
NRF2 and Key Transcriptional Targets in Melanoma Redox Manipulation



{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. NRF2/KEAP1/ARE Signaling
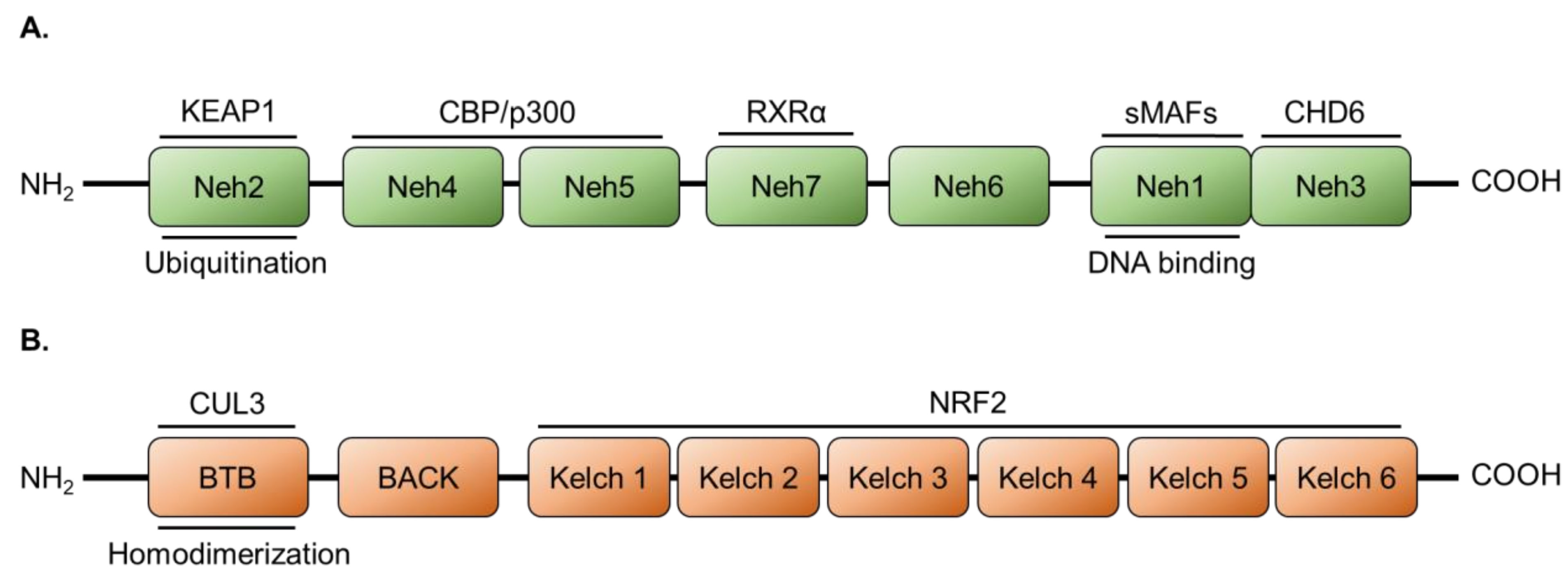
2.1. NRF2 Protein Structure
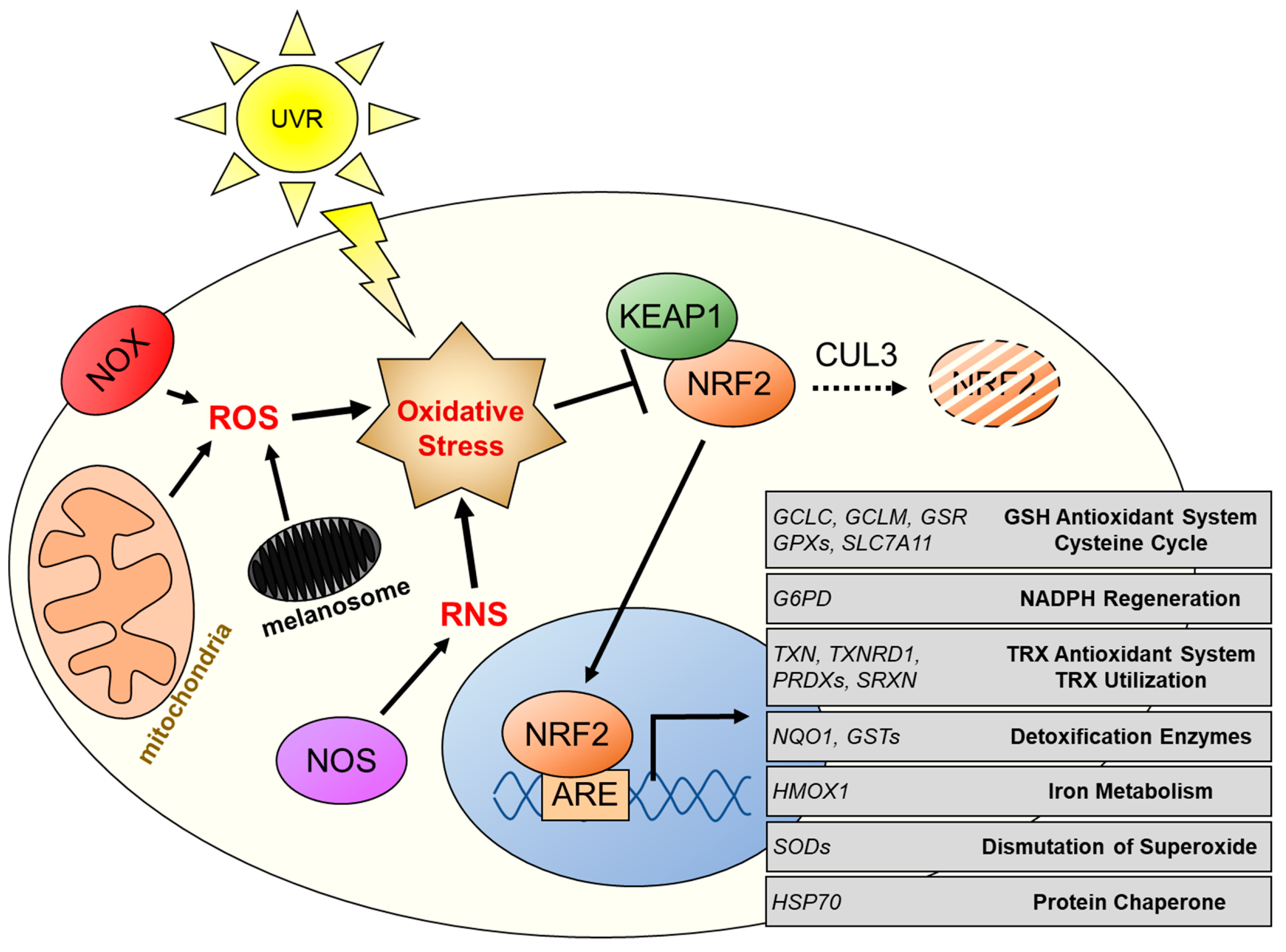
2.2. Regulation of NRF2 by KEAP1/CUL3
2.3. Activation of NRF2 in Melanomas
2.4. Expression and Regulation of NRF2 in Melanomas
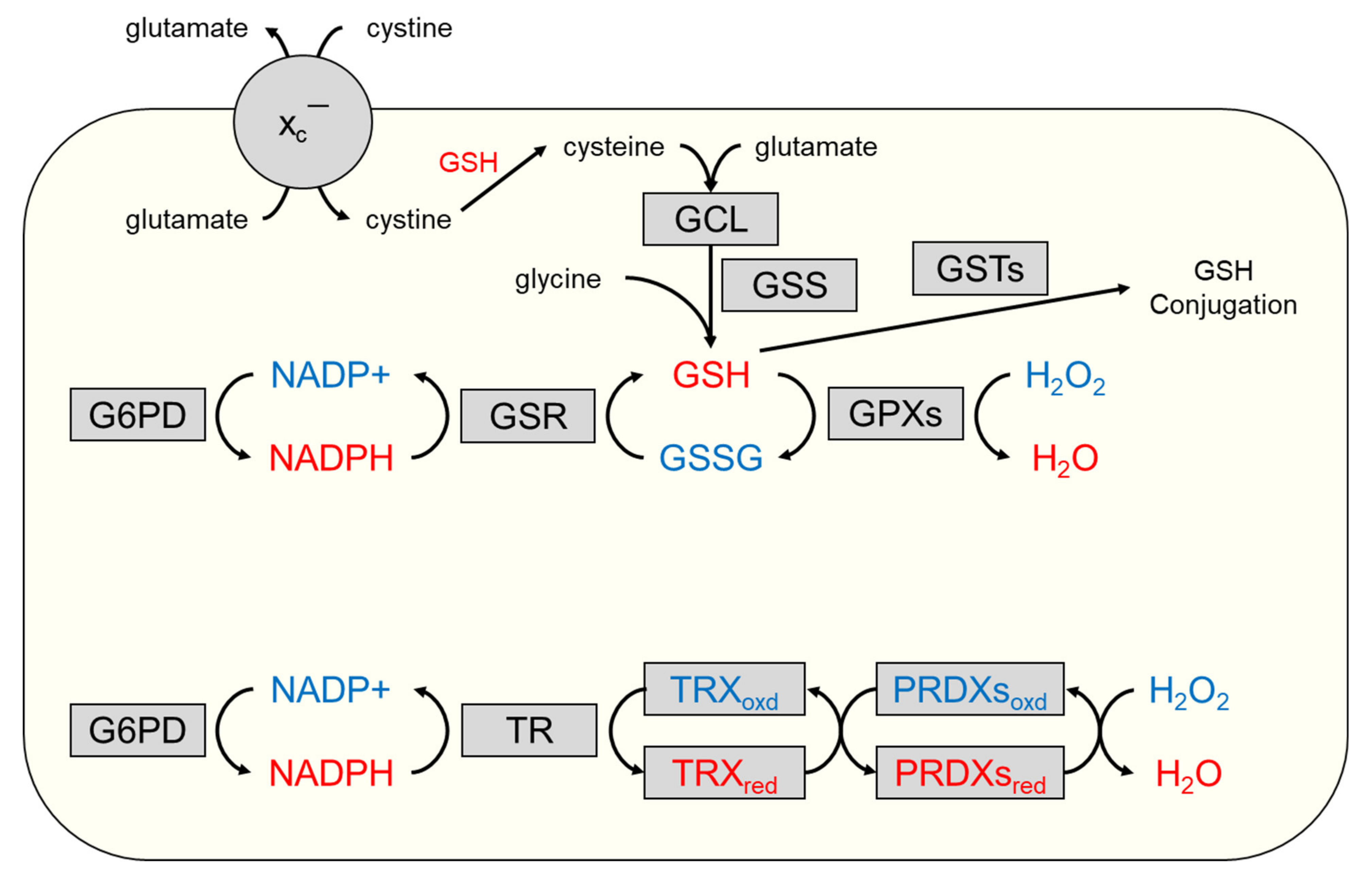
3. The Glutathione Antioxidant System
4. The Thioredoxin Antioxidant System
5. Cystine/Cysteine Cycling
6. Peroxiredoxins and Sulfiredoxin
7. NAD(P)H: Quinone Oxidoreductase 1
8. Heme Oxygenase 1
9. Heat Shock Protein 70
10. Superoxide Dismutase (SOD)
11. Clinical Applications of Exogenous Antioxidants
11.1. Vitamin A
11.2. Vitamin C
11.3. Vitamin E
11.4. Nicotinamide (Niacinamide)
11.5. Selenium
12. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Lim, H.W.; Collins, S.A.B.; Resneck, J.S., Jr.; Bolognia, J.L.; Hodge, J.A.; Rohrer, T.A.; Van Beek, M.J.; Margolis, D.J.; Sober, A.J.; Weinstock, M.A.; et al. The burden of skin disease in the United States. J. Am. Acad. Dermatol. 2017, 76, 958–972. [Google Scholar] [CrossRef] [Green Version]
- Islami, F.; Ward, E.M.; Sung, H.; Cronin, K.A.; Tangka, F.K.L.; Sherman, R.L.; Zhao, J.; Anderson, R.N.; Henley, S.J.; Yabroff, K.R.; et al. Annual Report to the Nation on the Status of Cancer, Part 1: National Cancer Statistics. J. Natl. Cancer Inst. 2021, 113, 1648–1669. [Google Scholar] [CrossRef]
- Lin, J.Y.; Fisher, D.E. Melanocyte biology and skin pigmentation. Nature 2007, 445, 843–850. [Google Scholar] [CrossRef]
- Boissy, R.E. Melanosome transfer to and translocation in the keratinocyte. Exp. Dermatol. 2003, 12 (Suppl. 2), S5–S12. [Google Scholar] [CrossRef] [PubMed]
- Mullenders, L.H.F. Solar UV damage to cellular DNA: From mechanisms to biological effects. Photochem. Photobiol. Sci. 2018, 17, 1842–1852. [Google Scholar] [CrossRef] [PubMed]
- Davis, E.J.; Johnson, D.B.; Sosman, J.A.; Chandra, S. Melanoma: What do all the mutations mean? Cancer 2018, 124, 3490–3499. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; van Akkooi, A.C.J.; Berking, C.; Griewank, K.G.; Gutzmer, R.; Hauschild, A.; Stang, A.; Roesch, A.; Ugurel, S. Melanoma. Lancet 2018, 392, 971–984. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, K.D.; Nogueira, L.; Mariotto, A.B.; Rowland, J.H.; Yabroff, K.R.; Alfano, C.M.; Jemal, A.; Kramer, J.L.; Siegel, R.L. Cancer treatment and survivorship statistics, 2019. CA Cancer J. Clin. 2019, 69, 363–385. [Google Scholar] [CrossRef] [Green Version]
- Domingues, B.; Lopes, J.M.; Soares, P.; Populo, H. Melanoma treatment in review. Immunotargets Ther. 2018, 7, 35–49. [Google Scholar] [CrossRef] [Green Version]
- Batus, M.; Waheed, S.; Ruby, C.; Petersen, L.; Bines, S.D.; Kaufman, H.L. Optimal management of metastatic melanoma: Current strategies and future directions. Am. J. Clin. Dermatol. 2013, 14, 179–194. [Google Scholar] [CrossRef] [Green Version]
- Van Zeijl, M.C.; van den Eertwegh, A.J.; Haanen, J.B.; Wouters, M.W. (Neo)adjuvant systemic therapy for melanoma. Eur. J. Surg. Oncol. 2017, 43, 534–543. [Google Scholar] [CrossRef]
- Garbe, C.; Peris, K.; Hauschild, A.; Saiag, P.; Middleton, M.; Bastholt, L.; Grob, J.J.; Malvehy, J.; Newton-Bishop, J.; Stratigos, A.J.; et al. Diagnosis and treatment of melanoma. European consensus-based interdisciplinary guideline—Update 2016. Eur. J. Cancer 2016, 63, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Christofyllakis, K.; Pföhler, C.; Bewarder, M.; Müller, C.S.L.; Thurner, L.; Rixecker, T.; Vogt, T.; Stilgenbauer, S.; Yordanova, K.; Kaddu-Mulindwa, D. Adjuvant therapy of high-risk (stages IIC-IV) malignant melanoma in the post interferon-alpha era: A systematic review and meta-analysis. Front. Oncol. 2021, 10, 637161. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.A.; Schuchter, L.M. Chemotherapy for Melanoma. Cancer Treat. Res. 2016, 167, 209–229. [Google Scholar] [CrossRef]
- Soengas, M.S.; Lowe, S.W. Apoptosis and melanoma chemoresistance. Oncogene 2003, 22, 3138–3151. [Google Scholar] [CrossRef] [Green Version]
- Flaherty, K.T. Targeting metastatic melanoma. Annu. Rev. Med. 2012, 63, 171–183. [Google Scholar] [CrossRef]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livingstone, E.; Zimmer, L.; Vaubel, J.; Schadendorf, D. BRAF, MEK and KIT inhibitors for melanoma: Adverse events and their management. Chin. Clin. Oncol. 2014, 3, 29. [Google Scholar] [CrossRef]
- Rizos, H.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Fung, C.; Hyman, J.; Haydu, L.E.; Mijatov, B.; Becker, T.M.; Boyd, S.C.; et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: Spectrum and clinical impact. Clin. Cancer Res. 2014, 20, 1965–1977. [Google Scholar] [CrossRef] [Green Version]
- Eroglu, Z.; Ribas, A. Combination Therapy with BRAF and MEK Inhibitors for Melanoma: Latest Evidence and Place in Therapy. Ther. Adv. Med. Oncol. 2016, 8, 48–56. [Google Scholar] [CrossRef] [Green Version]
- Flaherty, K.T.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N. Engl. J. Med. 2012, 367, 107–114. [Google Scholar] [CrossRef] [Green Version]
- Wright, C.J.M.; McCormack, P.L. Trametinib: First Global Approval. Drugs 2013, 73, 1245–1254. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Ascierto, P.A.; Dreno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandala, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karimkhani, C.; Gonzalez, R.; Dellavalle, R.P. A review of novel therapies for melanoma. Am. J. Clin. Dermatol. 2014, 15, 323–337. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: A multicentre, double-blind, phase 3 randomised controlled trial. Lancet 2015, 386, 444–451. [Google Scholar] [CrossRef]
- Pelster, M.S.; Amaria, R.N. Combined Targeted Therapy and Immunotherapy in Melanoma: A Review of the Impact on the Tumor Microenvironment and Outcomes of Early Clinical Trials. Ther. Adv. Med. Oncol. 2019, 11, 1758835919830826. [Google Scholar] [CrossRef] [Green Version]
- Van Breeschoten, J.; Wouters, M.W.J.M.; Hilarius, D.L.; Haanen, J.B.; Blank, C.U.; Aarts, M.J.B.; van den Berkmortel, F.W.P.J.; de Groot, J.-W.B.; Hospers, G.A.P.; Kapiteijn, E.; et al. First-Line BRAF/MEK Inhibitors versus Anti-PD-1 Monotherapy in BRAFV600-Mutant Advanced Melanoma Patients: A Propensity-Matched Survival Analysis. Br. J. Cancer 2021, 124, 1222–1230. [Google Scholar] [CrossRef]
- Gata, V.A.; Lisencu, C.I.; Vlad, C.I.; Piciu, D.; Irimie, A.; Achimas-Cadariu, P. Tumor infiltrating lymphocytes as a prognostic factor in malignant melanoma. Review of the literature. J. Buon. 2017, 22, 592–598. [Google Scholar] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasser, S.; Lim, L.H.K.; Cheung, F.S.G. The role of the tumour microenvironment in immunotherapy. Endocr. Relat. Cancer 2017, 24, T283–T295. [Google Scholar] [CrossRef] [PubMed]
- Khair, D.O.; Bax, H.J.; Mele, S.; Crescioli, S.; Pellizzari, G.; Khiabany, A.; Nakamura, M.; Harris, R.J.; French, E.; Hoffmann, R.M.; et al. Combining Immune Checkpoint Inhibitors: Established and Emerging Targets and Strategies to Improve Outcomes in Melanoma. Front. Immunol. 2019, 10, 453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Gide, T.N.; Wilmott, J.S.; Scolyer, R.A.; Long, G.V. Primary and Acquired Resistance to Immune Checkpoint Inhibitors in Metastatic Melanoma. Clin. Cancer Res. 2018, 24, 1260–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [Green Version]
- D’Orazio, J.; Jarrett, S.; Amaro-Ortiz, A.; Scott, T. UV radiation and the skin. Int. J. Mol. Sci. 2013, 14, 12222–12248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handy, D.E.; Loscalzo, J. Redox regulation of mitochondrial function. Antioxid. Redox Signal. 2012, 16, 1323–1367. [Google Scholar] [CrossRef]
- Denat, L.; Kadekaro, A.L.; Marrot, L.; Leachman, S.A.; Abdel-Malek, Z.A. Melanocytes as instigators and victims of oxidative stress. J. Invest. Dermatol. 2014, 134, 1512–1518. [Google Scholar] [CrossRef] [Green Version]
- Ryoo, I.G.; Kwak, M.K. Regulatory crosstalk between the oxidative stress-related transcription factor Nfe2l2/Nrf2 and mitochondria. Toxicol. Appl. Pharmacol. 2018, 359, 24–33. [Google Scholar] [CrossRef]
- Marrot, L.; Jones, C.; Perez, P.; Meunier, J.R. The significance of Nrf2 pathway in (photo)-oxidative stress response in melanocytes and keratinocytes of the human epidermis. Pigment. Cell Melanoma Res. 2008, 21, 79–88. [Google Scholar] [CrossRef]
- Cassidy, P.B.; Liu, T.; Florell, S.R.; Honeggar, M.; Leachman, S.A.; Boucher, K.M.; Grossman, D. A Phase II Randomized Placebo-Controlled Trial of Oral N-acetylcysteine for Protection of Melanocytic Nevi against UV-Induced Oxidative Stress In Vivo. Cancer Prev. Res. 2017, 10, 36–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpenter, E.L.; Wyant, M.B.; Indra, A.; Ito, S.; Wakamatsu, K.; Merrill, G.F.; Moos, P.J.; Cassidy, P.B.; Leachman, S.A.; Ganguli-Indra, G.; et al. Thioredoxin Reductase 1 Modulates Pigmentation and Photobiology of Murine Melanocytes in vivo. J. Invest. Dermatol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Meyskens, F.L., Jr.; McNulty, S.E.; Buckmeier, J.A.; Tohidian, N.B.; Spillane, T.J.; Kahlon, R.S.; Gonzalez, R.I. Aberrant redox regulation in human metastatic melanoma cells compared to normal melanocytes. Free Radic. Biol. Med. 2001, 31, 799–808. [Google Scholar] [CrossRef] [Green Version]
- Liu-Smith, F.; Dellinger, R.; Meyskens, F.L., Jr. Updates of reactive oxygen species in melanoma etiology and progression. Arch. Biochem. Biophys. 2014, 563, 51–55. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Abildgaard, C.; Guldberg, P. Molecular drivers of cellular metabolic reprogramming in melanoma. Trends Mol. Med. 2015, 21, 164–171. [Google Scholar] [CrossRef]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [Green Version]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Obrador, E.; Liu-Smith, F.; Dellinger, R.W.; Salvador, R.; Meyskens, F.L.; Estrela, J.M. Oxidative stress and antioxidants in the pathophysiology of malignant melanoma. Biol. Chem. 2019, 400, 589–612. [Google Scholar] [CrossRef] [Green Version]
- Carretero, J.; Obrador, E.; Anasagasti, M.J.; Martin, J.J.; Vidal-Vanaclocha, F.; Estrela, J.M. Growth-associated changes in glutathione content correlate with liver metastatic activity of B16 melanoma cells. Clin. Exp. Metastasis 1999, 17, 567–574. [Google Scholar] [CrossRef]
- Vene, R.; Castellani, P.; Delfino, L.; Lucibello, M.; Ciriolo, M.R.; Rubartelli, A. The cystine/cysteine cycle and GSH are independent and crucial antioxidant systems in malignant melanoma cells and represent druggable targets. Antioxid. Redox Signal. 2011, 15, 2439–2453. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, P.B.; Honeggar, M.; Poerschke, R.L.; White, K.; Florell, S.R.; Andtbacka, R.H.; Tross, J.; Anderson, M.; Leachman, S.A.; Moos, P.J. The role of thioredoxin reductase 1 in melanoma metabolism and metastasis. Pigment. Cell Melanoma Res. 2015, 28, 685–695. [Google Scholar] [CrossRef]
- Rotblat, B.; Grunewald, T.G.; Leprivier, G.; Melino, G.; Knight, R.A. Anti-oxidative stress response genes: Bioinformatic analysis of their expression and relevance in multiple cancers. Oncotarget 2013, 4, 2577–2590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bisevac, J.P.; Djukic, M.; Stanojevic, I.; Stevanovic, I.; Mijuskovic, Z.; Djuric, A.; Gobeljic, B.; Banovic, T.; Vojvodic, D. Association Between Oxidative Stress and Melanoma Progression. J. Med. Biochem. 2018, 37, 12–20. [Google Scholar] [CrossRef]
- Sander, C.S.; Hamm, F.; Elsner, P.; Thiele, J.J. Oxidative stress in malignant melanoma and non-melanoma skin cancer. Br. J. Dermatol. 2003, 148, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Garate, M.; Wani, A.A.; Li, G. The NAD(P)H:Quinone Oxidoreductase 1 induces cell cycle progression and proliferation of melanoma cells. Free Radic. Biol. Med. 2010, 48, 1601–1609. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wu, Y.; Bian, C.; Nisar, M.F.; Wang, M.; Hu, X.; Diao, Q.; Nian, W.; Wang, E.; Xu, W.; et al. Heme oxygenase 1 facilitates cell proliferation via the B-Raf-ERK signaling pathway in melanoma. Cell Commun. Signal. 2019, 17, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannavo, S.P.; Tonacci, A.; Bertino, L.; Casciaro, M.; Borgia, F.; Gangemi, S. The role of oxidative stress in the biology of melanoma: A systematic review. Pathol. Res. Pract. 2019, 215, 21–28. [Google Scholar] [CrossRef]
- Ramasamy, P.; Larkin, A.M.; Linge, A.; Tiernan, D.; McAree, F.; Horgan, N.; Moriarty, P.; Beatty, S.; Murphy, C.C.; Clynes, M.; et al. PRDX3 is associated with metastasis and poor survival in uveal melanoma. J. Clin. Pathol. 2020, 73, 408–412. [Google Scholar] [CrossRef]
- Lokaj, K.; Meierjohann, S.; Schutz, C.; Teutschbein, J.; Schartl, M.; Sickmann, A. Quantitative differential proteome analysis in an animal model for human melanoma. J. Proteome Res. 2009, 8, 1818–1827. [Google Scholar] [CrossRef] [PubMed]
- Le Gal, K.; Ibrahim, M.X.; Wiel, C.; Sayin, V.I.; Akula, M.K.; Karlsson, C.; Dalin, M.G.; Akyurek, L.M.; Lindahl, P.; Nilsson, J.; et al. Antioxidants can increase melanoma metastasis in mice. Sci. Transl. Med. 2015, 7, 308re8. [Google Scholar] [CrossRef]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igarashi, K.; Kataoka, K.; Itoh, K.; Hayashi, N.; Nishizawa, M.; Yamamoto, M. Regulation of transcription by dimerization of erythroid factor NF-E2 p45 with small Maf proteins. Nature 1994, 367, 568–572. [Google Scholar] [CrossRef] [PubMed]
- Katsuoka, F.; Yamamoto, M. Small Maf proteins (MafF, MafG, MafK): History, structure and function. Gene 2016, 586, 197–205. [Google Scholar] [CrossRef] [Green Version]
- Nioi, P.; Nguyen, T.; Sherratt, P.J.; Pickett, C.B. The carboxy-terminal Neh3 domain of Nrf2 is required for transcriptional activation. Mol. Cell Biol. 2005, 25, 10895–10906. [Google Scholar] [CrossRef] [Green Version]
- Nam, L.B.; Keum, Y.S. Binding partners of NRF2: Functions and regulatory mechanisms. Arch. Biochem. Biophys. 2019, 678, 108184. [Google Scholar] [CrossRef]
- Wang, H.; Liu, K.; Geng, M.; Gao, P.; Wu, X.; Hai, Y.; Li, Y.; Li, Y.; Luo, L.; Hayes, J.D.; et al. RXRalpha inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res. 2013, 73, 3097–3108. [Google Scholar] [CrossRef] [Green Version]
- Katoh, Y.; Itoh, K.; Yoshida, E.; Miyagishi, M.; Fukamizu, A.; Yamamoto, M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells 2001, 6, 857–868. [Google Scholar] [CrossRef]
- Ganner, A.; Pfeiffer, Z.C.; Wingendorf, L.; Kreis, S.; Klein, M.; Walz, G.; Neumann-Haefelin, E. The acetyltransferase p300 regulates NRF2 stability and localization. Biochem. Biophys. Res. Commun. 2020, 524, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Canning, P.; Sorrell, F.J.; Bullock, A.N. Structural basis of Keap1 interactions with Nrf2. Free Radic. Biol. Med. 2015, 88, 101–107. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghunath, A.; Sundarraj, K.; Nagarajan, R.; Arfuso, F.; Bian, J.; Kumar, A.P.; Sethi, G.; Perumal, E. Antioxidant response elements: Discovery, classes, regulation and potential applications. Redox Biol. 2018, 17, 297–314. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.A.; Kwak, M.K. The Nrf2 system as a potential target for the development of indirect antioxidants. Molecules 2010, 15, 7266–7291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jessen, C.; Kress, J.K.C.; Baluapuri, A.; Hufnagel, A.; Schmitz, W.; Kneitz, S.; Roth, S.; Marquardt, A.; Appenzeller, S.; Ade, C.P.; et al. The transcription factor NRF2 enhances melanoma malignancy by blocking differentiation and inducing COX2 expression. Oncogene 2020, 39, 6841–6855. [Google Scholar] [CrossRef]
- Kress, J.K.C.; Jessen, C.; Marquardt, A.; Hufnagel, A.; Meierjohann, S. NRF2 Enables EGFR Signaling in Melanoma Cells. Int. J. Mol. Sci. 2021, 22, 3803. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef]
- Ma, C.S.; Lv, Q.M.; Zhang, K.R.; Tang, Y.B.; Zhang, Y.F.; Shen, Y.; Lei, H.M.; Zhu, L. NRF2-GPX4/SOD2 axis imparts resistance to EGFR-tyrosine kinase inhibitors in non-small-cell lung cancer cells. Acta Pharmacol. Sin. 2021, 42, 613–623. [Google Scholar] [CrossRef]
- Choi, B.H.; Ryu, D.Y.; Ryoo, I.G.; Kwak, M.K. NFE2L2/NRF2 silencing-inducible miR-206 targets c-MET/EGFR and suppresses BCRP/ABCG2 in cancer cells. Oncotarget 2017, 8, 107188–107205. [Google Scholar] [CrossRef] [Green Version]
- Pizzimenti, S.; Ribero, S.; Cucci, M.A.; Grattarola, M.; Monge, C.; Dianzani, C.; Barrera, G.; Muzio, G. Oxidative Stress-Related Mechanisms in Melanoma and in the Acquired Resistance to Targeted Therapies. Antioxidants 2021, 10, 1942. [Google Scholar] [CrossRef]
- Liu, F.; Gomez Garcia, A.M.; Meyskens, F.L., Jr. NADPH oxidase 1 overexpression enhances invasion via matrix metalloproteinase-2 and epithelial-mesenchymal transition in melanoma cells. J. Invest. Dermatol. 2012, 132, 2033–2041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, T.; Kuang, Y.; Zhang, C.; Zhang, Z.; Chen, L.; Li, B.; Li, Y.; Wang, Y.; Yang, H.; Han, Q.; et al. Glucose-6-phosphate dehydrogenase and NADPH oxidase 4 control STAT3 activity in melanoma cells through a pathway involving reactive oxygen species, c-SRC and SHP2. Am. J. Cancer Res. 2015, 5, 1610–1620. [Google Scholar] [PubMed]
- Paudel, B.B.; Lewis, J.E.; Hardeman, K.N.; Hayford, C.E.; Robbins, C.J.; Stauffer, P.E.; Codreanu, S.G.; Sherrod, S.D.; McLean, J.A.; Kemp, M.L.; et al. An Integrative Gene Expression and Mathematical Flux Balance Analysis Identifies Targetable Redox Vulnerabilities in Melanoma Cells. Cancer Res. 2020, 80, 4565–4577. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Misner, B.; Ji, H.; Poulos, T.L.; Silverman, R.B.; Meyskens, F.L.; Yang, S. Targeting nitric oxide signaling with nNOS inhibitors as a novel strategy for the therapy and prevention of human melanoma. Antioxid. Redox Signal. 2013, 19, 433–447. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Rivera, E.; Jayaraman, P.; Parikh, F.; Davies, M.A.; Ekmekcioglu, S.; Izadmehr, S.; Milton, D.R.; Chipuk, J.E.; Grimm, E.A.; Estrada, Y.; et al. Inducible nitric oxide synthase drives mTOR pathway activation and proliferation of human melanoma by reversible nitrosylation of TSC2. Cancer Res. 2014, 74, 1067–1078. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Ren, F.; Qi, C.; Xu, L.; Fang, Y.; Liang, M.; Feng, J.; Chen, B.; Ning, W.; Cao, J. Intermittent hypoxia promotes melanoma lung metastasis via oxidative stress and inflammation responses in a mouse model of obstructive sleep apnea. Respir. Res. 2018, 19, 28. [Google Scholar] [CrossRef] [Green Version]
- Hamalainen, M.; Teppo, H.R.; Skarp, S.; Haapasaari, K.M.; Porvari, K.; Vuopala, K.; Kietzmann, T.; Karihtala, P. NRF1 and NRF2 mRNA and Protein Expression Decrease Early during Melanoma Carcinogenesis: An Insight into Survival and MicroRNAs. Oxid. Med. Cell Longev. 2019, 2019, 2647068. [Google Scholar] [CrossRef] [Green Version]
- Hintsala, H.R.; Jokinen, E.; Haapasaari, K.M.; Moza, M.; Ristimaki, A.; Soini, Y.; Koivunen, J.; Karihtala, P. Nrf2/Keap1 Pathway and Expression of Oxidative Stress Lesions 8-hydroxy-2′-deoxyguanosine and Nitrotyrosine in Melanoma. Anticancer Res. 2016, 36, 1497–1506. [Google Scholar]
- Weitzenbock, H.P.; Gschwendtner, A.; Wiesner, C.; Depke, M.; Schmidt, F.; Trautinger, F.; Hengstschlager, M.; Hundsberger, H.; Mikula, M. Proteome analysis of NRF2 inhibition in melanoma reveals CD44 up-regulation and increased apoptosis resistance upon vemurafenib treatment. Cancer Med. 2022, 11, 956–967. [Google Scholar] [CrossRef]
- Granados, K.; Poelchen, J.; Novak, D.; Utikal, J. Cellular Reprogramming-A Model for Melanoma Cellular Plasticity. Int. J. Mol. Sci. 2020, 21, 8274. [Google Scholar] [CrossRef] [PubMed]
- Goding, C.R. Commentary. A picture of Mitf in melanoma immortality. Oncogene 2011, 30, 2304–2306. [Google Scholar] [CrossRef]
- Miura, S.; Shibazaki, M.; Kasai, S.; Yasuhira, S.; Watanabe, A.; Inoue, T.; Kageshita, Y.; Tsunoda, K.; Takahashi, K.; Akasaka, T.; et al. A somatic mutation of the KEAP1 gene in malignant melanoma is involved in aberrant NRF2 activation and an increase in intrinsic drug resistance. J. Invest. Dermatol. 2014, 134, 553–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Chin, Y.E.; Zhang, D.D. Acetylation of Nrf2 by p300/CBP augments promoter-specific DNA binding of Nrf2 during the antioxidant response. Mol. Cell Biol. 2009, 29, 2658–2672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, W.M.; Baker, A.C.; Beroukhim, R.; Winckler, W.; Feng, W.; Marmion, J.M.; Laine, E.; Greulich, H.; Tseng, H.; Gates, C.; et al. Modeling genomic diversity and tumor dependency in malignant melanoma. Cancer Res. 2008, 68, 664–673. [Google Scholar] [CrossRef] [Green Version]
- Rotte, A.; Bhandaru, M.; Cheng, Y.; Sjoestroem, C.; Martinka, M.; Li, G. Decreased expression of nuclear p300 is associated with disease progression and worse prognosis of melanoma patients. PLoS ONE 2013, 8, e75405. [Google Scholar] [CrossRef] [Green Version]
- Hartman, M.L.; Czyz, M. MITF in melanoma: Mechanisms behind its expression and activity. Cell Mol. Life Sci. 2015, 72, 1249–1260. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; He, Y.; Robinson, V.; Yang, Z.; Hessler, P.; Lasko, L.M.; Lu, X.; Bhathena, A.; Lai, A.; Uziel, T.; et al. Targeting Lineage-specific MITF Pathway in Human Melanoma Cell Lines by A-485, the Selective Small-molecule Inhibitor of p300/CBP. Mol. Cancer Ther. 2018, 17, 2543–2550. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Wen, Y.; Zhang, X.; Zhao, F.; Lv, K.; Shi, J.H.; Shen, S.; Pan, X. Transcriptional network constituted of CBP, Ku70, NOX2, and BAX prevents the cell death of necrosis, paraptosis, and apoptosis in human melanoma. Cell Death Discov. 2021, 7, 40. [Google Scholar] [CrossRef]
- Chakravarti, N.; Lotan, R.; Diwan, A.H.; Warneke, C.L.; Johnson, M.M.; Prieto, V.G. Decreased expression of retinoid receptors in melanoma: Entailment in tumorigenesis and prognosis. Clin. Cancer Res. 2007, 13, 4817–4824. [Google Scholar] [CrossRef] [Green Version]
- Coleman, D.J.; Garcia, G.; Hyter, S.; Jang, H.S.; Chagani, S.; Liang, X.; Larue, L.; Ganguli-Indra, G.; Indra, A.K. Retinoid-X-receptors (alpha/beta) in melanocytes modulate innate immune responses and differentially regulate cell survival following UV irradiation. PLoS Genet. 2014, 10, e1004321. [Google Scholar] [CrossRef]
- Chorley, B.N.; Campbell, M.R.; Wang, X.; Karaca, M.; Sambandan, D.; Bangura, F.; Xue, P.; Pi, J.; Kleeberger, S.R.; Bell, D.A. Identification of novel NRF2-regulated genes by ChIP-Seq: Influence on retinoid X receptor alpha. Nucleic Acids Res. 2012, 40, 7416–7429. [Google Scholar] [CrossRef] [Green Version]
- Demary, K.; Wong, L.; Liou, J.S.; Faller, D.V.; Spanjaard, R.A. Redox control of retinoic acid receptor activity: A novel mechanism for retinoic acid resistance in melanoma cells. Endocrinology 2001, 142, 2600–2605. [Google Scholar] [CrossRef] [PubMed]
- Daud, A.I.; Wolchok, J.D.; Robert, C.; Hwu, W.J.; Weber, J.S.; Ribas, A.; Hodi, F.S.; Joshua, A.M.; Kefford, R.; Hersey, P.; et al. Programmed Death-Ligand 1 Expression and Response to the Anti-Programmed Death 1 Antibody Pembrolizumab in Melanoma. J. Clin. Oncol. 2016, 34, 4102–4109. [Google Scholar] [CrossRef] [PubMed]
- Thiem, A.; Hesbacher, S.; Kneitz, H.; di Primio, T.; Heppt, M.V.; Hermanns, H.M.; Goebeler, M.; Meierjohann, S.; Houben, R.; Schrama, D. IFN-gamma-induced PD-L1 expression in melanoma depends on p53 expression. J. Exp. Clin. Cancer Res. 2019, 38, 397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, B.; Tang, L.; Chen, S.; Yin, C.; Peng, S.; Li, X.; Liu, T.; Liu, W.; Han, C.; Stawski, L.; et al. Targeting the upstream transcriptional activator of PD-L1 as an alternative strategy in melanoma therapy. Oncogene 2018, 37, 4941–4954. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef] [Green Version]
- Mulcahy, R.T.; Wartman, M.A.; Bailey, H.H.; Gipp, J.J. Constitutive and beta-naphthoflavone-induced expression of the human gamma-glutamylcysteine synthetase heavy subunit gene is regulated by a distal antioxidant response element/TRE sequence. J. Biol. Chem. 1997, 272, 7445–7454. [Google Scholar] [CrossRef] [Green Version]
- Erickson, A.M.; Nevarea, Z.; Gipp, J.J.; Mulcahy, R.T. Identification of a variant antioxidant response element in the promoter of the human glutamate-cysteine ligase modifier subunit gene. Revision of the ARE consensus sequence. J. Biol. Chem. 2002, 277, 30730–30737. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.D.; Yang, H.; Whang, J.; Lu, S.C. Cloning and characterization of the human glutathione synthetase 5′-flanking region. Biochem. J. 2005, 390, 521–528. [Google Scholar] [CrossRef] [Green Version]
- Meredith, M.J.; Reed, D.J. Status of the mitochondrial pool of glutathione in the isolated hepatocyte. J. Biol. Chem. 1982, 257, 3747–3753. [Google Scholar] [CrossRef]
- Hwang, C.; Sinskey, A.J.; Lodish, H.F. Oxidized redox state of glutathione in the endoplasmic reticulum. Science 1992, 257, 1496–1502. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Rangasamy, T.; Thimmulappa, R.K.; Lee, H.; Osburn, W.O.; Brigelius-Flohe, R.; Kensler, T.W.; Yamamoto, M.; Biswal, S. Glutathione peroxidase 2, the major cigarette smoke-inducible isoform of GPX in lungs, is regulated by Nrf2. Am. J. Respir. Cell Mol. Biol. 2006, 35, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Harvey, C.J.; Thimmulappa, R.K.; Singh, A.; Blake, D.J.; Ling, G.; Wakabayashi, N.; Fujii, J.; Myers, A.; Biswal, S. Nrf2-regulated glutathione recycling independent of biosynthesis is critical for cell survival during oxidative stress. Free Radic. Biol. Med. 2009, 46, 443–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allocati, N.; Masulli, M.; Di Ilio, C.; Federici, L. Glutathione transferases: Substrates, inihibitors and pro-drugs in cancer and neurodegenerative diseases. Oncogenesis 2018, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Platz, A.; Jungnelius, U.; Grafstrom, E.; Lagerlof, B.; Mannervik, B.; Ringborg, U. Glutathione transferase P1-1 expression in human melanoma metastases: Correlation to N-RAS mutations and expression. Acta Oncol. 1995, 34, 759–765. [Google Scholar] [CrossRef]
- Suzuki, T.; Takagi, Y.; Osanai, H.; Li, L.; Takeuchi, M.; Katoh, Y.; Kobayashi, M.; Yamamoto, M. Pi class glutathione S-transferase genes are regulated by Nrf 2 through an evolutionarily conserved regulatory element in zebrafish. Biochem. J. 2005, 388, 65–73. [Google Scholar] [CrossRef] [Green Version]
- Funes, J.M.; Henderson, S.; Kaufman, R.; Flanagan, J.M.; Robson, M.; Pedley, B.; Moncada, S.; Boshoff, C. Oncogenic transformation of mesenchymal stem cells decreases Nrf2 expression favoring in vivo tumor growth and poorer survival. Mol. Cancer 2014, 13, 20. [Google Scholar] [CrossRef] [Green Version]
- Mougiakakos, D.; Okita, R.; Ando, T.; Durr, C.; Gadiot, J.; Ichikawa, J.; Zeiser, R.; Blank, C.; Johansson, C.C.; Kiessling, R. High expression of GCLC is associated with malignant melanoma of low oxidative phenotype and predicts a better prognosis. J. Mol. Med. 2012, 90, 935–944. [Google Scholar] [CrossRef]
- Franklin, C.C.; Backos, D.S.; Mohar, I.; White, C.C.; Forman, H.J.; Kavanagh, T.J. Structure, function, and post-translational regulation of the catalytic and modifier subunits of glutamate cysteine ligase. Mol. Aspects Med. 2009, 30, 86–98. [Google Scholar] [CrossRef] [Green Version]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef] [Green Version]
- Rocha, C.R.; Kajitani, G.S.; Quinet, A.; Fortunato, R.S.; Menck, C.F. NRF2 and glutathione are key resistance mediators to temozolomide in glioma and melanoma cells. Oncotarget 2016, 7, 48081–48092. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Zheng, Z.; Kim, K.Y.; Jin, X.; Roh, M.R.; Jin, Z. Hypermethylation and downregulation of glutathione peroxidase 3 are related to pathogenesis of melanoma. Oncol. Rep. 2016, 36, 2737–2744. [Google Scholar] [CrossRef] [Green Version]
- Yi, Z.; Jiang, L.; Zhao, L.; Zhou, M.; Ni, Y.; Yang, Y.; Yang, H.; Yang, L.; Zhang, Q.; Kuang, Y.; et al. Glutathione peroxidase 3 (GPX3) suppresses the growth of melanoma cells through reactive oxygen species (ROS)-dependent stabilization of hypoxia-inducible factor 1-alpha and 2-alpha. J. Cell Biochem. 2019, 120, 19124–19136. [Google Scholar] [CrossRef] [PubMed]
- Gao, K.; Lockwood, W.W.; Li, J.; Lam, W.; Li, G. Genomic analyses identify gene candidates for acquired irinotecan resistance in melanoma cells. Int. J. Oncol. 2008, 32, 1343–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J.; et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef]
- Ros-Bullon, M.R.; Sanchez-Pedreno, P.; Martinez-Liarte, J.H. Whole blood glutathione peroxidase activity in melanoma patients. Cancer Lett. 1999, 144, 25–30. [Google Scholar] [CrossRef]
- Czajkowski, R.; Czajkowska, A.; Drewa, T.; Olszewska, D.; Zegarski, W.; Piech, J.; Kowaliszyn, B.; Zegarska, B. Activity of antioxidant enzymes in melanoma patients. Int. J. Dermatol. 2013, 52, 1454–1456. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, J.; Zhang, X.; Chen, W. Glutathione reductase-mediated thiol oxidative stress suppresses metastasis of murine melanoma cells. Free Radic. Biol. Med. 2018, 129, 256–267. [Google Scholar] [CrossRef]
- Yang, H.C.; Wu, Y.H.; Yen, W.C.; Liu, H.Y.; Hwang, T.L.; Stern, A.; Chiu, D.T. The Redox Role of G6PD in Cell Growth, Cell Death, and Cancer. Cells 2019, 8, 1055. [Google Scholar] [CrossRef] [Green Version]
- Hu, T.; Zhang, C.; Tang, Q.; Su, Y.; Li, B.; Chen, L.; Zhang, Z.; Cai, T.; Zhu, Y. Variant G6PD levels promote tumor cell proliferation or apoptosis via the STAT3/5 pathway in the human melanoma xenograft mouse model. BMC Cancer 2013, 13, 251. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef]
- Chae, H.Z.; Chung, S.J.; Rhee, S.G. Thioredoxin-dependent peroxide reductase from yeast. J. Biol. Chem. 1994, 269, 27670–27678. [Google Scholar] [CrossRef]
- Saitoh, M.; Nishitoh, H.; Fujii, M.; Takeda, K.; Tobiume, K.; Sawada, Y.; Kawabata, M.; Miyazono, K.; Ichijo, H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998, 17, 2596–2606. [Google Scholar] [CrossRef] [Green Version]
- Holmgren, A.; Sengupta, R. The use of thiols by ribonucleotide reductase. Free Radic. Biol. Med. 2010, 49, 1617–1628. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K.; Murata, M.; Sachi, Y.; Nakamura, H.; Takeuchi, J.; Mori, K.; Yodoi, J. Distinct roles of thioredoxin in the cytoplasm and in the nucleus. A two-step mechanism of redox regulation of transcription factor NF-kappaB. J. Biol. Chem. 1999, 274, 27891–27897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muri, J.; Thut, H.; Feng, Q.; Kopf, M. Thioredoxin-1 distinctly promotes NF-kappaB target DNA binding and NLRP3 inflammasome activation independently of Txnip. eLife 2020, 9, e53627. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, F.; Soltani, A.; Ghahremanloo, A.; Javid, H.; Hashemy, S.I. The thioredoxin system and cancer therapy: A review. Cancer Chemother. Pharmacol. 2019, 84, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, H.J.; Karlenius, T.C.; Tonissen, K.F. Regulation of the human thioredoxin gene promoter and its key substrates: A study of functional and putative regulatory elements. Biochim. Biophys. Acta 2014, 1840, 303–314. [Google Scholar] [CrossRef]
- Wang, X.; Dong, H.; Li, Q.; Li, Y.; Hong, A. Thioredoxin induces Tregs to generate an immunotolerant tumor microenvironment in metastatic melanoma. Oncoimmunology 2015, 4, e1027471. [Google Scholar] [CrossRef] [Green Version]
- Meylan, P.; Pich, C.; Winkler, C.; Ginster, S.; Mury, L.; Sgandurra, M.; Dreos, R.; Frederick, D.T.; Hammond, M.; Boland, G.M.; et al. Low expression of the PPARgamma-regulated gene thioredoxin-interacting protein accompanies human melanoma progression and promotes experimental lung metastases. Sci. Rep. 2021, 11, 7847. [Google Scholar] [CrossRef] [PubMed]
- Balta, E.; Hardt, R.; Liang, J.; Kirchgessner, H.; Orlik, C.; Jahraus, B.; Hillmer, S.; Meuer, S.; Hubner, K.; Wabnitz, G.H.; et al. Spatial oxidation of L-plastin downmodulates actin-based functions of tumor cells. Nat. Commun. 2019, 10, 4073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sachweh, M.C.; Stafford, W.C.; Drummond, C.J.; McCarthy, A.R.; Higgins, M.; Campbell, J.; Brodin, B.; Arner, E.S.; Lain, S. Redox effects and cytotoxic profiles of MJ25 and auranofin towards malignant melanoma cells. Oncotarget 2015, 6, 16488–16506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannai, S. Exchange of cystine and glutamate across plasma membrane of human fibroblasts. J. Biol. Chem. 1986, 261, 2256–2263. [Google Scholar] [CrossRef]
- Conrad, M.; Sato, H. The oxidative stress-inducible cystine/glutamate antiporter, system x (c) (-): Cystine supplier and beyond. Amino Acids 2012, 42, 231–246. [Google Scholar] [CrossRef] [PubMed]
- Bassi, M.T.; Gasol, E.; Manzoni, M.; Pineda, M.; Riboni, M.; Martin, R.; Zorzano, A.; Borsani, G.; Palacin, M. Identification and characterisation of human xCT that co-expresses, with 4F2 heavy chain, the amino acid transport activity system xc. Pflugers. Arch. 2001, 442, 286–296. [Google Scholar] [CrossRef]
- Lin, W.; Wang, C.; Liu, G.; Bi, C.; Wang, X.; Zhou, Q.; Jin, H. SLC7A11/xCT in cancer: Biological functions and therapeutic implications. Am. J. Cancer Res. 2020, 10, 3106–3126. [Google Scholar]
- Sasaki, H.; Sato, H.; Kuriyama-Matsumura, K.; Sato, K.; Maebara, K.; Wang, H.; Tamba, M.; Itoh, K.; Yamamoto, M.; Bannai, S. Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J. Biol. Chem. 2002, 277, 44765–44771. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.S.; Jeong, B.S.; Wall, B.A.; Li, J.; Shan, N.L.; Wen, Y.; Goydos, J.S.; Chen, S. Participation of xCT in melanoma cell proliferation in vitro and tumorigenesis in vivo. Oncogenesis 2018, 7, 86. [Google Scholar] [CrossRef] [Green Version]
- Sato, M.; Onuma, K.; Domon, M.; Hasegawa, S.; Suzuki, A.; Kusumi, R.; Hino, R.; Kakihara, N.; Kanda, Y.; Osaki, M.; et al. Loss of the cystine/glutamate antiporter in melanoma abrogates tumor metastasis and markedly increases survival rates of mice. Int. J. Cancer 2020, 147, 3224–3235. [Google Scholar] [CrossRef]
- Wang, L.; Leite de Oliveira, R.; Huijberts, S.; Bosdriesz, E.; Pencheva, N.; Brunen, D.; Bosma, A.; Song, J.Y.; Zevenhoven, J.; Los-de Vries, G.T.; et al. An Acquired Vulnerability of Drug-Resistant Melanoma with Therapeutic Potential. Cell 2018, 173, 1413–1425.e14. [Google Scholar] [CrossRef] [Green Version]
- Rhee, S.G.; Woo, H.A.; Kil, I.S.; Bae, S.H. Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. J. Biol. Chem. 2012, 287, 4403–4410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Lee, S.; Lee, S.; Kang, S.W. 2-cys peroxiredoxins: Emerging hubs determining redox dependency of Mammalian signaling networks. Int. J. Cell Biol. 2014, 2014, 715867. [Google Scholar] [CrossRef] [Green Version]
- Brown, K.K.; Cox, A.G.; Hampton, M.B. Mitochondrial respiratory chain involvement in peroxiredoxin 3 oxidation by phenethyl isothiocyanate and auranofin. FEBS Lett. 2010, 584, 1257–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zito, E.; Melo, E.P.; Yang, Y.; Wahlander, A.; Neubert, T.A.; Ron, D. Oxidative protein folding by an endoplasmic reticulum-localized peroxiredoxin. Mol. Cell 2010, 40, 787–797. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Jang, H.H. The Role of Peroxiredoxin Family in Cancer Signaling. J. Cancer Prev. 2019, 24, 65–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manevich, Y.; Fisher, A.B. Peroxiredoxin 6, a 1-Cys peroxiredoxin, functions in antioxidant defense and lung phospholipid metabolism. Free Radic. Biol. Med. 2005, 38, 1422–1432. [Google Scholar] [CrossRef]
- Kim, Y.J.; Ahn, J.Y.; Liang, P.; Ip, C.; Zhang, Y.; Park, Y.M. Human prx1 gene is a target of Nrf2 and is up-regulated by hypoxia/reoxygenation: Implication to tumor biology. Cancer Res. 2007, 67, 546–554. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, I.; Mo, Y.; Gao, L.; Kazi, A.; Fisher, A.B.; Feinstein, S.I. Oxidant stress stimulates expression of the human peroxiredoxin 6 gene by a transcriptional mechanism involving an antioxidant response element. Free Radic. Biol. Med. 2009, 46, 146–153. [Google Scholar] [CrossRef]
- Woo, H.A.; Jeong, W.; Chang, T.S.; Park, K.J.; Park, S.J.; Yang, J.S.; Rhee, S.G. Reduction of cysteine sulfinic acid by sulfiredoxin is specific to 2-cys peroxiredoxins. J. Biol. Chem. 2005, 280, 3125–3128. [Google Scholar] [CrossRef] [Green Version]
- Soriano, F.X.; Leveille, F.; Papadia, S.; Higgins, L.G.; Varley, J.; Baxter, P.; Hayes, J.D.; Hardingham, G.E. Induction of sulfiredoxin expression and reduction of peroxiredoxin hyperoxidation by the neuroprotective Nrf2 activator 3H-1,2-dithiole-3-thione. J. Neurochem. 2008, 107, 533–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hintsala, H.R.; Soini, Y.; Haapasaari, K.M.; Karihtala, P. Dysregulation of redox-state-regulating enzymes in melanocytic skin tumours and the surrounding microenvironment. Histopathology 2015, 67, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Mishra, R.; Patel, H.; Abdulsalam, S.; Greis, K.D.; Kadekaro, A.L.; Merino, E.J.; Garrett, J.T. Utilization of Reactive Oxygen Species Targeted Therapy to Prolong the Efficacy of BRAF Inhibitors in Melanoma. J. Cancer 2018, 9, 4665–4676. [Google Scholar] [CrossRef]
- Furuta, J.; Nobeyama, Y.; Umebayashi, Y.; Otsuka, F.; Kikuchi, K.; Ushijima, T. Silencing of Peroxiredoxin 2 and aberrant methylation of 33 CpG islands in putative promoter regions in human malignant melanomas. Cancer Res. 2006, 66, 6080–6086. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.J.; Kang, D.H.; Choi, M.; Choi, Y.J.; Lee, J.Y.; Park, J.H.; Park, Y.J.; Lee, K.W.; Kang, S.W. Peroxiredoxin-2 represses melanoma metastasis by increasing E-Cadherin/beta-Catenin complexes in adherens junctions. Cancer Res. 2013, 73, 4744–4757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Q.; Jiang, H.; Matthews, C.P.; Colburn, N.H. Sulfiredoxin is an AP-1 target gene that is required for transformation and shows elevated expression in human skin malignancies. Proc. Natl. Acad. Sci. USA 2008, 105, 19738–19743. [Google Scholar] [CrossRef] [Green Version]
- Saei, A.A.; Gullberg, H.; Sabatier, P.; Beusch, C.M.; Johansson, K.; Lundgren, B.; Arvidsson, P.I.; Arner, E.S.J.; Zubarev, R.A. Comprehensive chemical proteomics for target deconvolution of the redox active drug auranofin. Redox Biol. 2020, 32, 101491. [Google Scholar] [CrossRef]
- Seong, I.; Kim, J. Gene expression regulation by agonist-independent constitutive signaling of melanocortin-1 receptor. Endocrinol. Metab. 2014, 29, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Chen, D.; Ma, K.; Wu, X.; Hao, H.; Jiang, S. NAD(P)H:Quinone Oxidoreductase 1 (NQO1) as a Therapeutic and Diagnostic Target in Cancer. J. Med. Chem. 2018, 61, 6983–7003. [Google Scholar] [CrossRef] [PubMed]
- Siegel, D.; Kepa, J.K.; Ross, D. NAD(P)H:quinone oxidoreductase 1 (NQO1) localizes to the mitotic spindle in human cells. PLoS ONE 2012, 7, e44861. [Google Scholar] [CrossRef] [Green Version]
- Rysava, A.; Cizkova, K.; Frankova, J.; Roubalova, L.; Ulrichova, J.; Vostalova, J.; Vrba, J.; Zalesak, B.; Rajnochova Svobodova, A. Effect of UVA radiation on the Nrf2 signalling pathway in human skin cells. J. Photochem. Photobiol. B 2020, 209, 111948. [Google Scholar] [CrossRef] [PubMed]
- Asher, G.; Lotem, J.; Cohen, B.; Sachs, L.; Shaul, Y. Regulation of p53 stability and p53-dependent apoptosis by NADH quinone oxidoreductase 1. Proc. Natl. Acad. Sci. USA 2001, 98, 1188–1193. [Google Scholar] [CrossRef]
- Moscovitz, O.; Tsvetkov, P.; Hazan, N.; Michaelevski, I.; Keisar, H.; Ben-Nissan, G.; Shaul, Y.; Sharon, M. A mutually inhibitory feedback loop between the 20S proteasome and its regulator, NQO1. Mol. Cell 2012, 47, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Shin, W.S.; Han, J.; Verwilst, P.; Kumar, R.; Kim, J.H.; Kim, J.S. Cancer Targeted Enzymatic Theranostic Prodrug: Precise Diagnosis and Chemotherapy. Bioconjug. Chem. 2016, 27, 1419–1426. [Google Scholar] [CrossRef]
- Favreau, L.V.; Pickett, C.B. Transcriptional regulation of the rat NAD(P)H:quinone reductase gene. Identification of regulatory elements controlling basal level expression and inducible expression by planar aromatic compounds and phenolic antioxidants. J. Biol. Chem. 1991, 266, 4556–4561. [Google Scholar] [CrossRef]
- Cheng, Y.; Li, J.; Martinka, M.; Li, G. The expression of NAD(P)H:quinone oxidoreductase 1 is increased along with NF-kappaB p105/p50 in human cutaneous melanomas. Oncol. Rep. 2010, 23, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, Z.; Li, G. Patient outcome prediction using multiple biomarkers in human melanoma: A clinicopathological study of 118 cases. Exp. Ther. Med. 2011, 2, 131–135. [Google Scholar] [CrossRef] [Green Version]
- Choi, T.Y.; Sohn, K.C.; Kim, J.H.; Kim, S.M.; Kim, C.H.; Hwang, J.S.; Lee, J.H.; Kim, C.D.; Yoon, T.J. Impact of NAD(P)H:quinone oxidoreductase-1 on pigmentation. J. Invest. Dermatol. 2010, 130, 784–792. [Google Scholar] [CrossRef] [Green Version]
- Chiang, S.K.; Chen, S.E.; Chang, L.C. A Dual Role of Heme Oxygenase-1 in Cancer Cells. Int. J. Mol. Sci. 2018, 20, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitti, M.; Piras, S.; Marinari, U.M.; Moretta, L.; Pronzato, M.A.; Furfaro, A.L. HO-1 Induction in Cancer Progression: A Matter of Cell Adaptation. Antioxidants 2017, 6, 29. [Google Scholar] [CrossRef]
- Maines, M.D.; Abrahamsson, P.A. Expression of heme oxygenase-1 (HSP32) in human prostate: Normal, hyperplastic, and tumor tissue distribution. Urology 1996, 47, 727–733. [Google Scholar] [CrossRef]
- Was, H.; Dulak, J.; Jozkowicz, A. Heme oxygenase-1 in tumor biology and therapy. Curr. Drug Targets 2010, 11, 1551–1570. [Google Scholar] [CrossRef]
- Tan, Q.; Wang, H.; Hu, Y.; Hu, M.; Li, X.; Aodengqimuge; Ma, Y.; Wei, C.; Song, L. Src/STAT3-dependent heme oxygenase-1 induction mediates chemoresistance of breast cancer cells to doxorubicin by promoting autophagy. Cancer Sci. 2015, 106, 1023–1032. [Google Scholar] [CrossRef]
- Chauveau, C.; Remy, S.; Royer, P.J.; Hill, M.; Tanguy-Royer, S.; Hubert, F.X.; Tesson, L.; Brion, R.; Beriou, G.; Gregoire, M.; et al. Heme oxygenase-1 expression inhibits dendritic cell maturation and proinflammatory function but conserves IL-10 expression. Blood 2005, 106, 1694–1702. [Google Scholar] [CrossRef] [Green Version]
- Tertil, M.; Skrzypek, K.; Florczyk, U.; Weglarczyk, K.; Was, H.; Collet, G.; Guichard, A.; Gil, T.; Kuzdzal, J.; Jozkowicz, A.; et al. Regulation and novel action of thymidine phosphorylase in non-small cell lung cancer: Crosstalk with Nrf2 and HO-1. PLoS ONE 2014, 9, e97070. [Google Scholar] [CrossRef] [PubMed]
- Prestera, T.; Talalay, P.; Alam, J.; Ahn, Y.I.; Lee, P.J.; Choi, A.M. Parallel induction of heme oxygenase-1 and chemoprotective phase 2 enzymes by electrophiles and antioxidants: Regulation by upstream antioxidant-responsive elements (ARE). Mol. Med. 1995, 1, 827–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Was, H.; Cichon, T.; Smolarczyk, R.; Rudnicka, D.; Stopa, M.; Chevalier, C.; Leger, J.J.; Lackowska, B.; Grochot, A.; Bojkowska, K.; et al. Overexpression of heme oxygenase-1 in murine melanoma: Increased proliferation and viability of tumor cells, decreased survival of mice. Am. J. Pathol. 2006, 169, 2181–2198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furfaro, A.L.; Ottonello, S.; Loi, G.; Cossu, I.; Piras, S.; Spagnolo, F.; Queirolo, P.; Marinari, U.M.; Moretta, L.; Pronzato, M.A.; et al. HO-1 downregulation favors BRAF(V600) melanoma cell death induced by Vemurafenib/PLX4032 and increases NK recognition. Int. J. Cancer 2020, 146, 1950–1962. [Google Scholar] [CrossRef] [PubMed]
- Di Biase, S.; Lee, C.; Brandhorst, S.; Manes, B.; Buonno, R.; Cheng, C.W.; Cacciottolo, M.; Martin-Montalvo, A.; de Cabo, R.; Wei, M.; et al. Fasting-Mimicking Diet Reduces HO-1 to Promote T Cell-Mediated Tumor Cytotoxicity. Cancer Cell 2016, 30, 136–146. [Google Scholar] [CrossRef] [Green Version]
- Richter, K.; Haslbeck, M.; Buchner, J. The heat shock response: Life on the verge of death. Mol. Cell 2010, 40, 253–266. [Google Scholar] [CrossRef]
- Vostakolaei, M.A.; Hatami-Baroogh, L.; Babaei, G.; Molavi, O.; Kordi, S.; Abdolalizadeh, J. Hsp70 in cancer: A double agent in the battle between survival and death. J. Cell Physiol. 2021, 236, 3420–3444. [Google Scholar] [CrossRef] [PubMed]
- Verbeke, P.; Clark, B.F.; Rattan, S.I. Reduced levels of oxidized and glycoxidized proteins in human fibroblasts exposed to repeated mild heat shock during serial passaging in vitro. Free Radic. Biol. Med. 2001, 31, 1593–1602. [Google Scholar] [CrossRef]
- Luo, Q.; Jiang, L.; Chen, G.; Feng, Y.; Lv, Q.; Zhang, C.; Qu, S.; Zhu, H.; Zhou, B.; Xiao, X. Constitutive heat shock protein 70 interacts with alpha-enolase and protects cardiomyocytes against oxidative stress. Free Radic. Res. 2011, 45, 1355–1365. [Google Scholar] [CrossRef] [PubMed]
- Lazarev, V.F.; Nikotina, A.D.; Mikhaylova, E.R.; Nudler, E.; Polonik, S.G.; Guzhova, I.V.; Margulis, B.A. Hsp70 chaperone rescues C6 rat glioblastoma cells from oxidative stress by sequestration of aggregating GAPDH. Biochem. Biophys. Res. Commun. 2016, 470, 766–771. [Google Scholar] [CrossRef]
- Gao, T.; Newton, A.C. The turn motif is a phosphorylation switch that regulates the binding of Hsp70 to protein kinase C. J. Biol. Chem. 2002, 277, 31585–31592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruchalski, K.; Mao, H.; Li, Z.; Wang, Z.; Gillers, S.; Wang, Y.; Mosser, D.D.; Gabai, V.; Schwartz, J.H.; Borkan, S.C. Distinct hsp70 domains mediate apoptosis-inducing factor release and nuclear accumulation. J. Biol. Chem. 2006, 281, 7873–7880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, D.V.; da Silva Nornberg, B.F.; Geracitano, L.A.; Barros, D.M.; Monserrat, J.M.; Marins, L.F. Induction of phase II enzymes and hsp70 genes by copper sulfate through the electrophile-responsive element (EpRE): Insights obtained from a transgenic zebrafish model carrying an orthologous EpRE sequence of mammalian origin. Fish. Physiol. Biochem. 2010, 36, 347–353. [Google Scholar] [CrossRef]
- Budina-Kolomets, A.; Webster, M.R.; Leu, J.I.; Jennis, M.; Krepler, C.; Guerrini, A.; Kossenkov, A.V.; Xu, W.; Karakousis, G.; Schuchter, L.; et al. HSP70 Inhibition Limits FAK-Dependent Invasion and Enhances the Response to Melanoma Treatment with BRAF Inhibitors. Cancer Res. 2016, 76, 2720–2730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, K.C.; Kim, D.S.; Choi, H.O.; Kim, K.H.; Chung, J.H.; Eun, H.C.; Lee, J.S.; Seo, J.S. Overexpression of HSP70 prevents ultraviolet B-induced apoptosis of a human melanoma cell line. Arch. Dermatol. Res. 2000, 292, 482–487. [Google Scholar] [CrossRef]
- Russo, A.; Cardile, V.; Avola, R.; Graziano, A.; Montenegro, I.; Said, B.; Madrid, A. Isocordoin analogues promote apoptosis in human melanoma cells via Hsp70. Phytother. Res. 2019, 33, 3242–3250. [Google Scholar] [CrossRef]
- Juhasz, K.; Thuenauer, R.; Spachinger, A.; Duda, E.; Horvath, I.; Vigh, L.; Sonnleitner, A.; Balogi, Z. Lysosomal rerouting of Hsp70 trafficking as a potential immune activating tool for targeting melanoma. Curr. Pharm. Des. 2013, 19, 430–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komarova, E.Y.; Suezov, R.V.; Nikotina, A.D.; Aksenov, N.D.; Garaeva, L.A.; Shtam, T.A.; Zhakhov, A.V.; Martynova, M.G.; Bystrova, O.A.; Istomina, M.S.; et al. Hsp70-containing extracellular vesicles are capable of activating of adaptive immunity in models of mouse melanoma and colon carcinoma. Sci. Rep. 2021, 11, 21314. [Google Scholar] [CrossRef] [PubMed]
- Altobelli, G.G.; Van Noorden, S.; Balato, A.; Cimini, V. Copper/Zinc Superoxide Dismutase in Human Skin: Current Knowledge. Front. Med. 2020, 7, 183. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.K.; St Clair, D.K. Manganese superoxide dismutase regulation and cancer. Free Radic. Biol. Med. 2012, 52, 2209–2222. [Google Scholar] [CrossRef]
- Griess, B.; Tom, E.; Domann, F.; Teoh-Fitzgerald, M. Extracellular superoxide dismutase and its role in cancer. Free Radic. Biol. Med. 2017, 112, 464–479. [Google Scholar] [CrossRef]
- Dong, J.; Sulik, K.K.; Chen, S.Y. Nrf2-mediated transcriptional induction of antioxidant response in mouse embryos exposed to ethanol in vivo: Implications for the prevention of fetal alcohol spectrum disorders. Antioxid. Redox Signal. 2008, 10, 2023–2033. [Google Scholar] [CrossRef] [Green Version]
- Dreger, H.; Westphal, K.; Weller, A.; Baumann, G.; Stangl, V.; Meiners, S.; Stangl, K. Nrf2-dependent upregulation of antioxidative enzymes: A novel pathway for proteasome inhibitor-mediated cardioprotection. Cardiovasc. Res. 2009, 83, 354–361. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.S.; Lee, S.Y.; Kim, D.H.; Kim, H.S. Protopanaxatriol Ginsenoside Rh1 Upregulates Phase II Antioxidant Enzyme Gene Expression in Rat Primary Astrocytes: Involvement of MAP Kinases and Nrf2/ARE Signaling. Biomol. Ther. 2016, 24, 33–39. [Google Scholar] [CrossRef] [Green Version]
- Nazarewicz, R.R.; Dikalova, A.; Bikineyeva, A.; Ivanov, S.; Kirilyuk, I.A.; Grigor’ev, I.A.; Dikalov, S.I. Does scavenging of mitochondrial superoxide attenuate cancer prosurvival signaling pathways? Antioxid. Redox Signal. 2013, 19, 344–349. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Gao, L.X.; Ma, X.Q.; Hu, F.X.; Li, C.M.; Lu, Z. Involvement of superoxide and nitric oxide in BRAF(V600E) inhibitor PLX4032-induced growth inhibition of melanoma cells. Integr. Biol. 2014, 6, 1211–1217. [Google Scholar] [CrossRef]
- Yuan, L.; Mishra, R.; Patel, H.; Alanazi, S.; Wei, X.; Ma, Z.; Garrett, J.T. BRAF Mutant Melanoma Adjusts to BRAF/MEK Inhibitors via Dependence on Increased Antioxidant SOD2 and Increased Reactive Oxygen Species Levels. Cancers 2020, 12, 1661. [Google Scholar] [CrossRef]
- Asgari, M.M.; Brasky, T.M.; White, E. Association of vitamin A and carotenoid intake with melanoma risk in a large prospective cohort. J. Invest. Dermatol. 2012, 132, 1573–1582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feskanich, D.; Willett, W.C.; Hunter, D.J.; Colditz, G.A. Dietary intakes of vitamins A, C, and E and risk of melanoma in two cohorts of women. Br. J. Cancer 2003, 88, 1381–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, I.M.; Cook, N.R.; Manson, J.E.; Buring, J.E.; Hennekens, C.H. Beta-carotene supplementation and incidence of cancer and cardiovascular disease: The Women’s Health Study. J. Natl. Cancer Inst. 1999, 91, 2102–2106. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.P.; Chu, R.X.; Liu, H. Vitamin A intake and risk of melanoma: A meta-analysis. PLoS ONE 2014, 9, e102527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mounessa, J.; Buntinx-Krieg, T.; Qin, R.; Dunnick, C.A.; Dellavalle, R.P. Primary and Secondary Chemoprevention of Malignant Melanoma. Am. J. Clin. Dermatol. 2016, 17, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Jeter, J.M.; Bowles, T.L.; Curiel-Lewandrowski, C.; Swetter, S.M.; Filipp, F.V.; Abdel-Malek, Z.A.; Geskin, L.J.; Brewer, J.D.; Arbiser, J.L.; Gershenwald, J.E.; et al. Chemoprevention agents for melanoma: A path forward into phase 3 clinical trials. Cancer 2019, 125, 18–44. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Yan, Y.; Ma, Y.N.; Yang, Y.X. Vitamin C at high concentrations induces cytotoxicity in malignant melanoma but promotes tumor growth at low concentrations. Mol. Carcinogen 2017, 56, 1965–1976. [Google Scholar] [CrossRef]
- Murray, J.C.; Burch, J.A.; Streilein, R.D.; Iannacchione, M.A.; Hall, R.P.; Pinnell, S.R. A topical antioxidant solution containing vitamins C and E stabilized by ferulic acid provides protection for human skin against damage caused by ultraviolet irradiation. J. Am. Acad. Dermatol. 2008, 59, 418–425. [Google Scholar] [CrossRef]
- Chhabra, G.; Ndiaye, M.A.; Garcia-Peterson, L.M.; Ahmad, N. Melanoma Chemoprevention: Current Status and Future Prospects. Photochem. Photobiol. 2017, 93, 975–989. [Google Scholar] [CrossRef] [Green Version]
- Hercberg, S.; Galan, P.; Preziosi, P.; Bertrais, S.; Mennen, L.; Malvy, D.; Roussel, A.M.; Favier, A.; Briancon, S. The SU.VI.MAX Study: A randomized, placebo-controlled trial of the health effects of antioxidant vitamins and minerals. Arch. Intern. Med. 2004, 164, 2335–2342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, J.H.; Seo, J.Y.; Lee, M.K.; Eun, H.C.; Lee, J.H.; Kang, S.; Fisher, G.J.; Voorhees, J.J. Ultraviolet modulation of human macrophage metalloelastase in human skin in vivo. J. Invest. Dermatol. 2002, 119, 507–512. [Google Scholar] [CrossRef] [Green Version]
- Minocha, R.; Damian, D.L.; Halliday, G.M. Melanoma and nonmelanoma skin cancer chemoprevention: A role for nicotinamide? Photodermatol. Photoimmunol. Photomed. 2018, 34, 5–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, A.C.; Martin, A.J.; Choy, B.; Fernandez-Penas, P.; Dalziell, R.A.; McKenzie, C.A.; Scolyer, R.A.; Dhillon, H.M.; Vardy, J.L.; Kricker, A.; et al. A Phase 3 Randomized Trial of Nicotinamide for Skin-Cancer Chemoprevention. N. Engl. J. Med. 2015, 373, 1618–1626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogoza-Janiszewska, E.; Malinska, K.; Baszuk, P.; Marciniak, W.; Derkacz, R.; Lener, M.; Jakubowska, A.; Cybulski, C.; Huzarski, T.; Masojc, B.; et al. Serum Selenium Level and 10-Year Survival after Melanoma. Biomedicines 2021, 9, 991. [Google Scholar] [CrossRef]
- Chen, Y.C.; Prabhu, K.S.; Mastro, A.M. Is Selenium a Potential Treatment for Cancer Metastasis? Nutrients 2013, 5, 1149–1168. [Google Scholar] [CrossRef] [Green Version]
- Evans, S.O.; Khairuddin, P.F.; Jameson, M.B. Optimising Selenium for Modulation of Cancer Treatments. Anticancer Res. 2017, 37, 6497–6509. [Google Scholar] [CrossRef] [Green Version]
- Rayman, M.P. Selenium and human health. Lancet 2012, 379, 1256–1268. [Google Scholar] [CrossRef]
- Rayman, M.P. Selenium intake, status, and health: A complex relationship. Hormones 2020, 19, 9–14. [Google Scholar] [CrossRef] [Green Version]
- Schott, M.; de Jel, M.M.; Engelmann, J.C.; Renner, P.; Geissler, E.K.; Bosserhoff, A.K.; Kuphal, S. Selenium-binding protein 1 is down-regulated in malignant melanoma. Oncotarget 2018, 9, 10445–10456. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Wong, Y.S. Selenocystine induces apoptosis of A375 human melanoma cells by activating ROS-mediated mitochondrial pathway and p53 phosphorylation. Cell Mol. Life Sci. 2008, 65, 2763–2775. [Google Scholar] [CrossRef] [PubMed]
- Poerschke, R.L.; Moos, P.J. Thioredoxin reductase 1 knockdown enhances selenazolidine cytotoxicity in human lung cancer cells via mitochondrial dysfunction. Biochem. Pharmacol. 2011, 81, 211–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonsson-Videsater, K.; Bjorkhem-Bergman, L.; Hossain, A.; Soderberg, A.; Eriksson, L.C.; Paul, C.; Rosen, A.; Bjornstedt, M. Selenite-induced apoptosis in doxorubicin-resistant cells and effects on the thioredoxin system. Biochem. Pharmacol. 2004, 67, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Marciel, M.P.; Khadka, V.S.; Deng, Y.; Kilicaslan, P.; Pham, A.; Bertino, P.; Lee, K.; Chen, S.; Glibetic, N.; Hoffmann, F.W.; et al. Correction: Selenoprotein K deficiency inhibits melanoma by reducing calcium flux required for tumor growth and metastasis. Oncotarget 2018, 9, 30937. [Google Scholar] [CrossRef]
- Ecker, A.; Barbosa, N.V.; Ardisson-Araujo, D. Accessing the transcriptional status of selenoproteins in skin cancer-derived cell lines. J. Trace Elem. Med. Biol. 2020, 60, 126476. [Google Scholar] [CrossRef]
- Deffuant, C.; Celerier, P.; Boiteau, H.L.; Litoux, P.; Dreno, B. Serum selenium in melanoma and epidermotropic cutaneous T-cell lymphoma. Acta Derm. Venereol. 1994, 74, 90–92. [Google Scholar] [CrossRef] [PubMed]
- Vinceti, M.; Rothman, K.J.; Bergomi, M.; Borciani, N.; Serra, L.; Vivoli, G. Excess melanoma incidence in a cohort exposed to high levels of environmental selenium. Cancer Epidemiol. Biomarkers Prev. 1998, 7, 853–856. [Google Scholar]
- Vinceti, M.; Filippini, T.; Del Giovane, C.; Dennert, G.; Zwahlen, M.; Brinkman, M.; Zeegers, M.P.; Horneber, M.; D’Amico, R.; Crespi, C.M. Selenium for preventing cancer. Cochrane Database Syst. Rev. 2018, 1, CD005195. [Google Scholar] [CrossRef]
- Algotar, A.M.; Stratton, M.S.; Ahmann, F.R.; Ranger-Moore, J.; Nagle, R.B.; Thompson, P.A.; Slate, E.; Hsu, C.H.; Dalkin, B.L.; Sindhwani, P.; et al. Phase 3 clinical trial investigating the effect of selenium supplementation in men at high-risk for prostate cancer. Prostate 2013, 73, 328–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karp, D.D.; Lee, S.J.; Keller, S.M.; Wright, G.S.; Aisner, S.; Belinsky, S.A.; Johnson, D.H.; Johnston, M.R.; Goodman, G.; Clamon, G.; et al. Randomized, double-blind, placebo-controlled, phase III chemoprevention trial of selenium supplementation in patients with resected stage I non-small-cell lung cancer: ECOG 5597. J. Clin. Oncol. 2013, 31, 4179–4187. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Wang, C.; Yu, W.; Fan, W.; Wang, S.; Shen, N.; Wu, P.; Li, X.; Wang, F. Selenium Exposure and Cancer Risk: An Updated Meta-analysis and Meta-regression. Sci. Rep. 2016, 6, 19213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corazo-Rozas, P.; Guerreschi, P.; André, F.; Gabert, P.; Lancel, S.; Dekiouk, S.; Fontaine, D.; Tardivel, M.; Savina, A.; Quesnel, B.; et al. Mitochondrial oxidative phosphorylation controls cancer cell’s life and death descisions upon exposure to MAPK inhibitors. Oncotarget 2016, 7, 39473–39485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpenter, E.L.; Chagani, S.; Nelson, D.; Cassidy, P.B.; Laws, M.; Ganguli-Indra, G.; Indra, A.K. Mitochondrial complex I inhibitor deguelin induces metabolic reprogramming and sensitizes vemurafenib-resistant BRAFV600E mutation bearing metastatic melanoma cells. Mol. Carcinog. 2019, 58, 1680–1690. [Google Scholar] [CrossRef] [Green Version]
- Vrijsen, S.; Besora-Casals, L.; van Veen, S.; Zielich, J.; van den Haute, C.; Hamouda, N.N.; Fischer, C.; Ghesquière, B.; Tournev, I.; Agostinis, P. ATP13A2-mediated endo-lysosomal polyamine export counters mitochondrial oxidative stress. Proc. Natl. Acad. Sci. USA 2020, 117, 31198–31207. [Google Scholar] [CrossRef]
- Diaconeasa, Z.; Ayvaz, H.; Ruginǎ, D.; Leopold, L.; Stǎnilǎ, A.; Socaciu, C.; Tăbăran, F.; Luput, L.; Mada, D.C.; Pintea, A.; et al. Melanoma inhibition by anthocyanins is associated with the reduction of oxidative stress biomarkers and changes in mitochondrial membrane potential. Plant. Foods Hum. Nutr. 2017, 72, 404–410. [Google Scholar] [CrossRef]
- Stevens, J.F.; Revel, J.S.; Maier, C.S. Mitochondira-centric review of polyphenol bioactivity in cancer models. Antioxid. Redox Signal. 2018, 29, 1589–1611. [Google Scholar] [CrossRef]
- Zhu, H.; Jia, Z.; Trush, M.A.; Li, Y.R. Nrf2 deficiency promotes melanoma growth and lung metastasis. React. Oxyg. Species 2016, 2, 308–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carpenter, E.L.; Becker, A.L.; Indra, A.K. NRF2 and Key Transcriptional Targets in Melanoma Redox Manipulation. Cancers 2022, 14, 1531. https://doi.org/10.3390/cancers14061531
Carpenter EL, Becker AL, Indra AK. NRF2 and Key Transcriptional Targets in Melanoma Redox Manipulation. Cancers. 2022; 14(6):1531. https://doi.org/10.3390/cancers14061531
Chicago/Turabian StyleCarpenter, Evan L., Alyssa L. Becker, and Arup K. Indra. 2022. "NRF2 and Key Transcriptional Targets in Melanoma Redox Manipulation" Cancers 14, no. 6: 1531. https://doi.org/10.3390/cancers14061531