplay between Fanconi Anemia and Homologous Recombination Pathways in Genome Integrity. Embo J. 2016, 35, 909–923. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W. DNA Single-Strand Break Repair and Human Genetic Disease. Trends Cell Biol. 2022, 32, 733–745. [Google Scholar] [CrossRef] [PubMed]
- Dawicki-McKenna, J.M.; Langelier, M.-F.; DeNizio, J.E.; Riccio, A.A.; Cao, C.D.; Karch, K.R.; McCauley, M.; Steffen, J.D.; Black, B.E.; Pascal, J.M. PARP-1 Activation Requires Local Unfolding of an Autoinhibitory Domain. Mol. Cell 2015, 60, 755–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sala, A.; Rocca, G.L.; Burgio, G.; Kotova, E.; Gesù, D.D.; Collesano, M.; Ingrassia, A.M.R.; Tulin, A.V.; Corona, D.F.V. The Nucleosome-Remodeling ATPase ISWI Is Regulated by Poly-ADP-Ribosylation. PLoS Biol. 2008, 6, e252. [Google Scholar] [CrossRef]
- Chou, D.M.; Adamson, B.; Dephoure, N.E.; Tan, X.; Nottke, A.C.; Hurov, K.E.; Gygi, S.P.; Colaiácovo, M.P.; Elledge, S.J. A Chromatin Localization Screen Reveals Poly (ADP Ribose)-Regulated Recruitment of the Repressive Polycomb and NuRD Complexes to Sites of DNA Damage. Proc. Natl. Acad. Sci. USA 2010, 107, 18475–18480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldecott, K.W. XRCC1 Protein; Form and Function. DNA Repair 2019, 81, 102664. [Google Scholar] [CrossRef]
- Wei, L.; Nakajima, S.; Hsieh, C.-L.; Kanno, S.; Masutani, M.; Levine, A.S.; Yasui, A.; Lan, L. Damage Response of XRCC1 at Sites of DNA Single Strand Breaks Is Regulated by Phosphorylation and Ubiquitylation after Degradation of Poly(ADP-Ribose). J. Cell Sci. 2013, 126, 4414–4423. [Google Scholar] [CrossRef] [Green Version]
- Breslin, C.; Hornyak, P.; Ridley, A.; Rulten, S.L.; Hanzlikova, H.; Oliver, A.W.; Caldecott, K.W. The XRCC1 Phosphate-Binding Pocket Binds Poly (ADP-Ribose) and Is Required for XRCC1 Function. Nucleic. Acids Res. 2015, 43, 6934–6944. [Google Scholar] [CrossRef] [Green Version]
- Demin, A.A.; Hirota, K.; Tsuda, M.; Adamowicz, M.; Hailstone, R.; Brazina, J.; Gittens, W.; Kalasova, I.; Shao, Z.; Zha, S.; et al. XRCC1 Prevents Toxic PARP1 Trapping during DNA Base Excision Repair. Mol. Cell 2021, 81, 3018–3030.e5. [Google Scholar] [CrossRef]
- Chaudhuri, A.R.; Nussenzweig, A. The Multifaceted Roles of PARP1 in DNA Repair and Chromatin Remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Caron, M.-C.; Sharma, A.K.; O’Sullivan, J.; Myler, L.R.; Ferreira, M.T.; Rodrigue, A.; Coulombe, Y.; Ethier, C.; Gagné, J.-P.; Langelier, M.-F.; et al. Poly(ADP-Ribose) Polymerase-1 Antagonizes DNA Resection at Double-Strand Breaks. Nat. Commun. 2019, 10, 2954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berti, M.; Chaudhuri, A.R.; Thangavel, S.; Gomathinayagam, S.; Kenig, S.; Vujanovic, M.; Odreman, F.; Glatter, T.; Graziano, S.; Mendoza-Maldonado, R.; et al. Human RECQ1 Promotes Restart of Replication Forks Reversed by DNA Topoisomerase I Inhibition. Nat. Struct. Mol. Biol. 2013, 20, 347–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haince, J.-F.; McDonald, D.; Rodrigue, A.; Déry, U.; Masson, J.-Y.; Hendzel, M.J.; Poirier, G.G. PARP1-Dependent Kinetics of Recruitment of MRE11 and NBS1 Proteins to Multiple DNA Damage Sites*. J. Biol. Chem. 2008, 283, 1197–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Zhang, H.; Xu, Z.; Tang, H.; Geng, A.; Cai, B.; Su, T.; Shi, J.; Jiang, C.; Tian, X.; et al. A PARP1–BRG1–SIRT1 Axis Promotes HR Repair by Reducing Nucleosome Density at DNA Damage Sites. Nucleic. Acids Res. 2019, 47, 8563–8580. [Google Scholar] [CrossRef]
- Bryant, H.E.; Petermann, E.; Schultz, N.; Jemth, A.-S.; Loseva, O.; Issaeva, N.; Johansson, F.; Fernandez, S.; McGlynn, P.; Helleday, T. PARP Is Activated at Stalled Forks to Mediate Mre11-Dependent Replication Restart and Recombination. EMBO J. 2009, 28, 2601–2615. [Google Scholar] [CrossRef] [Green Version]
- Murai, J.; Huang, S.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [Green Version]
- Groelly, F.J.; Fawkes, M.; Dagg, R.A.; Blackford, A.N.; Tarsounas, M. Targeting DNA Damage Response Pathways in Cancer. Nat. Rev. Cancer 2022, 23, 78–94. [Google Scholar] [CrossRef]
- Wang, N.; Yang, Y.; Jin, D.; Zhang, Z.; Shen, K.; Yang, J.; Chen, H.; Zhao, X.; Yang, L.; Lu, H. PARP Inhibitor Resistance in Breast and Gynecological Cancer: Resistance Mechanisms and Combination Therapy Strategies. Front. Pharm. 2022, 13, 967633. [Google Scholar] [CrossRef]
- Cervantes, R.B.; Stringer, J.R.; Shao, C.; Tischfield, J.A.; Stambrook, P.J. Embryonic Stem Cells and Somatic Cells Differ in Mutation Frequency and Type. Proc. Natl. Acad. Sci. USA 2002, 99, 3586–3590. [Google Scholar] [CrossRef] [Green Version]
- Cappell, S.D.; Chung, M.; Jaimovich, A.; Spencer, S.L.; Meyer, T. Irreversible APCCdh1 Inactivation Underlies the Point of No Return for Cell-Cycle Entry. Cell 2016, 166, 167–180. [Google Scholar] [CrossRef] [Green Version]
- Orford, K.W.; Scadden, D.T. Deconstructing Stem Cell Self-Renewal: Genetic Insights into Cell-Cycle Regulation. Nat. Rev. Genet. 2008, 9, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Boward, B.; Wu, T.; Dalton, S. Concise Review: Control of Cell Fate Through Cell Cycle and Pluripotency Networks. Stem Cells 2016, 34, 1427–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maynard, S.; Swistowska, A.M.; Lee, J.W.; Liu, Y.; Liu, S.; Cruz, A.B.D.; Rao, M.; de Souza-Pinto, N.C.; Zeng, X.; Bohr, V.A. Human Embryonic Stem Cells Have Enhanced Repair of Multiple Forms of DNA Damage. Stem Cells 2008, 26, 2266–2274. [Google Scholar] [CrossRef] [Green Version]
- Chlon, T.M.; Ruiz-Torres, S.; Maag, L.; Mayhew, C.N.; Wikenheiser-Brokamp, K.A.; Davies, S.M.; Mehta, P.; Myers, K.C.; Wells, J.M.; Wells, S.I. Overcoming Pluripotent Stem Cell Dependence on the Repair of Endogenous DNA Damage. Stem Cell Rep. 2016, 6, 44–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chlon, T.M.; Hoskins, E.E.; Mayhew, C.N.; Wikenheiser-Brokamp, K.A.; Davies, S.M.; Mehta, P.; Myers, K.C.; Wells, J.M.; Wells, S.I. High-Risk Human Papillomavirus E6 Protein Promotes Reprogramming of Fanconi Anemia Patient Cells through Repression of P53 but Does Not Allow for Sustained Growth of Induced Pluripotent Stem Cells. J. Virol. 2014, 88, 11315–11326. [Google Scholar] [CrossRef] [Green Version]
- Efroni, S.; Duttagupta, R.; Cheng, J.; Dehghani, H.; Hoeppner, D.J.; Dash, C.; Bazett-Jones, D.P.; Grice, S.L.; McKay, R.D.G.; Buetow, K.H.; et al. Global Transcription in Pluripotent Embryonic Stem Cells. Cell Stem Cell 2008, 2, 437–447. [Google Scholar] [CrossRef] [Green Version]
- Guzman-Ayala, M.; Sachs, M.; Koh, F.M.; Onodera, C.; Bulut-Karslioglu, A.; Lin, C.-J.; Wong, P.; Nitta, R.; Song, J.S.; Ramalho-Santos, M. Chd1 Is Essential for the High Transcriptional Output and Rapid Growth of the Mouse Epiblast. Development 2014, 142, 118–127. [Google Scholar] [CrossRef] [Green Version]
- Bulut-Karslioglu, A.; Jin, H.; Kim, Y.-K.; Cho, B.; Guzman-Ayala, M.; Williamson, A.J.K.; Hejna, M.; Stötzel, M.; Whetton, A.D.; Song, J.S.; et al. Chd1 Protects Genome Integrity at Promoters to Sustain Hypertranscription in Embryonic Stem Cells. Nat. Commun. 2021, 12, 4859. [Google Scholar] [CrossRef]
- Blakemore, D.; Vilaplana-Lopera, N.; Almaghrabi, R.; Gonzalez, E.; Moya, M.; Ward, C.; Murphy, G.; Gambus, A.; Petermann, E.; Stewart, G.S.; et al. MYBL2 and ATM Suppress Replication Stress in Pluripotent Stem Cells. Embo Rep. 2021, 22, e51120. [Google Scholar] [CrossRef]
- Chang, C.-H.; Zhang, M.; Rajapakshe, K.; Coarfa, C.; Edwards, D.; Huang, S.; Rosen, J.M. Mammary Stem Cells and Tumor-Initiating Cells Are More Resistant to Apoptosis and Exhibit Increased DNA Repair Activity in Response to DNA Damage. Stem Cell Rep. 2015, 5, 378–391. [Google Scholar] [CrossRef] [Green Version]
- Oliver, L.; Hue, E.; Séry, Q.; Lafargue, A.; Pecqueur, C.; Paris, F.; Vallette, F.M. Differentiation-Related Response to DNA Breaks in Human Mesenchymal Stem Cells. Stem Cells 2013, 31, 800–807. [Google Scholar] [CrossRef] [PubMed]
- Sotiropoulou, P.A.; Candi, A.; Mascré, G.; Clercq, S.D.; Youssef, K.K.; Lapouge, G.; Dahl, E.; Semeraro, C.; Denecker, G.; Marine, J.-C.; et al. Bcl-2 and Accelerated DNA Repair Mediates Resistance of Hair Follicle Bulge Stem Cells to DNA-Damage-Induced Cell Death. Nat. Cell Biol. 2010, 12, 572–582. [Google Scholar] [CrossRef] [PubMed]
- Catlin, S.N.; Busque, L.; Gale, R.E.; Guttorp, P.; Abkowitz, J.L. The Replication Rate of Human Hematopoietic Stem Cells in vivo. Blood 2011, 117, 4460–4466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohrin, M.; Bourke, E.; Alexander, D.; Warr, M.R.; Barry-Holson, K.; Beau, M.M.L.; Morrison, C.G.; Passegué, E. Hematopoietic Stem Cell Quiescence Promotes Error-Prone DNA Repair and Mutagenesis. Cell Stem Cell 2010, 7, 174–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longerich, S.; Li, J.; Xiong, Y.; Sung, P.; Kupfer, G.M. Stress and DNA Repair Biology of the Fanconi Anemia Pathway. Blood 2014, 124, 2812–2819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, D.; Lier, A.; Geiselhart, A.; Thalheimer, F.B.; Huntscha, S.; Sobotta, M.C.; Moehrle, B.; Brocks, D.; Bayindir, I.; Kaschutnig, P.; et al. Exit from Dormancy Provokes DNA-Damage-Induced Attrition in Haematopoietic Stem Cells. Nature 2015, 520, 549–552. [Google Scholar] [CrossRef]
- Biechonski, S.; Yassin, M.; Milyavsky, M. DNA-Damage Response in Hematopoietic Stem Cells: An Evolutionary Trade-off between Blood Regeneration and Leukemia Suppression. Carcinogenesis 2017, 38, 367–377. [Google Scholar] [CrossRef]
- Abad, E.; Graifer, D.; Lyakhovich, A. DNA Damage Response and Resistance of Cancer Stem Cells. Cancer Lett. 2020, 474, 106–117. [Google Scholar] [CrossRef]
- Abad, E.; Civit, L.; Potesil, D.; Zdrahal, Z.; Lyakhovich, A. Enhanced DNA Damage Response through RAD50 in Triple Negative Breast Cancer Resistant and Cancer Stem-like Cells Contributes to Chemoresistance. FEBS J. 2021, 288, 2184–2202. [Google Scholar] [CrossRef]
- Meyer, F.; Engel, A.M.; Krause, A.K.; Wagner, T.; Poole, L.; Dubrovska, A.; Peitzsch, C.; Rothkamm, K.; Petersen, C.; Borgmann, K. Efficient DNA Repair Mitigates Replication Stress Resulting in Less Immunogenic Cytosolic DNA in Radioresistant Breast Cancer Stem Cells. Front. Immunol. 2022, 13, 765284. [Google Scholar] [CrossRef]
- Azzoni, V.; Wicinski, J.; Macario, M.; Castagné, M.; Finetti, P.; Ambrosova, K.; Rouault, C.D.; Sergé, A.; Farina, A.; Agavnian, E.; et al. BMI1 Nuclear Location Is Critical for RAD51-Dependent Response to Replication Stress and Drives Chemoresistance in Breast Cancer Stem Cells. Cell Death Dis. 2022, 13, 96. [Google Scholar] [CrossRef] [PubMed]
- Karimi-Busheri, F.; Rasouli-Nia, A.; Mackey, J.R.; Weinfeld, M. Senescence Evasion by MCF-7 Human Breast Tumor-Initiating Cells. Breast Cancer Res. BCR 2010, 12, R31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.; Wang, Z.; Fong, C.; Liu, D.; Yip, T.; Au, S.; Zhu, G.; Yang, M. Chemoresistant Lung Cancer Stem Cells Display High DNA Repair Capability to Remove Cisplatin-induced DNA Damage. Brit. J. Pharm. 2017, 174, 302–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.; Tang, D. Prostate Cancer Stem-like Cells Proliferate Slowly and Resist Etoposide-Induced Cytotoxicity via Enhancing DNA Damage Response. Exp. Cell Res. 2014, 328, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma Stem Cells Promote Radioresistance by Preferential Activation of the DNA Damage Response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Ahmed, S.U.; Carruthers, R.; Gilmour, L.; Yildirim, S.; Watts, C.; Chalmers, A.J. Selective Inhibition of Parallel DNA Damage Response Pathways Optimizes Radiosensitization of Glioblastoma Stem-like Cells. Cancer Res. 2015, 75, 4416–4428. [Google Scholar] [CrossRef] [Green Version]
- Carruthers, R.D.; Ahmed, S.U.; Ramachandran, S.; Strathdee, K.; Kurian, K.M.; Hedley, A.; Gomez-Roman, N.; Kalna, G.; Neilson, M.P.; Gilmour, L.; et al. Replication Stress Drives Constitutive Activation of the DNA Damage Response and Radioresistance in Glioblastoma Stem-like Cells. Cancer Res. 2018, 78, 5060–5071. [Google Scholar] [CrossRef] [Green Version]
- Obara, E.A.A.; Aguilar-Morante, D.; Rasmussen, R.D.; Frias, A.; Vitting-Serup, K.; Lim, Y.C.; Elbæk, K.J.; Pedersen, H.; Vardouli, L.; Jensen, K.E.; et al. SPT6-Driven Error-Free DNA Repair Safeguards Genomic Stability of Glioblastoma Cancer Stem-like Cells. Nat. Commun. 2020, 11, 4709. [Google Scholar] [CrossRef]
- López, J.; Poitevin, A.; Mendoza-Martínez, V.; Pérez-Plasencia, C.; García-Carrancá, A. Cancer-Initiating Cells Derived from Established Cervical Cell Lines Exhibit Stem-Cell Markers and Increased Radioresistance. BMC Cancer 2012, 12, 48. [Google Scholar] [CrossRef] [Green Version]
- Ropolo, M.; Daga, A.; Griffero, F.; Foresta, M.; Casartelli, G.; Zunino, A.; Poggi, A.; Cappelli, E.; Zona, G.; Spaziante, R.; et al. Comparative Analysis of DNA Repair in Stem and Nonstem Glioma Cell Cultures. Mol. Cancer Res. 2009, 7, 383–392. [Google Scholar] [CrossRef] [Green Version]
- Ronco, C.; Martin, A.R.; Demange, L.; Benhida, R. ATM, ATR, CHK1, CHK2 and WEE1 Inhibitors in Cancer and Cancer Stem Cells. Medchemcomm 2016, 8, 295–319. [Google Scholar] [CrossRef] [PubMed]
- Signore, M.; Pelacchi, F.; di Martino, S.; Runci, D.; Biffoni, M.; Giannetti, S.; Morgante, L.; Majo, M.D.; Petricoin, E.F.; Stancato, L.; et al. Combined PDK1 and CHK1 Inhibition Is Required to Kill Glioblastoma Stem-like Cells in vitro and in vivo. Cell Death Dis. 2014, 5, e1223. [Google Scholar] [CrossRef]
- Bartucci, M.; Svensson, S.; Romania, P.; Dattilo, R.; Patrizii, M.; Signore, M.; Navarra, S.; Lotti, F.; Biffoni, M.; Pilozzi, E.; et al. Therapeutic Targeting of Chk1 in NSCLC Stem Cells during Chemotherapy. Cell Death Differ. 2012, 19, 768–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatesha, V.A.; Parsels, L.A.; Parsels, J.D.; Zhao, L.; Zabludoff, S.D.; Simeone, D.M.; Maybaum, J.; Lawrence, T.S.; Morgan, M.A. Sensitization of Pancreatic Cancer Stem Cells to Gemcitabine by Chk1 Inhibition. Neoplasia 2012, 14, 519–525. [Google Scholar] [CrossRef] [Green Version]
- Morgan, M.A.; Parsels, L.A.; Zhao, L.; Parsels, J.D.; Davis, M.A.; Hassan, M.C.; Arumugarajah, S.; Hylander-Gans, L.; Morosini, D.; Simeone, D.M.; et al. Mechanism of Radiosensitization by the Chk1/2 Inhibitor AZD7762 Involves Abrogation of the G2 Checkpoint and Inhibition of Homologous Recombinational DNA Repair. Cancer Res. 2010, 70, 4972–4981. [Google Scholar] [CrossRef] [Green Version]
- Gorecki, L.; Andrs, M.; Korabecny, J. Clinical Candidates Targeting the ATR–CHK1–WEE1 Axis in Cancer. Cancers 2021, 13, 795. [Google Scholar] [CrossRef] [PubMed]
- Sausville, E.; LoRusso, P.; Carducci, M.; Carter, J.; Quinn, M.F.; Malburg, L.; Azad, N.; Cosgrove, D.; Knight, R.; Barker, P.; et al. Phase I Dose-Escalation Study of AZD7762, a Checkpoint Kinase Inhibitor, in Combination with Gemcitabine in US Patients with Advanced Solid Tumors. Cancer Chemother. Pharmacol. 2014, 73, 539–549. [Google Scholar] [CrossRef] [Green Version]
- Bukhari, A.B.; Lewis, C.W.; Pearce, J.J.; Luong, D.; Chan, G.K.; Gamper, A.M. Inhibiting Wee1 and ATR Kinases Produces Tumor-Selective Synthetic Lethality and Suppresses Metastasis. J. Clin. Investig. 2019, 129, 1329–1344. [Google Scholar] [CrossRef] [Green Version]
- Sand, A.; Piacsek, M.; Donohoe, D.L.; Duffin, A.T.; Riddell, G.T.; Sun, C.; Tang, M.; Rovin, R.A.; Tjoe, J.A.; Yin, J. WEE1 Inhibitor, AZD1775, Overcomes Trastuzumab Resistance by Targeting Cancer Stem-like Properties in HER2-Positive Breast Cancer. Cancer Lett. 2020, 472, 119–131. [Google Scholar] [CrossRef]
- McGrail, D.J.; Lin, C.C.-J.; Dai, H.; Mo, W.; Li, Y.; Stephan, C.; Davies, P.; Lu, Z.; Mills, G.B.; Lee, J.-S.; et al. Defective Replication Stress Response Is Inherently Linked to the Cancer Stem Cell Phenotype. Cell Rep. 2018, 23, 2095–2106. [Google Scholar] [CrossRef] [Green Version]
- Manic, G.; Signore, M.; Sistigu, A.; Russo, G.; Corradi, F.; Siteni, S.; Musella, M.; Vitale, S.; Angelis, M.L.D.; Pallocca, M.; et al. CHK1-Targeted Therapy to Deplete DNA Replication-Stressed, P53-Deficient, Hyperdiploid Colorectal Cancer Stem Cells. Gut 2018, 67, 903. [Google Scholar] [CrossRef] [PubMed]
- Franco, S.D.; Parrino, B.; Gaggianesi, M.; Pantina, V.D.; Bianca, P.; Nicotra, A.; Mangiapane, L.R.; Iacono, M.L.; Ganduscio, G.; Veschi, V.; et al. CHK1 Inhibitor Sensitizes Resistant Colorectal Cancer Stem Cells to Nortopsentin. Iscience 2021, 24, 102664. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Pan, W.; Xing, Y.; Xiao, Y.; Chen, J.; Xu, Z. Recent Advances in DDR (DNA Damage Response) Inhibitors for Cancer Therapy. Eur. J. Med. Chem. 2022, 230, 114109. [Google Scholar] [CrossRef]
- Frosina, G.; Ravetti, J.L.; Corvò, R.; Fella, M.; Garrè, M.L.; Levrero, F.; Marcello, D.; Marubbi, D.; Morana, G.; Mussap, M.; et al. Faithful Animal Modelling of Human Glioma by Using Primary Initiating Cells and Its Implications for Radiosensitization Therapy. Sci. Rep. 2018, 8, 14191. [Google Scholar] [CrossRef] [Green Version]
- Durant, S.T.; Zheng, L.; Wang, Y.; Chen, K.; Zhang, L.; Zhang, T.; Yang, Z.; Riches, L.; Trinidad, A.G.; Fok, J.H.L.; et al. The Brain-Penetrant Clinical ATM Inhibitor AZD1390 Radiosensitizes and Improves Survival of Preclinical Brain Tumor Models. Sci. Adv. 2018, 4, eaat1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarrar, A.; Lotti, F.; DeVecchio, J.; Ferrandon, S.; Gantt, G.; Mace, A.; Karagkounis, G.; Orloff, M.; Venere, M.; Hitomi, M.; et al. Poly(ADP-Ribose) Polymerase Inhibition Sensitizes Colorectal Cancer-Initiating Cells to Chemotherapy. Stem Cells 2019, 37, 42–53. [Google Scholar] [CrossRef] [Green Version]
- Quiñonero, F.; Cepero, A.; Urbano, D.; Muñoz-Gámez, J.A.; Martín-Guerrero, S.M.; Martín-Oliva, D.; Prados, J.; Melguizo, C.; Ortiz, R. Identification of PARP-1 in Cancer Stem Cells of Gastrointestinal Cancers: A Preliminary Study. J. Biosci. 2021, 46, 6. [Google Scholar] [CrossRef]
- Cong, K.; Peng, M.; Kousholt, A.N.; Lee, W.T.C.; Lee, S.; Nayak, S.; Krais, J.; VanderVere-Carozza, P.S.; Pawelczak, K.S.; Calvo, J.; et al. Replication Gaps Are a Key Determinant of PARP Inhibitor Synthetic Lethality with BRCA Deficiency. Mol. Cell 2021, 81, 3128–3144. [Google Scholar] [CrossRef]
- Manic, G.; Musella, M.; Corradi, F.; Sistigu, A.; Vitale, S.; Rehim, S.S.A.; Mattiello, L.; Malacaria, E.; Galassi, C.; Signore, M.; et al. Control of Replication Stress and Mitosis in Colorectal Cancer Stem Cells through the Interplay of PARP1, MRE11 and RAD51. Cell Death Differ. 2021, 28, 2060–2082. [Google Scholar] [CrossRef]
- Kim, H.; Xu, H.; George, E.; Hallberg, D.; Kumar, S.; Jagannathan, V.; Medvedev, S.; Kinose, Y.; Devins, K.; Verma, P.; et al. Combining PARP with ATR Inhibition Overcomes PARP Inhibitor and Platinum Resistance in Ovarian Cancer Models. Nat. Commun. 2020, 11, 3726. [Google Scholar] [CrossRef]
- Fang, Y.; McGrail, D.J.; Sun, C.; Labrie, M.; Chen, X.; Zhang, D.; Ju, Z.; Vellano, C.P.; Lu, Y.; Li, Y.; et al. Sequential Therapy with PARP and WEE1 Inhibitors Minimizes Toxicity While Maintaining Efficacy. Cancer Cell 2019, 35, 851–867.e7. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Bernier, C.; Kaiser, B.; Fournier, S.; Li, L.; Desjardins, J.; Skeldon, A.; Rimkunas, V.; Veloso, A.; Young, J.T.F.; et al. Guiding ATR and PARP Inhibitor Combinationswith Chemogenomic Screens. Cell Rep. 2022, 40, 111081. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-Gómez, G.; Díaz-Chávez, J.; Chávez-Blanco, A.; Gonzalez-Fiero, A.; Jiménez-Salazar, J.E.; Damián-Matsumura, P.; Gómez-Quiroz, L.E.; Dueñas-González, A. Nicotinamide Sensitizes Human Breast Cancer Cells to the Cytotoxic Effects of Radiation and Cisplatin. Oncol. Rep. 2015, 33, 721–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikas, I.P.; Paschou, S.A.; Ryu, H.S. The Role of Nicotinamide in Cancer Chemoprevention and Therapy. Biomolecules 2020, 10, 477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaanders, J.H.; Bussink, J.; Kogel, A.J. van der ARCON: A Novel Biology-Based Approach in Radiotherapy. Lancet Oncol. 2002, 3, 728–737. [Google Scholar] [CrossRef]
- Wang, W.; Yang, C.; Wang, T.; Deng, H. Complex Roles of Nicotinamide N-Methyltransferase in Cancer Progression. Cell Death Dis. 2022, 13, 267. [Google Scholar] [CrossRef]
- Pozzi, V.; Salvolini, E.; Lucarini, G.; Salvucci, A.; Campagna, R.; Rubini, C.; Sartini, D.; Emanuelli, M. Cancer Stem Cell Enrichment Is Associated with Enhancement of Nicotinamide N-methyltransferase Expression. Iubmb. Life 2020, 72, 1415–1425. [Google Scholar] [CrossRef]
- Jung, J.; Kim, L.J.Y.; Wang, X.; Wu, Q.; Sanvoranart, T.; Hubert, C.G.; Prager, B.C.; Wallace, L.C.; Jin, X.; Mack, S.C.; et al. Nicotinamide Metabolism Regulates Glioblastoma Stem Cell Maintenance. JCI Insight 2017, 2, e90019. [Google Scholar] [CrossRef] [Green Version]
- Parsons, R.B.; Facey, P.D. Nicotinamide N-Methyltransferase: An Emerging Protagonist in Cancer Macro(r)Evolution. Biomolecules 2021, 11, 1418. [Google Scholar] [CrossRef]
- Li, X.-Y.; Pi, Y.-N.; Chen, Y.; Zhu, Q.; Xia, B.-R. Nicotinamide N-Methyltransferase: A Promising Biomarker and Target for Human Cancer Therapy. Front. Oncol. 2022, 12, 894744. [Google Scholar] [CrossRef]
- Liu, Y.; Burness, M.L.; Martin-Trevino, R.; Guy, J.; Bai, S.; Harouaka, R.; Brooks, M.D.; Shang, L.; Fox, A.; Luther, T.K.; et al. RAD51 Mediates Resistance of Cancer Stem Cells to PARP Inhibition in Triple-Negative Breast Cancer. Clin. Cancer Res. 2017, 23, 514–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruíz, G.; Valencia-González, H.A.; León-Galicia, I.; García-Villa, E.; García-Carrancá, A.; Gariglio, P. Inhibition of RAD51 by SiRNA and Resveratrol Sensitizes Cancer Stem Cells Derived from HeLa Cell Cultures to Apoptosis. Stem Cells Int. 2018, 2018, 2493869. [Google Scholar] [CrossRef] [PubMed]
- Mattiello, L.; Rehim, S.S.A.; Musella, M.; Sistigu, A.; Guarracino, A.; Vitale, S.; Corradi, F.; Galassi, C.; Sperati, F.; Manic, G.; et al. The Targeting of MRE11 or RAD51 Sensitizes Colorectal Cancer Stem Cells to CHK1 Inhibition. Cancers 2021, 13, 1957. [Google Scholar] [CrossRef]
- Turdo, A.; Gaggianesi, M.; Franco, S.D.; Veschi, V.; D’Accardo, C.; Porcelli, G.; Iacono, M.L.; Pillitteri, I.; Verona, F.; Militello, G.; et al. Effective Targeting of Breast Cancer Stem Cells by Combined Inhibition of Sam68 and Rad51. Oncogene 2022, 41, 2196–2209. [Google Scholar] [CrossRef]
- Budke, B.; Lv, W.; Kozikowski, A.P.; Connell, P.P. Recent Developments Using Small Molecules to Target RAD51: How to Best Modulate RAD51 for Anticancer Therapy? Chemmedchem 2016, 11, 2468–2473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mason, J.M.; Logan, H.L.; Budke, B.; Wu, M.; Pawlowski, M.; Weichselbaum, R.R.; Kozikowski, A.P.; Bishop, D.K.; Connell, P.P. The RAD51-Stimulatory Compound RS-1 Can Exploit the RAD51 Overexpression That Exists in Cancer Cells and Tumors. Cancer Res. 2014, 74, 3546–3555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, D.E.; Francis-Newton, N.J.; Marsh, M.E.; Coyne, A.G.; Fischer, G.; Moschetti, T.; Bayly, A.R.; Sharpe, T.D.; Haas, K.T.; Barber, L.; et al. A Small-Molecule Inhibitor of the BRCA2-RAD51 Interaction Modulates RAD51 Assembly and Potentiates DNA Damage-Induced Cell Death. Cell Chem. Biol. 2021, 28, 835–847.e5. [Google Scholar] [CrossRef]
- Timme, C.R.; Rath, B.H.; O’Neill, J.W.; Camphausen, K.; Tofilon, P.J. The DNA-PK Inhibitor VX-984 Enhances the Radiosensitivity of Glioblastoma Cells Grown in vitro and as Orthotopic Xenografts. Mol. Cancer Ther. 2018, 17, 1207–1216. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, F.W.; Finlay, M.R.V.; Ting, A.K.T.; Beattie, D.; Lamont, G.M.; Fallan, C.; Wrigley, G.L.; Schimpl, M.; Howard, M.R.; Williamson, B.; et al. The Discovery of 7-Methyl-2-[(7-Methyl[1,2,4]Triazolo[1,5-a]Pyridin-6-Yl)Amino]-9-(Tetrahydro-2H-pyran-4-Yl)-7,9-Dihydro-8H-purin-8-One (AZD7648), a Potent and Selective DNA-Dependent Protein Kinase (DNA-PK) Inhibitor. J. Med. Chem. 2020, 63, 3461–3471. [Google Scholar] [CrossRef] [Green Version]
- Zenke, F.T.; Zimmermann, A.; Sirrenberg, C.; Dahmen, H.; Kirkin, V.; Pehl, U.; Grombacher, T.; Wilm, C.; Fuchss, T.; Amendt, C.; et al. Pharmacologic Inhibitor of DNA-PK, M3814, Potentiates Radiotherapy and Regresses Human Tumors in Mouse Models. Mol. Cancer 2020, 19, 1091–1101. [Google Scholar] [CrossRef] [Green Version]
- Ciszewski, W.M.; Tavecchio, M.; Dastych, J.; Curtin, N.J. DNA-PK Inhibition by NU7441 Sensitizes Breast Cancer Cells to Ionizing Radiation and Doxorubicin. Breast Cancer Res. Treat. 2014, 143, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, H.; Liu, T.; Huang, M.; Butter, P.-P.; Li, C.; Zhang, L.; Kao, G.D.; Gong, Y.; Maity, A.; et al. Temporal DNA-PK Activation Drives Genomic Instability and Therapy Resistance in Glioma Stem Cells. JCI Insight 2018, 3, e98096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, X.; Huang, Z.; Zhai, K.; Huang, Q.; Tao, W.; Kim, L.; Wu, Q.; Almasan, A.; Yu, J.S.; Li, X.; et al. Inhibiting DNA-PK Induces Glioma Stem Cell Differentiation and Sensitizes Glioblastoma to Radiation in Mice. Sci. Transl. Med. 2021, 13, eabc7275. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.-C.; Wang, I.-H. Enhanced Delivery of Etoposide across the Blood–Brain Barrier to Restrain Brain Tumor Growth Using Melanotransferrin Antibody- and Tamoxifen-Conjugated Solid Lipid Nanoparticles. J. Drug Target 2016, 24, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Bradley, R.K.; Anczuków, O. RNA Splicing Dysregulation and the Hallmarks of Cancer. Nat. Rev. Cancer 2023, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Read, A.; Natrajan, R. Splicing Dysregulation as a Driver of Breast Cancer. Endocr.-Relat. Cancer 2018, 25, R467–R478. [Google Scholar] [CrossRef] [Green Version]
- Kechavarzi, B.; Janga, S.C. Dissecting the Expression Landscape of RNA-Binding Proteins in Human Cancers. Genome Biol. 2014, 15, R14. [Google Scholar] [CrossRef] [Green Version]
- Wan, L.; Deng, M.; Zhang, H. SR Splicing Factors Promote Cancer via Multiple Regulatory Mechanisms. Genes 2022, 13, 1659. [Google Scholar] [CrossRef]
- Li, H.; Liu, J.; Shen, S.; Dai, D.; Cheng, S.; Dong, X.; Sun, L.; Guo, X. Pan-cancer Analysis of Alternative Splicing Regulator Heterogeneous Nuclear Ribonucleoproteins (HnRNPs) Family and Their Prognostic Potential. J. Cell Mol. Med. 2020, 24, 11111–11119. [Google Scholar] [CrossRef]
- Crews, L.A.; Balaian, L.; Delos Santos, N.P.; Leu, H.S.; Court, A.C.; Lazzari, E.; Sadarangani, A.; Zipeto, M.A.; La Clair, J.J.; Villa, R.; et al. RNA Splicing Modulation Selectively Impairs Leukemia Stem Cell Maintenance in Secondary Human AML. Cell Stem Cell 2016, 19, 599–612. [Google Scholar] [CrossRef] [Green Version]
- Smeets, M.F.; Tan, S.Y.; Xu, J.J.; Anande, G.; Unnikrishnan, A.; Chalk, A.M.; Taylor, S.R.; Pimanda, J.E.; Wall, M.; Purton, L.E.; et al. Srsf2 P95H Initiates Myeloid Bias and Myelodysplastic/Myeloproliferative Syndrome from Hemopoietic Stem Cells. Blood 2018, 132, 608–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; et al. Frequent Pathway Mutations of Splicing Machinery in Myelodysplasia. Nature 2011, 478, 64–69. [Google Scholar] [CrossRef]
- Guo, L.; Ke, H.; Zhang, H.; Zou, L.; Yang, Q.; Lu, X.; Zhao, L.; Jiao, B. TDP43 Promotes Stemness of Breast Cancer Stem Cells through CD44 Variant Splicing Isoforms. Cell Death Dis. 2022, 13, 428. [Google Scholar] [CrossRef] [PubMed]
- Watermann, D.O.; Tang, Y.; zur Hausen, A.; Jäger, M.; Stamm, S.; Stickeler, E. Splicing Factor Tra2-Β1 Is Specifically Induced in Breast Cancer and Regulates Alternative Splicing of the CD44 Gene. Cancer Res. 2006, 66, 4774–4780. [Google Scholar] [CrossRef] [Green Version]
- Best, A.; Dagliesh, C.; Ehrmann, I.; Kheirollahi-Kouhestani, M.; Tyson-Capper, A.; Elliott, D.J. Expression of Tra2β in Cancer Cells as a Potential Contributory Factor to Neoplasia and Metastasis. Int. J. Cell Biol. 2013, 2013, 843781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghigna, C.; Giordano, S.; Shen, H.; Benvenuto, F.; Castiglioni, F.; Comoglio, P.M.; Green, M.R.; Riva, S.; Biamonti, G. Cell Motility Is Controlled by SF2/ASF through Alternative Splicing of the Ron Protooncogene. Mol. Cell 2005, 20, 881–890. [Google Scholar] [CrossRef]
- Zhang, Y.; Fang, Y.; Ma, L.; Xu, J.; Lv, C.; Deng, L.; Zhu, G. LINC00857 Regulated by ZNF460 Enhances the Expression of CLDN12 by Sponging MiR-150-5p and Recruiting SRSF1 for Alternative Splicing to Promote Epithelial-Mesenchymal Transformation of Pancreatic Adenocarcinoma Cells. Rna Biol. 2022, 19, 548–559. [Google Scholar] [CrossRef]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR Substrate Analysis Reveals Extensive Protein Networks Responsive to DNA Damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Leva, V.; Giuliano, S.; Bardoni, A.; Camerini, S.; Crescenzi, M.; Lisa, A.; Biamonti, G.; Montecucco, A. Phosphorylation of SRSF1 Is Modulated by Replicational Stress. Nucleic Acids Res. 2012, 40, 1106–1117. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.; Zheng, X.; Chen, H.; Guo, Y.; Jiang, H.; He, X.; Zhu, X.; Zheng, Y. Splicing Function of Mitotic Regulators Links R-Loop–Mediated DNA Damage to Tumor Cell Killing. J. Cell Biol. 2015, 209, 235–246. [Google Scholar] [CrossRef] [Green Version]
- Matos, D.A.; Zhang, J.-M.; Ouyang, J.; Nguyen, H.D.; Genois, M.-M.; Zou, L. ATR Protects the Genome against R Loops through a MUS81-Triggered Feedback Loop. Mol. Cell 2020, 77, 514–527.e4. [Google Scholar] [CrossRef] [PubMed]
- Adamson, B.; Smogorzewska, A.; Sigoillot, F.D.; King, R.W.; Elledge, S.J. A Genome-Wide Homologous Recombination Screen Identifies the RNA-Binding Protein RBMX as a Component of the DNA Damage Response. Nat. Cell Biol. 2012, 14, 318–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lappin, K.M.; Barros, E.M.; Jhujh, S.S.; Irwin, G.W.; McMillan, H.; Liberante, F.G.; Latimer, C.; LaBonte, M.J.; Mills, K.I.; Harkin, D.P.; et al. Cancer-associated sf3b1 mutations confer a brca-like cellular phenotype and synthetic lethality to parp inhibitors. Cancer Res. 2022, 82, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, J.-Y.; Huang, Y.-J.; Gu, Y.; Qiu, J.; Qian, H.; Shao, C.; Zhang, X.; Hu, J.; Li, H.; et al. The Augmented R-Loop Is a Unifying Mechanism for Myelodysplastic Syndromes Induced by High-Risk Splicing Factor Mutations. Mol. Cell 2018, 69, 412–425.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.; Ahmed, D.; Dolatshad, H.; Tatwavedi, D.; Schulze, U.; Sanchi, A.; Ryley, S.; Dhir, A.; Carpenter, L.; Watt, S.M.; et al. SF3B1 Mutations Induce R-Loop Accumulation and DNA Damage in MDS and Leukemia Cells with Therapeutic Implications. Leukemia 2020, 34, 2525–2530. [Google Scholar] [CrossRef] [PubMed]
- Alors-Perez, E.; Blázquez-Encinas, R.; Alcalá, S.; Viyuela-García, C.; Pedraza-Arevalo, S.; Herrero-Aguayo, V.; Jiménez-Vacas, J.M.; Mafficini, A.; Sánchez-Frías, M.E.; Cano, M.T.; et al. Dysregulated Splicing Factor SF3B1 Unveils a Dual Therapeutic Vulnerability to Target Pancreatic Cancer Cells and Cancer Stem Cells with an Anti-Splicing Drug. J. Exp. Clin. Cancer Res. 2021, 40, 382. [Google Scholar] [CrossRef]
- Best, A.; James, K.; Dalgliesh, C.; Hong, E.; Kheirolahi-Kouhestani, M.; Curk, T.; Xu, Y.; Danilenko, M.; Hussain, R.; Keavney, B.; et al. Human Tra2 Proteins Jointly Control a CHEK1 Splicing Switch among Alternative and Constitutive Target Exons. Nat. Commun. 2014, 5, 4760. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Zhan, P.; Feng, S.; Ji, H.; Tian, W.; Wang, M.; Cheng, C.; Song, B. SRSF6 Regulates Alternative Splicing of Genes Involved in DNA Damage Response and DNA Repair in HeLa Cells. Oncol. Rep. 2020, 44, 1851–1862. [Google Scholar] [CrossRef]
- Ye, Y.; Yu, F.; Li, Z.; Xie, Y.; Yu, X. RNA Binding Protein Serine/Arginine Splicing Factor 1 Promotes the Proliferation, Migration and Invasion of Hepatocellular Carcinoma by Interacting with RecQ Protein-like 4 MRNA. Bioengineered 2021, 12, 6144–6154. [Google Scholar] [CrossRef]
- Luong, T.T.; Bernstein, K.A. Role and Regulation of the RECQL4 Family during Genomic Integrity Maintenance. Genes 2021, 12, 1919. [Google Scholar] [CrossRef]
- Król, S.K.; Kaczmarczyk, A.; Wojnicki, K.; Wojtas, B.; Gielniewski, B.; Grajkowska, W.; Kotulska, K.; Szczylik, C.; Czepko, R.; Banach, M.; et al. Aberrantly Expressed RECQL4 Helicase Supports Proliferation and Drug Resistance of Human Glioma Cells and Glioma Stem Cells. Cancers 2020, 12, 2919. [Google Scholar] [CrossRef]
- Chiang, K.; Zielinska, A.E.; Shaaban, A.M.; Sanchez-Bailon, M.P.; Jarrold, J.; Clarke, T.L.; Zhang, J.; Francis, A.; Jones, L.J.; Smith, S.; et al. PRMT5 Is a Critical Regulator of Breast Cancer Stem Cell Function via Histone Methylation and FOXP1 Expression. Cell Rep. 2017, 21, 3498–3513. [Google Scholar] [CrossRef] [Green Version]
- Hamard, P.-J.; Santiago, G.E.; Liu, F.; Karl, D.L.; Martinez, C.; Man, N.; Mookhtiar, A.K.; Duffort, S.; Greenblatt, S.; Verdun, R.E.; et al. PRMT5 Regulates DNA Repair by Controlling the Alternative Splicing of Histone-Modifying Enzymes. Cell Rep. 2018, 24, 2643–2657. [Google Scholar] [CrossRef] [Green Version]
- Sachamitr, P.; Ho, J.C.; Ciamponi, F.E.; Ba-Alawi, W.; Coutinho, F.J.; Guilhamon, P.; Kushida, M.M.; Cavalli, F.M.G.; Lee, L.; Rastegar, N.; et al. PRMT5 Inhibition Disrupts Splicing and Stemness in Glioblastoma. Nat. Commun. 2021, 12, 979. [Google Scholar] [CrossRef]
- Bezzi, M.; Teo, S.X.; Muller, J.; Mok, W.C.; Sahu, S.K.; Vardy, L.A.; Bonday, Z.Q.; Guccione, E. Regulation of Constitutive and Alternative Splicing by PRMT5 Reveals a Role for Mdm4 Pre-MRNA in Sensing Defects in the Spliceosomal Machinery. Genes Dev. 2013, 27, 1903–1916. [Google Scholar] [CrossRef] [Green Version]
- Thandapani, P.; O’Connor, T.R.; Bailey, T.L.; Richard, S. Defining the RGG/RG Motif. Mol. Cell 2013, 50, 613–623. [Google Scholar] [CrossRef] [Green Version]
- Lorton, B.M.; Shechter, D. Cellular Consequences of Arginine Methylation. Cell Mol. Life Sci. 2019, 76, 2933–2956. [Google Scholar] [CrossRef]
- Friesen, W.J.; Massenet, S.; Paushkin, S.; Wyce, A.; Dreyfuss, G. SMN, the Product of the Spinal Muscular Atrophy Gene, Binds Preferentially to Dimethylarginine-Containing Protein Targets. Mol. Cell 2001, 7, 1111–1117. [Google Scholar] [CrossRef]
- Radzisheuskaya, A.; Shliaha, P.V.; Grinev, V.; Lorenzini, E.; Kovalchuk, S.; Shlyueva, D.; Gorshkov, V.; Hendrickson, R.C.; Jensen, O.N.; Helin, K. PRMT5 Methylome Profiling Uncovers a Direct Link to Splicing Regulation in Acute Myeloid Leukemia. Nat. Struct. Mol. Biol. 2019, 26, 999–1012. [Google Scholar] [CrossRef]
- Maron, M.I.; Casill, A.D.; Gupta, V.; Roth, J.S.; Sidoli, S.; Query, C.C.; Gamble, M.J.; Shechter, D. Type I and II PRMTs Inversely Regulate Post-Transcriptional Intron Detention through Sm and CHTOP Methylation. Elife 2022, 11, e72867. [Google Scholar] [CrossRef]
- Clarke, T.L.; Sanchez-Bailon, M.P.; Chiang, K.; Reynolds, J.J.; Herrero-Ruiz, J.; Bandeiras, T.M.; Matias, P.M.; Maslen, S.L.; Skehel, J.M.; Stewart, G.S.; et al. PRMT5-Dependent Methylation of the TIP60 Coactivator RUVBL1 Is a Key Regulator of Homologous Recombination. Mol. Cell 2017, 65, 900–916. [Google Scholar] [CrossRef]
- Jarrold, J.; Davies, C.C. PRMTs and Arginine Methylation: Cancer’s Best-Kept Secret? Trends Mol. Med. 2019, 25, 993–1009. [Google Scholar] [CrossRef]
- Fong, J.Y.; Pignata, L.; Goy, P.-A.; Kawabata, K.C.; Lee, S.C.-W.; Koh, C.M.; Musiani, D.; Massignani, E.; Kotini, A.G.; Penson, A.; et al. Therapeutic Targeting of RNA Splicing Catalysis through Inhibition of Protein Arginine Methylation. Cancer Cell 2019, 36, 194–209.e9. [Google Scholar] [CrossRef]
- Dewaele, M.; Tabaglio, T.; Willekens, K.; Bezzi, M.; Teo, S.X.; Low, D.H.P.; Koh, C.M.; Rambow, F.; Fiers, M.; Rogiers, A.; et al. Antisense Oligonucleotide–Mediated MDM4 Exon 6 Skipping Impairs Tumor Growth. J. Clin. Investig. 2016, 126, 68–84. [Google Scholar] [CrossRef]
- Olivieri, M.; Cho, T.; Álvarez-Quilón, A.; Li, K.; Schellenberg, M.J.; Zimmermann, M.; Hustedt, N.; Rossi, S.E.; Adam, S.; Melo, H.; et al. A Genetic Map of the Response to DNA Damage in Human Cells. Cell 2020, 182, 481–496.e21. [Google Scholar] [CrossRef]
- Pollard, S.M.; Yoshikawa, K.; Clarke, I.D.; Danovi, D.; Stricker, S.; Russell, R.; Bayani, J.; Head, R.; Lee, M.; Bernstein, M.; et al. Glioma Stem Cell Lines Expanded in Adherent Culture Have Tumor-Specific Phenotypes and Are Suitable for Chemical and Genetic Screens. Cell Stem Cell 2009, 4, 568–580. [Google Scholar] [CrossRef] [Green Version]
- MacLeod, G.; Bozek, D.A.; Rajakulendran, N.; Monteiro, V.; Ahmadi, M.; Steinhart, Z.; Kushida, M.M.; Yu, H.; Coutinho, F.J.; Cavalli, F.M.G.; et al. Genome-Wide CRISPR-Cas9 Screens Expose Genetic Vulnerabilities and Mechanisms of Temozolomide Sensitivity in Glioblastoma Stem Cells. Cell Rep. 2019, 27, 971–986.e9. [Google Scholar] [CrossRef] [Green Version]
- Rogers, Z.N.; McFarland, C.D.; Winters, I.P.; Naranjo, S.; Chuang, C.-H.; Petrov, D.; Winslow, M.M. A Quantitative and Multiplexed Approach to Uncover the Fitness Landscape of Tumor Suppression in vivo. Nat. Methods 2017, 14, 737–742. [Google Scholar] [CrossRef] [Green Version]
- Lewis, C.W.; Bukhari, A.B.; Xiao, E.J.; Choi, W.-S.; Smith, J.D.; Homola, E.; Mackey, J.R.; Campbell, S.D.; Gamper, A.M.; Chan, G.K. Upregulation of Myt1 Promotes Acquired Resistance of Cancer Cells to Wee1 Inhibition. Cancer Res. 2019, 79, 5971–5985. [Google Scholar] [CrossRef] [Green Version]
- Willms, R.J.; Zeng, J.; Campbell, S.D. Myt1 Kinase Couples Mitotic Cell Cycle Exit with Differentiation in Drosophila. Cell Rep. 2020, 33, 108400. [Google Scholar] [CrossRef]
- Lee, J.; Taylor, C.A.; Barnes, K.M.; Shen, A.; Stewart, E.V.; Chen, A.; Xiang, Y.K.; Bao, Z.; Shen, K. A Myt1 Family Transcription Factor Defines Neuronal Fate by Repressing Non-Neuronal Genes. Elife 2019, 8, e46703. [Google Scholar] [CrossRef]
- Lloyd, R.L.; Urban, V.; Muñoz-Martínez, F.; Ayestaran, I.; Thomas, J.C.; de Renty, C.; O’Connor, M.J.; Forment, J.V.; Galanty, Y.; Jackson, S.P. Loss of Cyclin C or CDK8 Provides ATR Inhibitor Resistance by Suppressing Transcription-Associated Replication Stress. Nucleic Acids Res. 2021, 49, gkab628. [Google Scholar] [CrossRef]
- Feng, W.; Simpson, D.A.; Carvajal-Garcia, J.; Price, B.A.; Kumar, R.J.; Mose, L.E.; Wood, R.D.; Rashid, N.; Purvis, J.E.; Parker, J.S.; et al. Genetic Determinants of Cellular Addiction to DNA Polymerase Theta. Nat. Commun. 2019, 10, 4286. [Google Scholar] [CrossRef] [Green Version]
- Dai, C.-H.; Chen, P.; Li, J.; Lan, T.; Chen, Y.-C.; Qian, H.; Chen, K.; Li, M.-Y. Co-Inhibition of Pol θ and HR Genes Efficiently Synergize with Cisplatin to Suppress Cisplatin-Resistant Lung Cancer Cells Survival. Oncotarget 2016, 7, 65157–65170. [Google Scholar] [CrossRef] [Green Version]
- Rao, X.; Xing, B.; Wu, Z.; Bin, Y.; Chen, Y.; Xu, Y.; Zhou, D.; Zhou, X.; Wu, C.; Ye, W.; et al. Targeting Polymerase θ Impairs Tumorigenesis and Enhances Radiosensitivity in Lung Adenocarcinoma. Cancer Sci. 2023. [Google Scholar] [CrossRef]
- Zhou, J.; Gelot, C.; Pantelidou, C.; Li, A.; Yücel, H.; Davis, R.E.; Färkkilä, A.; Kochupurakkal, B.; Syed, A.; Shapiro, G.I.; et al. A First-in-Class Polymerase Theta Inhibitor Selectively Targets Homologous-Recombination-Deficient Tumors. Nat. Cancer 2021, 2, 598–610. [Google Scholar] [CrossRef]
- Bennett, C.F.; Krainer, A.R.; Cleveland, D.W. Antisense Oligonucleotide Therapies for Neurodegenerative Diseases. Annu. Rev. Neurosci. 2019, 42, 385–406. [Google Scholar] [CrossRef]
- Lu, S.X.; Neef, E.D.; Thomas, J.D.; Sabio, E.; Rousseau, B.; Gigoux, M.; Knorr, D.A.; Greenbaum, B.; Elhanati, Y.; Hogg, S.J.; et al. Pharmacologic Modulation of RNA Splicing Enhances Anti-Tumor Immunity. Cell 2021, 184, 4032–4047.e31. [Google Scholar] [CrossRef]
- Shen, R.; Liu, D.; Wang, X.; Guo, Z.; Sun, H.; Song, Y.; Wang, D. DNA Damage and Activation of CGAS/STING Pathway Induce Tumor Microenvironment Remodeling. Front. Cell Dev. Biol. 2022, 9, 828657. [Google Scholar] [CrossRef]
- Pantelidou, C.; Sonzogni, O.; Taveira, M.D.O.; Mehta, A.K.; Kothari, A.; Wang, D.; Visal, T.; Li, M.K.; Pinto, J.; Castrillon, J.A.; et al. PARP Inhibitor Efficacy Depends on CD8+ T-Cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discov. 2019, 9, 722–737. [Google Scholar] [CrossRef] [Green Version]
- Yao, L.-C.; Aryee, K.-E.; Cheng, M.; Kaur, P.; Keck, J.G.; Brehm, M.A. Target Identification and Validation in Drug Discovery, Methods and Protocols. Methods Mol. Biol. 2019, 1953, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Lyu, Y.; Yang, Y.-G.; Hu, Z. Humanized Rodent Models for Cancer Research. Front. Oncol. 2020, 10, 1696. [Google Scholar] [CrossRef] [PubMed]
- Fiorini, E.; Veghini, L.; Corbo, V. Modeling Cell Communication in Cancer With Organoids: Making the Complex Simple. Front. Cell Dev. Biol. 2020, 8, 166. [Google Scholar] [CrossRef] [Green Version]
- Kola, P.; Nagesh, P.K.B.; Roy, P.K.; Deepak, K.; Reis, R.L.; Kundu, S.C.; Mandal, M. Innovative Nanotheranostics: Smart Nanoparticles Based Approach to Overcome Breast Cancer Stem Cells Mediated Chemo- and Radioresistances. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2023, e1876. [Google Scholar] [CrossRef]
- Ning, S.-T.; Lee, S.-Y.; Wei, M.-F.; Peng, C.-L.; Lin, S.Y.-F.; Tsai, M.-H.; Lee, P.-C.; Shih, Y.-H.; Lin, C.-Y.; Luo, T.-Y.; et al. Targeting Colorectal Cancer Stem-Like Cells with Anti-CD133 Antibody-Conjugated SN-38 Nanoparticles. ACS Appl. Mater. Interfaces 2016, 8, 17793–17804. [Google Scholar] [CrossRef] [PubMed]
- El-Sahli, S.; Hua, K.; Sulaiman, A.; Chambers, J.; Li, L.; Farah, E.; McGarry, S.; Liu, D.; Zheng, P.; Lee, S.-H.; et al. A Triple-Drug Nanotherapy to Target Breast Cancer Cells, Cancer Stem Cells, and Tumor Vasculature. Cell Death Dis. 2021, 12, 8. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strategy | Enriched Cells (%) | Reference |
|---|---|---|
| EpCAM+CD24lo/−CD44+Lin− | 0.60 | [3] |
| CD24lo/−CD44+ | 1.32–5.00 | [22] |
| CD133+ | 2.00–5.90 | [22] |
| EpCAM+CD49f+ | Unknown | [21] |
| CD29+CD24hiLin− (murine) | 5.00–10.00 | [20] |
| ALDH1+ | 3.00–10.00 | [24] |
| CD24+/lowSca-1−CD49fhi (murine) | 20.00–30.00 | [13] |
| ABCG2hi | 0.2–5.00 | [23] |
| DDR Pathway | Mechanism | DNA Damaging Agent(s) | CSCs | References |
|---|---|---|---|---|
| HR | Increased RAD50 (MRN) expression | Mitomycin | BCSCs (tumoursphere) | [139] |
| HR RS response | ATR-CHK1 axis | IR, hydroxyurea | BCSCs (ALDH1+) | [140] |
| HR RS response | BMI1-RAD51 axis | Cisplatin | BCSCs (ALDH1+) | [141] |
| HR ICL repair MMR | Reduced intracellular [cisplatin]; increased gene expression (PMS2, ERCC1, MLH1, MSH2) | Cisplatin | Lung CSCs (chemoresistant cell line) | [143] |
| DSB repair | Enhanced CHK1 activation | Etoposide, docetaxel | Prostate CSCs (tumoursphere) | [144] |
| SSB repair DSB repair | Enhanced CHK1/2 activation | IR | GSCs (CD133+) | [145,146] |
| RS response | ATR-CHK1 axis; constitutive RS | IR | GSCs (CD133+) | [147] |
| HR | SPT6-mediated HR | IR | GSCs (CD133+) | [148] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gillespie, M.S.; Ward, C.M.; Davies, C.C. DNA Repair and Therapeutic Strategies in Cancer Stem Cells. Cancers 2023, 15, 1897. https://doi.org/10.3390/cancers15061897
Gillespie MS, Ward CM, Davies CC. DNA Repair and Therapeutic Strategies in Cancer Stem Cells. Cancers. 2023; 15(6):1897. https://doi.org/10.3390/cancers15061897
Chicago/Turabian StyleGillespie, Matthew S., Ciara M. Ward, and Clare C. Davies. 2023. "DNA Repair and Therapeutic Strategies in Cancer Stem Cells" Cancers 15, no. 6: 1897. https://doi.org/10.3390/cancers15061897