
Advances in Molecular Pathophysiology and Targeted Therapy for Cushing’s Disease

Abstract
:Simple Summary
Abstract
1. Introduction
2. Molecular Pathophysiology
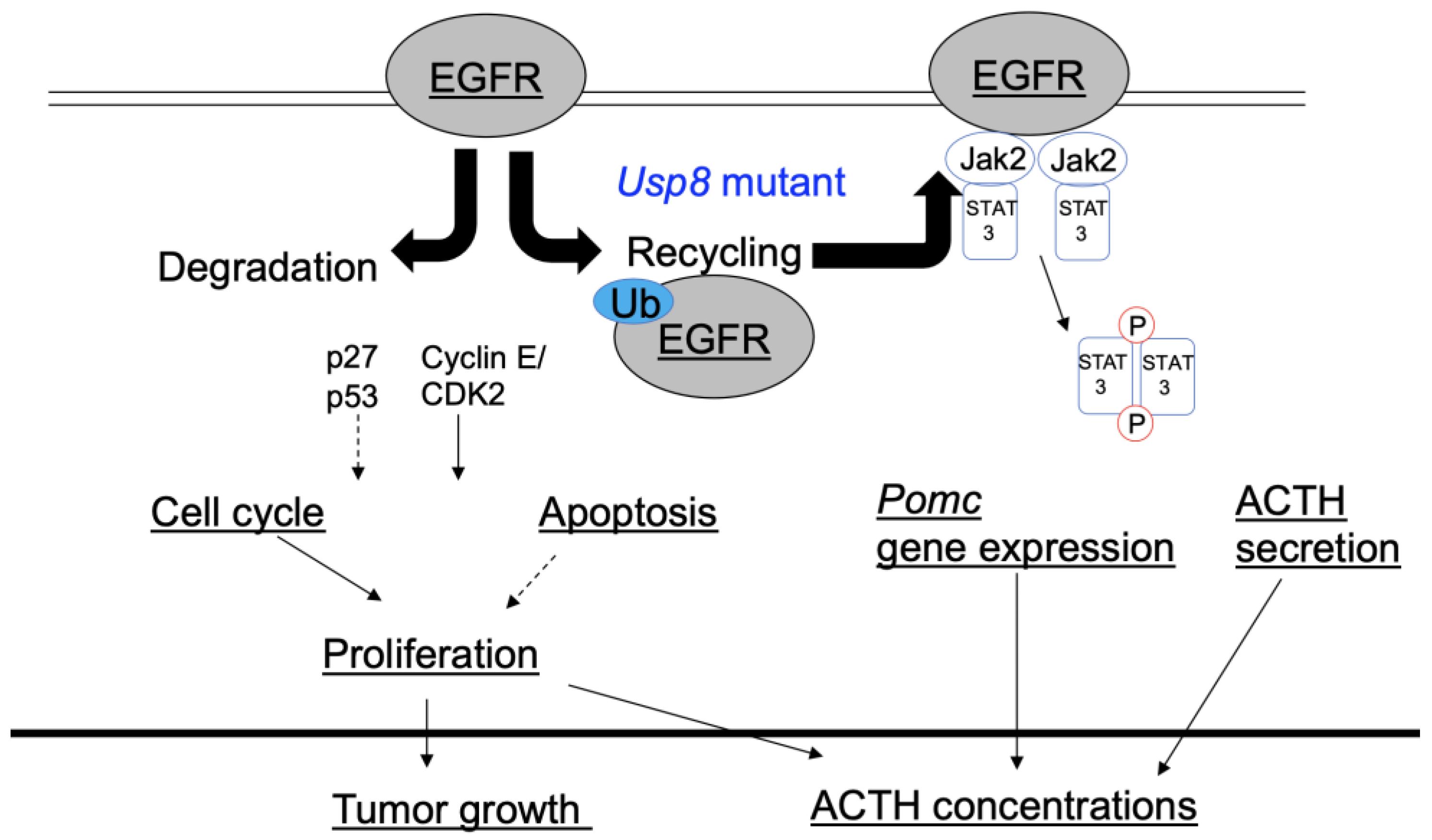
2.1. Mutations in the Ubiquitin-Specific Protease 8 (USP8) Gene and Epidermal Growth Factor Receptor (EGFR) Expression
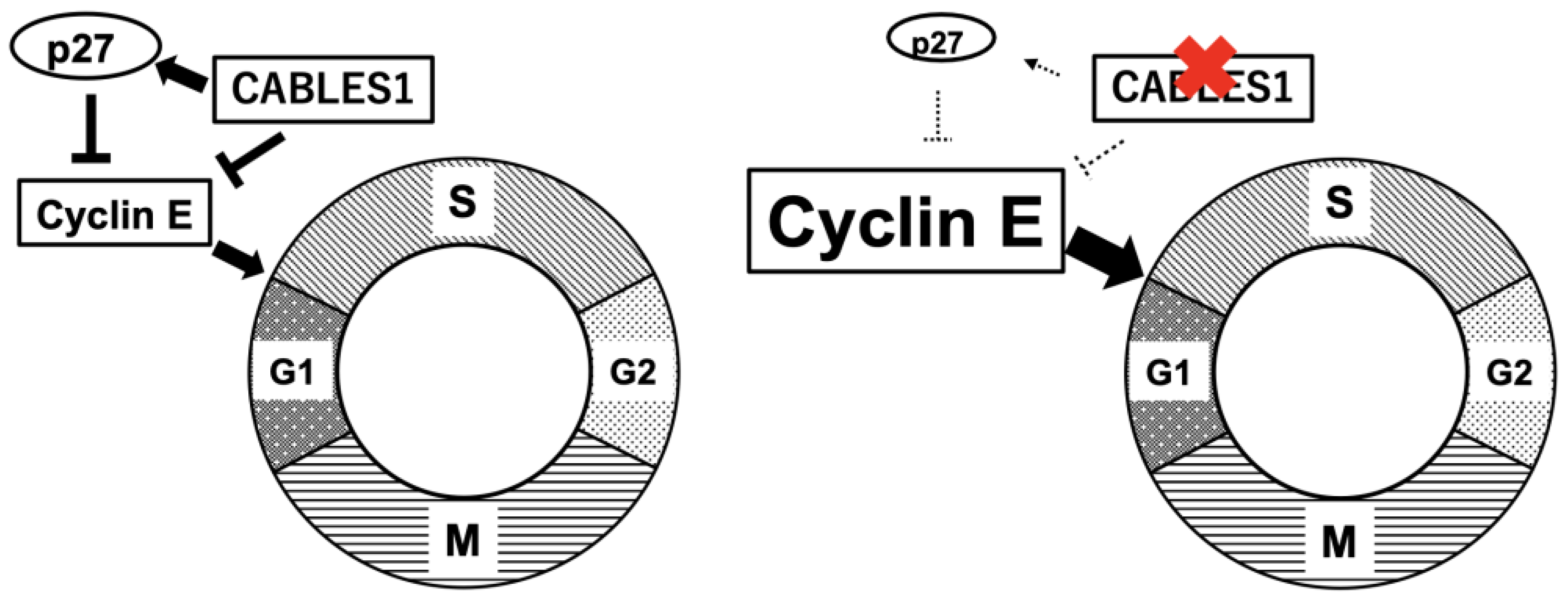
2.2. Cell Cycle Regulator
2.3. Genetic Familial Syndromes, Oncogenes, and Tumor Suppressor Genes
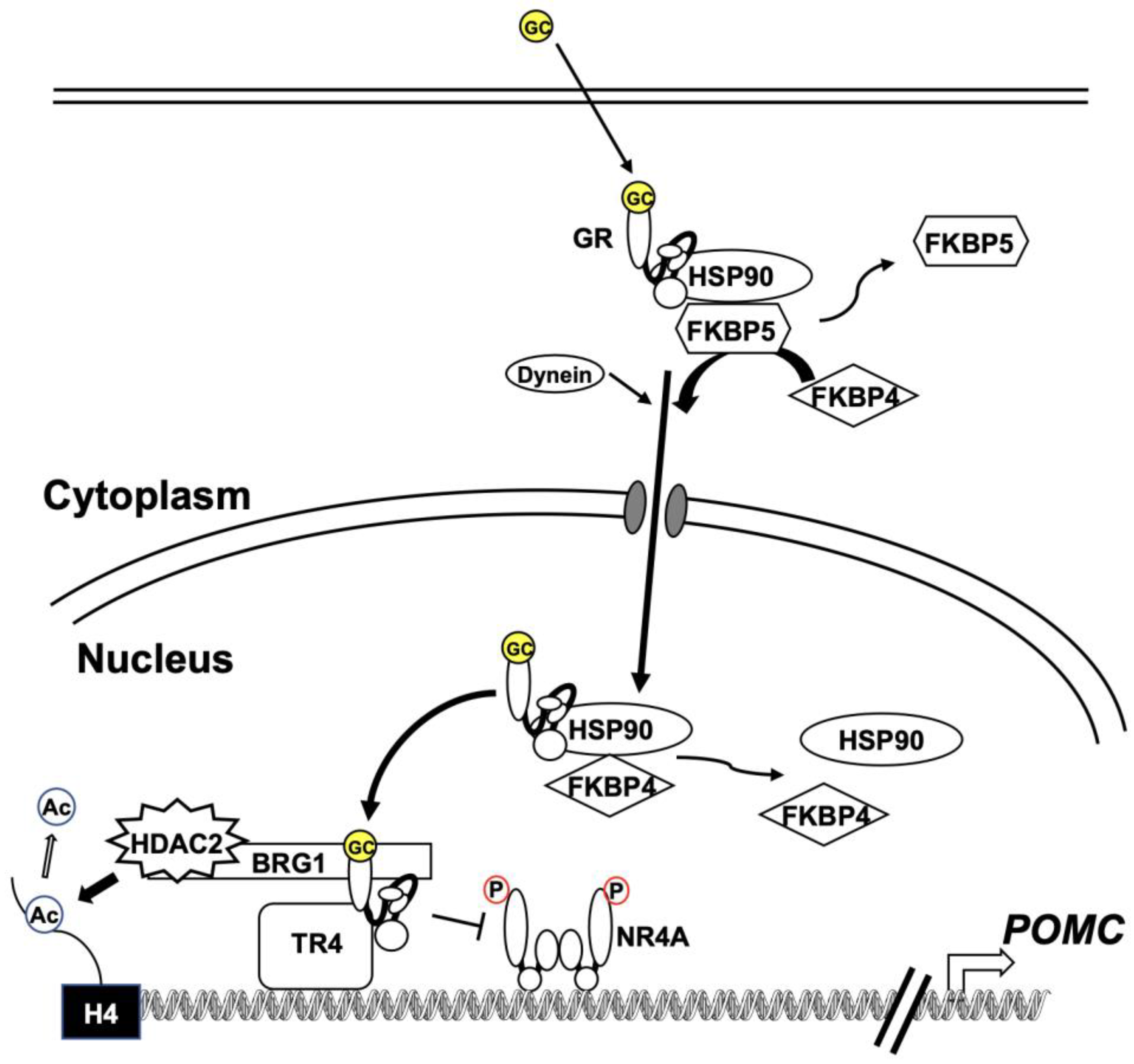
2.4. Glucocorticoid Resistance
3. The Practice of Treatment
3.1. Surgery
3.2. Radiotherapy
3.3. Medical Therapy
3.3.1. Clinical Management of Targeted Pituitary Therapy
Pasireotide
Cabergoline
Temozolomide
3.3.2. Clinical Management of Targeted Adrenal Gland Therapy (Summary in Table 1)
Ketoconazole

{kind=link}
{kind=link}
{kind=link}
| Drug Name | Interact with CYP Types | Dose Range | Adverse Effects | References |
|---|---|---|---|---|
| Ketoconazole | CYP17A1, 11A1, 11B1, 3A4 | 400–1200 mg | Cytolitic hepatitis, mild gastrointestinal upset, male hypogonadism, and QT prolongation | [73,74] |
| Metyrapone | CYP11B1, 11B2 | 500–3000 mg | Gastrointestinal upset, dizziness, nausea, hypokalemia, liver damage, and hirsutism | [75,76] |
| Osilodrostat | CYP11B1, 11B2 | 2–30 mg | QT prolomgation, fatigue, appetite loss, headache, nausea, dizziness, hypokalemia, hypertension, and hirsutism | [77,78] |
| Mitotane | CYP3A4 | 500–3000 mg | Gastrointestinal and neurological disorders, hypercholesterolemia, and hypothyroidism | [79] |
| Mifepristone | None | 300–1200 mg | Hypokalemia, hypertension, edema, metrorragia, and endometrial hyperplasia | [80,81] |
Metyrapone
Osilodrostat
Mitotane
Mifepristone
3.3.3. Advances in Novel Potential Therapy (Summary in Table 2)
USP8 Inhibitors
| Specific Inhibitors | Drug Name | Dose Range | Possible Adverse Effects | References |
|---|---|---|---|---|
| USP8 | DUBs-IN-2 | Unknown | Not reported | [13,14] |
| Tyrosine kinase or EGFR | Gefitinib, lapatinib, and SD-1029 | Unknown | Interstitial pneumonia, severe diarrhea, and hepatitis | [9,12,85,86] |
| HSP90 | Silibinin and CCT018159 | Unknown | Severe diarrhea, and eye disorders | [41,87] |
| Histone deacetylase | Trichostatin A, suberoylanilide hydroxamic acid, romidepsin, and tubastatin A | Unknown | Dehydration, hyperglycemia, and thrombocytopenia | [88,89,90,91,92] |
| Cyclin-dependent kinase | R-roscovitine | Unknown | Pancytopenia, nausea, and diarrhea | [19,20] |
| ACTH neutralizing antibody | ALD1613 | Unknown | Not reported | [93] |
| Immune checkpoint inhibitors | Ipilimumab and nivolumab | 1 and 3 mg/kg BW | Immune-related adverse events | [94] |
Tyrosine Kinase or EGFR Inhibitors
HSP90 Inhibitors
Histone Deacetylase Inhibitors
Cyclin-Dependent Kinase Inhibitors
ACTH-Neutralizing Antibody
Immunotherapy
4. Discussion and Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Reincke, M.; Sbiera, S.; Hayakawa, A.; Theodoropoulou, M.; Osswald, A.; Beuschlein, F.; Meitinger, T.; Mizuno-Yamasaki, E.; Kawaguchi, K.; Saeki, Y.; et al. Mutations in the deubiquitinase gene USP8 cause Cushing’s disease. Nat. Genet. 2015, 47, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Perez-Rivas, L.G.; Theodoropoulou, M.; Ferraù, F.; Nusser, C.; Kawaguchi, K.; Stratakis, C.A.; Faucz, F.R.; Wildemberg, L.E.; Assié, G.; Beschorner, R.; et al. The gene of the ubiquitin-specific protease 8 is frequently mutated in adenomas causing Cushing’s disease. J. Clin. Endocrinol. Metab. 2015, 100, E997–E1004. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.Y.; Song, Z.J.; Chen, J.H.; Wang, Y.F.; Li, S.Q.; Zhou, L.F.; Mao, Y.; Li, Y.M.; Hu, R.G.; Zhang, Z.Y.; et al. Recurrent gain-of-function USP8 mutations in Cushing’s disease. Cell Res. 2015, 25, 306–317. [Google Scholar] [CrossRef]
- Jordan, S.; Lidhar, K.; Korbonits, M.; Lowe, D.G.; Grossman, A.B. Cyclin D and cyclin E expression in normal and adenomatous pituitary. Eur. J. Endocrinol. 2000, 143, R1–R6. [Google Scholar] [CrossRef] [Green Version]
- Lidhar, K.; Korbonits, M.; Jordan, S.; Khalimova, Z.; Kaltsas, G.; Lu, X.; Clayton, R.N.; Jenkins, P.J.; Monson, J.P.; Besser, G.M.; et al. Low expression of the cell cycle inhibitor p27Kip1 in normal corticotroph cells, corticotroph tumors, and malignant pituitary tumors. J. Clin. Endocrinol. Metab. 1999, 84, 3823–3830. [Google Scholar] [CrossRef] [PubMed]
- Roussel-Gervais, A.; Bilodeau, S.; Vallette, S.; Berthelet, F.; Lacroix, A.; Figarella-Branger, D.; Brue, T.; Drouin, J. Cooperation between cyclin E and p27(Kip1) in pituitary tumorigenesis. Mol. Endocrinol. 2010, 24, 1835–1845. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, K.; Inoshita, N.; Kawaguchi, K.; Ibrahim Ardisasmita, A.; Suzuki, H.; Fukuhara, N.; Okada, M.; Nishioka, H.; Takeuchi, Y.; Komada, M.; et al. The USP8 mutational status may predict drug susceptibility in corticotroph adenomas of Cushing’s disease. Eur. J. Endocrinol. 2016, 174, 213–226. [Google Scholar] [CrossRef] [Green Version]
- Cohen, M.; Persky, R.; Stegemann, R.; Hernández-Ramírez, L.C.; Zeltser, D.; Lodish, M.B.; Chen, A.; Keil, M.F.; Tatsi, C.; Faucz, F.R.; et al. Germline USP8 mutation associated with pediatric Cushing disease and other clinical features: A new syndrome. J. Clin. Endocrinol. Metab. 2019, 104, 4676–4682. [Google Scholar] [CrossRef]
- Fukuoka, H.; Cooper, O.; Ben-Shlomo, A.; Mamelak, A.; Ren, S.G.; Bruyette, D.; Melmed, S. EGFR as a therapeutic target for human, canine, and mouse ACTH-secreting pituitary adenomas. J. Clin. Investig. 2011, 121, 4712–4721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Feng, M.; Dai, C.; Bao, X.; Deng, K.; Yao, Y.; Wang, R. Expression of EGFR in pituitary corticotroph adenomas and its relationship with tumor behavior. Front. Endocrinol. 2019, 10, 785. [Google Scholar] [CrossRef]
- Araki, T.; Liu, X.; Kameda, H.; Tone, Y.; Fukuoka, H.; Tone, M.; Melmed, S. EGFR Induces E2F1-mediated corticotroph tumorigenesis. J. Endocr. Soc. 2017, 1, 127–143. [Google Scholar] [CrossRef] [Green Version]
- Asari, Y.; Kageyama, K.; Sugiyama, A.; Kogawa, H.; Niioka, K.; Daimon, M. Lapatinib decreases the ACTH production and proliferation of corticotroph tumor cells. Endocr. J. 2019, 66, 515–522. [Google Scholar] [CrossRef] [Green Version]
- Kageyama, K.; Asari, Y.; Sugimoto, Y.; Niioka, K.; Daimon, M. Ubiquitin-specific protease 8 inhibitor suppresses adrenocorticotropic hormone production and corticotroph tumor cell proliferation. Endocr. J. 2020, 67, 177–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treppiedi, D.; Di Muro, G.; Marra, G.; Barbieri, A.M.; Mangili, F.; Catalano, R.; Serban, A.; Ferrante, E.; Locatelli, M.; Lania, A.G.; et al. USP8 inhibitor RA-9 reduces ACTH release and cell growth in tumor corticotrophs. Endocr. Relat. Cancer 2021, 28, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Sesta, A.; Cassarino, M.F.; Cavagnini, F.; Pecori Giraldi, F.P. Role of the ubiquitin/proteasome system on ACTH turnover in rat corticotropes. Endocrine 2018, 61, 511–517. [Google Scholar] [CrossRef] [Green Version]
- Sesta, A.; Cassarino, M.F.; Terreni, M.; Ambrogio, A.G.; Libera, L.; Bardelli, D.; Lasio, G.; Losa, M.; Pecori Giraldi, F. Ubiquitin-specific protease 8 mutant corticotrope adenomas present unique secretory and molecular features and shed light on the role of ubiquitylation on ACTH processing. Neuroendocrinology 2020, 110, 119–129. [Google Scholar] [CrossRef]
- Chen, J.; Jian, X.; Deng, S.; Ma, Z.; Shou, X.; Shen, Y.; Zhang, Q.; Song, Z.; Li, Z.; Peng, H.; et al. Identification of recurrent USP48 and BRAF mutations in Cushing’s disease. Nat. Commun. 2018, 9, 3171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sbiera, S.; Perez-Rivas, L.G.; Taranets, L.; Weigand, I.; Flitsch, J.; Graf, E.; Monoranu, C.M.; Saeger, W.; Hagel, C.; Honegger, J.; et al. Driver mutations in USP8 wild-type Cushing’s disease. Neuro. Oncol. 2019, 21, 1273–1283. [Google Scholar] [CrossRef]
- Liu, N.A.; Jiang, H.; Ben-Shlomo, A.; Wawrowsky, K.; Fan, X.M.; Lin, S.; Melmed, S. Targeting zebrafish and murine pituitary corticotroph tumors with a cyclin-dependent kinase (CDK) inhibitor. Proc. Natl. Acad. Sci. USA 2011, 108, 8414–8419. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.A.; Araki, T.; Cuevas-Ramos, D.; Hong, J.; Ben-Shlomo, A.; Tone, Y.; Tone, M.; Melmed, S. Cyclin E-mediated human proopiomelanocortin regulation as a therapeutic target for Cushing disease. J. Clin. Endocrinol. Metab. 2015, 100, 2557–2564. [Google Scholar] [CrossRef]
- Roussel-Gervais, A.; Couture, C.; Langlais, D.; Takayasu, S.; Balsalobre, A.; Rueda, B.R.; Zukerberg, L.R.; Figarella-Branger, D.; Brue, T.; Drouin, J. The Cables1 gene in glucocorticoid regulation of pituitary corticotrope growth and Cushing disease. J. Clin. Endocrinol. Metab. 2016, 101, 513–522. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Ramírez, L.C.; Gam, R.; Valdés, N.; Lodish, M.B.; Pankratz, N.; Balsalobre, A.; Gauthier, Y.; Faucz, F.R.; Trivellin, G.; Chittiboina, P.; et al. Loss-of-function mutations in the CABLES1 gene are a novel cause of Cushing’s disease. Endocr. Relat. Cancer 2017, 24, 379–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foth, M.; McMahon, M. Autophagy inhibition in BRAF-driven cancers. Cancers 2021, 13, 3498. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drouin, J. 60 years of POMC: Transcriptional and epigenetic regulation of POMC gene expression. J. Mol. Endocrinol. 2016, 56, T99–T112. [Google Scholar] [CrossRef] [Green Version]
- Abraham, A.P.; Pai, R.; Beno, D.L.; Chacko, G.; Asha, H.S.; Rajaratnam, S.; Kapoor, N.; Thomas, N.; Chacko, A.G. USP8, USP48, and BRAF mutations differ in their genotype-phenotype correlation in Asian Indian patients with Cushing’s disease. Endocrine 2022, 75, 549–559. [Google Scholar] [CrossRef]
- Treppiedi, D.; Barbieri, A.M.; Di Muro, G.; Marra, G.; Mangili, F.; Catalano, R.; Esposito, E.; Ferrante, E.; Serban, A.L.; Locatelli, M.; et al. Genetic profiling of a cohort of Italian patients with ACTH-secreting pituitary tumors and characterization of a novel USP8 gene variant. Cancers 2021, 13, 4022. [Google Scholar] [CrossRef] [PubMed]
- Sbiera, S.; Kunz, M.; Weigand, I.; Deutschbein, T.; Dandekar, T.; Fassnacht, M. The new genetic landscape of Cushing’s disease: Deubiquitinases in the spotlight. Cancers 2019, 11, 1761. [Google Scholar] [CrossRef] [Green Version]
- Nieman, L.K. Molecular derangements and the diagnosis of ACTH-Dependent Cushing’s syndrome. Endocr. Rev. 2021, 43, 852–877. [Google Scholar] [CrossRef]
- Dichek, H.L.; Nieman, L.K.; Oldfield, E.H.; Pass, H.I.; Malley, J.D.; Cutler, G.B., Jr. A comparison of the standard high dose dexamethasone suppression test and the overnight 8-mg dexamethasone suppression test for the differential diagnosis of adrenocorticotropin-dependent Cushing’s syndrome. J. Clin. Endocrinol. Metab. 1994, 78, 418–422. [Google Scholar] [CrossRef]
- Lamberts, S.W. Glucocorticoid receptors and Cushing’s disease. Mol. Cell. Endocrinol. 2002, 197, 69–72. [Google Scholar] [CrossRef]
- Karl, M.; Lamberts, S.W.; Koper, J.W.; Katz, D.A.; Huizenga, N.E.; Kino, T.; Haddad, B.R.; Hughes, M.R.; Chrousos, G.P. Cushing’s disease preceded by generalized glucocorticoid resistance: Clinical consequences of a novel, dominant-negative glucocorticoid receptor mutation. Proc. Assoc. Am. Physicians 1996, 108, 296–307. [Google Scholar]
- Song, Z.J.; Reitman, Z.J.; Ma, Z.Y.; Chen, J.H.; Zhang, Q.L.; Shou, X.F.; Huang, C.X.; Wang, Y.F.; Li, S.Q.; Mao, Y.; et al. The genome-wide mutational landscape of pituitary adenomas. Cell Res. 2016, 26, 1255–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tateno, T.; Izumiyama, H.; Doi, M.; Yoshimoto, T.; Shichiri, M.; Inoshita, N.; Oyama, K.; Yamada, S.; Hirata, Y. Differential gene expression in ACTH -secreting and non-functioning pituitary tumors. Eur. J. Endocrinol. 2007, 157, 717–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korbonits, M.; Bujalska, I.; Shimojo, M.; Nobes, J.; Jordan, S.; Grossman, A.B.; Stewart, P.M. Expression of 11 beta-hydroxysteroid dehydrogenase isoenzymes in the human pituitary: Induction of the type 2 enzyme in corticotropinomas and other pituitary tumors. J. Clin. Endocrinol. Metab. 2001, 86, 2728–2733. [Google Scholar] [CrossRef] [Green Version]
- Martens, C.; Bilodeau, S.; Maira, M.; Gauthier, Y.; Drouin, J. Protein–protein interactions and transcriptional antagonism between the subfamily of NGFI-B/Nur77 orphan nuclear receptors and glucocorticoid receptor. Mol. Endocrinol. 2005, 19, 885–897. [Google Scholar] [CrossRef]
- Bilodeau, S.; Vallette-Kasic, S.; Gauthier, Y.; Figarella-Branger, D.; Brue, T.; Berthelet, F.; Lacroix, A.; Batista, D.; Stratakis, C.; Hanson, J.; et al. Role of Brg1 and HDAC2 in GR trans-repression of the pituitary POMC gene and misexpression in Cushing disease. Genes Dev. 2006, 20, 2871–2886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picard, D.; Khursheed, B.; Garabedian, M.J.; Fortin, M.G.; Lindquist, S.; Yamamoto, K.R. Reduced levels of hsp90 compromise steroid receptor action in vivo. Nature 1990, 348, 166–168. [Google Scholar] [CrossRef]
- Tiwari, M.; Oasa, S.; Yamamoto, J.; Mikuni, S.; Kinjo, M. A quantitative study of internal and external interactions of homodimeric glucocorticoid receptor using fluorescence cross-correlation spectroscopy in a live cell. Sci. Rep. 2017, 7, 4336. [Google Scholar] [CrossRef] [Green Version]
- Davies, T.H.; Ning, Y.M.; Sánchez, E.R. A new first step in activation of steroid receptors: Hormone-induced switching of FKBP51 and FKBP52 immunophilins. J. Biol. Chem. 2002, 277, 4597–4600. [Google Scholar] [CrossRef] [Green Version]
- Riebold, M.; Kozany, C.; Freiburger, L.; Sattler, M.; Buchfelder, M.; Hausch, F.; Stalla, G.K.; Paez-Pereda, M.A. A C-terminal HSP90 inhibitor restores glucocorticoid sensitivity and relieves a mouse allograft model of Cushing disease. Nat. Med. 2015, 21, 276–280. [Google Scholar] [CrossRef]
- Resmini, E.; Santos, A.; Aulinas, A.; Webb, S.M.; Vives-Gilabert, Y.; Cox, O.; Wand, G.; Lee, R.S. Reduced DNA methylation of FKBP5 in Cushing’s syndrome. Endocrine 2016, 54, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Bancos, I.; Hatipoglu, B.A.; Yuen, K.C.J.; Chandramohan, L.; Chaudhari, S.; Moraitis, A.G. Evaluation of FKBP5 as a cortisol activity biomarker in patients with ACTH-dependent Cushing syndrome. J. Clin. Transl. Endocrinol. 2021, 24, 100256. [Google Scholar] [CrossRef]
- Kageyama, K.; Iwasaki, Y.; Watanuki, Y.; Niioka, K.; Daimon, M. Differential effects of Fkbp4 and Fkbp5 on regulation of the proopiomelanocortin gene in murine AtT-20 corticotroph cells. Int. J. Mol. Sci. 2021, 22, 5724. [Google Scholar] [CrossRef]
- Du, L.; Bergsneider, M.; Mirsadraei, L.; Young, S.H.; Jonker, J.W.; Downes, M.; Yong, W.H.; Evans, R.M.; Heaney, A.P. Evidence for orphan nuclear receptor TR4 in the etiology of Cushing disease. Proc. Natl. Acad. Sci. USA 2013, 110, 8555–8560. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Du, L.; Heaney, A.P. Testicular receptor-4: Novel regulator of glucocorticoid resistance. J. Clin. Endocrinol. Metab. 2016, 101, 3123–3133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baussart, B.; Gaillard, S. Pituitary surgery for Cushing’s disease. Acta Neurochir. 2021, 163, 3155–3159. [Google Scholar] [CrossRef] [PubMed]
- Biller, B.M.; Grossman, A.B.; Stewart, P.M.; Melmed, S.; Bertagna, X.; Bertherat, J.; Buchfelder, M.; Colao, A.; Hermus, A.R.; Hofland, L.J.; et al. Treatment of adrenocorticotropin-dependent Cushing’s syndrome: A consensus statement. J. Clin. Endocrinol. Metab. 2008, 93, 2454–2462. [Google Scholar] [CrossRef] [Green Version]
- Oki, Y. Medical management of functioning pituitary adenoma: An update. Neurol. Med. Chir. 2014, 54, 958–965. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Nakao, T.; Ogawa, W.; Fukuoka, H. Aggressive Cushing’s disease: Molecular pathology and its therapeutic approach. Front. Endocrinol. 2021, 12, 650791. [Google Scholar] [CrossRef]
- Gheorghiu, M.L. Updates in the outcomes of radiation therapy for Cushing’s disease. Best Pract. Res. Clin. Endocrinol. Metab. 2021, 35, 101514. [Google Scholar] [CrossRef]
- Colao, A.; Petersenn, S.; Newell-Price, J.; Findling, J.W.; Gu, F.; Maldonado, M.; Schoenherr, U.; Mills, D.; Salgado, L.R.; Biller, B.M.; et al. A 12-month phase 3 study of pasireotide in Cushing’s disease. N. Engl. J. Med. 2012, 366, 914–924. [Google Scholar] [CrossRef] [Green Version]
- Lacroix, A.; Gu, F.; Gallardo, W.; Pivonello, R.; Yu, Y.; Witek, P.; Boscaro, M.; Salvatori, R.; Yamada, M.; Tauchmanova, L.; et al. Efficacy and safety of once-monthly pasireotide in Cushing’s disease: A 12 month clinical trial. Lancet Diabetes Endocrinol. 2018, 6, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, A.; Gu, F.; Schopohl, J.; Kandra, A.; Pedroncelli, A.M.; Jin, L.; Pivonello, R. Pasireotide treatment significantly reduces tumor volume in patients with Cushing’s disease: Results from a Phase 3 study. Pituitary 2020, 23, 203–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treppiedi, D.; Giardino, E.; Catalano, R.; Mangili, F.; Vercesi, P.; Sala, E.; Locatelli, M.; Arosio, M.; Spada, A.; Mantovani, G.; et al. Somatostatin analogs regulate tumor corticotrophs growth by reducing ERK1/2 activity. Mol. Cell. Endocrinol. 2019, 483, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Theodoropoulou, M.; Stalla, G.K. Somatostatin receptors: From signaling to clinical practice. Front. Neuroendocrinol. 2013, 34, 228–252. [Google Scholar] [CrossRef] [PubMed]
- Albani, A.; Perez-Rivas, L.G.; Tang, S.; Simon, J.; Lucia, K.E.; Colón-Bolea, P.; Schopohl, J.; Roeber, S.; Buchfelder, M.; Rotermund, R.; et al. Improved pasireotide response in USP8 mutant corticotroph tumours in vitro. Endocr. Relat. Cancer 2022, 29, 503–511. [Google Scholar] [CrossRef]
- Boscaro, M.; Bertherat, J.; Findling, J.; Fleseriu, M.; Atkinson, A.B.; Petersenn, S.; Schopohl, J.; Snyder, P.; Hughes, G.; Trovato, A.; et al. Extended treatment of Cushing’s disease with pasireotide: Results from a 2-year, Phase II study. Pituitary 2014, 17, 320–326. [Google Scholar] [CrossRef] [Green Version]
- Silverstein, J.M. Hyperglycemia induced by pasireotide in patients with Cushing’s disease or acromegaly. Pituitary 2016, 19, 536–543. [Google Scholar] [CrossRef] [Green Version]
- Henry, R.R.; Ciaraldi, T.P.; Armstrong, D.; Burke, P.; Ligueros-Saylan, M.; Mudaliar, S. Hyperglycemia associated with pasireotide: Results from a mechanistic study in healthy volunteers. J. Clin. Endocrinol. Metab. 2013, 98, 3446–3453. [Google Scholar] [CrossRef] [Green Version]
- Chisholm, C.; Greenberg, G.R. Somatostatin-28 regulates GLP-1 secretion via somatostatin receptor subtype 5 in rat intestinal cultures. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E311–E317. [Google Scholar] [CrossRef] [PubMed]
- Kumar, U.; Sasi, R.; Suresh, S.; Patel, A.; Thangaraju, M.; Metrakos, P.; Patel, S.C.; Patel, Y.C. Subtype-selective expression of the five somatostatin receptors (hSSTR1-5) in human pancreatic islet cells: A quantitative double-label immunohistochemical analysis. Diabetes 1999, 48, 77–85. [Google Scholar] [CrossRef]
- Samson, S.L.; Gu, F.; Feldt-Rasmussen, U.; Zhang, S.; Yu, Y.; Witek, P.; Kalra, P.; Pedroncelli, A.M.; Pultar, P.; Jabbour, N.; et al. Managing pasireotide-associated hyperglycemia: A randomized, open-label, Phase IV study. Pituitary 2021, 24, 887–903. [Google Scholar] [CrossRef] [PubMed]
- Palui, R.; Sahoo, J.; Kamalanathan, S.; Kar, S.S.; Selvarajan, S.; Durgia, H. Effect of cabergoline monotherapy in Cushing’s disease: An individual participant data meta-analysis. J. Endocrinol. Investig. 2018, 41, 1445–1455. [Google Scholar] [CrossRef] [PubMed]
- Pivonello, R.; De Martino, M.C.; Cappabianca, P.; De Leo, M.; Faggiano, A.; Lombardi, G.; Hofland, L.J.; Lamberts, S.W.; Colao, A. The medical treatment of Cushing’s disease: Effectiveness of chronic treatment with the dopamine agonist cabergoline in patients unsuccessfully treated by surgery. J. Clin. Endocrinol. Metab. 2009, 94, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Auriemma, R.S.; Pivonello, R.; Ferreri, L.; Priscitelli, P.; Colao, A. Cabergoline use for pituitary tumors and valvular disorders. Endocrinol. Metab. Clin. N. Am. 2015, 44, 89–97. [Google Scholar] [CrossRef]
- Stiles, C.E.; Lloyd, G.; Bhattacharyya, S.; Steeds, R.P.; Boomla, K.; Bestwick, J.P.; Drake, W.M. Incidence of cabergoline-associated valvulopathy in primary care patients with prolactinoma using hard cardiac endpoints. J. Clin. Endocrinol. Metab. 2021, 106, e711–e720. [Google Scholar] [CrossRef]
- Florio, T.; Barbieri, F.; Spaziante, R.; Zona, G.; Hofland, L.J.; van Koetsveld, P.M.; Feelders, R.A.; Stalla, G.K.; Theodoropoulou, M.; Culler, M.D.; et al. Efficacy of a dopamine-somatostatin chimeric molecule, BIM-23A760, in the control of cell growth from primary cultures of human non-functioning pituitary adenomas: A multi-center study. Endocr. Relat. Cancer 2008, 15, 583–596. [Google Scholar] [CrossRef] [Green Version]
- Günther, T.; Tulipano, G.; Dournaud, P.; Bousquet, C.; Csaba, Z.; Kreienkamp, H.J.; Lupp, A.; Korbonits, M.; Castaño, J.P.; Wester, H.J.; et al. International union of basic and clinical pharmacology. CV. Somatostatin Receptors: Structure, Function, Ligands, and New Nomenclature. Pharmacol. Rev. 2018, 70, 763–835. [Google Scholar] [CrossRef] [Green Version]
- Annamalai, A.K.; Dean, A.F.; Kandasamy, N.; Kovacs, K.; Burton, H.; Halsall, D.J.; Shaw, A.S.; Antoun, N.M.; Cheow, H.K.; Kirollos, R.W.; et al. Temozolomide responsiveness in aggressive corticotroph tumours: A case report and review of the literature. Pituitary 2012, 15, 276–287. [Google Scholar] [CrossRef]
- Hirohata, T.; Asano, K.; Ogawa, Y.; Takano, S.; Amano, K.; Isozaki, O.; Iwai, Y.; Sakata, K.; Fukuhara, N.; Nishioka, H.; et al. DNA mismatch repair protein (MSH6) correlated with the responses of atypical pituitary adenomas and pituitary carcinomas to temozolomide: The national cooperative study by the Japan Society for Hypothalamic and Pituitary Tumors. J. Clin. Endocrinol. Metab. 2013, 98, 1130–1136. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Gander, M.; Leyvraz, S.; Newlands, E. Current and future developments in the use of temozolomide for the treatment of brain tumours. Lancet Oncol. 2001, 2, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Ferriere, A.; Tabarin, A. Cushing’s disease. Presse Med. 2021, 50, 104091. [Google Scholar] [CrossRef]
- Varlamov, E.V.; Han, A.J.; Fleseriu, M. Updates in adrenal steroidogenesis inhibitors for Cushing’s syndrome—A practical guide. Best Pract. Res. Clin. Endocrinol. Metab. 2021, 35, 101490. [Google Scholar] [CrossRef]
- Daniel, E.; Aylwin, S.; Mustafa, O.; Ball, S.; Munir, A.; Boelaert, K.; Chortis, V.; Cuthbertson, D.J.; Daousi, C.; Rajeev, S.P.; et al. Effectiveness of metyrapone in treating Cushing’s syndrome: A retrospective multicenter study in 195 patients. J. Clin. Endocrinol. Metab. 2015, 100, 4146–4154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleseriu, M.; Auchus, R.; Bancos, I.; Ben-Shlomo, A.; Bertherat, J.; Biermasz, N.R.; Boguszewski, C.L.; Bronstein, M.D.; Buchfelder, M.; Carmichael, J.D.; et al. Consensus on diagnosis and management of Cushing’s disease: A guideline update. Lancet Diabetes Endocrinol. 2021, 9, 847–875. [Google Scholar] [CrossRef] [PubMed]
- Pivonello, R.; Fleseriu, M.; Newell-Price, J.; Bertagna, X.; Findling, J.; Shimatsu, A.; Gu, F.; Auchus, R.; Leelawattana, R.; Lee, E.J.; et al. Efficacy and safety of osilodrostat in patients with Cushing’s disease (LINC 3): A multicentre phase III study with a double-blind, randomised withdrawal phase. Lancet Diabetes Endocrinol. 2020, 8, 748–761. [Google Scholar] [CrossRef]
- Gadelha, M.; Bex, M.; Feelders, R.A.; Heaney, A.P.; Auchus, R.J.; Gilis-Januszewska, A.; Witek, P.; Belaya, Z.; Yu, Y.; Liao, Z.; et al. Randomized trial of osilodrostat for the treatment of Cushing disease. J. Clin. Endocrinol. Metab. 2022, 107, e2882–e2895. [Google Scholar] [CrossRef]
- Hinojosa-Amaya, J.M.; Cuevas-Ramos, D.; Fleseriu, M. Medical management of Cushing’s syndrome: Current and emerging treatments. Drugs 2019, 79, 935–956. [Google Scholar] [CrossRef]
- Guarda, F.J.; Findling, J.; Yuen, K.C.J.; Fleseriu, M.; Nachtigall, L.B. Mifepristone increases thyroid hormone requirements in patients with central hypothyroidism: A multicenter study. J. Endocr. Soc. 2019, 3, 1707–1714. [Google Scholar] [CrossRef]
- Fleseriu, M.; Findling, J.W.; Koch, C.A.; Schlaffer, S.M.; Buchfelder, M.; Gross, C. Changes in plasma ACTH levels and corticotroph tumor size in patients with Cushing’s disease during long-term treatment with the glucocorticoid receptor antagonist mifepristone. J. Clin. Endocrinol. Metab. 2014, 99, 3718–3727. [Google Scholar] [CrossRef]
- Saini, N.; Mahindra, A. Therapeutic strategies for the treatment of multiple myeloma. Discov. Med. 2013, 15, 251–258. [Google Scholar] [PubMed]
- Jeong, C.H. Inhibition of ubiquitin-specific peptidase 8 suppresses growth of gefitinib-resistant non-small cell lung cancer cells by inducing apoptosis. J. Cancer Prev. 2015, 20, 57–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, M.; Vallese, S.; Peretto, I.; Jacq, X.; Rain, J.C.; Colland, F.; Guedat, P. Synthesis and biological evaluation of 9-oxo-9H-indeno[1,2-b]pyrazine-2,3-dicarbonitrile analogues as potential inhibitors of deubiquitinating enzymes. ChemMedChem 2010, 5, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Seshacharyulu, P.; Ponnusamy, M.P.; Haridas, D.; Jain, M.; Ganti, A.K.; Batra, S.K. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 15–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asari, Y.; Kageyama, K.; Nakada, Y.; Tasso, M.; Takayasu, S.; Niioka, K.; Ishigame, N.; Daimon, M. Inhibitory effects of a selective Jak2 inhibitor on adrenocorticotropic hormone production and proliferation of corticotroph tumor att20 cells. Onco Targets Ther. 2017, 10, 4329–4338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiyama, A.; Kageyama, K.; Murasawa, S.; Ishigame, N.; Niioka, K.; Daimon, M. Inhibition of heat shock protein 90 decreases ACTH production and cell proliferation in AtT-20 cells. Pituitary 2015, 18, 542–553. [Google Scholar] [CrossRef] [PubMed]
- Nakada, Y.; Kageyama, K.; Sugiyama, A.; Desaki, R.; Takayasu, S.; Niioka, K.; Murasawa, S.; Ishigame, N.; Asari, Y.; Iwasaki, Y.; et al. Inhibitory effects of trichostatin A on adrenocorticotropic hormone production and proliferation of corticotroph tumor AtT-20 cells. Endocr. J. 2015, 62, 1083–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Chatain, G.P.; Bugarini, A.; Wang, X.; Maric, D.; Walbridge, S.; Zhuang, Z.; Chittiboina, P. Histone deacetylase inhibitor SAHA is a promising treatment of Cushing disease. J. Clin. Endocrinol. Metab. 2017, 102, 2825–2835. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Cheng, H.; Kwan, W.; Lubieniecka, J.M.; Nielsen, T.O. Histone deacetylase inhibitors induce growth arrest, apoptosis, and differentiation in clear cell sarcoma models. Mol. Cancer Ther. 2008, 7, 1751–1761. [Google Scholar] [CrossRef] [Green Version]
- Hagiwara, R.; Kageyama, K.; Niioka, K.; Takayasu, S.; Tasso, M.; Daimon, M. Involvement of histone deacetylase 1/2 in adrenocorticotropic hormone synthesis and proliferation of corticotroph tumor AtT-20 cells. Peptides 2021, 136, 170441. [Google Scholar] [CrossRef]
- Hagiwara, R.; Kageyama, K.; Iwasaki, Y.; Niioka, K.; Daimon, M. Effects of tubastatin A on adrenocorticotropic hormone synthesis and proliferation of AtT-20 corticotroph tumor cells. Endocr. J. 2022, 69, 1053–1060. [Google Scholar] [CrossRef]
- Feldhaus, A.L.; Anderson, K.; Dutzar, B.; Ojala, E.; McNeill, P.D.; Fan, P.; Mulligan, J.; Marzolf, S.; Karasek, C.; Scalley-Kim, M.; et al. ALD1613, a novel long-acting monoclonal antibody to control ACTH-driven pharmacology. Endocrinology 2017, 158, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duhamel, C.; Ilie, M.D.; Salle, H.; Nassouri, A.S.; Gaillard, S.; Deluche, E.; Assaker, R.; Mortier, L.; Cortet, C.; Raverot, G. Immunotherapy in corticotroph and lactotroph aggressive tumors and carcinomas: Two case reports and a review of the literature. J. Pers. Med. 2020, 10, 88. [Google Scholar] [CrossRef]
- Theodoropoulou, M.; Arzberger, T.; Gruebler, Y.; Jaffrain-Rea, M.L.; Schlegel, J.; Schaaf, L.; Petrangeli, E.; Losa, M.; Stalla, G.K.; Pagotto, U. Expression of epidermal growth factor receptor in neoplastic pituitary cells: Evidence for a role in corticotropinoma cells. J. Endocrinol. 2004, 183, 385–394. [Google Scholar] [CrossRef] [Green Version]
- Andl, C.D.; Mizushima, T.; Oyama, K.; Bowser, M.; Nakagawa, H.; Rustgi, A.K. EGFR-induced cell migration is mediated predominantly by the JAK-STAT pathway in primary esophageal keratinocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G1227–G1237. [Google Scholar] [CrossRef]
- Zheng, Q.; Han, L.; Dong, Y.; Tian, J.; Huang, W.; Liu, Z.; Jia, X.; Jiang, T.; Zhang, J.; Li, X.; et al. JAK2/STAT3 targeted therapy suppresses tumor invasion via disruption of the EGFRvIII/JAK2/STAT3 axis and associated focal adhesion in EGFRvIII-expressing glioblastoma. Neuro Oncol. 2014, 16, 1229–1243. [Google Scholar] [CrossRef] [Green Version]
- Xu, A.W.; Ste-Marie, L.; Kaelin, C.B.; Barsh, G.S. Inactivation of signal transducer and activator of transcription 3 in proopiomelanocortin (POMC) neurons causes decreased POMC expression, mild obesity, and defects in compensatory refeeding. Endocrinology 2007, 148, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Bousquet, C.; Zatelli, M.C.; Melmed, S. Direct regulation of pituitary proopiomelanocortin by STAT3 provides a novel mechanism for immuno-neuroendocrine interfacing. J. Clin. Investig. 2000, 106, 1417–1425. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Schwab, J.H.; Schoenfeld, A.J.; Hornicek, F.J.; Wood, K.B.; Nielsen, G.P.; Choy, E.; Mankin, H.; Duan, Z. A novel target for treatment of chordoma: Signal transducers and activators of transcription 3. Mol. Cancer Ther. 2009, 8, 2597–2605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.T. Dual HDAC and PI3K Inhibitor: A novel potential therapeutic option in Cushing disease. J. Clin. Endocrinol. Metab. 2021, 106, e1036–e1038. [Google Scholar] [CrossRef] [PubMed]
- Reisine, T.; Guild, S. Activators of protein kinase C and cyclic AMP-dependent protein kinase regulate intracellular calcium levels through distinct mechanisms in mouse anterior pituitary tumor cells. Mol. Pharmacol. 1987, 32, 488–496. [Google Scholar] [PubMed]
- Vyas, S.; Bishop, J.F.; Gehlert, D.R.; Patel, J. Effects of protein kinase C down-regulation on secretory events and proopiomelanocortin gene expression in anterior pituitary tumor (AtT-20) cells. J. Neurochem. 1990, 54, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Muşat, M.; Vax, V.V.; Borboli, N.; Gueorguiev, M.; Bonner, S.; Korbonits, M.; Grossman, A.B. Cell cycle dysregulation in pituitary oncogenesis. Front. Horm. Res. 2004, 32, 34–62. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, K.; Sugiyama, A.; Murasawa, S.; Asari, Y.; Niioka, K.; Oki, Y.; Daimon, M. Aphidicolin inhibits cell proliferation via the p53-GADD45β pathway in AtT-20 cells. Endocr. J. 2015, 62, 645–654. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takayasu, S.; Kageyama, K.; Daimon, M. Advances in Molecular Pathophysiology and Targeted Therapy for Cushing’s Disease. Cancers 2023, 15, 496. https://doi.org/10.3390/cancers15020496
Takayasu S, Kageyama K, Daimon M. Advances in Molecular Pathophysiology and Targeted Therapy for Cushing’s Disease. Cancers. 2023; 15(2):496. https://doi.org/10.3390/cancers15020496
Chicago/Turabian StyleTakayasu, Shinobu, Kazunori Kageyama, and Makoto Daimon. 2023. "Advances in Molecular Pathophysiology and Targeted Therapy for Cushing’s Disease" Cancers 15, no. 2: 496. https://doi.org/10.3390/cancers15020496