
Engineering Induced Pluripotent Stem Cells for Cancer Immunotherapy

 , , ,
, , ,  ,
,  , ,
, ,  and
and
Abstract
:Simple Summary
Abstract
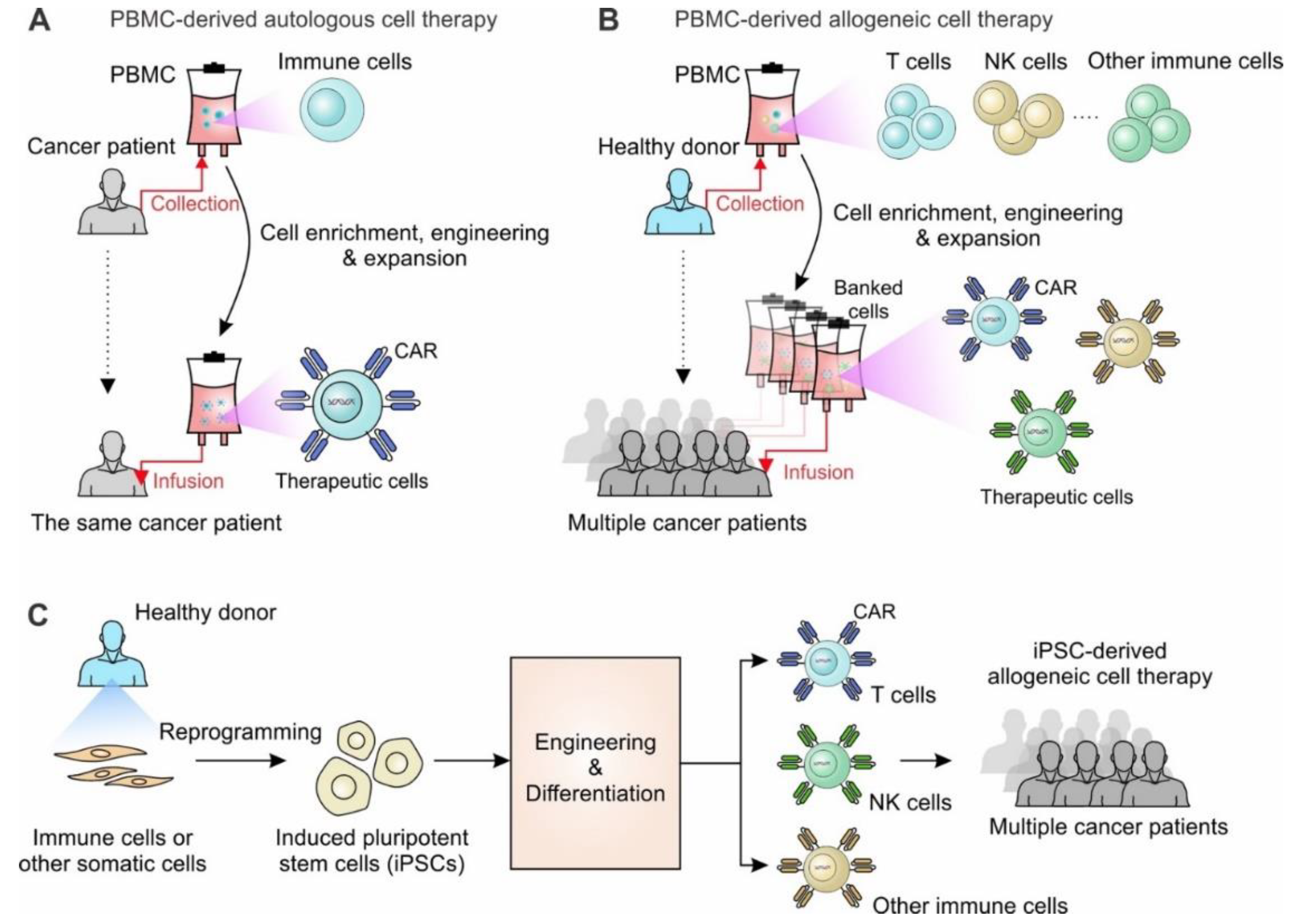
1. Introduction
2. iPSC Technology: Advances and Opportunities
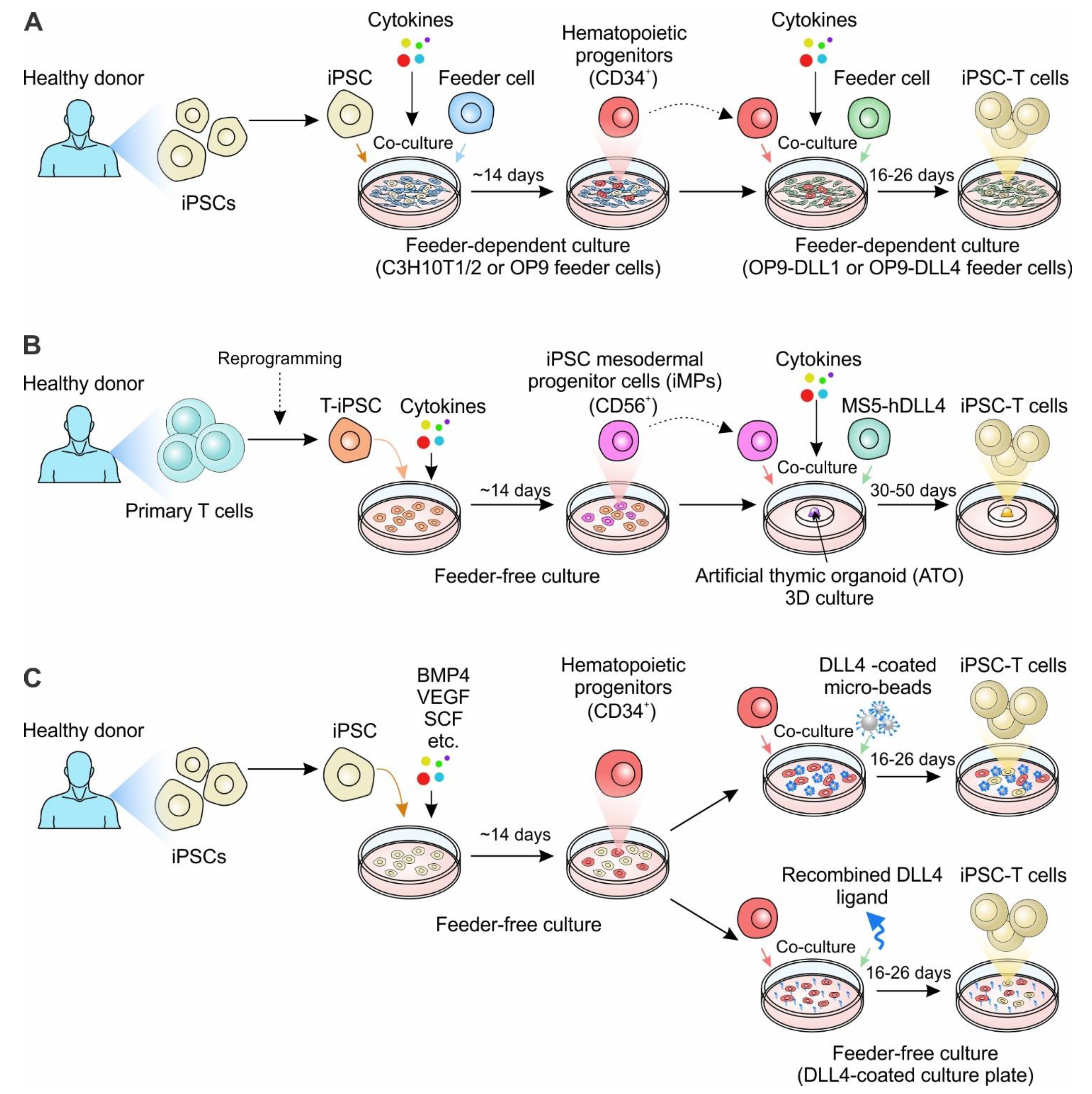
3. Engineering iPSCs for T Cell-Based Therapy
3.1. Engineering T-iPSCs
3.2. Engineering iPSCs in Combination with TCR Transgene Overexpression
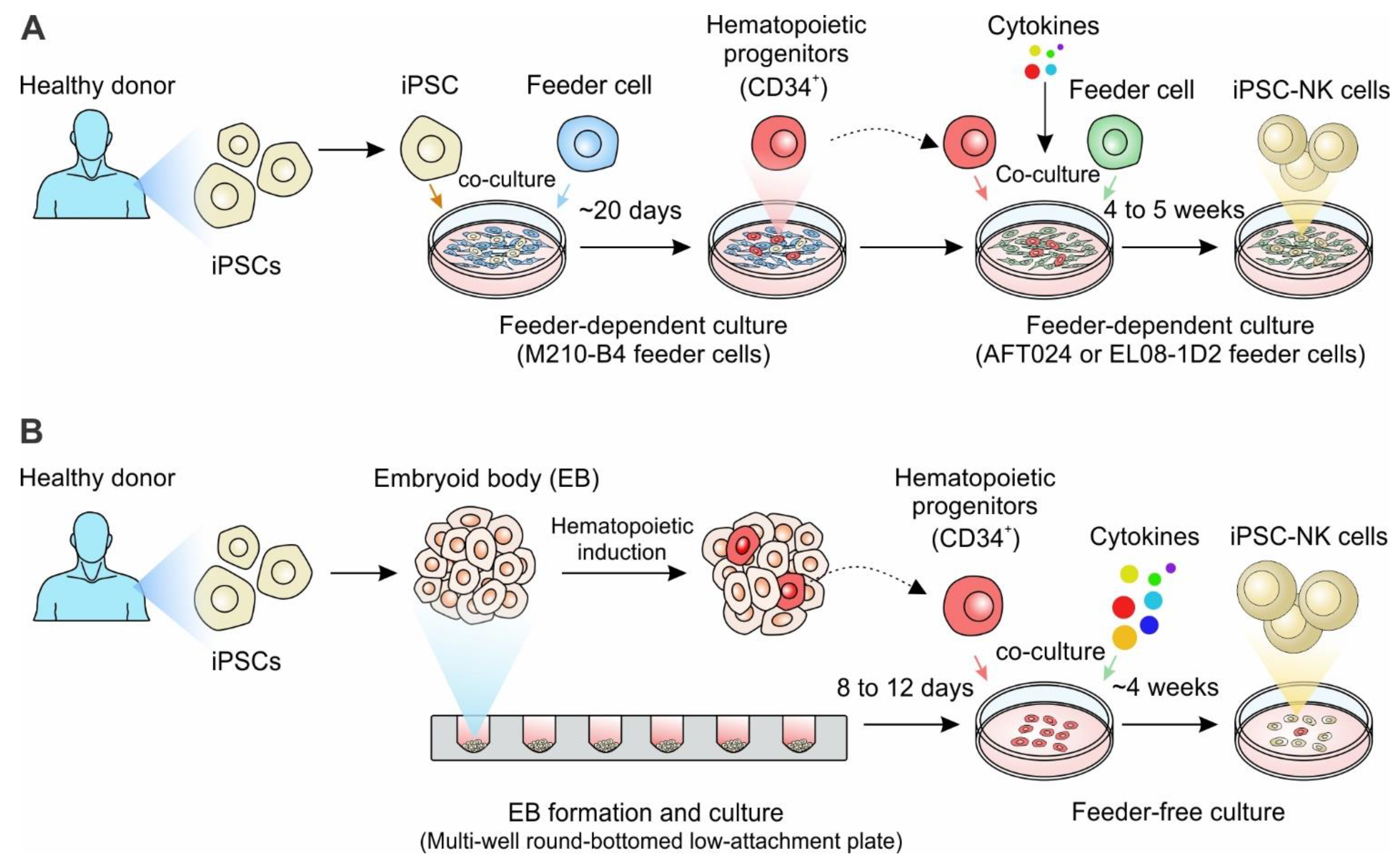
4. Engineering iPSCs for NK Cell-Based Therapy
4.1. NK Cells as a Promising Alternative to T Cells for Cellular Therapy
4.2. Generation of iPSC-NK Cells
4.3. iPSC-NK Cells in Cell Therapy
5. Engineering iPSCs for Other Immune Cell-Based Therapy
5.1. iPSC-Engineered Macrophage Cell Therapy
5.2. iPSC-Engineered MAIT Cell Therapy
5.3. iPSC-Engineered γδ T Cell Therapy
5.4. iPSC-Engineered iNKT Cell Therapy
6. Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ji, H.H.; Tang, X.W.; Dong, Z.; Song, L.; Jia, Y.T. Adverse Event Profiles of Anti-CTLA-4 and Anti-PD-1 Monoclonal Antibodies Alone or in Combination: Analysis of Spontaneous Reports Submitted to FAERS. Clin. Drug Investig. 2019, 39, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Lisi, L.; Lacal, P.M.; Martire, M.; Navarra, P.; Graziani, G. Clinical experience with CTLA-4 blockade for cancer immunotherapy: From the monospecific monoclonal antibody ipilimumab to probodies and bispecific molecules targeting the tumor microenvironment. Pharmacol. Res. 2022, 175, 105997. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zheng, P. How Does an Anti-CTLA-4 Antibody Promote Cancer Immunity? Trends Immunol. 2018, 39, 953–956. [Google Scholar] [CrossRef] [PubMed]
- Lei, Q.; Wang, D.; Sun, K.; Wang, L.; Zhang, Y. Resistance Mechanisms of Anti-PD1/PDL1 Therapy in Solid Tumors. Front. Cell Dev. Biol. 2020, 8, 672. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhang, L.; Yu, J.; Zhang, Y.; Pang, X.; Ma, C.; Shen, M.; Ruan, S.; Wasan, H.S.; Qiu, S. Clinical efficacy and safety of anti-PD-1/PD-L1 inhibitors for the treatment of advanced or metastatic cancer: A systematic review and meta-analysis. Sci. Rep. 2020, 10, 2083. [Google Scholar] [CrossRef] [Green Version]
- Botticelli, A.; Cirillo, A.; Strigari, L.; Valentini, F.; Cerbelli, B.; Scagnoli, S.; Cerbelli, E.; Zizzari, I.G.; Rocca, C.D.; D’Amati, G.; et al. Anti-PD-1 and Anti-PD-L1 in Head and Neck Cancer: A Network Meta-Analysis. Front. Immunol. 2021, 12, 705096. [Google Scholar] [CrossRef]
- Ottaviano, G.; Chiesa, R.; Feuchtinger, T.; Vickers, M.A.; Dickinson, A.; Gennery, A.R.; Veys, P.; Todryk, S. Adoptive T Cell Therapy Strategies for Viral Infections in Patients Receiving Haematopoietic Stem Cell Transplantation. Cells 2019, 8, 47. [Google Scholar] [CrossRef] [Green Version]
- Kaeuferle, T.; Krauss, R.; Blaeschke, F.; Willier, S.; Feuchtinger, T. Strategies of adoptive T -cell transfer to treat refractory viral infections post allogeneic stem cell transplantation. J. Hematol. Oncol. 2019, 12, 13. [Google Scholar] [CrossRef] [Green Version]
- Parajuli, S.; Jorgenson, M.; Meyers, R.O.; Djamali, A.; Galipeau, J. Role of Virus-Specific T Cell Therapy for Cytomegalovirus and BK Infections in Kidney Transplant Recipients. Kidney360 2021, 2, 905–915. [Google Scholar] [CrossRef]
- O’Reilly, R.J.; Prockop, S.; Hasan, A.N.; Koehne, G.; Doubrovina, E. Virus-specific T-cell banks for ‘off the shelf’ adoptive therapy of refractory infections. Bone Marrow Transplant. 2016, 51, 1163–1172. [Google Scholar] [CrossRef] [Green Version]
- Morotti, M.; Albukhari, A.; Alsaadi, A.; Artibani, M.; Brenton, J.D.; Curbishley, S.M.; Dong, T.; Dustin, M.L.; Hu, Z.; McGranahan, N.; et al. Promises and challenges of adoptive T-cell therapies for solid tumours. Br. J. Cancer 2021, 124, 1759–1776. [Google Scholar] [CrossRef] [PubMed]
- Rohaan, M.W.; Wilgenhof, S.; Haanen, J. Adoptive cellular therapies: The current landscape. Virchows Arch. 2019, 474, 449–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, R.S.; Rezvani, K. Chimeric Antigen Receptor Expressing Natural Killer Cells for the Immunotherapy of Cancer. Front. Immunol. 2018, 9, 283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Margolin, K. Tumor-infiltrating lymphocytes in melanoma. Curr. Oncol. Rep. 2012, 14, 468–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van den Berg, J.H.; Heemskerk, B.; van Rooij, N.; Gomez-Eerland, R.; Michels, S.; van Zon, M.; de Boer, R.; Bakker, N.A.M.; Jorritsma-Smit, A.; van Buuren, M.M.; et al. Tumor infiltrating lymphocytes (TIL) therapy in metastatic melanoma: Boosting of neoantigen-specific T cell reactivity and long-term follow-up. J. Immunother. Cancer 2020, 8, e000848. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sun, J.; Chen, K.; Ma, P.; Lei, Q.; Xing, S.; Cao, Z.; Sun, S.; Yu, Z.; Liu, Y.; et al. Perspectives of tumor-infiltrating lymphocyte treatment in solid tumors. BMC Med. 2021, 19, 140. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Watkins, R.; Vilgelm, A.E. Cell Therapy With TILs: Training and Taming T Cells to Fight Cancer. Front. Immunol. 2021, 12, 690499. [Google Scholar] [CrossRef]
- Tsimberidou, A.M.; Van Morris, K.; Vo, H.H.; Eck, S.; Lin, Y.F.; Rivas, J.M.; Andersson, B.S. T-cell receptor-based therapy: An innovative therapeutic approach for solid tumors. J. Hematol. Oncol. 2021, 14, 102. [Google Scholar] [CrossRef]
- Manfredi, F.; Cianciotti, B.C.; Potenza, A.; Tassi, E.; Noviello, M.; Biondi, A.; Ciceri, F.; Bonini, C.; Ruggiero, E. TCR Redirected T Cells for Cancer Treatment: Achievements, Hurdles, and Goals. Front. Immunol. 2020, 11, 1689. [Google Scholar] [CrossRef]
- Zhao, Q.; Jiang, Y.; Xiang, S.; Kaboli, P.J.; Shen, J.; Zhao, Y.; Wu, X.; Du, F.; Li, M.; Cho, C.H.; et al. Engineered TCR-T Cell Immunotherapy in Anticancer Precision Medicine: Pros and Cons. Front. Immunol. 2021, 12, 658753. [Google Scholar] [CrossRef]
- Poncette, L.; Chen, X.; Lorenz, F.K.; Blankenstein, T. Effective NY-ESO-1-specific MHC II-restricted T cell receptors from antigen-negative hosts enhance tumor regression. J. Clin. Investig. 2019, 129, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Bethune, M.T.; Li, X.H.; Yu, J.; McLaughlin, J.; Cheng, D.; Mathis, C.; Moreno, B.H.; Woods, K.; Knights, A.J.; Garcia-Diaz, A.; et al. Isolation and characterization of NY-ESO-1-specific T cell receptors restricted on various MHC molecules. Proc. Natl. Acad. Sci. USA 2018, 115, E10702–E10711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leahy, A.B.; Newman, H.; Li, Y.; Liu, H.; Myers, R.; DiNofia, A.; Dolan, J.G.; Callahan, C.; Baniewicz, D.; Devine, K.; et al. CD19-targeted chimeric antigen receptor T-cell therapy for CNS relapsed or refractory acute lymphocytic leukaemia: A post-hoc analysis of pooled data from five clinical trials. Lancet Haematol. 2021, 8, e711–e722. [Google Scholar] [CrossRef]
- Spiegel, J.Y.; Patel, S.; Muffly, L.; Hossain, N.M.; Oak, J.; Baird, J.H.; Frank, M.J.; Shiraz, P.; Sahaf, B.; Craig, J.; et al. CAR T cells with dual targeting of CD19 and CD22 in adult patients with recurrent or refractory B cell malignancies: A phase 1 trial. Nat. Med. 2021, 27, 1419–1431. [Google Scholar] [CrossRef] [PubMed]
- Halim, L.; Maher, J. CAR T-cell immunotherapy of B-cell malignancy: The story so far. Ther. Adv. Vaccines Immunother. 2020, 8, 2515135520927164. [Google Scholar] [CrossRef]
- Guedan, S.; Calderon, H.; Posey, A.D., Jr.; Maus, M.V. Engineering and Design of Chimeric Antigen Receptors. Mol. Ther. Methods Clin. Dev. 2019, 12, 145–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindner, S.E.; Johnson, S.M.; Brown, C.E.; Wang, L.D. Chimeric antigen receptor signaling: Functional consequences and design implications. Sci. Adv. 2020, 6, eaaz3223. [Google Scholar] [CrossRef]
- Jayaraman, J.; Mellody, M.P.; Hou, A.J.; Desai, R.P.; Fung, A.W.; Pham, A.H.T.; Chen, Y.Y.; Zhao, W. CAR-T design: Elements and their synergistic function. EBioMedicine 2020, 58, 102931. [Google Scholar] [CrossRef]
- Ponterio, E.; De Maria, R.; Haas, T.L. Identification of Targets to Redirect CAR T Cells in Glioblastoma and Colorectal Cancer: An Arduous Venture. Front. Immunol. 2020, 11, 565631. [Google Scholar] [CrossRef]
- Sanber, K.; Savani, B.; Jain, T. Graft-versus-host disease risk after chimeric antigen receptor T-cell therapy: The diametric opposition of T cells. Br. J. Haematol. 2021, 195, 660–668. [Google Scholar] [CrossRef]
- Liu, P.; Liu, M.; Lyu, C.; Lu, W.; Cui, R.; Wang, J.; Li, Q.; Mou, N.; Deng, Q.; Yang, D. Acute Graft-Versus-Host Disease After Humanized Anti-CD19-CAR T Therapy in Relapsed B-ALL Patients After Allogeneic Hematopoietic Stem Cell Transplant. Front. Oncol. 2020, 10, 573822. [Google Scholar] [CrossRef] [PubMed]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gonen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Zhang, Y.; Cheng, C.; Cheng, A.W.; Zhang, X.; Li, N.; Xia, C.; Wei, X.; Liu, X.; Wang, H. CRISPR-Cas9-mediated multiplex gene editing in CAR-T cells. Cell Res. 2017, 27, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Albinger, N.; Hartmann, J.; Ullrich, E. Current status and perspective of CAR-T and CAR-NK cell therapy trials in Germany. Gene Ther. 2021, 28, 513–527. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.A.; Miller, J.S. Exploring the NK cell platform for cancer immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 85–100. [Google Scholar] [CrossRef]
- Liu, S.; Galat, V.; Galat, Y.; Lee, Y.K.A.; Wainwright, D.; Wu, J. NK cell-based cancer immunotherapy: From basic biology to clinical development. J. Hematol. Oncol. 2021, 14, 7. [Google Scholar] [CrossRef]
- Minetto, P.; Guolo, F.; Pesce, S.; Greppi, M.; Obino, V.; Ferretti, E.; Sivori, S.; Genova, C.; Lemoli, R.M.; Marcenaro, E. Harnessing NK Cells for Cancer Treatment. Front. Immunol. 2019, 10, 2836. [Google Scholar] [CrossRef] [Green Version]
- Islam, R.; Pupovac, A.; Evtimov, V.; Boyd, N.; Shu, R.; Boyd, R.; Trounson, A. Enhancing a Natural Killer: Modification of NK Cells for Cancer Immunotherapy. Cells 2021, 10, 1058. [Google Scholar] [CrossRef]
- Kim, H.S.; Kim, J.Y.; Seol, B.; Song, C.L.; Jeong, J.E.; Cho, Y.S. Directly reprogrammed natural killer cells for cancer immunotherapy. Nat. Biomed. Eng. 2021, 5, 1360–1376. [Google Scholar] [CrossRef]
- Caldwell, K.J.; Gottschalk, S.; Talleur, A.C. Allogeneic CAR Cell Therapy-More Than a Pipe Dream. Front. Immunol. 2020, 11, 618427. [Google Scholar] [CrossRef]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ‘Off-the-shelf’ allogeneic CAR T cells: Development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, D.I.; Le Nours, J.; Andrews, D.M.; Uldrich, A.P.; Rossjohn, J. Unconventional T Cell Targets for Cancer Immunotherapy. Immunity 2018, 48, 453–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Lai, Y.S.; Li, Y.; Blum, R.H.; Kaufman, D.S. Concise Review: Human Pluripotent Stem Cells to Produce Cell-Based Cancer Immunotherapy. Stem Cells 2018, 36, 134–145. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, S. Pluripotent Stem Cell-Based Cell Therapy-Promise and Challenges. Cell Stem Cell 2020, 27, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: A decade of progress. Nat. Rev. Drug Discov. 2017, 16, 115–130. [Google Scholar] [CrossRef]
- Huang, C.Y.; Liu, C.L.; Ting, C.Y.; Chiu, Y.T.; Cheng, Y.C.; Nicholson, M.W.; Hsieh, P.C.H. Human iPSC banking: Barriers and opportunities. J. Biomed. Sci. 2019, 26, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, G.; Zhang, Y. Embryonic stem cell and induced pluripotent stem cell: An epigenetic perspective. Cell Res. 2013, 23, 49–69. [Google Scholar] [CrossRef] [Green Version]
- Vizcardo, R.; Masuda, K.; Yamada, D.; Ikawa, T.; Shimizu, K.; Fujii, S.; Koseki, H.; Kawamoto, H. Regeneration of human tumor antigen-specific T cells from iPSCs derived from mature CD8(+) T cells. Cell Stem Cell 2013, 12, 31–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, T.; Kaneko, S.; Kawana-Tachikawa, A.; Tajima, Y.; Goto, H.; Zhu, D.; Nakayama-Hosoya, K.; Iriguchi, S.; Uemura, Y.; Shimizu, T.; et al. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell 2013, 12, 114–126. [Google Scholar] [CrossRef] [Green Version]
- Themeli, M.; Kloss, C.C.; Ciriello, G.; Fedorov, V.D.; Perna, F.; Gonen, M.; Sadelain, M. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat. Biotechnol. 2013, 31, 928–933. [Google Scholar] [CrossRef]
- Wang, B.; Iriguchi, S.; Waseda, M.; Ueda, N.; Ueda, T.; Xu, H.; Minagawa, A.; Ishikawa, A.; Yano, H.; Ishi, T.; et al. Generation of hypoimmunogenic T cells from genetically engineered allogeneic human induced pluripotent stem cells. Nat. Biomed. Eng. 2021, 5, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Blum, R.H.; Bjordahl, R.; Gaidarova, S.; Rogers, P.; Lee, T.T.; Abujarour, R.; Bonello, G.B.; Wu, J.; Tsai, P.F.; et al. Pluripotent stem cell-derived NK cells with high-affinity noncleavable CD16a mediate improved antitumor activity. Blood 2020, 135, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Kaufman, D.S. An Improved Method to Produce Clinical-Scale Natural Killer Cells from Human Pluripotent Stem Cells. Methods Mol. Biol. 2019, 2048, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Ni, Z.; Knorr, D.A.; Kaufman, D.S. Hematopoietic and nature killer cell development from human pluripotent stem cells. Methods Mol. Biol. 2013, 1029, 33–41. [Google Scholar] [CrossRef]
- Capsomidis, A.; Benthall, G.; Van Acker, H.H.; Fisher, J.; Kramer, A.M.; Abeln, Z.; Majani, Y.; Gileadi, T.; Wallace, R.; Gustafsson, K.; et al. Chimeric Antigen Receptor-Engineered Human Gamma Delta T Cells: Enhanced Cytotoxicity with Retention of Cross Presentation. Mol. Ther. 2018, 26, 354–365. [Google Scholar] [CrossRef] [Green Version]
- Kitayama, S.; Zhang, R.; Liu, T.Y.; Ueda, N.; Iriguchi, S.; Yasui, Y.; Kawai, Y.; Tatsumi, M.; Hirai, N.; Mizoro, Y.; et al. Cellular Adjuvant Properties, Direct Cytotoxicity of Re-differentiated Valpha24 Invariant NKT-like Cells from Human Induced Pluripotent Stem Cells. Stem Cell Rep. 2016, 6, 213–227. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Tian, L.; Dai, X.; Yu, H.; Wang, J.; Lei, A.; Zhu, M.; Xu, J.; Zhao, W.; Zhu, Y.; et al. Pluripotent stem cell-derived CAR-macrophage cells with antigen-dependent anti-cancer cell functions. J. Hematol. Oncol. 2020, 13, 153. [Google Scholar] [CrossRef]
- Ackermann, M.; Rafiei Hashtchin, A.; Manstein, F.; Carvalho Oliveira, M.; Kempf, H.; Zweigerdt, R.; Lachmann, N. Continuous human iPSC-macrophage mass production by suspension culture in stirred tank bioreactors. Nat. Protoc. 2022, 17, 513–539. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.W.; Bhattarai, N. CAR-T Cell Therapy: Mechanism, Management, and Mitigation of Inflammatory Toxicities. Front. Immunol. 2021, 12, 693016. [Google Scholar] [CrossRef] [PubMed]
- Maeda, T.; Nagano, S.; Kashima, S.; Terada, K.; Agata, Y.; Ichise, H.; Ohtaka, M.; Nakanishi, M.; Fujiki, F.; Sugiyama, H.; et al. Regeneration of Tumor-Antigen-Specific Cytotoxic T Lymphocytes from iPSCs Transduced with Exogenous TCR Genes. Mol. Ther. Methods Clin. Dev. 2020, 19, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, H.; Masuda, K.; Nagano, S. Regeneration of antigen-specific T cells by using induced pluripotent stem cell (iPSC) technology. Int. Immunol. 2021, 33, 827–833. [Google Scholar] [CrossRef]
- Maeda, T.; Nagano, S.; Ichise, H.; Kataoka, K.; Yamada, D.; Ogawa, S.; Koseki, H.; Kitawaki, T.; Kadowaki, N.; Takaori-Kondo, A.; et al. Regeneration of CD8alphabeta T Cells from T-cell-Derived iPSC Imparts Potent Tumor Antigen-Specific Cytotoxicity. Cancer Res. 2016, 76, 6839–6850. [Google Scholar] [CrossRef] [Green Version]
- Minagawa, A.; Yoshikawa, T.; Yasukawa, M.; Hotta, A.; Kunitomo, M.; Iriguchi, S.; Takiguchi, M.; Kassai, Y.; Imai, E.; Yasui, Y.; et al. Enhancing T Cell Receptor Stability in Rejuvenated iPSC-Derived T Cells Improves Their Use in Cancer Immunotherapy. Cell Stem Cell 2018, 23, 850–858.e4. [Google Scholar] [CrossRef] [Green Version]
- Iriguchi, S.; Yasui, Y.; Kawai, Y.; Arima, S.; Kunitomo, M.; Sato, T.; Ueda, T.; Minagawa, A.; Mishima, Y.; Yanagawa, N.; et al. A clinically applicable and scalable method to regenerate T-cells from iPSCs for off-the-shelf T-cell immunotherapy. Nat. Commun. 2021, 12, 430. [Google Scholar] [CrossRef]
- Trotman-Grant, A.C.; Mohtashami, M.; De Sousa Casal, J.; Martinez, E.C.; Lee, D.; Teichman, S.; Brauer, P.M.; Han, J.; Anderson, M.K.; Zuniga-Pflucker, J.C. DL4-mubeads induce T cell lineage differentiation from stem cells in a stromal cell-free system. Nat. Commun. 2021, 12, 5023. [Google Scholar] [CrossRef]
- Wang, Z.; McWilliams-Koeppen, H.P.; Reza, H.; Ostberg, J.R.; Chen, W.; Wang, X.; Huynh, C.; Vyas, V.; Chang, W.C.; Starr, R.; et al. 3D-organoid culture supports differentiation of human CAR(+) iPSCs into highly functional CAR T cells. Cell Stem Cell 2022, 29, 515–527.e8. [Google Scholar] [CrossRef]
- Sadeqi Nezhad, M.; Abdollahpour-Alitappeh, M.; Rezaei, B.; Yazdanifar, M.; Seifalian, A.M. Induced Pluripotent Stem Cells (iPSCs) Provide a Potentially Unlimited T Cell Source for CAR-T Cell Development and Off-the-Shelf Products. Pharm. Res. 2021, 38, 931–945. [Google Scholar] [CrossRef]
- Karagiannis, P.; Kim, S.I. iPSC-Derived Natural Killer Cells for Cancer Immunotherapy. Mol. Cells 2021, 44, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Bagheri, Y.; Barati, A.; Aghebati-Maleki, A.; Aghebati-Maleki, L.; Yousefi, M. Current progress in cancer immunotherapy based on natural killer cells. Cell Biol. Int. 2021, 45, 2–17. [Google Scholar] [CrossRef] [PubMed]
- Goldenson, B.H.; Zhu, H.; Wang, Y.M.; Heragu, N.; Bernareggi, D.; Ruiz-Cisneros, A.; Bahena, A.; Ask, E.H.; Hoel, H.J.; Malmberg, K.J.; et al. Umbilical Cord Blood and iPSC-Derived Natural Killer Cells Demonstrate Key Differences in Cytotoxic Activity and KIR Profiles. Front. Immunol. 2020, 2435. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.J.; Liu, C.L.; Kuo, M.L.; Shen, C.N.; Shen, C.R. An Alternative Cell Therapy for Cancers: Induced Pluripotent Stem Cell (iPSC)-Derived Natural Killer Cells. Biomedicines 2021, 9, 1323. [Google Scholar] [CrossRef]
- Ruggeri, L.; Capanni, M.; Urbani, E.; Perruccio, K.; Shlomchik, W.D.; Tosti, A.; Posati, S.; Rogaia, D.; Frassoni, F.; Aversa, F.; et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science 2002, 295, 2097–2100. [Google Scholar] [CrossRef] [Green Version]
- Lupo, K.B.; Matosevic, S. Natural Killer Cells as Allogeneic Effectors in Adoptive Cancer Immunotherapy. Cancers 2019, 11, 769. [Google Scholar] [CrossRef] [Green Version]
- Dolstra, H.; Roeven, M.W.H.; Spanholtz, J.; Hangalapura, B.N.; Tordoir, M.; Maas, F.; Leenders, M.; Bohme, F.; Kok, N.; Trilsbeek, C.; et al. Successful Transfer of Umbilical Cord Blood CD34(+) Hematopoietic Stem and Progenitor-derived NK Cells in Older Acute Myeloid Leukemia Patients. Clin. Cancer Res. 2017, 23, 4107–4118. [Google Scholar] [CrossRef] [Green Version]
- Romee, R.; Rosario, M.; Berrien-Elliott, M.M.; Wagner, J.A.; Jewell, B.A.; Schappe, T.; Leong, J.W.; Abdel-Latif, S.; Schneider, S.E.; Willey, S.; et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci. Transl. Med. 2016, 8, 357ra123. [Google Scholar] [CrossRef] [Green Version]
- Michel, T.; Ollert, M.; Zimmer, J. A Hot Topic: Cancer Immunotherapy and Natural Killer Cells. Int. J. Mol. Sci. 2022, 23, 797. [Google Scholar] [CrossRef]
- Heipertz, E.L.; Zynda, E.R.; Stav-Noraas, T.E.; Hungler, A.D.; Boucher, S.E.; Kaur, N.; Vemuri, M.C. Current Perspectives on "Off-The-Shelf" Allogeneic NK and CAR-NK Cell Therapies. Front. Immunol. 2021, 12, 732135. [Google Scholar] [CrossRef]
- Lu, H.; Zhao, X.Y.; Li, Z.Y.; Hu, Y.; Wang, H.F. From CAR-T Cells to CAR-NK Cells: A Developing Immunotherapy Method for Hematological Malignancies. Front. Oncol. 2021, 11, 720501. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.Z.; Dong, H.; Liang, Y.; Ham, J.D.; Rizwan, R.; Chen, J.Z. CAR-NK cells: A promising cellular immunotherapy for cancer. Ebiomedicine 2020, 59, 102975. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.L.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Kerbauy, L.N.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Daher, M.; Basar, R.; Gokdemir, E.; Baran, N.; Uprety, N.; Cortes, A.K.N.; Mendt, M.; Kerbauy, L.N.; Banerjee, P.P.; Shanley, M.; et al. Targeting a cytokine checkpoint enhances the fitness of armored cord blood CAR-NK cells. Blood 2021, 137, 624–636. [Google Scholar] [CrossRef]
- Wrona, E.; Borowiec, M.; Potemski, P. CAR-NK Cells in the Treatment of Solid Tumors. Int. J. Mol. Sci. 2021, 22, 5899. [Google Scholar] [CrossRef]
- Marofi, F.; Saleh, M.M.; Rahman, H.S.; Suksatan, W.; Al-Gazally, M.E.; Abdelbasset, W.K.; Thangavelu, L.; Yumashev, A.V.; Hassanzadeh, A.; Yazdanifar, M.; et al. CAR-engineered NK cells; a promising therapeutic option for treatment of hematological malignancies. Stem Cell Res. Ther. 2021, 12, 374. [Google Scholar] [CrossRef]
- Woll, P.S.; Grzywacz, B.; Tian, X.H.; Marcus, R.K.; Knorr, D.A.; Verneris, M.R.; Kaufman, D.S. Human embryonic stem cells differentiate into a homogeneous population of natural killer cells with potent in vivo antitumor activity. Blood 2009, 113, 6094–6101. [Google Scholar] [CrossRef]
- Ni, Z.Y.; Knorr, D.A.; Clouser, C.L.; Hexum, M.K.; Southern, P.; Mansky, L.M.; Park, I.H.; Kaufman, D.S. Human Pluripotent Stem Cells Produce Natural Killer Cells That Mediate Anti-HIV-1 Activity by Utilizing Diverse Cellular Mechanisms. J. Virol. 2011, 85, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Knorr, D.A.; Ni, Z.; Hermanson, D.; Hexum, M.K.; Bendzick, L.; Cooper, L.J.; Lee, D.A.; Kaufman, D.S. Clinical-scale derivation of natural killer cells from human pluripotent stem cells for cancer therapy. Stem Cells Transl. Med. 2013, 2, 274–283. [Google Scholar] [CrossRef]
- Euchner, J.; Sprissler, J.; Cathomen, T.; Furst, D.; Schrezenmeier, H.; Debatin, K.M.; Schwarz, K.; Felgentreff, K. Natural Killer Cells Generated From Human Induced Pluripotent Stem Cells Mature to CD56(bright)CD16(+)NKp80(+/−) In-Vitro and Express KIR2DL2/DL3 and KIR3DL1. Front. Immunol. 2021, 12, 640672. [Google Scholar] [CrossRef]
- Ng, E.S.; Davis, R.; Stanley, E.G.; Elefanty, A.G. A protocol describing the use of a recombinant protein-based, animal product-free medium (APEL) for human embryonic stem cell differentiation as spin embryoid bodies. Nat. Protoc. 2008, 3, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Bock, A.M.; Knorr, D.; Kaufman, D.S. Development, expansion, and in vivo monitoring of human NK cells from human embryonic stem cells (hESCs) and and induced pluripotent stem cells (iPSCs). J. Vis. Exp. 2013, 74, e50337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermanson, D.L.; Ni, Z.Y.; Knorr, D.A.; Bendzick, L.; Pribyl, L.J.; Geller, M.; Kaufman, D.S. Functional Chimeric Antigen Receptor-Expressing Natural Killer Cells Derived From Human Pluripotent Stem Cells. Blood 2013, 122, 896. [Google Scholar] [CrossRef]
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell 2018, 23, 181–192.e185. [Google Scholar] [CrossRef] [Green Version]
- Bjordahl, R.; Gaidarova, S.; Goodridge, J.P.; Mahmood, S.; Bonello, G.; Robinson, M.; Ruller, C.; Pribadi, M.; Lee, T.; Abujarour, R.; et al. FT576: A Novel Multiplexed Engineered Off-the-Shelf Natural Killer Cell Immunotherapy for the Dual-Targeting of CD38 and Bcma for the Treatment of Multiple Myeloma. Blood 2019, 134, 3214. [Google Scholar] [CrossRef]
- Goodridge, J.P.; Bjordahl, R.; Mahmood, S.; Reiser, J.; Gaidarova, S.; Blum, R.; Cichocki, F.; Chu, H.Y.; Bonello, G.; Lee, T.; et al. FT576: Multi-Specific Off-the-Shelf CAR-NK Cell Therapy Engineered for Enhanced Persistence, Avoidance of Self-Fratricide and Optimized Mab Combination Therapy to Prevent Antigenic Escape and Elicit a Deep and Durable Response in Multiple Myeloma. Blood 2020, 136, 4–5. [Google Scholar] [CrossRef]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef]
- Weiss, G.; Schaible, U.E. Macrophage defense mechanisms against intracellular bacteria. Immunol. Rev. 2015, 264, 182–203. [Google Scholar] [CrossRef] [Green Version]
- Takiguchi, H.; Yang, C.X.; Yang, C.W.T.; Sahin, B.; Whalen, B.A.; Milne, S.; Akata, K.; Yamasaki, K.; Yang, J.S.W.; Cheung, C.Y.; et al. Macrophages with reduced expressions of classical M1 and M2 surface markers in human bronchoalveolar lavage fluid exhibit pro-inflammatory gene signatures. Sci. Rep. 2021, 11, 8282. [Google Scholar] [CrossRef]
- Arango Duque, G.; Descoteaux, A. Macrophage cytokines: Involvement in immunity and infectious diseases. Front. Immunol. 2014, 5, 491. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Biswas, S.K.; Galdiero, M.R.; Sica, A.; Locati, M. Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 2013, 229, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, B.C.; Nakata, K.; Bonella, F.; Campo, I.; Griese, M.; Hamilton, J.; Wang, T.; Morgan, C.; Cottin, V.; McCarthy, C. Pulmonary alveolar proteinosis. Nat. Rev. Dis. Primers 2019, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Mouton, A.J.; Lindsey, M.L. Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl. Res. 2018, 191, 15–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galloway, D.A.; Phillips, A.E.M.; Owen, D.R.J.; Moore, C.S. Phagocytosis in the Brain: Homeostasis and Disease. Front. Immunol. 2019, 10, 790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [Green Version]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef]
- Chen, Y.; Yu, Z.; Tan, X.; Jiang, H.; Xu, Z.; Fang, Y.; Han, D.; Hong, W.; Wei, W.; Tu, J. CAR-macrophage: A new immunotherapy candidate against solid tumors. Biomed. Pharmacother. 2021, 139, 111605. [Google Scholar] [CrossRef]
- Lyadova, I.; Gerasimova, T.; Nenasheva, T. Macrophages Derived From Human Induced Pluripotent Stem Cells: The Diversity of Protocols, Future Prospects, and Outstanding Questions. Front. Cell Dev. Biol. 2021, 9, 640703. [Google Scholar] [CrossRef]
- Bosshart, H.; Heinzelmann, M. THP-1 cells as a model for human monocytes. Ann. Transl. Med. 2016, 4, 438. [Google Scholar] [CrossRef] [Green Version]
- Carpenedo, R.L.; Sargent, C.Y.; McDevitt, T.C. Rotary suspension culture enhances the efficiency, yield, and homogeneity of embryoid body differentiation. Stem Cells 2007, 25, 2224–2234. [Google Scholar] [CrossRef] [PubMed]
- Spelke, D.P.; Ortmann, D.; Khademhosseini, A.; Ferreira, L.; Karp, J.M. Methods for embryoid body formation: The microwell approach. Methods Mol. Biol. 2011, 690, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.W.; Hsiao, Y.H.; Chen, C.C.; Yet, S.F.; Hsu, C.H. A PDMS-Based Microfluidic Hanging Drop Chip for Embryoid Body Formation. Molecules 2016, 21, 882. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, C.; Hale, C.; Mukhopadhyay, S. A Simple Multistep Protocol for Differentiating Human Induced Pluripotent Stem Cells into Functional Macrophages. Methods Mol. Biol. 2018, 1784, 13–28. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Yakala, G.K.; van den Hil, F.E.; Cochrane, A.; Mummery, C.L.; Orlova, V.V. Differentiation and Functional Comparison of Monocytes and Macrophages from hiPSCs with Peripheral Blood Derivatives. Stem Cell Rep. 2019, 12, 1282–1297. [Google Scholar] [CrossRef]
- Ackermann, M.; Kempf, H.; Hetzel, M.; Hesse, C.; Hashtchin, A.R.; Brinkert, K.; Schott, J.W.; Haake, K.; Kuhnel, M.P.; Glage, S.; et al. Bioreactor-based mass production of human iPSC-derived macrophages enables immunotherapies against bacterial airway infections. Nat. Commun. 2018, 9, 5088. [Google Scholar] [CrossRef]
- Toubal, A.; Nel, I.; Lotersztajn, S.; Lehuen, A. Mucosal-associated invariant T cells and disease. Nat. Rev. Immunol. 2019, 19, 643–657. [Google Scholar] [CrossRef] [PubMed]
- Dias, J.; Boulouis, C.; Gorin, J.B.; van den Biggelaar, R.; Lal, K.G.; Gibbs, A.; Loh, L.; Gulam, M.Y.; Sia, W.R.; Bari, S.; et al. The CD4(−)CD8(−) MAIT cell subpopulation is a functionally distinct subset developmentally related to the main CD8(+) MAIT cell pool. Proc. Natl. Acad. Sci. USA 2018, 115, E11513–E11522. [Google Scholar] [CrossRef] [Green Version]
- Vacchini, A.; Chancellor, A.; Spagnuolo, J.; Mori, L.; De Libero, G. MR1-Restricted T Cells Are Unprecedented Cancer Fighters. Front. Immunol. 2020, 11, 751. [Google Scholar] [CrossRef]
- Won, E.J.; Ju, J.K.; Cho, Y.N.; Jin, H.M.; Park, K.J.; Kim, T.J.; Kwon, Y.S.; Kee, H.J.; Kim, J.C.; Kee, S.J.; et al. Clinical relevance of circulating mucosal-associated invariant T cell levels and their anti-cancer activity in patients with mucosal-associated cancer. Oncotarget 2016, 7, 76274–76290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, L.; Lin, Y.; Zheng, W.; Hong, S.; Tang, X.; Zhao, P.; Li, M.; Ni, J.; Li, C.; Wang, L.; et al. Circulating and tumor-infiltrating mucosal associated invariant T (MAIT) cells in colorectal cancer patients. Sci. Rep. 2016, 6, 20358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaler, C.R.; Tun-Abraham, M.E.; Skaro, A.I.; Khazaie, K.; Corbett, A.J.; Mele, T.; Hernandez-Alejandro, R.; Haeryfar, S.M.M. Mucosa-associated invariant T cells infiltrate hepatic metastases in patients with colorectal carcinoma but are rendered dysfunctional within and adjacent to tumor microenvironment. Cancer Immunol. Immunother. 2017, 66, 1563–1575. [Google Scholar] [CrossRef] [PubMed]
- Duan, M.; Goswami, S.; Shi, J.Y.; Wu, L.J.; Wang, X.Y.; Ma, J.Q.; Zhang, Z.; Shi, Y.; Ma, L.J.; Zhang, S.; et al. Activated and Exhausted MAIT Cells Foster Disease Progression and Indicate Poor Outcome in Hepatocellular Carcinoma. Clin. Cancer Res. 2019, 25, 3304–3316. [Google Scholar] [CrossRef] [Green Version]
- Tourret, M.; Talvard-Balland, N.; Lambert, M.; Ben Youssef, G.; Chevalier, M.F.; Bohineust, A.; Yvorra, T.; Morin, F.; Azarnoush, S.; Lantz, O.; et al. Human MAIT cells are devoid of alloreactive potential: Prompting their use as universal cells for adoptive immune therapy. J. Immunother. Cancer 2021, 9, e003123. [Google Scholar] [CrossRef]
- Petley, E.V.; Koay, H.F.; Henderson, M.A.; Sek, K.; Todd, K.L.; Keam, S.P.; Lai, J.; House, I.G.; Li, J.; Zethoven, M.; et al. MAIT cells regulate NK cell-mediated tumor immunity. Nat. Commun. 2021, 12, 4746. [Google Scholar] [CrossRef]
- Dusseaux, M.; Martin, E.; Serriari, N.; Peguillet, I.; Premel, V.; Louis, D.; Milder, M.; Le Bourhis, L.; Soudais, C.; Treiner, E.; et al. Human MAIT cells are xenobiotic-resistant, tissue-targeted, CD161hi IL-17-secreting T cells. Blood 2011, 117, 1250–1259. [Google Scholar] [CrossRef]
- Kawaguchi, K.; Umeda, K.; Hiejima, E.; Iwai, A.; Mikami, M.; Nodomi, S.; Saida, S.; Kato, I.; Hiramatsu, H.; Yasumi, T.; et al. Influence of post-transplant mucosal-associated invariant T cell recovery on the development of acute graft-versus-host disease in allogeneic bone marrow transplantation. Int. J. Hematol. 2018, 108, 66–75. [Google Scholar] [CrossRef]
- Bhattacharyya, A.; Hanafi, L.A.; Sheih, A.; Golob, J.L.; Srinivasan, S.; Boeckh, M.J.; Pergam, S.A.; Mahmood, S.; Baker, K.K.; Gooley, T.A.; et al. Graft-Derived Reconstitution of Mucosal-Associated Invariant T Cells after Allogeneic Hematopoietic Cell Transplantation. Biol. Blood Marrow Transplant. 2018, 24, 242–251. [Google Scholar] [CrossRef] [Green Version]
- Reantragoon, R.; Corbett, A.J.; Sakala, I.G.; Gherardin, N.A.; Furness, J.B.; Chen, Z.; Eckle, S.B.; Uldrich, A.P.; Birkinshaw, R.W.; Patel, O.; et al. Antigen-loaded MR1 tetramers define T cell receptor heterogeneity in mucosal-associated invariant T cells. J. Exp. Med. 2013, 210, 2305–2320. [Google Scholar] [CrossRef]
- Wakao, H.; Yoshikiyo, K.; Koshimizu, U.; Furukawa, T.; Enomoto, K.; Matsunaga, T.; Tanaka, T.; Yasutomi, Y.; Yamada, T.; Minakami, H.; et al. Expansion of functional human mucosal-associated invariant T cells via reprogramming to pluripotency and redifferentiation. Cell Stem Cell 2013, 12, 546–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayday, A.C.; Saito, H.; Gillies, S.D.; Kranz, D.M.; Tanigawa, G.; Eisen, H.N.; Tonegawa, S. Structure, organization, and somatic rearrangement of T cell gamma genes. Cell 1985, 40, 259–269. [Google Scholar] [CrossRef]
- Born, W.; Miles, C.; White, J.; O’Brien, R.; Freed, J.H.; Marrack, P.; Kappler, J.; Kubo, R.T. Peptide sequences of T-cell receptor delta and gamma chains are identical to predicted X and gamma proteins. Nature 1987, 330, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Carding, S.R.; Egan, P.J. Gammadelta T cells: Functional plasticity and heterogeneity. Nat. Rev. Immunol. 2002, 2, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Pistoia, V.; Tumino, N.; Vacca, P.; Veneziani, I.; Moretta, A.; Locatelli, F.; Moretta, L. Human gammadelta T-Cells: From Surface Receptors to the Therapy of High-Risk Leukemias. Front. Immunol. 2018, 9, 984. [Google Scholar] [CrossRef] [Green Version]
- Kunkele, K.P.; Wesch, D.; Oberg, H.H.; Aichinger, M.; Supper, V.; Baumann, C. Vgamma9Vdelta2 T Cells: Can We Re-Purpose a Potent Anti-Infection Mechanism for Cancer Therapy? Cells 2020, 9, 829. [Google Scholar] [CrossRef] [Green Version]
- Kunzmann, V.; Bauer, E.; Wilhelm, M. Gamma/delta T-cell stimulation by pamidronate. N. Engl. J. Med. 1999, 340, 737–738. [Google Scholar] [CrossRef]
- Kabelitz, D.; Serrano, R.; Kouakanou, L.; Peters, C.; Kalyan, S. Cancer immunotherapy with gammadelta T cells: Many paths ahead of us. Cell. Mol. Immunol. 2020, 17, 925–939. [Google Scholar] [CrossRef]
- Handgretinger, R.; Schilbach, K. The potential role of gammadelta T cells after allogeneic HCT for leukemia. Blood 2018, 131, 1063–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yazdanifar, M.; Barbarito, G.; Bertaina, A.; Airoldi, I. gammadelta T Cells: The Ideal Tool for Cancer Immunotherapy. Cells 2020, 9, 1305. [Google Scholar] [CrossRef]
- Simoes, A.E.; Di Lorenzo, B.; Silva-Santos, B. Molecular Determinants of Target Cell Recognition by Human gammadelta T Cells. Front. Immunol. 2018, 9, 929. [Google Scholar] [CrossRef] [Green Version]
- Correia, D.V.; Lopes, A.; Silva-Santos, B. Tumor cell recognition by gammadelta T lymphocytes: T-cell receptor vs. NK-cell receptors. Oncoimmunology 2013, 2, e22892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Yang, W.; Pan, M.; Scully, E.; Girardi, M.; Augenlicht, L.H.; Craft, J.; Yin, Z. Gamma delta T cells provide an early source of interferon gamma in tumor immunity. J. Exp. Med. 2003, 198, 433–442. [Google Scholar] [CrossRef] [Green Version]
- Moser, B.; Brandes, M. Gammadelta T cells: An alternative type of professional APC. Trends Immunol. 2006, 27, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.A.; Golovchenko, N.B.; Edelblum, K.L. gammadelta T cell migration: Separating trafficking from surveillance behaviors at barrier surfaces. Immunol. Rev. 2020, 298, 165–180. [Google Scholar] [CrossRef] [PubMed]
- Braza, M.S.; Klein, B.; Fiol, G.; Rossi, J.F. gammadelta T-cell killing of primary follicular lymphoma cells is dramatically potentiated by GA101, a type II glycoengineered anti-CD20 monoclonal antibody. Haematologica 2011, 96, 400–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deniger, D.C.; Switzer, K.; Mi, T.; Maiti, S.; Hurton, L.; Singh, H.; Huls, H.; Olivares, S.; Lee, D.A.; Champlin, R.E.; et al. Bispecific T-cells expressing polyclonal repertoire of endogenous gammadelta T-cell receptors and introduced CD19-specific chimeric antigen receptor. Mol. Ther. 2013, 21, 638–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makkouk, A.; Yang, X.C.; Barca, T.; Lucas, A.; Turkoz, M.; Wong, J.T.S.; Nishimoto, K.P.; Brodey, M.M.; Tabrizizad, M.; Gundurao, S.R.Y.; et al. Off-the-shelf Vdelta1 gamma delta T cells engineered with glypican-3 (GPC-3)-specific chimeric antigen receptor (CAR) and soluble IL-15 display robust antitumor efficacy against hepatocellular carcinoma. J. Immunother. Cancer 2021, 9, e003441. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, K.P.; Barca, T.; Azameera, A.; Makkouk, A.; Romero, J.M.; Bai, L.; Brodey, M.M.; Kennedy-Wilde, J.; Shao, H.; Papaioannou, S.; et al. Allogeneic CD20-targeted gammadelta T cells exhibit innate and adaptive antitumor activities in preclinical B-cell lymphoma models. Clin. Transl. Immunol. 2022, 11, e1373. [Google Scholar] [CrossRef]
- Rozenbaum, M.; Meir, A.; Aharony, Y.; Itzhaki, O.; Schachter, J.; Bank, I.; Jacoby, E.; Besser, M.J. Gamma-Delta CAR-T Cells Show CAR-Directed and Independent Activity Against Leukemia. Front. Immunol. 2020, 11, 1347. [Google Scholar] [CrossRef]
- Schietinger, A.; Greenberg, P.D. Tolerance and exhaustion: Defining mechanisms of T cell dysfunction. Trends Immunol. 2014, 35, 51–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, D.; Koyanagi-Aoi, M.; Taniguchi-Ikeda, M.; Yoshida, Y.; Azuma, T.; Aoi, T. The Generation of Human gammadeltaT Cell-Derived Induced Pluripotent Stem Cells from Whole Peripheral Blood Mononuclear Cell Culture. Stem Cells Transl. Med. 2018, 7, 34–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohta, R.; Niwa, A.; Taniguchi, Y.; Suzuki, N.M.; Toga, J.; Yagi, E.; Saiki, N.; Nishinaka-Arai, Y.; Okada, C.; Watanabe, A.; et al. Laminin-guided highly efficient endothelial commitment from human pluripotent stem cells. Sci. Rep. 2016, 6, 35680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, J.; Tang, S.Y.; Wang, S. Derivation of mimetic gammadelta T cells endowed with cancer recognition receptors from reprogrammed gammadelta T cell. PLoS ONE 2019, 14, e0216815. [Google Scholar] [CrossRef]
- Berzins, S.P.; Smyth, M.J.; Baxter, A.G. Presumed guilty: Natural killer T cell defects and human disease. Nat. Rev. Immunol. 2011, 11, 131–142. [Google Scholar] [CrossRef]
- Brennan, P.J.; Brigl, M.; Brenner, M.B. Invariant natural killer T cells: An innate activation scheme linked to diverse effector functions. Nat. Rev. Immunol. 2013, 13, 101–117. [Google Scholar] [CrossRef]
- Beekman, E.M.; Porcelli, S.A.; Morita, C.T.; Behar, S.M.; Furlong, S.T.; Brenner, M.B. Recognition of a Lipid Antigen by Cd1-Restricted Alpha-Beta(+) T-Cells. Nature 1994, 372, 691–694. [Google Scholar] [CrossRef]
- Fujii, S.I.; Hemmi, H.; Steinman, R.M. Innate V alpha 14(+) natural killer T cells mature dendritic cells, leading to strong adaptive immunity. Immunol. Rev. 2007, 220, 183–198. [Google Scholar] [CrossRef]
- Mavers, M.; Maas-Bauer, K.; Negrin, R.S. Invariant Natural Killer T Cells As Suppressors of Graft-versus-Host Disease in Allogeneic Hematopoietic Stem Cell Transplantation. Front. Immunol. 2017, 8, 900. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.R.; Zhou, Y.; Kim, Y.J.; Zhu, Y.N.; Ma, F.Y.; Yu, J.J.; Wang, Y.C.; Chen, X.H.; Li, Z.; Zeng, S.; et al. Development of allogeneic HSC-engineered iNKT cells for off-the-shelf cancer immunotherapy. Cell Rep. Med. 2021, 2, 100449. [Google Scholar] [CrossRef]
- Zhu, Y.N.; Smith, D.J.; Zhou, Y.; Li, Y.R.; Yu, J.J.; Lee, D.; Wang, Y.C.; Di Biase, S.; Wang, X.; Hardoy, C.; et al. Development of Hematopoietic Stem Cell-Engineered Invariant Natural Killer T Cell Therapy for Cancer. Cell Stem Cell 2019, 25, 542–557.e9. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.R.; Zhou, Y.; Kramer, A.; Yang, L. Engineering stem cells for cancer immunotherapy. Trends Cancer 2021, 7, 1059–1073. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Li, Y.R.; Zeng, S.; Yang, L. Methods for Studying Mouse and Human Invariant Natural Killer T Cells. Methods Mol. Biol. 2021, 2388, 35–57. [Google Scholar] [CrossRef] [PubMed]
- Watarai, H.; Fujii, S.; Koseki, H.; Taniguchi, M. Murine induced pluripotent stem cells can be derived from and differentiate into natural killer T cells. Differentiation 2010, 80, S45. [Google Scholar] [CrossRef]
- Motohashi, S.; Iinuma, T.; Kurokawa, T.; Koseki, H. Application of iPS Cell-Derived NKT Cells to Cancer Immunotherapy. Gan Kagaku Ryoho 2020, 47, 1411–1414. [Google Scholar]
- Serwold, T.; Hochedlinger, K.; Inlay, M.A.; Jaenisch, R.; Weissman, I.L. Early TCR expression and aberrant T cell development in mice with endogenous prerearranged T cell receptor genes. J. Immunol. 2007, 179, 928–938. [Google Scholar] [CrossRef] [Green Version]
- Ando, M.; Kinoshita, S.; Furukawa, Y.; Ando, J.; Nakauchi, H.; Brenner, M.K. Improving the safety of iPSC-derived T cell therapy. In Molecular Players in iPSC Technology; Academic Press: Cambridge, MA, USA, 2022; pp. 95–115. [Google Scholar] [CrossRef]
- Hou, A.J.; Chen, L.C.; Chen, Y.Y. Navigating CAR-T cells through the solid-tumour microenvironment. Nat. Rev. Drug Discov. 2021, 20, 531–550. [Google Scholar] [CrossRef]
- Kankeu Fonkoua, L.A.; Sirpilla, O.; Sakemura, R.; Siegler, E.L.; Kenderian, S.S. CAR T cell therapy and the tumor microenvironment: Current challenges and opportunities. Mol. Ther.-Oncolytics 2022, 25, 69–77. [Google Scholar] [CrossRef]
- Parihar, R.; Rivas, C.; Huynh, M.; Omer, B.; Lapteva, N.; Metelitsa, L.S.; Gottschalk, S.M.; Rooney, C.M. NK Cells Expressing a Chimeric Activating Receptor Eliminate MDSCs and Rescue Impaired CAR-T Cell Activity against Solid Tumors. Cancer Immunol. Res. 2019, 7, 363–375. [Google Scholar] [CrossRef]
- Giraldo, N.A.; Sanchez-Salas, R.; Peske, J.D.; Vano, Y.; Becht, E.; Petitprez, F.; Validire, P.; Ingels, A.; Cathelineau, X.; Fridman, W.H.; et al. The clinical role of the TME in solid cancer. Br. J. Cancer 2019, 120, 45–53. [Google Scholar] [CrossRef]
- Wang, H.; Kaur, G.; Sankin, A.I.; Chen, F.; Guan, F.; Zang, X. Immune checkpoint blockade and CAR-T cell therapy in hematologic malignancies. J. Hematol. Oncol. 2019, 12, 59. [Google Scholar] [CrossRef] [PubMed]
- Narayan, V.; Barber-Rotenberg, J.S.; Jung, I.Y.; Lacey, S.F.; Rech, A.J.; Davis, M.M.; Hwang, W.T.; Lal, P.; Carpenter, E.L.; Maude, S.L.; et al. PSMA-targeting TGFbeta-insensitive armored CAR T cells in metastatic castration-resistant prostate cancer: A phase 1 trial. Nat. Med. 2022, 28, 724–734. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Boesteanu, A.C.; Binder, Z.A.; Xu, C.; Reid, R.A.; Rodriguez, J.L.; Cook, D.R.; Thokala, R.; Blouch, K.; McGettigan-Croce, B.; et al. Checkpoint Blockade Reverses Anergy in IL-13Ralpha2 Humanized scFv-Based CAR T Cells to Treat Murine and Canine Gliomas. Mol. Ther.-Oncolytics 2018, 11, 20–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Clinical Trial | Description | Cell Product | Malignancies | Company |
|---|---|---|---|---|
| NCT03841110 | FT500 in combination with checkpoint inhibitors against solid tumors | iPSC-NK | Solid tumor | Fate Pharmaceutics |
| NCT04630769, NCT04023071 | FT516 and IL2 with Enoblituzumab for ovarian cancer; FT516 combination with CD20-directed monoclonal antibodies | iPSC-NK (non-cleavable CD16 Fc receptor) | Ovarian cancer; Advanced B-cell lymphoma | Fate Pharmaceutics |
| NCT04555811; NCT04245722 | FT596 with Rituximab as relapse prevention after autologous HSCT for NHL; FT596 as a monotherapy and in combination with anti-CD20 monoclonal antibodies | iPSC-NK(hnCD16, IL15RF, CAR-19) | B cell lymphoma (BCL); Chronic lymphocytic leukemia (CLL) | Fate Pharmaceutics |
| NCT04714372; NCT05069935; NCT04614636 | FT538 in combination with Daratumumab in acute myeloid leukemia (AML); FT538 in combination with monoclonal antibodies in advance solid tumors; FT538 in subjects with advanced hematologic malignancies | iPSC-NK (hnCD16, IL15RF + CD38KO) | AML, MM, solid tumors | Fate Pharmaceutics |
| NCT05182073 | FT576 in subjects with multiple myeloma (MM) | iPSC-NK (hnCD16, IL15RF + CD38KO, CAR-BCMA) | MM | Fate Pharmaceutics |
| NCT04629729 | FT819 in subjects with B-cell malignancies | iPSC-T (CAR-19, TCR-KO) | BCL, CLL, ALL | Fate Pharmaceutics |
| NCT03190278 | Study evaluating safety and efficacy of UCART123 | Allogeneic T-cells expressing anti-CD123 CAR | Relapsed/refractory acute myeloid leukemia (AML) | Cellectis |
| NCAT041500497 | Phase 1 study of UCART22 in patients with R/R CD22+ BALL | Allogeneic T cells expressing anti-CD22 CAR | Relapsed or refractory CD22 + B-cell acute lymphoblastic leukemia | Cellectis |
| NCT04142619 | Study evaluating safety of and efficacy of UCART targeting CS1 in patients with R/R MM | Allogeneic T cells expressing anti-CS1 CAR | Relapsed/refractory MM | Cellectis |
| NCT02735083; NCT02808442; NCT02746952 | Study of UCART19 in patients with R/R BALL | Allogeneic T cells expressing anti-CD19 CAR | R/R BALL | Cellectis |
| NCT04416984 | Safety and efficacy of ALLO-501A anti-CD19 allogeneic CAR T cels in adults with R/R LBCL | Allogeneic T cells expressing anti-CD19 CAR, CD52 KO, TCR KO | R/R LBCL, R/R NHL | Cellectis/Allogene |
| NCT04093596 | Safety and efficacy of ALLO-715 BCMA allogeneic T cells in adults with R/R MM | Allogeneic T cells expressing anti-BCMA CAR, CD52 KO, TCR KO | R/R MM | Cellectis/Allogene |
| NCT04696731 | Safety and efficacy of ALLO-316 in subjects with advanced or metastatic clear cell RCC | Allogeneic T cells targeting CD70 | RCC | Allogene |
| NCT04991948; NCT03692429 | Study of Pembrolizumab treatment after CYAD-101 with FOLFOX reconditioning in mCRC | Allogeneic T cells targeting NKG2DL | mCRC | Celyad |
| NCT04613557 | Safety, activity and cell kinetics of CYAD-211 in patients with R/R MM | Allogeneic T cells targeting BCMA | r/r MM | Celyad |
| NCT03769467; NCT04554914; NCT03394365; NCT02822495; NCT03131934; NCT01192464 | Therapeutic effects of Tebelecleucel in subjects with diseases | EBV-CTL (Tabelecleucel, or tab-cel) | EBV-induced lymphomas and other diseases | Atara |
| NCT03283826 | Phase 1/2 study to evaluate the safety and efficacy of ATA188 in subjects with progressive MS | EBV-CTL | Progressive MS | Atara |
| NCT05252403; NCT03389035 | Residual disease driven strategy for CARCIK-CD19 (CMN-005) in adults/pediatric BCP-ALL | Allogeneic CARCIK-CD19 | ALL | Coimmune |
| NCT04735471; NCT04911478 | A study of ADI-001 in B cell malignancies | Allogeneic CD20-targeted gd T cells | B cell maliganacy | Adicet Bio |
| Cell Therapy | Pros | Cons |
|---|---|---|
| Conventional T cell | Abundant source Easy to expand in vitro Scalable and standardized quality controls for manufacturing | Time-consuming and costly MHC dependent T cell exhaustion GVHD Low ability of trafficking and infiltrating into solid tumors |
| NK cell | No need for previous antigen priming Multiple innate activating receptors that can mediate killing MHC independent No GVHD | Low persistence in the absence of cytokine Low number in patients Low ability of trafficking and infiltrating into solid tumors |
| iNKT cell | Innate and adaptive features Invariant TCR recognizes lipid antigens presented by CD1d No GVHD Low toxicities | Low number in patients May have immunosuppressive properties (Th2, Th17) Limited clinical data with CAR-iNKT cells |
| γδT cell | Innate and adaptive features MHC independent No GVHD Low toxicities | Extremely low number in patients May have immunosuppressive properties (γδ T17, Vδ1 γδ T cells, γδ Treg) Limited clinical data with CAR- γδT cells |
| MAIT cell | Solid tumor-infiltrating capacity Direct anti-tumor cytotoxicity both in vitro and in vivo Resistant to tumor antigen escape | Unclear mechanisms in suppressing tumor Lack of clinical data with CAR-MAIT cells |
| Macrophage cell | Penetration into solid tumors Phagocytosis of tumor cells and innate immune response No GVHD Cross present antigens to αβ T cells | Poor proliferation both in vitro and in vivo May have immunosuppressive properties (M2) Limited clinical data with CAR-macrophage cells |
| Publications | Final Products (Immune Cell Types) | Start Material | iPSC Genetic Modification | Feeder or Feeder-Free | Overall Procedure Time | Major Components in Culture Medium |
|---|---|---|---|---|---|---|
| Nishimura et al., 2013 [49] | Conventional αβ T cells | CD3+ PBMC T cells for a healthy donor; HIV-1-specific CD8+ T cells | NA | Feeder: C3H10T1/2, OP9-DL1 | 33–40 days | VEGF, SCF, FLT-3L, IL-7, IL-15 |
| Themeli et al., 2013 [50] | Conventional αβ T cells | Peripheral blood T lymphocytes (PBL) from a healthy donor | 19CAR-engineering | Half feeder-free; Half feeder: OP9-DLL1 | ~30 days | BMP-4, FGF, VEGF, SCF, FLT-3L, IL-3, IL-7 |
| Vizcardo et al., 2013 [48] | Conventional αβ T cells | JKF6 cells (MART-1 specific TILs) | NA | Feeder: OP9, OP9-DLL1 | ~40 days | SCF, FLT-3L, IL-7, IL-2 |
| Maeda et al., 2016 [65] | Conventional αβ T cells | LMP2-specific CTLs from a healthy donor | NA | Feeder: OP9, OP9-DLL1 | ~6–8 weeks | IL-7, FLT-3L, SCF, IL-2, IL-21 |
| Minagawa et al., 2018 [66] | Conventional αβ T cells | GPC3-specific CTLs from GPC3 peptide-vaccinated patients; Monocyte-derived HLA-typed iPSCs | RAG2 knockout; WT1-TCR transduction | Feeder: C3H10T1/2, OP9-DLL1 | NA | FGF, VEGF, SCF, IL-7, FLT-3L, IL-15, |
| Maeda et al., 2020 [63] | Conventional αβ T cells | Monocytes derived iPSCs from the HLA-homo donor | WT1-TCR transduction | Feeder: OP9, OP9-DLL1 | ~36 days | FGF, IL-7, FLT-3L, SCF, IL-7, IL-21 |
| Iriguchi et al., 2021 [67] | Conventional αβ T cells | Peripheral blood T cells; HIV-1-specific CTLs; RAG2-deleted GPC3 T-iPSCs | NA | Feeder-free | ~42 days | CHIR99021, BMP4, FGF, VEGF, SCF, TPO, FLT-3L, SDF1α |
| Trotman-Grant et al., 2021 [68] | Conventional αβ T cells | Human IPS11- and STIPS cell lines | NA | Feeder-free | ~42 days | BMP4, FGF, VEGF, FLT-3L, SCF, IL-7 |
| Wang et al., 2021 [51] | Conventional αβ T cells | Human iPSC line: GPC3-16-1 (generated from CTLs) | PVR knockout; HLA-E transduction; B2M knockout; CIITA knockout | Feeder: OP9-DLL1 | NA | CHIR99201, FGF, VEGF, BMP4, SCF, FLT-3L, TPO, IL-7, IL-15 |
| Wang et al., 2022 [69] | Conventional αβ T cells | iPSC clones from CD62L+ T cells | 19CAR-engineering | Feeder: MS5-DLL4 | ~51–64 days | BMP4, VEGF, FGF, EGM-2, SB-431542, SCF, FLT3, IL-7, TPO, IL-2, IL-7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.; Li, M.; Zhou, K.; Brown, J.; Tsao, T.; Cen, X.; Husman, T.; Bajpai, A.; Dunn, Z.S.; Yang, L. Engineering Induced Pluripotent Stem Cells for Cancer Immunotherapy. Cancers 2022, 14, 2266. https://doi.org/10.3390/cancers14092266
Zhou Y, Li M, Zhou K, Brown J, Tsao T, Cen X, Husman T, Bajpai A, Dunn ZS, Yang L. Engineering Induced Pluripotent Stem Cells for Cancer Immunotherapy. Cancers. 2022; 14(9):2266. https://doi.org/10.3390/cancers14092266
Chicago/Turabian StyleZhou, Yang, Miao Li, Kuangyi Zhou, James Brown, Tasha Tsao, Xinjian Cen, Tiffany Husman, Aarushi Bajpai, Zachary Spencer Dunn, and Lili Yang. 2022. "Engineering Induced Pluripotent Stem Cells for Cancer Immunotherapy" Cancers 14, no. 9: 2266. https://doi.org/10.3390/cancers14092266