
Hepatitis Virus and Hepatocellular Carcinoma: Recent Advances


Abstract
:Simple Summary
Abstract
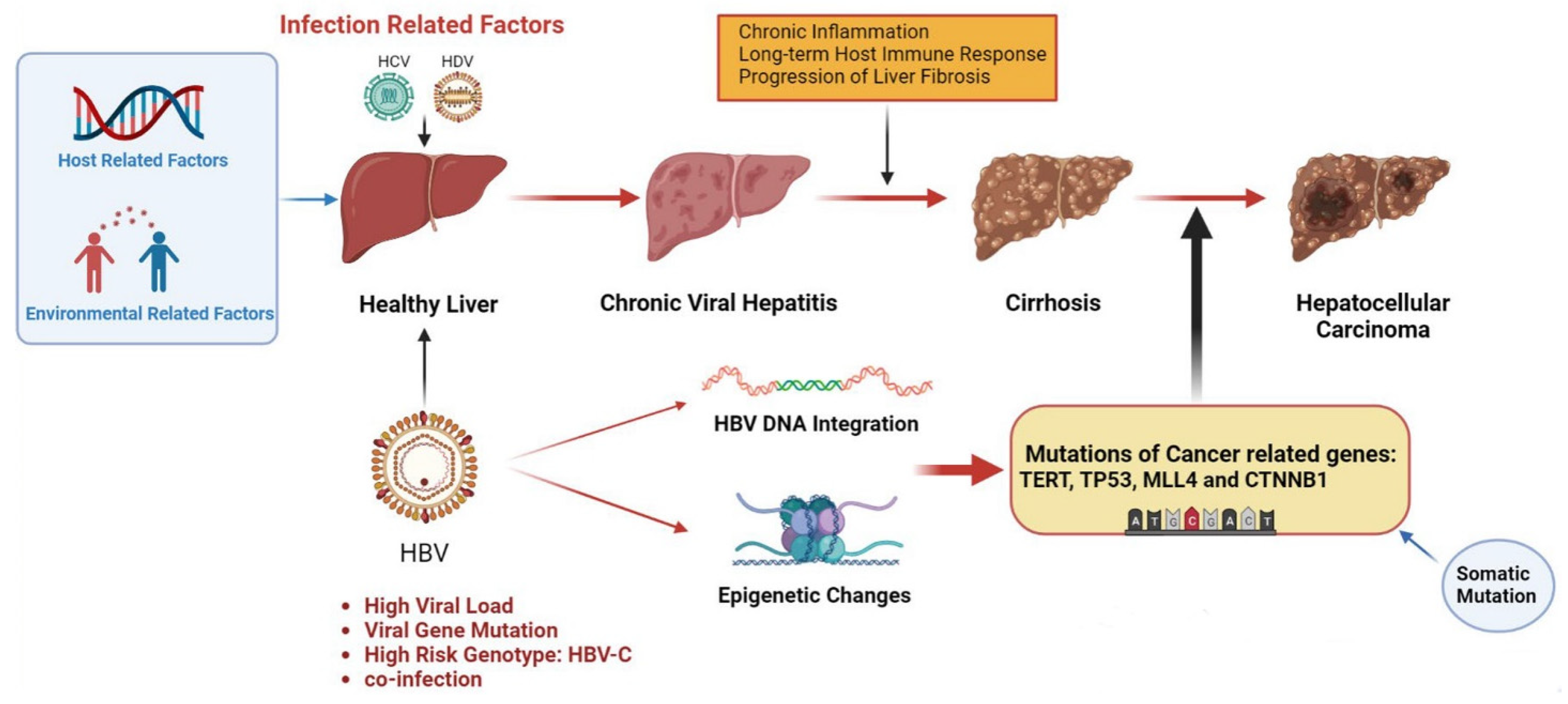
1. Introduction
2. Hepatitis-B-Virus-Induced Hepatocellular Carcinoma
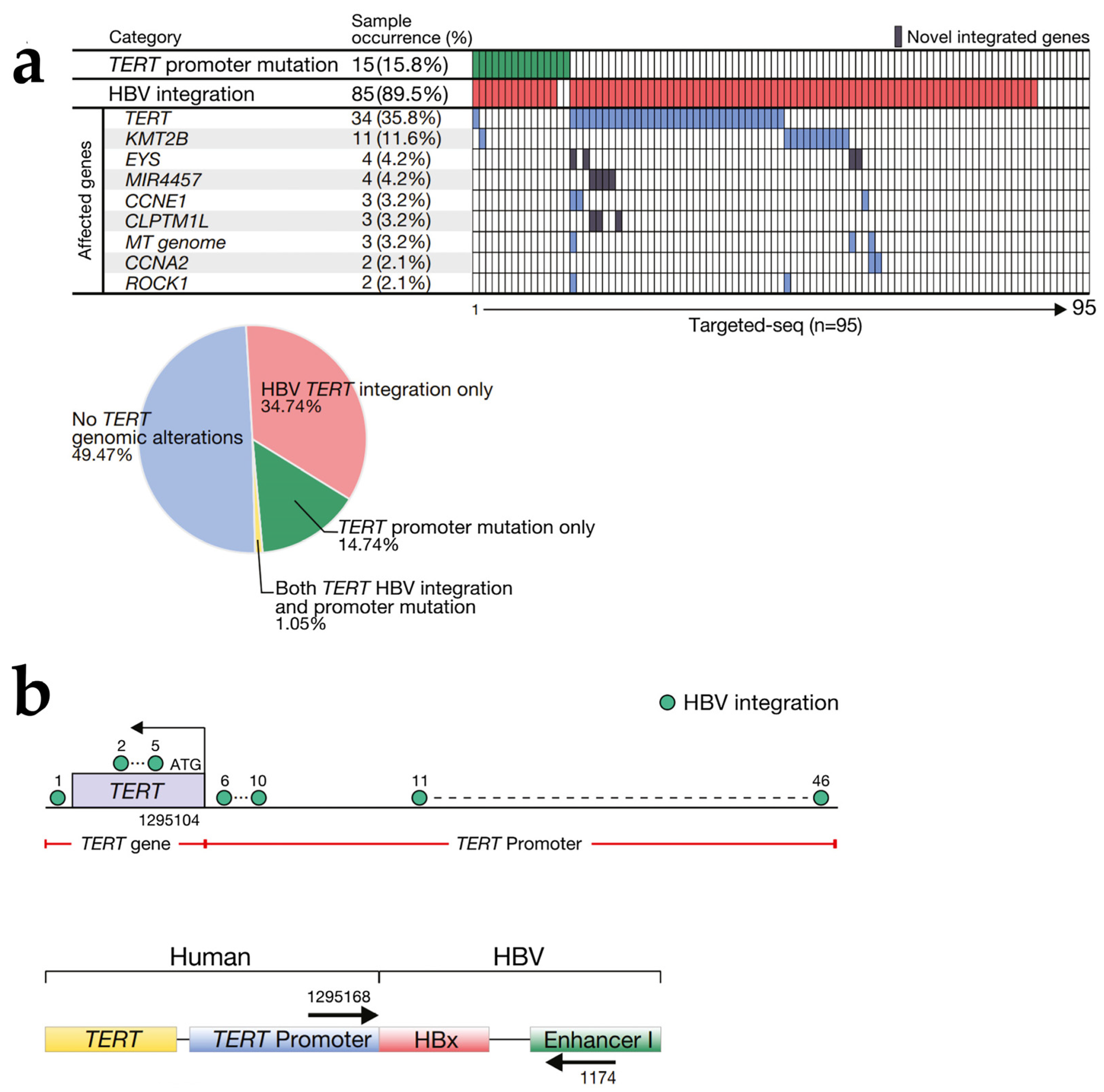
2.1. HBV DNA Integration
2.2. Epigenetic Changes
2.3. HBV Gene Mutation
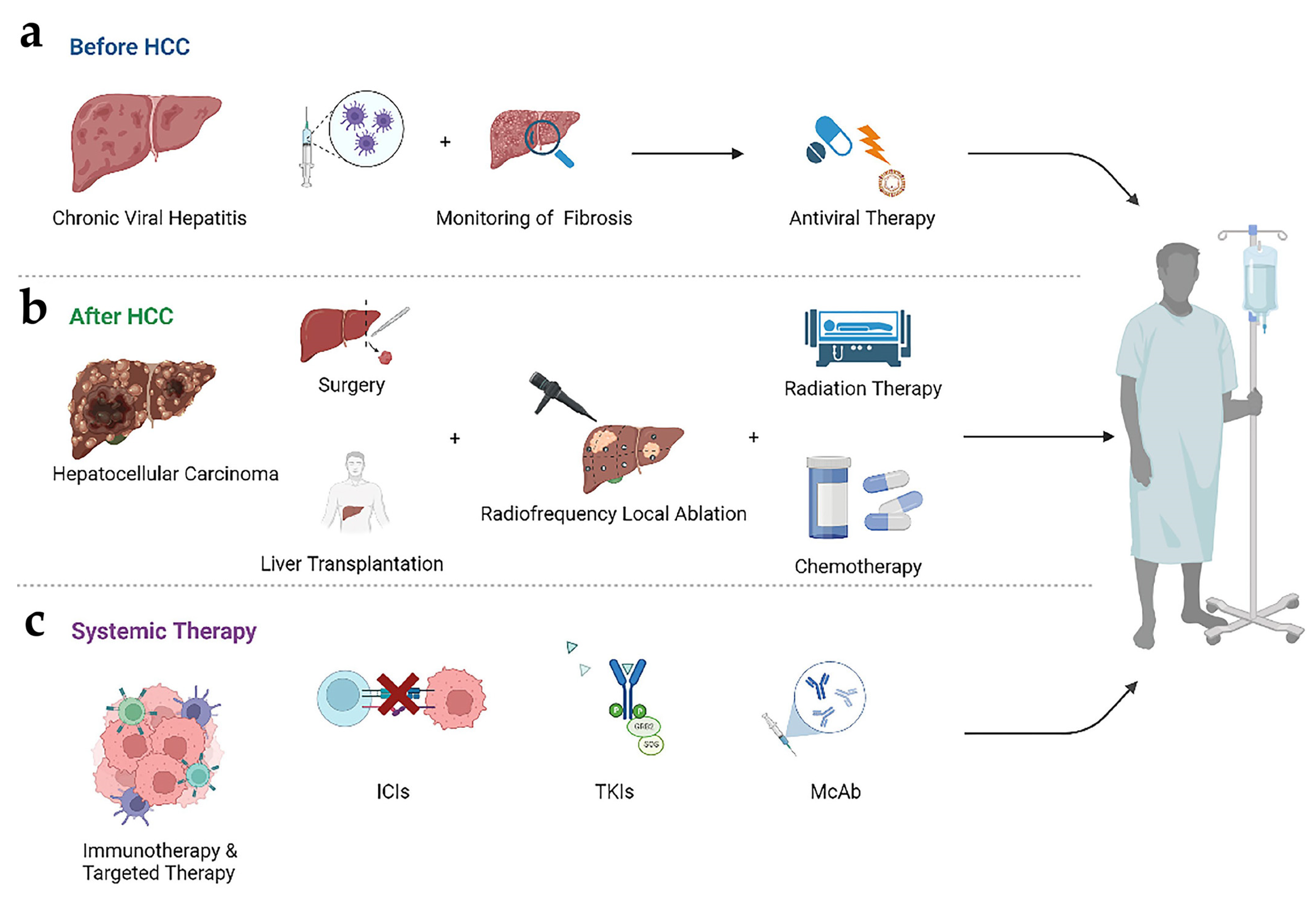
2.4. Progress in Treatment

3. Hepatitis-C-Virus-Induced Hepatocellular Carcinoma
4. Hepatitis-D-Virus-Induced Hepatocellular Carcinoma
5. Hepatitis E Virus and Hepatocellular Carcinoma
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef]
- Dhanasekaran, R.; Nault, J.-C.; Roberts, L.R.; Zucman-Rossi, J. Genomic Medicine and Implications for Hepatocellular Carcinoma Prevention and Therapy. Gastroenterology 2019, 156, 492–509. [Google Scholar] [CrossRef]
- Ninio, L.; Nissani, A.; Meirson, T.; Domovitz, T.; Genna, A.; Twafra, S.; Srikanth, K.D.; Dabour, R.; Avraham, E.; Davidovich, A.; et al. Hepatitis C Virus Enhances the Invasiveness of Hepatocellular Carcinoma via EGFR-Mediated Invadopodia Formation and Activation. Cells 2019, 8, 1395. [Google Scholar] [CrossRef] [Green Version]
- Sagnelli, E.; Macera, M.; Russo, A.; Coppola, N.; Sagnelli, C. Epidemiological and etiological variations in hepatocellular carcinoma. Infection 2020, 48, 7–17. [Google Scholar] [CrossRef]
- Yau, T.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Kang, Y.-K.; Hou, M.-M.; Numata, K.; Yeo, W.; Chopra, A.; Ikeda, M.; et al. Nivolumab in advanced hepatocellular carcinoma: Sorafenib-experienced Asian cohort analysis. J. Hepatol. 2019, 71, 543–552. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Zhang, L.; Liu, W.; Ding, Y.; Yin, J.; Ren, R.; Li, Q.; Chen, Y.; Shen, J.; Tan, X.; et al. Trends in cancer mortality in China from 2004 to 2018: A nationwide longitudinal study. Cancer Commun. 2021, 41, 1024–1036. [Google Scholar] [CrossRef]
- Koshiol, J.; Argirion, I.; Liu, Z.; Kim Lam, T.; O’Brien, T.R.; Yu, K.; McGlynn, K.A.; Petrick, J.L.; Pinto, L.; Chen, C.-J.; et al. Immunologic markers and risk of hepatocellular carcinoma in hepatitis B virus- and hepatitis C virus-infected individuals. Aliment. Pharmacol. Ther. 2021, 54, 833–842. [Google Scholar] [CrossRef]
- Kanwal, F.; Kramer, J.; Asch, S.M.; Chayanupatkul, M.; Cao, Y.; El-Serag, H.B. Risk of Hepatocellular Cancer in HCV Patients Treated with Direct-Acting Antiviral Agents. Gastroenterology 2017, 153, 996–1005.e1. [Google Scholar] [CrossRef] [Green Version]
- Yi, S.-W.; Choi, J.-S.; Yi, J.-J.; Lee, Y.-H.; Han, K.J. Risk factors for hepatocellular carcinoma by age, sex, and liver disorder status: A prospective cohort study in Korea. Cancer 2018, 124, 2748–2757. [Google Scholar] [CrossRef]
- Wait, S.; Kell, E.; Hamid, S.; Muljono, D.H.; Sollano, J.; Mohamed, R.; Shah, S.; Mamun Al, M.; Abbas, Z.; Johnston, J.; et al. Hepatitis B and hepatitis C in southeast and southern Asia: Challenges for governments. Lancet Gastroenterol. Hepatol. 2016, 1, 248–255. [Google Scholar] [CrossRef] [PubMed]
- EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [CrossRef] [PubMed] [Green Version]
- Marrero, J.A.; Kulik, L.M.; Sirlin, C.B.; Zhu, A.X.; Finn, R.S.; Abecassis, M.M.; Roberts, L.R.; Heimbach, J.K. Diagnosis, Staging, and Management of Hepatocellular Carcinoma: 2018 Practice Guidance by the American Association for the Study of Liver Diseases. Hepatology 2018, 68, 723–750. [Google Scholar] [CrossRef] [Green Version]
- Zheng, B.; Liu, X.-L.; Fan, R.; Bai, J.; Wen, H.; Du, L.-T.; Jiang, G.-Q.; Wang, C.-Y.; Fan, X.-T.; Ye, Y.-N.; et al. The Landscape of Cell-Free HBV Integrations and Mutations in Cirrhosis and Hepatocellular Carcinoma Patients. Clin. Cancer Res. 2021, 27, 3772–3783. [Google Scholar] [CrossRef]
- Jia, L.; Gao, Y.; He, Y.; Hooper, J.D.; Yang, P. HBV induced hepatocellular carcinoma and related potential immunotherapy. Pharmacol. Res. 2020, 159, 104992. [Google Scholar] [CrossRef]
- D’Souza, S.; Lau, K.C.; Coffin, C.S.; Patel, T.R. Molecular mechanisms of viral hepatitis induced hepatocellular carcinoma. World J. Gastroenterol. 2020, 26, 5759–5783. [Google Scholar] [CrossRef]
- Péneau, C.; Imbeaud, S.; La Bella, T.; Hirsch, T.Z.; Caruso, S.; Calderaro, J.; Paradis, V.; Blanc, J.-F.; Letouzé, E.; Nault, J.-C.; et al. Hepatitis B virus integrations promote local and distant oncogenic driver alterations in hepatocellular carcinoma. Gut 2022, 71, 616–626. [Google Scholar] [CrossRef]
- Ekpanyapong, S.; Reddy, K.R. Hepatitis C virus therapy in advanced liver disease: Outcomes and challenges. United Eur. Gastroenterol. J. 2019, 7, 642–650. [Google Scholar] [CrossRef] [Green Version]
- Khatun, M.; Ray, R.B. Mechanisms Underlying Hepatitis C Virus-Associated Hepatic Fibrosis. Cells 2019, 8, 1249. [Google Scholar] [CrossRef] [Green Version]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Koh, C.; Heller, T.; Glenn, J.S. Pathogenesis of and New Therapies for Hepatitis D. Gastroenterology 2019, 156, 461–476.e1. [Google Scholar] [CrossRef] [PubMed]
- Puigvehí, M.; Moctezuma-Velázquez, C.; Villanueva, A.; Llovet, J.M. The oncogenic role of hepatitis delta virus in hepatocellular carcinoma. JHEP Rep. 2019, 1, 120–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef]
- Zucman-Rossi, J.; Villanueva, A.; Nault, J.-C.; Llovet, J.M. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology 2015, 149, 1226–1239.e4. [Google Scholar] [CrossRef] [Green Version]
- Letouzé, E.; Shinde, J.; Renault, V.; Couchy, G.; Blanc, J.-F.; Tubacher, E.; Bayard, Q.; Bacq, D.; Meyer, V.; Semhoun, J.; et al. Mutational signatures reveal the dynamic interplay of risk factors and cellular processes during liver tumorigenesis. Nat. Commun. 2017, 8, 1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathkar, P.P.; Chen, X.; Sulovari, A.; Li, D. Characterization of Hepatitis B Virus Integrations Identified in Hepatocellular Carcinoma Genomes. Viruses 2021, 13, 245. [Google Scholar] [CrossRef]
- Debing, Y.; Moradpour, D.; Neyts, J.; Gouttenoire, J. Update on hepatitis E virology: Implications for clinical practice. J. Hepatol. 2016, 65, 200–212. [Google Scholar] [CrossRef] [Green Version]
- Walker, C.M.; Feng, Z.; Lemon, S.M. Reassessing immune control of hepatitis A virus. Curr. Opin. Virol. 2015, 11, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Shin, E.-C.; Sung, P.S.; Park, S.-H. Immune responses and immunopathology in acute and chronic viral hepatitis. Nat. Rev. Immunol. 2016, 16, 509–523. [Google Scholar] [CrossRef]
- Borentain, P.; Colson, P.; Bolon, E.; Gauchez, P.; Coso, D.; Gérolami, R. Hepatocellular carcinoma complicating hepatitis E virus-related cirrhosis. Hepatology 2018, 67, 446–448. [Google Scholar] [CrossRef]
- Kamar, N.; Selves, J.; Mansuy, J.-M.; Ouezzani, L.; Péron, J.-M.; Guitard, J.; Cointault, O.; Esposito, L.; Abravanel, F.; Danjoux, M.; et al. Hepatitis E virus and chronic hepatitis in organ-transplant recipients. N. Engl. J. Med. 2008, 358, 811–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gérolami, R.; Moal, V.; Colson, P. Chronic hepatitis E with cirrhosis in a kidney-transplant recipient. N. Engl. J. Med. 2008, 358, 859–860. [Google Scholar] [CrossRef] [Green Version]
- Hoan, N.X.; Tong, H.V.; Hecht, N.; Sy, B.T.; Marcinek, P.; Meyer, C.G.; Song, L.H.; Toan, N.L.; Kurreck, J.; Kremsner, P.G.; et al. Hepatitis E Virus Superinfection and Clinical Progression in Hepatitis B Patients. EBioMedicine 2015, 2, 2080–2086. [Google Scholar] [CrossRef] [Green Version]
- Tseng, T.-C.; Liu, C.-J.; Chang, C.T.; Su, T.-H.; Yang, W.-T.; Tsai, C.-H.; Chen, C.-L.; Yang, H.-C.; Liu, C.-H.; Chen, P.-J.; et al. HEV superinfection accelerates disease progression in patients with chronic HBV infection and increases mortality in those with cirrhosis. J. Hepatol. 2020, 72, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Salerno, D.; Chiodo, L.; Alfano, V.; Floriot, O.; Cottone, G.; Paturel, A.; Pallocca, M.; Plissonnier, M.-L.; Jeddari, S.; Belloni, L.; et al. Hepatitis B protein HBx binds the DLEU2 lncRNA to sustain cccDNA and host cancer-related gene transcription. Gut 2020, 69, 2016–2024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J. Hepatol. 2017, 67, 370–398. [CrossRef] [PubMed] [Green Version]
- Akinyemiju, T.; Abera, S.; Ahmed, M.; Alam, N.; Alemayohu, M.A.; Allen, C.; Al-Raddadi, R.; Alvis-Guzman, N.; Amoako, Y.; Artaman, A.; et al. The Burden of Primary Liver Cancer and Underlying Etiologies From 1990 to 2015 at the Global, Regional, and National Level: Results From the Global Burden of Disease Study 2015. JAMA Oncol. 2017, 3, 1683–1691. [Google Scholar] [CrossRef]
- Stanaway, J.D.; Flaxman, A.D.; Naghavi, M.; Fitzmaurice, C.; Vos, T.; Abubakar, I.; Abu-Raddad, L.J.; Assadi, R.; Bhala, N.; Cowie, B.; et al. The global burden of viral hepatitis from 1990 to 2013: Findings from the Global Burden of Disease Study 2013. Lancet 2016, 388, 1081–1088. [Google Scholar] [CrossRef] [Green Version]
- Chang, M.-H.; You, S.-L.; Chen, C.-J.; Liu, C.-J.; Lai, M.-W.; Wu, T.-C.; Wu, S.-F.; Lee, C.-M.; Yang, S.-S.; Chu, H.-C.; et al. Long-term Effects of Hepatitis B Immunization of Infants in Preventing Liver Cancer. Gastroenterology 2016, 151, 472–480.e1. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-L.; Yang, J.-Y.; Lin, S.-F.; Sun, C.-A.; Bai, C.-H.; You, S.-L.; Chen, C.-J.; Kao, J.-H.; Chen, P.-J.; Chen, D.-S. Slow decline of hepatitis B burden in general population: Results from a population-based survey and longitudinal follow-up study in Taiwan. J. Hepatol. 2015, 63, 354–363. [Google Scholar] [CrossRef]
- Tu, T.; Zhang, H.; Urban, S. Hepatitis B Virus DNA Integration: In Vitro Models for Investigating Viral Pathogenesis and Persistence. Viruses 2021, 13, 180. [Google Scholar] [CrossRef] [PubMed]
- Sze, K.M.-F.; Ho, D.W.-H.; Chiu, Y.-T.; Tsui, Y.-M.; Chan, L.-K.; Lee, J.M.-F.; Chok, K.S.-H.; Chan, A.C.-Y.; Tang, C.-N.; Tang, V.W.-L.; et al. Hepatitis B Virus-Telomerase Reverse Transcriptase Promoter Integration Harnesses Host ELF4, Resulting in Telomerase Reverse Transcriptase Gene Transcription in Hepatocellular Carcinoma. Hepatology 2021, 73, 23–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raimondo, G.; Locarnini, S.; Pollicino, T.; Levrero, M.; Zoulim, F.; Lok, A.S. Update of the statements on biology and clinical impact of occult hepatitis B virus infection. J. Hepatol. 2019, 71, 397–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diogo Dias, J.; Sarica, N.; Neuveut, C. Early Steps of Hepatitis B Life Cycle: From Capsid Nuclear Import to cccDNA Formation. Viruses 2021, 13, 757. [Google Scholar] [CrossRef] [PubMed]
- Ligat, G.; Schuster, C.; Baumert, T.F. Hepatitis B Virus Core Variants, Liver Fibrosis, and Hepatocellular Carcinoma. Hepatology 2019, 69, 5–8. [Google Scholar] [CrossRef] [Green Version]
- Levrero, M.; Zucman-Rossi, J. Mechanisms of HBV-induced hepatocellular carcinoma. J. Hepatol. 2016, 64, S84–S101. [Google Scholar] [CrossRef]
- Guerrieri, F.; Belloni, L.; Pediconi, N.; Levrero, M. Molecular mechanisms of HBV-associated hepatocarcinogenesis. Semin. Liver Dis. 2013, 33, 147–156. [Google Scholar] [CrossRef]
- Álvarez, E.G.; Demeulemeester, J.; Otero, P.; Jolly, C.; García-Souto, D.; Pequeño-Valtierra, A.; Zamora, J.; Tojo, M.; Temes, J.; Baez-Ortega, A.; et al. Aberrant integration of Hepatitis B virus DNA promotes major restructuring of human hepatocellular carcinoma genome architecture. Nat. Commun. 2021, 12, 6910. [Google Scholar] [CrossRef]
- An, P.; Xu, J.; Yu, Y.; Winkler, C.A. Host and Viral Genetic Variation in HBV-Related Hepatocellular Carcinoma. Front. Genet. 2018, 9, 261. [Google Scholar] [CrossRef]
- Hayashi, S.; Khan, A.; Simons, B.C.; Homan, C.; Matsui, T.; Ogawa, K.; Kawashima, K.; Murakami, S.; Takahashi, S.; Isogawa, M.; et al. An Association Between Core Mutations in Hepatitis B Virus Genotype F1b and Hepatocellular Carcinoma in Alaskan Native People. Hepatology 2019, 69, 19–33. [Google Scholar] [CrossRef]
- Ringelhan, M.; McKeating, J.A.; Protzer, U. Viral hepatitis and liver cancer. Philos. Trans. R Soc. Lond. B Biol. Sci. 2017, 372, 20160274. [Google Scholar] [CrossRef] [Green Version]
- Tu, T.; Budzinska, M.A.; Shackel, N.A.; Urban, S. HBV DNA Integration: Molecular Mechanisms and Clinical Implications. Viruses 2017, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Caballero, A.; Tabernero, D.; Buti, M.; Rodriguez-Frias, F. Hepatitis B virus: The challenge of an ancient virus with multiple faces and a remarkable replication strategy. Antiviral. Res. 2018, 158, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Sung, W.-K.; Zheng, H.; Li, S.; Chen, R.; Liu, X.; Li, Y.; Lee, N.P.; Lee, W.H.; Ariyaratne, P.N.; Tennakoon, C.; et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 2012, 44, 765–769. [Google Scholar] [CrossRef] [PubMed]
- Mak, L.-Y.; Wong, D.K.-H.; Pollicino, T.; Raimondo, G.; Hollinger, F.B.; Yuen, M.-F. Occult hepatitis B infection and hepatocellular carcinoma: Epidemiology, virology, hepatocarcinogenesis and clinical significance. J. Hepatol. 2020, 73, 952–964. [Google Scholar] [CrossRef]
- Budzinska, M.A.; Shackel, N.A.; Urban, S.; Tu, T. Cellular Genomic Sites of Hepatitis B Virus DNA Integration. Genes 2018, 9, 365. [Google Scholar] [CrossRef] [Green Version]
- Nault, J.-C.; Ningarhari, M.; Rebouissou, S.; Zucman-Rossi, J. The role of telomeres and telomerase in cirrhosis and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 544–558. [Google Scholar] [CrossRef]
- Tu, T.; Budzinska, M.A.; Vondran, F.W.R.; Shackel, N.A.; Urban, S. Hepatitis B Virus DNA Integration Occurs Early in the Viral Life Cycle in an Infection Model via Sodium Taurocholate Cotransporting Polypeptide-Dependent Uptake of Enveloped Virus Particles. J. Virol. 2018, 92, e02007-17. [Google Scholar] [CrossRef] [Green Version]
- Wooddell, C.I.; Yuen, M.-F.; Chan, H.L.-Y.; Gish, R.G.; Locarnini, S.A.; Chavez, D.; Ferrari, C.; Given, B.D.; Hamilton, J.; Kanner, S.B.; et al. RNAi-based treatment of chronically infected patients and chimpanzees reveals that integrated hepatitis B virus DNA is a source of HBsAg. Sci. Transl. Med. 2017, 9, eaan0241. [Google Scholar] [CrossRef] [Green Version]
- Lau, C.-C.; Sun, T.; Ching, A.K.K.; He, M.; Li, J.-W.; Wong, A.M.; Co, N.N.; Chan, A.W.H.; Li, P.-S.; Lung, R.W.M.; et al. Viral-human chimeric transcript predisposes risk to liver cancer development and progression. Cancer Cell 2014, 25, 335–349. [Google Scholar] [CrossRef]
- Ignatiadis, M.; Sledge, G.W.; Jeffrey, S.S. Liquid biopsy enters the clinic—Implementation issues and future challenges. Nat. Rev. Clin. Oncol. 2021, 18, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.L.; Pectasides, E.; Hanna, G.J.; Parsons, H.A.; Choudhury, A.D.; Oxnard, G.R. Circulating tumor DNA in advanced solid tumors: Clinical relevance and future directions. CA Cancer J. Clin. 2021, 71, 176–190. [Google Scholar] [CrossRef] [PubMed]
- Moss, J.; Magenheim, J.; Neiman, D.; Zemmour, H.; Loyfer, N.; Korach, A.; Samet, Y.; Maoz, M.; Druid, H.; Arner, P.; et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat. Commun. 2018, 9, 5068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hlady, R.A.; Zhao, X.; Pan, X.; Yang, J.D.; Ahmed, F.; Antwi, S.O.; Giama, N.H.; Patel, T.; Roberts, L.R.; Liu, C.; et al. Genome-wide discovery and validation of diagnostic DNA methylation-based biomarkers for hepatocellular cancer detection in circulating cell free DNA. Theranostics 2019, 9, 7239–7250. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Li, J.; Gassa, A.; Buchner, D.; Alakus, H.; Dong, Q.; Ren, N.; Liu, M.; Odenthal, M.; Stippel, D.; et al. Circulating tumor DNA as an emerging liquid biopsy biomarker for early diagnosis and therapeutic monitoring in hepatocellular carcinoma. Int. J. Biol. Sci. 2020, 16, 1551–1562. [Google Scholar] [CrossRef]
- Yue, Y.; Liu, J.; He, C. RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev. 2015, 29, 1343–1355. [Google Scholar] [CrossRef] [Green Version]
- An, Y.; Duan, H. The role of m6A RNA methylation in cancer metabolism. Mol. Cancer 2022, 21, 14. [Google Scholar] [CrossRef]
- Imam, H.; Khan, M.; Gokhale, N.S.; McIntyre, A.B.R.; Kim, G.-W.; Jang, J.Y.; Kim, S.-J.; Mason, C.E.; Horner, S.M.; Siddiqui, A. -methyladenosine modification of hepatitis B virus RNA differentially regulates the viral life cycle. Proc. Natl. Acad. Sci. USA 2018, 115, 8829–8834. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Zhu, M.; Pan, R.; Fang, T.; Cao, Y.-Y.; Chen, S.; Zhao, X.; Lei, C.-Q.; Guo, L.; Chen, Y.; et al. The tumor suppressor PTEN has a critical role in antiviral innate immunity. Nat. Immunol. 2016, 17, 241–249. [Google Scholar] [CrossRef]
- Chen, L.; Liu, S.; Tao, Y. Regulating tumor suppressor genes: Post-translational modifications. Signal Transduct. Target Ther. 2020, 5, 90. [Google Scholar] [CrossRef]
- Xie, P.; Peng, Z.; Chen, Y.; Li, H.; Du, M.; Tan, Y.; Zhang, X.; Lu, Z.; Cui, C.-P.; Liu, C.H.; et al. Neddylation of PTEN regulates its nuclear import and promotes tumor development. Cell Res. 2021, 31, 291–311. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.-W.; Imam, H.; Khan, M.; Mir, S.A.; Kim, S.-J.; Yoon, S.K.; Hur, W.; Siddiqui, A. HBV-Induced Increased N6 Methyladenosine Modification of PTEN RNA Affects Innate Immunity and Contributes to HCC. Hepatology 2021, 73, 533–547. [Google Scholar] [CrossRef]
- Huang, Z.; Zhou, J.-K.; Peng, Y.; He, W.; Huang, C. The role of long noncoding RNAs in hepatocellular carcinoma. Mol. Cancer 2020, 19, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarfaraz, N.; Somarowthu, S.; Bouchard, M.J. The Interplay of Long Non-Coding RNAs and Hepatitis B Virus. J. Med. Virol. 2022, 95, e28058. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Chen, L.; Gu, J.; Zhang, H.; Yuan, J.; Lian, Q.; Lv, G.; Wang, S.; Wu, Y.; Yang, Y.-C.T.; et al. Recurrently deregulated lncRNAs in hepatocellular carcinoma. Nat. Commun. 2017, 8, 14421. [Google Scholar] [CrossRef] [Green Version]
- Guerrieri, F.; Belloni, L.; D’Andrea, D.; Pediconi, N.; Le Pera, L.; Testoni, B.; Scisciani, C.; Floriot, O.; Zoulim, F.; Tramontano, A.; et al. Genome-wide identification of direct HBx genomic targets. BMC Genom. 2017, 18, 184. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, D.; Deb, M.; Kar, S.; Pradhan, N.; Parbin, S.; Kirtana, R.; Singh, S.P.; Suma, S.G.; Roy, A.; Manna, S.; et al. Dissecting miRNA facilitated physiology and function in human breast cancer for therapeutic intervention. Semin. Cancer Biol. 2021, 72, 46–64. [Google Scholar] [CrossRef]
- Li, G.; Zhang, W.; Gong, L.; Huang, X. MicroRNA 125a-5p Inhibits Cell Proliferation and Induces Apoptosis in Hepatitis B Virus-Related Hepatocellular Carcinoma by Downregulation of ErbB3. Oncol. Res. 2019, 27, 449–458. [Google Scholar] [CrossRef]
- Chen, W.; Jiang, J.; Gong, L.; Shu, Z.; Xiang, D.; Zhang, X.; Bi, K.; Diao, H. Hepatitis B virus P protein initiates glycolytic bypass in HBV-related hepatocellular carcinoma via a FOXO3/miRNA-30b-5p/MINPP1 axis. J. Exp. Clin. Cancer Res. 2021, 40, 1. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, Z.; Wang, Y.; Fang, M.; Zhou, J.; Li, Y.; Dai, E.; Feng, Z.; Wang, H.; Yang, Z.; et al. Using Quasispecies Patterns of Hepatitis B Virus to Predict Hepatocellular Carcinoma with Deep Sequencing and Machine Learning. J. Infect. Dis. 2021, 223, 1887–1896. [Google Scholar] [CrossRef]
- Tseng, T.-C.; Liu, C.-J.; Yang, H.-C.; Chen, C.-L.; Yang, W.-T.; Tsai, C.-S.; Kuo, S.F.-T.; Verbree, F.C.; Su, T.-H.; Wang, C.-C.; et al. Higher proportion of viral basal core promoter mutant increases the risk of liver cirrhosis in hepatitis B carriers. Gut 2015, 64, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Lazarevic, I.; Banko, A.; Miljanovic, D.; Cupic, M. Immune-Escape Hepatitis B Virus Mutations Associated with Viral Reactivation upon Immunosuppression. Viruses 2019, 11, 778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pu, R.; Liu, W.; Zhou, X.; Chen, X.; Hou, X.; Cai, S.; Chen, L.; Wu, J.; Yang, F.; Tan, X.; et al. The Effects and Underlying Mechanisms of Hepatitis B Virus X Gene Mutants on the Development of Hepatocellular Carcinoma. Front. Oncol. 2022, 12, 836517. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yan, Q.; Gong, L.; Xu, H.; Liu, B.; Fang, X.; Yu, D.; Li, L.; Wei, T.; Wang, Y.; et al. C-terminal truncated HBx initiates hepatocarcinogenesis by downregulating TXNIP and reprogramming glucose metabolism. Oncogene 2021, 40, 1147–1161. [Google Scholar] [CrossRef]
- Ren, F.; Li, W.; Zhao, S.; Wang, L.; Wang, Q.; Li, M.; Xiang, A.; Guo, Y. A3G-induced mutations show a low prevalence and exhibit plus-strand regional distribution in hepatitis B virus DNA from patients with non-hepatocellular carcinoma (HCC) and HCC. J. Med. Virol. 2021, 93, 3672–3678. [Google Scholar] [CrossRef]
- Yan, L.; Deng, Y.; Zhou, J.; Zhao, H.; Wang, G. Serum YKL-40 as a biomarker for liver fibrosis in chronic hepatitis B patients with normal and mildly elevated ALT. Infection 2018, 46, 385–393. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Zhao, H.; Zhou, J.; Yan, L.; Wang, G. Complement 5a is an indicator of significant fibrosis and earlier cirrhosis in patients chronically infected with hepatitis B virus. Infection 2017, 45, 75–81. [Google Scholar] [CrossRef] [Green Version]
- Llovet, J.M.; De Baere, T.; Kulik, L.; Haber, P.K.; Greten, T.F.; Meyer, T.; Lencioni, R. Locoregional therapies in the era of molecular and immune treatments for hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 293–313. [Google Scholar] [CrossRef]
- Losic, B.; Craig, A.J.; Villacorta-Martin, C.; Martins-Filho, S.N.; Akers, N.; Chen, X.; Ahsen, M.E.; von Felden, J.; Labgaa, I.; DʹAvola, D.; et al. Intratumoral heterogeneity and clonal evolution in liver cancer. Nat. Commun. 2020, 11, 291. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Kao, E.; Gao, X.; Sandig, H.; Limmer, K.; Pavon-Eternod, M.; Jones, T.E.; Landry, S.; Pan, T.; Weitzman, M.D.; et al. Codon-usage-based inhibition of HIV protein synthesis by human schlafen 11. Nature 2012, 491, 125–128. [Google Scholar] [CrossRef]
- Zhou, C.; Liu, C.; Liu, W.; Chen, W.; Yin, Y.; Li, C.-W.; Hsu, J.L.; Sun, J.; Zhou, Q.; Li, H.; et al. SLFN11 inhibits hepatocellular carcinoma tumorigenesis and metastasis by targeting RPS4X via mTOR pathway. Theranostics 2020, 10, 4627–4643. [Google Scholar] [CrossRef]
- Jiang, Y.; Sun, A.; Zhao, Y.; Ying, W.; Sun, H.; Yang, X.; Xing, B.; Sun, W.; Ren, L.; Hu, B.; et al. Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. Nature 2019, 567, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Tsai, T.-Y.; Huang, M.-T.; Sung, P.-S.; Peng, C.-Y.; Tao, M.-H.; Yang, H.-I.; Chang, W.-C.; Yang, A.-S.; Yu, C.-M.; Lin, Y.-P.; et al. SIGLEC-3 (CD33) serves as an immune checkpoint receptor for HBV infection. J. Clin. Invest. 2021, 131, e141965. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.S.Y.; Covert, E.; Wilson, E.; Kottilil, S. Chronic Hepatitis B Infection: A Review. JAMA 2018, 319, 1802–1813. [Google Scholar] [CrossRef] [PubMed]
- Global prevalence and genotype distribution of hepatitis C virus infection in 2015: A modelling study. Lancet Gastroenterol. Hepatol. 2017, 2, 161–176. [CrossRef] [Green Version]
- Ko, K.; Nagashima, S.; Yamamoto, C.; Takahashi, K.; Matsuo, J.; Ohisa, M.; Akita, T.; Matyakubov, J.; Mirzaev, U.; Katayama, K.; et al. Eighteen-year follow-up cohort study on hepatitis B and C virus infections related long-term prognosis among hemodialysis patients in Hiroshima. J. Med. Virol. 2020, 92, 3436–3447. [Google Scholar] [CrossRef]
- Groves, J.; Dodd, R.Y.; Foster, G.A.; Stramer, S.L. Genotype Distribution and Demographic Characteristics of Hepatitis C Virus NAT Yield Cases among US Blood Donors. Clin. Infect. Dis. 2022, 75, 1714–1722. [Google Scholar] [CrossRef]
- Piconese, S.; Cammarata, I.; Barnaba, V. Viral hepatitis, inflammation, and cancer: A lesson for autoimmunity. J Autoimmun. 2018, 95, 58–68. [Google Scholar] [CrossRef]
- Stephenson, A.A.; Cao, S.; Taggart, D.J.; Vyavahare, V.P.; Suo, Z. Design, synthesis, and evaluation of liver-specific gemcitabine prodrugs for potential treatment of hepatitis C virus infection and hepatocellular carcinoma. Eur. J. Med. Chem. 2021, 213, 113135. [Google Scholar] [CrossRef]
- Scheel, T.K.H.; Rice, C.M. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat. Med. 2013, 19, 837–849. [Google Scholar] [CrossRef]
- Nahon, P.; Layese, R.; Bourcier, V.; Cagnot, C.; Marcellin, P.; Guyader, D.; Pol, S.; Larrey, D.; De Lédinghen, V.; Ouzan, D.; et al. Incidence of Hepatocellular Carcinoma After Direct Antiviral Therapy for HCV in Patients with Cirrhosis Included in Surveillance Programs. Gastroenterology 2018, 155, 1436–1450.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beste, L.A.; Green, P.; Berry, K.; Belperio, P.; Ioannou, G.N. Hepatitis C-Related Hepatocellular Carcinoma Incidence in the Veterans Health Administration After Introduction of Direct-Acting Antivirals. JAMA 2020, 324, 1003–1005. [Google Scholar] [CrossRef]
- Akuta, N.; Suzuki, F.; Sezaki, H.; Kobayashi, M.; Fujiyama, S.; Kawamura, Y.; Hosaka, T.; Kobayashi, M.; Saitoh, S.; Suzuki, Y.; et al. Complex Association of Virus- and Host-Related Factors with Hepatocellular Carcinoma Rate following Hepatitis C Virus Clearance. J. Clin. Microbiol. 2019, 57, e01463-18. [Google Scholar] [CrossRef] [Green Version]
- Rinaldi, L.; Perrella, A.; Guarino, M.; De Luca, M.; Piai, G.; Coppola, N.; Pafundi, P.C.; Ciardiello, F.; Fasano, M.; Martinelli, E.; et al. Incidence and risk factors of early HCC occurrence in HCV patients treated with direct acting antivirals: A prospective multicentre study. J. Transl. Med. 2019, 17, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinaldi, L.; Nevola, R.; Franci, G.; Perrella, A.; Corvino, G.; Marrone, A.; Berretta, M.; Morone, M.V.; Galdiero, M.; Giordano, M.; et al. Risk of Hepatocellular Carcinoma after HCV Clearance by Direct-Acting Antivirals Treatment Predictive Factors and Role of Epigenetics. Cancers 2020, 12, 1351. [Google Scholar] [CrossRef] [PubMed]
- Blackard, J.T.; Sherman, K.E. Hepatitis B virus (HBV) reactivation-The potential role of direct-acting agents for hepatitis C virus (HCV). Rev. Med. Virol. 2018, 28, e1984. [Google Scholar] [CrossRef]
- Liu, C.-J.; Chuang, W.-L.; Sheen, I.S.; Wang, H.-Y.; Chen, C.-Y.; Tseng, K.-C.; Chang, T.-T.; Massetto, B.; Yang, J.C.; Yun, C.; et al. Efficacy of Ledipasvir and Sofosbuvir Treatment of HCV Infection in Patients Coinfected with HBV. Gastroenterology 2018, 154, 989–997. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.J.; Sheen, I.S.; Chen, C.Y.; Chuang, W.L.; Wang, H.Y.; Tseng, K.C.; Chang, T.T.; Yang, J.; Massetto, B.; Suri, V.; et al. Ledipasvir/Sofosbuvir for Patients Coinfected with Chronic Hepatitis C and Hepatitis B in Taiwan: Follow-up at 108 Weeks Posttreatment. Clin. Infect. Dis. 2022, 75, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Mutz, P.; Metz, P.; Lempp, F.A.; Bender, S.; Qu, B.; Schöneweis, K.; Seitz, S.; Tu, T.; Restuccia, A.; Frankish, J.; et al. HBV Bypasses the Innate Immune Response and Does Not Protect HCV From Antiviral Activity of Interferon. Gastroenterology 2018, 154, 1791–1804.e22. [Google Scholar] [CrossRef] [Green Version]
- Bersoff-Matcha, S.J.; Cao, K.; Jason, M.; Ajao, A.; Jones, S.C.; Meyer, T.; Brinker, A. Hepatitis B Virus Reactivation Associated with Direct-Acting Antiviral Therapy for Chronic Hepatitis C Virus: A Review of Cases Reported to the U.S. Food and Drug Administration Adverse Event Reporting System. Ann. Intern. Med. 2017, 166, 792–798. [Google Scholar] [CrossRef]
- Wang, M.; Wang, Y.; Feng, X.; Wang, R.; Wang, Y.; Zeng, H.; Qi, J.; Zhao, H.; Li, N.; Cai, J.; et al. Contribution of hepatitis B virus and hepatitis C virus to liver cancer in China north areas: Experience of the Chinese National Cancer Center. Int. J. Infect. Dis. 2017, 65, 15–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Wang, L.; Zhang, H.; Ou, W.; Li, D.; Feng, Y.; Zhuang, H.; Shao, Y. Changing Epidemiology of Hepatitis B Virus and Hepatitis C Virus Coinfection in a Human Immunodeficiency Virus-Positive Population in China: Results From the Third and Fourth Nationwide Molecular Epidemiologic Surveys. Clin. Infect. Dis. 2021, 73, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Fairley, C.K.; Sasadeusz, J.; He, J.; Wei, X.; Zeng, H.; Jing, J.; Mao, L.; Chen, X.; Zhang, L. HBV, HCV, and HBV/HCV co-infection among HIV-positive patients in Hunan province, China: Regimen selection, hepatotoxicity, and antiretroviral therapy outcome. J. Med. Virol. 2018, 90, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Mücke, M.M.; Backus, L.I.; Mücke, V.T.; Coppola, N.; Preda, C.M.; Yeh, M.-L.; Tang, L.S.Y.; Belperio, P.S.; Wilson, E.M.; Yu, M.-L.; et al. Hepatitis B virus reactivation during direct-acting antiviral therapy for hepatitis C: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2018, 3, 172–180. [Google Scholar] [CrossRef]
- Sagnelli, E.; Coppola, N.; Marrocco, C.; Onofrio, M.; Sagnelli, C.; Coviello, G.; Scolastico, C.; Filippini, P. Hepatitis C virus superinfection in hepatitis B virus chronic carriers: A reciprocal viral interaction and a variable clinical course. J. Clin. Virol. 2006, 35, 317–320. [Google Scholar] [CrossRef]
- Lempp, F.A.; Ni, Y.; Urban, S. Hepatitis delta virus: Insights into a peculiar pathogen and novel treatment options. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 580–589. [Google Scholar] [CrossRef]
- Stockdale, A.J.; Kreuels, B.; Henrion, M.Y.R.; Giorgi, E.; Kyomuhangi, I.; de Martel, C.; Hutin, Y.; Geretti, A.M. The global prevalence of hepatitis D virus infection: Systematic review and meta-analysis. J. Hepatol. 2020, 73, 523–532. [Google Scholar] [CrossRef]
- Koh, C.; Canini, L.; Dahari, H.; Zhao, X.; Uprichard, S.L.; Haynes-Williams, V.; Winters, M.A.; Subramanya, G.; Cooper, S.L.; Pinto, P.; et al. Oral prenylation inhibition with lonafarnib in chronic hepatitis D infection: A proof-of-concept randomised, double-blind, placebo-controlled phase 2A trial. Lancet Infect. Dis. 2015, 15, 1167–1174. [Google Scholar] [CrossRef] [Green Version]
- Offensperger, W.B.; Offensperger, S.; Walter, E.; Blum, H.E.; Gerok, W. Suramin prevents duck hepatitis B virus infection in vivo. Antimicrob. Agents Chemother. 1993, 37, 1539–1542. [Google Scholar] [CrossRef] [Green Version]
- Bazinet, M.; Pântea, V.; Cebotarescu, V.; Cojuhari, L.; Jimbei, P.; Albrecht, J.; Schmid, P.; Le Gal, F.; Gordien, E.; Krawczyk, A.; et al. Safety and efficacy of REP 2139 and pegylated interferon alfa-2a for treatment-naive patients with chronic hepatitis B virus and hepatitis D virus co-infection (REP 301 and REP 301-LTF): A non-randomised, open-label, phase 2 trial. Lancet Gastroenterol. Hepatol. 2017, 2, 877–889. [Google Scholar] [CrossRef]
- Todt, D.; Friesland, M.; Moeller, N.; Praditya, D.; Kinast, V.; Brüggemann, Y.; Knegendorf, L.; Burkard, T.; Steinmann, J.; Burm, R.; et al. Robust hepatitis E virus infection and transcriptional response in human hepatocytes. Proc. Natl. Acad. Sci. USA 2020, 117, 1731–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Z.; de Man, R.A.; Kamar, N.; Pan, Q. Chronic hepatitis E: Advancing research and patient care. J. Hepatol. 2022, 77, 1109–1123. [Google Scholar] [CrossRef] [PubMed]
- Himmelsbach, K.; Bender, D.; Hildt, E. Life cycle and morphogenesis of the hepatitis E virus. Emerg. Microbes. Infect. 2018, 7, 196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behrendt, P.; Friesland, M.; Wißmann, J.-E.; Kinast, V.; Stahl, Y.; Praditya, D.; Hueffner, L.; Nörenberg, P.M.; Bremer, B.; Maasoumy, B.; et al. Hepatitis E virus is highly resistant to alcohol-based disinfectants. J. Hepatol. 2022, 76, 1062–1069. [Google Scholar] [CrossRef]
- Chapuy-Regaud, S.; Dubois, M.; Plisson-Chastang, C.; Bonnefois, T.; Lhomme, S.; Bertrand-Michel, J.; You, B.; Simoneau, S.; Gleizes, P.-E.; Flan, B.; et al. Characterization of the lipid envelope of exosome encapsulated HEV particles protected from the immune response. Biochimie 2017, 141, 70–79. [Google Scholar] [CrossRef]
- Wedemeyer, H.; Pischke, S.; Manns, M.P. Pathogenesis and treatment of hepatitis e virus infection. Gastroenterology 2012, 142, 1388–1397.e1. [Google Scholar] [CrossRef]
- Cordes, A.K.; Goudeva, L.; Lütgehetmann, M.; Wenzel, J.J.; Behrendt, P.; Wedemeyer, H.; Heim, A. Risk of transfusion-transmitted hepatitis E virus infection from pool-tested platelets and plasma. J. Hepatol. 2022, 76, 46–52. [Google Scholar] [CrossRef]
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y.; et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2095–2128. [Google Scholar] [CrossRef]
- Clemente-Casares, P.; Ramos-Romero, C.; Ramirez-Gonzalez, E.; Mas, A. Hepatitis E Virus in Industrialized Countries: The Silent Threat. Biomed. Res. Int. 2016, 2016, 9838041. [Google Scholar] [CrossRef]
- Boccia, D.; Guthmann, J.-P.; Klovstad, H.; Hamid, N.; Tatay, M.; Ciglenecki, I.; Nizou, J.-Y.; Nicand, E.; Guerin, P.J. High mortality associated with an outbreak of hepatitis E among displaced persons in Darfur, Sudan. Clin. Infect. Dis. 2006, 42, 1679–1684. [Google Scholar] [CrossRef]
- EASL Clinical Practice Guidelines on hepatitis E virus infection. J. Hepatol. 2018, 68, 1256–1271. [CrossRef] [PubMed]
- Buescher, G.; Ozga, A.-K.; Lorenz, E.; Pischke, S.; May, J.; Addo, M.M.; Horvatits, T. Hepatitis E seroprevalence and viremia rate in immunocompromised patients: A systematic review and meta-analysis. Liver Int. 2021, 41, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Forner, A.; Llovet, J.M.; Bruix, J. Hepatocellular carcinoma. Lancet 2012, 379, 1245–1255. [Google Scholar] [CrossRef]
- Colson, P.; Borentain, P.; Gérolami, R. Hepatitis E virus as an agent of hepatocellular carcinoma. Int. J. Infect Dis. 2019, 80, 62–63. [Google Scholar] [CrossRef] [Green Version]
- Bai, M.-J.; Zhou, N.; Dong, W.; Li, G.-X.; Cong, W.; Zhu, X.-Q. Seroprevalence and risk factors of hepatitis E virus infection in cancer patients in eastern China. Int. J. Infect. Dis. 2018, 71, 42–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amougou Atsama, M.; Atangana, P.J.A.; Noah Noah, D.; Moundipa, P.F.; Pineau, P.; Njouom, R. Hepatitis E virus infection as a promoting factor for hepatocellular carcinoma in Cameroon: Preliminary Observations. Int. J. Infect. Dis. 2017, 64, 4–8. [Google Scholar] [CrossRef] [Green Version]
- Xue, M.; Lin, X.; Lin, Q.-X.; Pu, X.; Liu, J.; Li, X.-F.; Hou, J.; Liu, X.; Chen, R. Association between hepatitis B and E virus infection and hepatocellular carcinoma risk. Int. J. Cancer 2021, 148, 2974–2981. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Hepatitis Virus | Mechanisms | Type | References |
|---|---|---|---|
| HBV | HBV integrations promote local and distant oncogenic driver alterations (TERT, TP53, MYC) in HCC * | DNA integration | [17] |
| HBV integrations in multiple cancer-associated genes (such as TERT, MLL4, and CCNE1) * | DNA integration | [26] | |
| HBV-TERT promoter integration harnesses host ELF4 resulting in TERT gene transcription in HCC * | DNA integration | [42] | |
| HBV-induced increased N6 methyladenosine modification of PTEN RNA affects innate immunity and contributes to HCC * | Epigenetic Changes | [72] | |
| Hepatitis B protein HBx binds the DLEU2 lncRNA to sustain cccDNA and host cancer-related gene transcription * | Epigenetic Changes | [35] | |
| HBV P protein initiates glycolytic bypass in HCC via a FOXO3/miRNA-30b-5p/MINPP1 axis * | Epigenetic Changes | [79] | |
| Core mutations in HBV genotype F1b | Gene Mutation | [50] | |
| HBV reverse transcriptase gene mutation | Gene Mutation | [80] | |
| HBV X gene mutants | Gene Mutation | [83] | |
| A3G-induced HBV DNA mutations * | Gene Mutation | [85] | |
| Low expression of SLFN11 in HCC * | Abnormal expression of specific protein | [91] | |
| High expression of SOAT1* | Abnormal expression of specific protein | [92] | |
| HCV | Chronic inflammatory response and hepatic fibrosis | Immune and inflammatory response | [98] |
| DAA therapy induces HBV reactivation and HCC development (to be further demonstrated) * | Therapeutic influence | [104,106] | |
| HDV | Interactions between HDV and HBV and the development of chronic viral hepatitis fibrosis and cirrhosis | HBV co-infection | [22] |
| Activation of STAT-3 or oxidative-stress-induced activation of NF-κB promotes inflammatory reaction * | Inflammatory reaction | [116] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, C.; Jiang, X.; Li, M.; Luo, Y. Hepatitis Virus and Hepatocellular Carcinoma: Recent Advances. Cancers 2023, 15, 533. https://doi.org/10.3390/cancers15020533
Shen C, Jiang X, Li M, Luo Y. Hepatitis Virus and Hepatocellular Carcinoma: Recent Advances. Cancers. 2023; 15(2):533. https://doi.org/10.3390/cancers15020533
Chicago/Turabian StyleShen, Chen, Xin Jiang, Mei Li, and Yao Luo. 2023. "Hepatitis Virus and Hepatocellular Carcinoma: Recent Advances" Cancers 15, no. 2: 533. https://doi.org/10.3390/cancers15020533