
The Role of Ferric Nitrilotriacetate in Renal Carcinogenesis and Cell Death: From Animal Models to Clinical Implications


Department of Pathology and Biological Responses, Graduate School of Medicine, Nagoya University, 65 Tsurumai-cho, Showa-Ku, Nagoya 466-8550, Japan
Cancers 2022, 14(6), 1495; https://doi.org/10.3390/cancers14061495
Submission received: 12 February 2022
/
Revised: 8 March 2022
/
Accepted: 13 March 2022
/
Published: 15 March 2022
(This article belongs to the Special Issue Iron and Cancer)
Abstract
:Simple Summary
Iron is essential for cellular growth and survival. As a consequence, iron deficiency causes pleiotropic effects on the organism, while iron overload is also deleterious by means of oxidative tissue injury, which causes hepatic cirrhosis, diabetes mellitus, and cardiomyopathy in humans. Non-heme iron comprises ferric ion (Fe(III)), which is much more prominent in the transferrin, ferritin, or labile iron pool than the ferrous ion (Fe(II)); in contrast, ferrous ion yields more reactive oxygen species (ROS) than ferric ion does. In rodents, ferric nitrilotriacetate (Fe-NTA) elicits hepatic and renal oxidized lipids via a glutathione-cycle-dependent iron reduction that eventually causes renal cell carcinoma (RCC). In addition to iron-mediated carcinogenesis, ferroptosis is triggered by the iron-dependent accumulation of lipid peroxidation to lethal levels. Here, the mechanisms of iron- and ROS-mediated RCC and the therapeutic possibility of ferroptosis are discussed.
Abstract
Iron is essential for cellular growth, and various ferroproteins and heme-containing proteins are involved in a myriad of cellular functions, such as DNA synthesis, oxygen transport, and catalytic reactions. As a consequence, iron deficiency causes pleiotropic effects, such as hypochromic microcytic anemia and growth disturbance, while iron overload is also deleterious by oxidative injury. To prevent the generation of iron-mediated reactive oxygen species (ROS), ferritin is synthesized to store excess iron in cells that are consistent with the clinical utility of the serum ferritin concentration to monitor the therapeutic effect of iron-chelation. Among the animal models exploring iron-induced oxidative stress, ferric nitrilotriacetate (Fe-NTA) was shown to initiate hepatic and renal lipid peroxidation and the development of renal cell carcinoma (RCC) after repeated intraperitoneal injections of Fe-NTA. Here, current understanding of Fe-NTA-induced oxidative stress mediated by glutathione-cycle-dependent iron reduction and the molecular mechanisms of renal carcinogenesis are summarized in combination with a summary of the relationship between the pathogenesis of human RCC and iron metabolism. In addition to iron-mediated carcinogenesis, the ferroptosis that is triggered by the iron-dependent accumulation of lipid peroxidation and is implicated in the carcinogenesis is discussed.
1. Introduction
Iron is the most abundant transition metal in the body. Iron-containing proteins utilize iron at catalytic sites and facilitate oxygen transport, oxygen metabolism, energy metabolism, DNA synthesis, and repair [1]. Iron deficiency causes impaired growth and cellular functions; thus, mammals develop recycling systems to keep iron in the body. The absorption of dietary iron is tightly regulated at the duodenum through divalent metal transporter 1 (DMT1). Indeed, a previous study showed that Dmt1-deficient mice were unable to survive without red blood cell transfusion [2] and that the mutation of human DMT1 causes hypochromic microcytic anemia. The concentration of chelatable iron within body fluids is normally maintained at a low level (less than 1 μM), while iron overloading conditions elevate the redox-active and chelatable non-transferrin-bound iron concentration to 1–20 μM, causing damage to several tissues by the induced reactive oxygen species (ROS) [3]. Iron-induced ROS exacerbate multiple diseases, such as neurological diseases (Alzheimer’s, Huntington’s, and Parkinson disease’s), cardiovascular system diseases (atherosclerosis by excess lipid peroxidation and hereditary hemochromatosis (HH)-induced cardiomyopathy), hepatocellular carcinoma (HCC) (HH, hepatitis B virus (HBV), or hepatitis C virus (HCV)), or asbestos-related asbestosis, lung cancer, and malignant mesothelioma (MM) [4]. With the current advances that have been made in nutrition, iron deficiency is quite rare, except in women before menopause and in patients who suffer from gastrointestinal bleeding or eating disorders. In this article, investigations of the iron-triggered oxidative stress that causes carcinogenesis or cell death, namely ferroptosis, are reviewed.
2. Iron-Induced Renal Carcinogenesis
2.1. Ferric Nitrilotriacetate (Fe-NTA) Induces Renal Oxidative Injury and Eventually Leads to Renal Cell Carcinoma (RCC)
Animal models of Fe-NTA injections were first developed as an experimental iron overload model that caused diabetes mellitus by means of iron loading to pancreatic endocrine cells [5]. The use of NTA enables ferric ions to be maintained in a mono- or oligo-state in solution at neutral pH by forming a weak iron complex [6] and a short-lived intermediate complex of NTA-ferric ion-transferrin (Tf) [7]. The peak iron concentration in serum after a single intraperitoneal injection of 10 mgFe/kg body weight was observed at 1 h and returned to untreated levels after 24 h [6]. A single Fe-NTA injection also elevated hepatic lipid peroxidation and the release of serum hepatic injury markers, such as aspartate aminotransferase (AST) and alanine aminotransferase (ALT) [6]. In 1982, RCC development was observed in male Wistar rats after repeated injections of Fe-NTA [8]. Oral administration of Fe-NTA did not induce RCC [8], which is consistent with the tight regulation of intestinal iron absorption [9]. Despite the high incidence and early development of Fe-NTA-induced RCC, HCC is rare in wild-type (Wt) littermates of rats and mice [8]. Indeed, the combination of a single intraperitoneal injection of N-diethylnitrosamine (DEN) (100 mg/kg body weight), which is used for the initiator of HCC development [10], and repeated intraperitoneal injections of Fe-NTA elevated the incidence of RCC, while no HCC was developed with the injection of DEN alone or with the combined injections of DEN and Fe-NTA in male ddY mice [11] or male Wistar rats [12], suggesting that the liver is more robust against Fe-NTA-induced oxidative stress than the kidney.
Repeated intraperitoneal injections of copper-NTA, which induced hepatic cirrhosis, hemolytic anemia, and renal tubular necrosis [13], led to a higher incidence of RCC (12/32) than HCC (1/32) and hepatic sarcoma (1/32), while these cancers developed more slowly than Fe-NTA-induced RCC did in male Wistar rats [14]. In another study, 100% (13/13) of the Long–Evans Cinnamon (LEC) rats, which harbor an Atp7b mutation, developed necrotizing hepatitis in addition to developing HCC (13/13) and RCC (4/13) at 111–120 weeks [15]. These HCC and renal tubular injuries were attenuated by a copper-chelating drug, D-penicillamine [15], indicating that copper is critical for these diseases, while Wilson’s disease patients, who carry ATP7B mutations, with increased copper accumulations and ROS in the mitochondria develop neurological, psychiatric, ophthalmological, and hepatic symptoms without frequent renal involvement [16]. On the other hand, aluminum-NTA, which causes acute tubular necrosis and regenerative epithelial cells, did not induce RCC in rats [17].
Among the metal chelators, NTA initiated lipid peroxidation more efficiently than diethylenetriaminepentaacetate (DTPA), ethylenediamine-N, N, N′, N′-tetraacetate (EDTA), ADP, and deferoxamine (DFO) did when chelated with iron [18]. The standard redox potentials range from +0.59 V for Fe-NTA [19], +0.03 V for Fe-DTPA, +0.11 V for ionic form of iron, +0.12 V for Fe-EDTA, +0.10 V for Fe-ADP, −0.45 V for Fe-DFO, to −0.40 V for Fe-Tf at pH 7.0 [20], indicating the highly oxidizing potency of Fe-NTA. Indeed, the intraperitoneal injection of Fe-NTA elevated the renal 2-thiobarbituric acid reactive substances (TBARS) more than when the same ferric chloride dosage without a chelator was applied, while Fe-EDTA or Fe-DFO did not induce a significant increase in the renal TBARS in male Wistar rats [21]. On the other hand, EDTA, DTPA, and citrate with iron (molar ratio 1:1) elicited a catalytic reduction of H2O2 that disappeared when these chelators were present at concentrations that were over a hundred times greater than the iron concentration [22], indicating that the substrate or ferric–chelator complex ratio are important in such assessments. In addition to NTA, ethylenediamine-N,N′-diacetate (EDDA) with ferric ion induced renal lipid peroxidation and RCC [23], and Fe-iminodiacetate (IDA) also elicited renal lipid peroxidation and proximal tubular necrosis, while N-(2-hydroxyethyl)iminodiacetate (HIDA), ethylenediamine-N,N′-dipropionate (EDDP), and N,N′-bis(2-hydroxybenzyl)ethylendiamine–N,N′-diacetate (HBED) with ferric ions were ineffective in initiating renal lipid peroxidation in male Wistar rats [24]. While in sera, the iron concentration was not significantly different between the initiating lipid peroxidation group (Fe-NTA and Fe-EDDA pH 7.4; and Fe-IDA pH 5.2, 6.2, and 7.2) and the absence of the lipid peroxidation group (Fe-EDTA pH 7.4 and Fe-IDA pH 8.2) in Wistar rats, indicating the importance of the iron–chelator complex as an origin of ROS [24]. The origin of different iron chelate complexes in ROS generation is hypothesized as being the result of Fe-NTA and Fe-IDA forming a μ-oxo dimer iron at a neutral pH by means of crystal X-ray analysis [24,25].
2.2. Mechanisms of Fe-NTA-Induced Renal Oxidative Injury
After the intraperitoneal injection of Fe-NTA, Fe-NTA is delivered to renal proximal tubules through systemic circulation and glomerular filtration. At the renal proximal tubular lumen, Fe-NTA is reduced to ferrous-NTA by cysteine or cysteinylglycine, which are yielded by the glutathione (GSH) cycle (Figure 1). Although GSH has a redox potential similar to that of N-acetyl-cysteine (NAC) [20], GSH does not trigger the reduction of Fe-NTA at the same efficiency as cysteine or cysteinylglycine [26]. In brush border membrane, γ-glutamyl transpeptidase (γ-GTP)-mediated GSH cycling is presumed to maximize the re-absorption of filtrated plasma GSH based on the fact that the enzymatic activity of γ-GTP is the highest in the renal proximal tubular cells and the inhibitor or genetic impairment of γ-GTP increased the urinary excretion of GSH [27]. Pretreatment with AT-125, which inhibits γ-GTP, or butionine sulfoximine (BSO), which inhibits γ-glutamylcysteine synthetase (γ-GCS), attenuates Fe-NTA-induced renal lipid peroxidation, which explains why the distal tubules are not damaged severely by Fe-NTA [28]. The mechanism of Fe-NTA-induced oxidative injury is hypothesized to form a binuclear Fe-O-Fe unit structure (μ-oxo dimer iron), which oxidizes the surrounding biomolecules [29]. Indeed, the modification of the glutamate α-COO− ligand in GSH allowed the reduction of Fe-NTA and the elevation of lipid peroxidation in rat liver microsomes, suggesting the iron reducing capacity of GSH depends on liganding more than the redox potential and thiol pKa [26], and thiolate-ligated iron(II) sites have been shown to be essential for the function of many metalloenzymes [30], indicating the importance of μ-oxo dimer iron during redox reactions. Notably, Fe-NTA generated alkyl and peroxyl radicals in the renal tissue in an electron paramagnetic resonance (EPR) study [31]; thus, a ferrous-NTA-catalyzed Fenton reaction is presumed to trigger renal carcinogenesis [32]. These results indicate the importance of the GSH cycle on the brush border membrane in the renal proximal tubules. In addition to these catalytic reactions, γ-GTP was overexpressed in hepatic preneoplastic lesions in rats who had been treated with chemical carcinogens, indicating the tumor promotional role of γ-GTP by allowing the salvaging of extracellular GSH [33].
The castrated male mice and female mice all survived, while all of the male mice died from repeated Fe-NTA injections in the A/J strain, indicating the sex differences against Fe-NTA-induced oxidative stress [37]. Indeed, the administration of testosterone in castrated male mice and female mice elevated renal lipid peroxidation to levels that were as high as those seen in male mice after Fe-NTA injection [37]. In contrast, estriol attenuated Fe-NTA-induced renal lipid peroxidation in male ddY mice [38]. Furthermore, castration and estradiol suppressed Fe-NTA-induced renal carcinogenesis; conversely, ovariectomy and testosterone increased the frequency of renal carcinogenesis in Wistar rats [39]. Furthermore, the half-life of GSH in males was shorter than that observed in females, something that can be partially explained by the increased γ-GTP activity in males [40,41]. These renal and hepatic GSH cycles were delayed by castration and estradiol treatments in male mice, while testosterone accelerated the cycles in female C57BL/6 mice [40]. However, the intravenous injection of estrogen 5 min prior to intraperitoneal Fe-NTA injection was ineffective in protecting male mice, indicating that estradiol did not attenuate Fe-NTA by direct chelating or scavenging ROS [38]. These results indicate that feminization is critical to protect the liver and kidney from Fe-NTA-induced oxidative injury via an altered GSH cycle and/or feminized protein expression.
Recently, the male-specific expression of the cystine transporter Slc7a13 was observed in a murine kidney model and was shown to potentially be able to modulate the renal GSH cycle, although perfect palindrome estrogen- and androgen-responsive elements were not detected in Slc7a13 [36]. Among the various cystine transporters, Slc3a1 forms a heterodimer for the amino acid transport system and showed its highest expression in the non-neoplastic kidney and intestine [42]. Further, SLC7A11, which forms a heterodimer with SLC3A2 and is known as a xCT (cystine/glutamate antiporter), has higher expression in RCC than in the non-neoplastic kidney and has been shown to be related to a worse prognosis in RCC [43]. The loss of Slc7a11-dependent cystine uptake increased the susceptibility to ROS in Slc7a11-deleted mice, while renal stones, which are frequently caused by cystinuria, were not seen [44], suggesting xCT is not critical in reabsorption of filtrated cystine. These results are consistent with previous results indicating that cystinuria is caused by the mutation of either SLC3A1 or SLC7A9 [34] (Figure 1). Furthermore, the kidney-specific ablation of the murine androgen receptor (AR) caused increased ammonia excretion and increased expression of the Na+-K+-2Cl− cotransporter (NKCC2) and phosphoenolpyruvate carboxykinase (PEPCK), while it triggered the suppression of renal size, Na+/H+ exchanger isoform 3 (NHE3), and Na+-bicarbonate cotransporter 1 (NBCe1) [45]. Future studies may reveal feminization-induced protection against iron-induced oxidative stress.
2.3. Molecular Mechanisms of Fe-NTA-Induced Carcinogenesis in Rodents Compared to Human RCC
The von Hippel–Lindau (VHL) mutation shows that it is able to increase susceptibility to hemangioblastoma in the retina or central nervous system, neuroendocrine tumors (notably pheochromocytoma), and familial or sporadic clear-cell RCC (ccRCC) at a high frequency in humans [46]. This Vhl mutation was not detected in Fe-NTA-induced RCCs [47,48]. Furthermore, no H-, K-, or N-ras gene mutations (codon 12, 13, or 61) were detected via direct sequencing in the 12 Fe-NTA-induced RCCs [49] and in the 16 RCCs [50] with low incidences of p53 mutations, showing rates of (1/12) and (2/16), respectively. While low mutation frequencies of Vhl, ras, and p53 were detected in the Fe-NTA-induced RCC model, the combined deletion of Vhl, p53, and Rb1 in the renal tubular cells caused ccRCC in mice [51]. The single or double conditional knock out (cKO) of Vhl and/or p53 and Rb1 in the renal tubular cells showed only the development of cysts and microscopic neoplasms, indicating the importance of the simultaneous disruption of the p53 and G1/S cell cycle checkpoint [51,52]. Meanwhile, the double cKO of Vhl and BRCA1-associated protein 1 (Bap1) developed ccRCC faster than the double cKO of Vhl and polybromo 1 (Pbrm1), which encodes the BRG1-associated factor 180 (BAF180), a component of the SWI/SNF-B chromatin-remodeling complex [53]. The absence of RCC development by means of murine single Vhl disruption is presumed to be due to the fact that murine Vhl is located on chromosome (chr) 6, whereas Bap1 and Pbrm1 are located on chr 14, causing the avoidance of the simultaneous loss of Vhl and Bap1 or Pbrm1, while BAP1, PBRM1, and VHL are located on chr 3p21 and 3p25 and are frequently co-deleted in ccRCC [53].
The VHL complex, which comprises pVHL, elongin B, elongin C, Cullin-2, and RBX1, degrades hypoxia-inducible factor (HIF) 1α and 2α by ubiquitination once the HIFs are hydroxylated by the α-ketoglutarate-dependent prolyl hydroxylases (PHDs) (Figure 2). In VHL-mutated ccRCCs, both HIF1α and HIF2α are important, due to the frequent HIF1A deletions (40–55%); furthermore, a murine model demonstrated the sufficient inhibition of ccRCC pathogenesis by Hif2α degradation and the necessity for the ccRCC formation by Hif2α stabilization [46]. More recently, the inhibition of the HIF2α and HIF1β heterodimer by therapeutic small molecules has been shown to effectively suppress ccRCC progression in patients [54].
The tuberous sclerosis complex (TSC) is caused by a germline loss-of-function mutations of either the TSC1 or TSC2 gene develops a wide range of tumors in the brain (subependymal giant cell astrocytoma and cerebral cortical tuber), heart (rhabdomyoma), kidney (angiomyolipoma, cyst, and RCC), lung (lymphangioleiomyomatosis), or skin (angiofibroma) [55]. In a hereditary RCC Eker rat model, Tsc2 was shown to be the causative gene; however, the Tsc2 mutation was only detected in 3% (1/34) of Fe-NTA-induced RCC [48]. On the other hand, biallelic Tsc2 mutations were observed in N-ethyl-N-hydroxyethylnitrosamine (EHEN) with KBrO3-induced RCCs (2/3) and DEN-induced RCCs (3/5) [56]. These results indicate that different renal carcinogens, which generate ROS, confer selective genes. In Tsc2-mutated Eker rats, the homozygous deletion (HD) of Cdkn2a was detected in the cell lines (5/7 or 14/24) and was not detected in the primary RCCs [57,58]. These results suggest that the loss of Cdkn2a plays a tumor progressive role in vitro in Tsc2-mutated RCC [58]. Meanwhile, in Fe-NTA-induced RCC, the methylation of the promoter region or the loss of heterozygosity (LOH), or HD was detected in Cdkn2a (21/39) and Cdkn2b (12/39), respectively [59]. The allelic loss of Cdkn2a was detected by fluorescent in situ hybridization (FISH) in male Wistar rats after receiving Fe-NTA injections for 3 weeks [60]. These results suggest that the loss of Cdkn2a/2b is triggered at the early tumor initiative stage. Indeed, germline mutations in CDKN2B predisposed to ccRCC, while the copy number abnormalities in the CDKN2A/2B region were detected in approximately 15% of sporadic ccRCC cases [61], indicating the presence of a common carcinogenic pathway in rodents and human RCC. In addition to the inactivation of Cdkn2a/2b, Met amplification was significantly associated with increased tumor size in Fe-NTA-induced RCC (Figure 2). Furthermore, transforming growth factor α (Tgf-α), which is induced by Hif and activates epidermal growth factor receptor (Egfr) signaling, was expressed in Fe-NTA-induced rat RCC [62]. While male Wt rats developed Fe-NTA-induced RCC, nearly 90% of Wistar strain or Brown Norway/Fischer344 (BN/F344) F1 hybrid rats during a two-year observation period [32], male Wt mice developed RCC in the A/J strain (18/29), C57BL/6 strain (1/14) [63], and ddY strain (7/20) [11], indicating the difference in the genomic instability against Fe-NTA-induced oxidative injury between the mouse strains and rats [64].
Fe-NTA-induced renal oxidative stress induced the activation of nuclear factor erythroid 2-related factor 2 (Nrf2), which elevated the antioxidative phase II metabolizing enzymes, such as GSH-S-transferases (Gst), NADPH quinone oxidoreductase 1 (Nqo1), heme oxygenase-1 (Hmox1), and γ-Gcs [65,66] (Figure 2). While Hmox1 protects cells from ROS, the release of ferrous ions from heme may elevate lipid peroxidation and cellular injury; thus, the role of Hmox1 in the iron overloaded condition requires further investigation. Not limited to ROS, Nrf2 is also activated by several electrophiles, such as fumarate or curcumin. Curcumin induced phase II metabolizing enzymes [67] as well as attenuated Fe-NTA-induced ROS by direct scavenging that suppress renal carcinogenesis in male ddY mice [11,68]. In addition to curcumin, dietary vitamin E prevented Fe-NTA-induced lipid peroxidation [69] and significantly suppressed Fe-NTA-induced renal carcinogenesis in male Wistar rats [70] (Figure 1). Meanwhile, phlebotomy did not suppress the incidence of Fe-NTA-induced RCC in male Wistar rats [71]. In systemic Dmt1-IRE (iron-responsive element) transgenic mice, the Fe-NTA-induced renal damage was not obviously elevated [72]. Future studies may reveal the role of iron-induced oxidative damage in renal carcinogenesis that could contribute to identifying chemopreventive compounds for RCC.
2.4. Iron Modulates the Expressions of HIF-α by PHDs and HIF2α by IRE/Iron Regulatory Protein (IRP)
The dietary iron efflux into systemic circulation is transported by ferroportin (FPN, SLC40A1), which is internalized and degraded by hepcidin when excess iron or inflammation are sensed in the liver, which regulates systemic iron storage [1]. The translation of HIF2α mRNA is regulated by IRE/iron regulatory protein 1 or 2 (IRP1/2) through IRE in the 5′ untranslated region (UTR), while HIF1α does not contain IRE in mRNA. Thus, iron deficiency elevated the protein expression level of HIF1α and 2α via suppressed PHDs; in contrast, HIF2α was decreased by elevated IRE/IRP activity [46] (Figure 3). In murine embryonal fibroblasts (MEF) from Irp1 KO mice, the Hif2α protein was expressed in both iron-sufficient and DFO-treated conditions and was higher than expression levels in the Wt littermates, indicating that Irp1/2 and Phds regulate Hif2α protein expression separately [73]. The intestine-specific ablation of Hif2α decreased the mRNA levels of Dmt1(IRE) and Fpn in the duodenum, hepcidin in the liver, and iron concentration in serum and the liver [74]. Conversely, the pharmacological inhibition of PHDs, which are clinically prescribed for renal anemia to activate HIF signaling, increased erythropoietin and DMT1, while PHD expression was induced by the HIFs to form a negative feedback loop to prevent the persistent activation of HIF signaling [75]. In addition to DMT1, ZRT/IRT-like protein (ZIP) 14 exhibited iron uptake, while the intestine-specific ablation of Dmt1 caused a lethal iron-deficiency that was restored by the intraperitoneal injection of iron, indicating that Dmt1 is essential for intestinal iron absorption [76]. The loss of IRE/IRPs-dependent translational suppression increases the levels of protein expression in FPN, the ferritin heavy chain (FTH), and the ferritin light chain (FTL) that have IRE in the 5′ UTR region of the mRNA and decreases intracellular labile iron by exporting iron to outside of the cell or by storing it in proteins to reduce iron-induced cellular injury. When HIF2α is mutated to be stabilized by inhibiting the hydroxylation of proline and the subsequent binding to VHL, multiple paraganglioma, duodenal somatostatinoma, and polycythemia are developed [77]. Furthermore, a gain of function in HIF2α developed vascular malformation in patients and mouse models that may have been caused by the failure of elevated oxygen tension-induced normal venous regression [78]. These results demonstrate that the continuous activation of HIF2α signaling is not associated with the iron overload phenotype; thus, further study may reveal the interaction of iron and HIF signaling in RCCs. Based on the evidences of iron metabolism, the clinical trial of iron-chelating therapy by DFO is effective in hematological neoplasms that are relatively addicted to iron supply for tumor growth. Furthermore, the clinical trial of DFO for cancer therapy demonstrated partial response in HCC, while only a 7% overall response rate (ORR, the percentage of patients with a predefined reduction in tumor size for a certain duration) was observed in metastatic RCC [1], which may have been caused by the increased HIF-α in the absence of active PHDs. From the viewpoint of iron-mediated PHDs activation, elevated iron in RCC may be useful for a therapeutic modality.
2.5. Disruption in Succinate Dehydrogenase (SDH) or Fumarate Hydratase (FH) Causes RCC by Inhibiting PHDs
In addition to VHL, which causes familial RCC, the mutation of SDH predisposes one to oncocytic RCC and gastrointestinal stromal tumors (GIST) with a lower frequency than paraganglioma and phenochromocytoma. SDH, which oxidizes succinate to fumarate with the reduction of FAD+ to FADH2 as complex II in the mitochondrial respiratory chain, is assembled by SDHA, SDHB, SDHC, and SDHD subunits [79]. Among the SDH subunits, SDHB is mutated most frequently, followed by SDHC and SDHD, in hereditary SDH-deficient RCC, while germline mutations in SDHA have been described to cause neurodegenerative diseases, such as Leigh syndrome without neoplastic disorder [80]. In addition to congenital disease, the acquired SDHA mutation caused oncocytic RCCs without SDHB/C/D mutations [81]. Among SDHB mutations, Fe-S cluster mutations in the L(I)YR motif amino acid residues were occupied for 50% of the SDHB-mutated RCC and GIST, suggesting the importance of the L(I)YR motif in renal cell carcinogenesis [79]. In addition to SDH, the germline inactivation of FH that catalyzes fumarate to malate in the TCA cycle and suppresses AMP-activated protein kinase (AMPK) signaling increases the susceptibility to papillary RCC (type 2) and leiomyomatosis in the skin and uterus [73]. The elevation of succinate or fumarate caused by a metabolic shift in the TCA cycle inhibits the PHDs that activate HIF signaling [82]. In SDH- or FH-deficient RCCs, the protein expression of HIF1α but not HIF2α was more predominant than ccRCC expression [73,79].
2.6. Ischemia Reperfusion and Iron-Mediated ROS Induces Renal Medullary Carcinoma
Sickle cell nephropathy, which causes iron overload in the kidney, is characterized by impaired urinary concentration ability, proteinuria, hematuria, chronic tubulointerstitial nephritis, hemolysis, and increased susceptibility to acute kidney injury (AKI), urinary tract infections, and medullary carcinoma due to altered renal hemodynamics, such as the perfusion paradox, which includes medullary hypoperfusion and renal and/or cortical hyperperfusion, and unique ischemia–reperfusion injury (IRI) with repeated multifocal and microvascular occlusions with subsequent infarction [83]. Renal medullary carcinoma (RMC), which was originally described in 1995 and comprises less than 0.5% of all RCCs, is predominantly detected in young African Americans with the sickle cell trait or sickle hemoglobinopathies [84]. RMC is characterized by a near complete loss of SMARCB1 expression, which is a component of the SWI/SNF chromatin remodeling complex and harbors fewer genetic alterations than other RCCs (cc, ch, and papillary). No VHL, PBRM1, BAP1, TP53, or CDKN2A mutations have been reported [85], indicating that IRI and iron overload-induced ROS confer SMARCB1.
2.7. Histopathology of RCC
The histopathologies of ccRCC (VHL disease or BAP1-associated tumor predisposition syndrome), SDH-mutated oncocytic RCC, and hereditary papillary RCC (constitutive activation of MET (type 1) or FH-mutated hereditary leiomyomatosis and RCC (type 2)) represents a single predominant histology, while TSC-mutated RCC represents heterogeneous components that show a striking contrast (Table 1) [55,86]. In human chromophobe (ch) RCC, which is caused by the germline mutation of Tsc1/2 or FLCN or PTEN, the most frequent mutations were detected in p53 and PTEN, followed by a loss of expression in CDKN2A [87]. In addition to these established associations of causative genes with characteristic histopathology, the mutation of the microphthalmia-associated transcription factor (MiTF), which consists of the basic helix–loop–helix family of transcription factors with TFE3 and TFEB, was shown to cause familial renal cancer and cutaneous melanoma [88]. These results suggest that chromophobe histopathology favors the activation of the mTOR pathway (Table 1).
2.8. Iron-Induced Peritoneal Malignant Mesothelioma (MM) in Rats
In addition to RCCs, the repeated intraperitoneal injection of Fe-NTA causes iron deposition on the surface that is susceptible to peritoneal MM development. Indeed, the iron saccharate that accumulated on the serosal surface caused MM development in peritoneum and tunica vaginalis without RCC and pleural MM in male Wistar rats. This mesothelial carcinogenesis was accelerated by simultaneous NTA treatment [90], indicating the importance of elevated chelated and/or mobile iron in the local peritoneal fluid by NTA. The development of MM via the intraperitoneal injection of iron saccharate was predominantly observed in male (16/24) but not female (1/30) BN/F344 F1 hybrid rats [91]. Furthermore, the HD of Cdkn2a/2b was detected in iron-saccharate-induced sarcomatoid mesothelioma (SM) (4/5) and epithelioid mesothelioma (EM) (0/6) in BN/F344 F1 hybrid rats [91], and EM (1/5) was observed in male Wistar rats [92]. In three Fe-NTA-induced peritoneal MM, no ras, p53, Vhl, or Tsc2 mutations were detected [48,49], while in human MM, the most frequent genetic mutations were observed in CDKN2A/2B, NF2 (neurofibromatosis 2), and BAP1 [4,93]. Epidemiological studies indicate that the onset of MM is associated with the exposure of asbestos fibers. Among them, crocidolite and amosite, which contain higher iron in the fiber are more carcinogenic than chrysotile, suggesting that iron accelerates mesothelial injury and carcinogenesis via ROS production [4]. In a rat model of intraperitoneal injection of crocidolite, iron reductions by an iron chelator, deferasirox [94], and by phlebotomy [95] prevented the MM developments, indicating the importance of iron storage in the body. Indeed, phlebotomy is useful in hereditary hemochromatosis patients as well as in HCV-infected patients, as it can suppress hepatitis activity and HCC development [96]. Meanwhile, MM cells that harbor BAP1 or NF2 mutations are vulnerable to iron-dependent cell death [4,93].
3. Ferroptosis
3.1. Concept of Ferroptosis
Ferroptosis is established to dictate the nonapoptotic cell death that is triggered by the iron-dependent accumulation of oxidized lipids, including polyunsaturated fatty acids (PUFAs) to lethal levels [97], and is implicated in the pathogenesis of neurodegenerative diseases (Alzheimer’s, Huntington’s, and Parkinson’s diseases), brain attacks (stroke, intracerebral hemorrhage, and traumatic brain injury), and IRI [98]. Ferroptosis was not originally affected in the necroptotic signaling pathway by receptor-interacting protein kinases (RIPK) 1 and 3, while ferroptosis shares a typical morphology with necrosis, along with small mitochondria, a decreased crista, a condensed inner membrane, and a ruptured outer membrane [99]. Indeed, iron triggers the phospholipids (PL)-PUFA-OOH (hydroperoxide), -OO• (peroxyl radical), and -O• (alkoxyl radical), which are scavenged by α-tocopherol (α-Toc) to form α-Toc• (Figure 4A); thus, long-term iron overload is presumed to be a soil for carcinogenesis by acquiring ferroptosis-resistance [4]. Indeed, PL-PUFA-OOH were significantly elevated in the red blood cells of Alzheimer’s disease patients [100], suggesting that the systemic accumulation of lipid hydroperoxides accelerates cerebral damage. In addition to these pathogenic involvements of these diseases, ferroptosis is an eliminator of potent tumor-initiating cells and tumor cells, indicating a promising therapeutic approach for cancers. When considering the induction of ferroptosis in cancer therapy, ferroptosis increases the risk of neuronal cell death in the central nervous system, potentially aggravating neurological symptoms [101]. Indeed, iron accumulation that is visualized by magnetic resonance imaging (MRI) at the substantia nigra was associated with the disease severity in Parkinson’s disease patients, suggesting the therapeutic potential of iron-chelating therapy [102].
3.2. The Mechanisms of Ferroptosis
Ferroptosis is initiated by specific enzymatic pathways or iron-catalyzed free radical chain reactions that are suppressed by direct ROS scavenging, such as vitamin E, coenzyme Q10 (CoQ10), and ferrostatin-1, or the supplementation of GSH sources, such as NAC and cystine, iron-chelating, such as DFO, or the inhibition of glutaminolysis, lipoxygenases (LOXs), acyl-CoA synthetase long chain family member 4 (ACSL4), or lysophosphatidylcholine acyltransferase 3 (LPCAT3) [98]. Conversely, ferroptosis is induced through the inhibition of a cystine/glutamate antiporter (SLC3A2/SLC7A11 heterodimer), also known as xCT with erastin or glutamate, depleting GSH with BSO, or inhibiting glutathione peroxidase 4 (GPX4) with (1S,3R)-RSL3 (RSL3) [98]. Among the eight distinct isoforms of GPXs, only GPX4 catalyzes detoxifying phospholipid hydroperoxide (PL-OOH) at the cellular membrane, which inhibits membrane instability and permeabilization as follows: PL-OOH + 2 GSH → PL-OH (phospholipid alcohol) + GSSG + H2O, while the other GPXs reduce H2O2 and small fatty acid hydroperoxides, including soluble free fatty acid peroxides [104,105,106]. Furthermore, L-glutamine (Gln), which is hydrolyzed by glutaminase to Glu, induced ferroptosis that was ameliorated by inhibiting transaminase from converting Glu to α-ketoglutarate [107]. Indeed, α-ketoglutarate, succinate, fumarate, and malate promoted cystine-deprivation-induced ferroptosis via the hyperpolarization of the mitochondrial membrane potential [108], indicating that the activated TCA cycle causes ferroptosis by the means of dysregulated mitochondrial respiration when fueled by glutaminolysis. In addition to these metabolites, the basal NADPH level was correlated with sensitivity to ferroptosis inducers [109]. NADPH contributes to the following reactions: GSSG+ 2 NADPH → 2 GSH + 2 NADP+ by GSH reductase, CoQ10H•/α-Toc + NADPH → CoQ10H2/α-Toc + NADP+ by ferroptosis suppressor protein 1 (FSP1), and BH2 (dihydrobiopterin) + 2 NADPH → BH4 + 2 NADP+ by dihydrofolate reductase (DHFR) in combination with guanosine triphosphate cyclohydrolase 1 (GCH1), the rate-limiting synthetic enzyme for BH4 that scavenges ROS and suppresses ferroptosis in a GPX4-independent manner [93]. Taken together, the disrupted homeostasis of iron, amino acids, thiols, and PUFAs with oxygenating machinery is critical in ferroptotic cascades (Figure 4B).
3.3. Ferroptosis in the Kidney and RCC
The cKO of Gpx4, which is expressed at higher levels in the proximal tubular cells than it is in the glomeruli, caused AKI with increased phosphatidylcholine (PC)-, phsphatidylethanolamine (PE)-, and cardiolipin (CL)-esterified linoleic acid (LA, C18:2), arachidonic acid (AA, C20:4), and docosahexaenoic acid (DHA, C22:6) oxidations, while Gpx4 KO mice showed embryonic lethality [105,110]. Other studies focusing on Gpx4 cKO mice disclosed that PE-AA and PE-adrenoyl (AdA, C22:4) acyls transduce ferroptotic death signaling in the MEFs and kidney [103]. In AKIs, renal tubular death that is mediated by caspases-mediated apoptosis and inflammasome-mediated pyroptosis is detected less frequently than necrosis and ferroptosis [111]. The amelioration of renal IRI by the ferroptosis inhibitor was enhanced by the combination of the necrosis inhibitor that suppressed RIPK, indicating the interaction of these signaling pathways [112], while the replacement of the active selenocysteine in Gpx4 did not become sensitized to cisplatin-induced AKI, indicating that impaired Gpx4 did not aggravate all of the AKIs [113].
In the ccRCC cell lines, erastin and BSO inhibited cellular growth, indicating the importance of GSH biosynthesis and the GPX-dependent detoxifying pathway [114]. Indeed, the tumoricidal activity of erastin was associated with the SLC7A11 expression levels [115]. Furthermore, in HLRCC-patient-derived RCC cell lines harboring the FH mutation, the increased proliferation with resistance to cystine-starvation-induced ferroptosis was canceled by the forced expression of Wt FH [108], suggesting that the induction of ferroptosis contributes to RCC suppression.
3.4. Induction of Oxidized Lipids and RCC by Fe-NTA
Fe-NTA generated a wide range of C2–12 aldehydes and C7–12 acyloins, metabolites of saturated aldehydes in the renal and hepatic tissues of male Wistar rats [116]. Among these oxidized lipids, a single injection of Fe-NTA elevated 4-hydroxy-2-nonenal (HNE) at the highest fold change (27.3-fold) and malondialdehyde (MDA), the most abundant aldehyde in the kidney [116]. A single Fe-NTA injection also decreased PUFAs, such as AA and DHA to 70–80%, while linoleic acid was not significantly oxidized in male Wistar rats [117]. In line with previous reports indicating that the supplementation of AA enhanced RSL-3-induced ferroptosis [103], the supplementation of dietary DHA enhanced Fe-NTA-induced renal oxidative injury, which could be restored by α-Toc supplementation in mice [118]. These results indicate that dietary lipids modified Fe-NTA-induced oxidative stress (Figure 4A). In addition to elevated lipid peroxidations, Fe-NTA decreased ferroptosis-suppressive substrates, such as GSH and NADPH [119]. Simultaneously, Fe-NTA activated Nrf2, which suppresses ferroptosis by inducing detoxifying enzymes [104]; thus, the final redox balance in the microenvironments may affect the fates of cells. Furthermore, the Gpx4 heteroKO mice prolonged their life spans by increasing the susceptibility to cell death compared to Wt mice [120]. Indeed, the A/J strain, which develops higher Fe-NTA-induced RCC, yielded lower oxidative DNA damage, lipid peroxidation, and ferroptosis than the C57/BL6 strain, indicating the role of ferroptosis in cancer prevention by eliminating mutation-prone cells [63].
An epidemiological study to assess the risk of RCC onset provided no clear association between the dietary intake of ascorbate or vitamin E and RCC risk [121]; recently, an umbrella review of meta-analyses focusing on the prevention of RCC in dietary compounds indicated no high-quality evidence or moderate-quality evidence of vitamin C intake and low-quality evidence of α-Toc and calcium intake [122], indicating the difficulty of translating the preventive clinical measures from basic research. In human trials for chemoprevention by antioxidative compounds on asbestos-exposed workers or smokers, the number of lung cancer patients increased after supplemental β-carotene with retinol, which is presumed to have a toxic pro-oxidant effect that contaminates breakdown products or affects uptake modulation for other carotenoids [20]. In addition to these factors, this tumor promotional effect of β-carotene may be initiated by the escape of mutated cells from ferroptosis (Figure 4B).
4. Conclusions
In mammals, the tight regulation of intestinal iron absorption from the diet inhibits iron-mediated tissue injury. Meanwhile, in experimental models, the intraperitoneal injection of Fe-NTA initiates oxidative injury by the GSH-cycle-dependent reduction of ferric to ferrous ion. Furthermore, the repeated injection of Fe-NTA causes the inactivation of Cdkn2a/2b in renal proximal tubules that finally develop renal carcinogenesis in rodents. In contrast to iron-induced carcinogenesis, iron-dependent cell death is established as ferroptosis. Ferroptosis forms a “wave of death” in renal tubular cells, such as free-radical-initiated propagative chain reactions. Indeed, a multi-target drug combining iron chelation, ROS-scavenging, and anti-inflammatory effects is proposed to restore ischemic injury by terminating ferroptosis [102]. In addition, the ferroptosis induction that is based on the understanding of redox biology is an emerging and promising modality for cancer therapy. Further study is warranted to explore the role of ferroptosis in clinical medicine.
Funding
This work was funded by JSPS KAKENHI grant number 21K06968.
Acknowledgments
The author wish to thank Toyokuni (Department of Pathology and Biological Responses, Graduate School of Medicine, Nagoya University) for helpful discussions.
Conflicts of Interest
The author declares no conflict of interest.
Abbreviations
| AKI | acute kidney injury |
| BN/F344 | Brown Norway/Fischer344 |
| DFO | deferoxamine |
| DMT1 | divalent metal transporter 1 |
| Fe-NTA | ferric nitrilotriacetate |
| GSH | glutathione |
| GPX4 | glutathione peroxidase 4 |
| HCC | hepatocellular carcinoma |
| HD | homozygous deletion |
| IRE | iron-responsive element |
| IRI | ischemia-reperfusion injury |
| IRP | iron-regulatory protein |
| LOH | loss of heterozygosity |
| MM | malignant mesothelioma |
| RCC | renal cell carcinoma |
| ROS | reactive oxygen species |
| Wt | wild-type |
References
- Torti, S.V.; Manz, D.H.; Paul, B.T.; Blanchette-Farra, N.; Torti, F.M. Iron and Cancer. Annu. Rev. Nutr. 2018, 38, 97–125. [Google Scholar] [CrossRef] [PubMed]
- Gunshin, H.; Fujiwara, Y.; Custodio, A.O.; Direnzo, C.; Robine, S.; Andrews, N.C. Slc11a2 is required for intestinal iron absorption and erythropoiesis but dispensable in placenta and liver. J. Clin. Investig. 2005, 115, 1258–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badu-Boateng, C.; Naftalin, R.J. Ascorbate and ferritin interactions: Consequences for iron release in vitro and in vivo and implications for inflammation. Free Radic. Biol. Med. 2019, 133, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Toyokuni, S.; Kong, Y.; Cheng, Z.; Sato, K.; Hayashi, S.; Ito, F.; Jiang, L.; Yanatori, I.; Okazaki, Y.; Akatsuka, S. Carcinogenesis as Side Effects of Iron and Oxygen Utilization: From the Unveiled Truth toward Ultimate Bioengineering. Cancers 2020, 12, 3320. [Google Scholar] [CrossRef]
- Awai, M.; Narasaki, M.; Yamanoi, Y.; Seno, S. Induction of diabetes in animals by parenteral administration of ferric nitrilotriacetate. A model of experimental hemochromatosis. Am. J. Pathol. 1979, 95, 663–673. [Google Scholar]
- Awai, M. Experimental iron overload using ferric nitrilotriacetate (Fe3+-NTA): A proposal model of the diseases in man caused by iron overload. Nihon Ketsueki Gakkai Zasshi 1986, 49, 1650–1659. [Google Scholar]
- Bates, G.W.; Wernicke, J. The kinetics and mechanism of iron(3) exchange between chelates and transferrin. IV. The reaction of transferrin with iron(3) nitrilotriacetate. J. Biol. Chem. 1971, 246, 3679–3685. [Google Scholar] [CrossRef]
- Okada, S. Iron-induced tissue damage and cancer: The role of reactive oxygen species-free radicals. Pathol. Int. 1996, 46, 311–332. [Google Scholar] [CrossRef]
- Nemeth, E.; Ganz, T. Hepcidin-Ferroportin Interaction Controls Systemic Iron Homeostasis. Int. J. Mol. Sci. 2021, 22, 6493. [Google Scholar] [CrossRef]
- Uehara, T.; Pogribny, I.P.; Rusyn, I. The DEN and CCl4-Induced Mouse Model of Fibrosis and Inflammation-Associated Hepatocellular Carcinoma. Curr. Protoc. Pharmacol. 2014, 66, 14–30. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, M.; Shah, M.D.; Vun-Sang, S.; Okazaki, Y.; Okada, S. The therapeutic potential of curcumin in alleviating N-diethylnitrosamine and iron nitrilotriacetate induced renal cell tumours in mice via inhibition of oxidative stress: Implications for cancer chemoprevention. Biomed. Pharmacother. 2021, 139, 111636. [Google Scholar] [CrossRef]
- Athar, M.; Iqbal, M. Ferric nitrilotriacetate promotes N-diethylnitrosamine-induced renal tumorigenesis in the rat: Implications for the involvement of oxidative stress. Carcinogenesis 1998, 19, 1133–1139. [Google Scholar] [CrossRef] [Green Version]
- Toyokuni, S.; Okada, S.; Hamazaki, S.; Fujioka, M.; Li, J.L.; Midorikawa, O. Cirrhosis of the liver induced by cupric nitrilotriacetate in Wistar rats. An experimental model of copper toxicosis. Am. J. Pathol. 1989, 134, 1263–1274. [Google Scholar]
- Toyokuni, S.; Tanaka, T.; Nishiyama, Y.; Okamoto, K.; Nakashima, Y.; Hamazaki, S.; Okada, S.; Hiai, H. Induction of renal cell carcinoma in male Wistar rats treated with cupric nitrilotriacetate. Lab. Investig. 1996, 75, 239–248. [Google Scholar]
- Kitaura, K.; Chone, Y.; Satake, N.; Akagi, A.; Ohnishi, T.; Suzuki, Y.; Izumi, K. Role of copper accumulation in spontaneous renal carcinogenesis in Long-Evans Cinnamon rats. Jpn. J. Cancer Res. 1999, 90, 385–392. [Google Scholar] [CrossRef]
- Shribman, S.; Poujois, A.; Bandmann, O.; Czlonkowska, A.; Warner, T.T. Wilson’s disease: Update on pathogenesis, biomarkers and treatments. J. Neurol. Neurosurg. Psychiatry 2021, 92, 1053–1061. [Google Scholar] [CrossRef]
- Ebina, Y.; Okada, S.; Hamazaki, S.; Ogino, F.; Li, J.L.; Midorikawa, O. Nephrotoxicity and renal cell carcinoma after use of iron- and aluminum-nitrilotriacetate complexes in rats. J. Natl. Cancer Inst. 1986, 76, 107–113. [Google Scholar]
- Hamazaki, S.; Okada, S.; Li, J.L.; Toyokuni, S.; Midorikawa, O. Oxygen reduction and lipid peroxidation by iron chelates with special reference to ferric nitrilotriacetate. Arch. Biochem. Biophys. 1989, 272, 10–17. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, C.; Wang, X.; Marshall, M.J.; Zachara, J.M.; Rosso, K.M.; Dupuis, M.; Fredrickson, J.K.; Heald, S.; Shi, L. Kinetics of reduction of Fe(III) complexes by outer membrane cytochromes MtrC and OmcA of Shewanella oneidensis MR-1. Appl. Environ. Microbiol. 2008, 74, 6746–6755. [Google Scholar] [CrossRef] [Green Version]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine, 5th ed.; Oxford University Press: Croydon, UK, 2015. [Google Scholar]
- Liu, M.; Okada, S.; Kawabata, T. Radical-promoting “free” iron level in the serum of rats treated with ferric nitrilotriacetate: Comparison with other iron chelate complexes. Acta Med. Okayama 1991, 45, 401–408. [Google Scholar] [CrossRef]
- Engelmann, M.D.; Bobier, R.T.; Hiatt, T.; Cheng, I.F. Variability of the Fenton reaction characteristics of the EDTA, DTPA, and citrate complexes of iron. Biometals 2003, 16, 519–527. [Google Scholar] [CrossRef]
- Liu, M.; Okada, S. Induction of free radicals and tumors in the kidneys of Wistar rats by ferric ethylenediamine-N,N′-diacetate. Carcinogenesis 1994, 15, 2817–2821. [Google Scholar] [CrossRef]
- Mizuno, R.; Kawabata, T.; Sutoh, Y.; Nishida, Y.; Okada, S. Oxidative renal tubular injuries induced by aminocarboxylate-type iron (III) coordination compounds as candidate renal carcinogens. Biometals 2006, 19, 675–683. [Google Scholar] [CrossRef]
- Nishida, Y.; Yoshizawa, K.; Akamatsu, T. Preparation of Iron(III) Complex with Nitrilotriacetic Acid and Origin of Its Unique Reactivity. Chem. Lett. 1991, 20, 1521–1524. [Google Scholar] [CrossRef]
- Spear, N.; Aust, S.D. Thiol-mediated NTA-Fe(III) reduction and lipid peroxidation. Arch. Biochem. Biophys. 1994, 312, 198–202. [Google Scholar] [CrossRef]
- Bachhawat, A.K.; Thakur, A.; Kaur, J.; Zulkifli, M. Glutathione transporters. Biochim. Biophys. Acta 2013, 1830, 3154–3164. [Google Scholar] [CrossRef]
- Okada, S.; Minamiyama, Y.; Hamazaki, S.; Toyokuni, S.; Sotomatsu, A. Glutathione cycle dependency of ferric nitrilotriacetate-induced lipid peroxidation in mouse proximal renal tubules. Arch. Biochem. Biophys. 1993, 301, 138–142. [Google Scholar] [CrossRef]
- Nishida, Y.; Ito, Y.; Satoh, T. Origin of renal proximal tubular injuries by Fe(III)-nta chelate. Z. Nat. C J. Biosci. 2007, 62, 608–612. [Google Scholar] [CrossRef] [Green Version]
- Ekanayake, D.M.; Pham, D.; Probst, A.L.; Miller, J.R.; Popescu, C.V.; Fiedler, A.T. Electronic structures and spectroscopic signatures of diiron intermediates generated by O2 activation of nonheme iron(II)-thiolate complexes. Dalton Trans. 2021, 50, 14432–14443. [Google Scholar] [CrossRef]
- Toyokuni, S.; Masumizu, T.; Ozeki, M.; Kondo, S.; Hiroyasu, M.; Kohno, M.; Hiai, H. An electron spin resonance study on alkylperoxyl radical in thin-sliced renal tissues from ferric nitrilotriacetate-treated rats: The effect of alpha-tocopherol feeding. Free Radic. Res. 2001, 35, 245–255. [Google Scholar] [CrossRef]
- Akatsuka, S.; Yamashita, Y.; Ohara, H.; Liu, Y.T.; Izumiya, M.; Abe, K.; Ochiai, M.; Jiang, L.; Nagai, H.; Okazaki, Y.; et al. Fenton reaction induced cancer in wild type rats recapitulates genomic alterations observed in human cancer. PLoS ONE 2012, 7, e43403. [Google Scholar] [CrossRef] [PubMed]
- Corti, A.; Belcastro, E.; Dominici, S.; Maellaro, E.; Pompella, A. The dark side of gamma-glutamyltransferase (GGT): Pathogenic effects of an ‘antioxidant’ enzyme. Free Radic. Biol. Med. 2020, 160, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Broer, S. Amino acid transport across mammalian intestinal and renal epithelia. Physiol. Rev. 2008, 88, 249–286. [Google Scholar] [CrossRef] [PubMed]
- Bhutia, Y.D.; Ganapathy, V. Glutamine transporters in mammalian cells and their functions in physiology and cancer. Biochim. Biophys. Acta 2016, 1863, 2531–2539. [Google Scholar] [CrossRef] [PubMed]
- Nagamori, S.; Wiriyasermkul, P.; Guarch, M.E.; Okuyama, H.; Nakagomi, S.; Tadagaki, K.; Nishinaka, Y.; Bodoy, S.; Takafuji, K.; Okuda, S.; et al. Novel cystine transporter in renal proximal tubule identified as a missing partner of cystinuria-related plasma membrane protein rBAT/SLC3A1. Proc. Natl. Acad. Sci. USA 2016, 113, 775–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.L.; Okada, S.; Hamazaki, S.; Deng, I.L.; Midorikawa, O. Sex differences in ferric nitrilotriacetate-induced lipid peroxidation and nephrotoxicity in mice. Biochim. Biophys. Acta 1988, 963, 82–87. [Google Scholar] [CrossRef]
- Toyokuni, S.; Okada, S.; Hamazaki, S.; Minamiyama, Y.; Yamada, Y.; Liang, P.; Fukunaga, Y.; Midorikawa, O. Combined histochemical and biochemical analysis of sex hormone dependence of ferric nitrilotriacetate-induced renal lipid peroxidation in ddY mice. Cancer Res. 1990, 50, 5574–5580. [Google Scholar]
- Deguchi, J.; Miyamoto, M.; Okada, S. Sex hormone-dependent renal cell carcinogenesis induced by ferric nitrilotriacetate in Wistar rats. Jpn. J. Cancer Res. 1995, 86, 1068–1071. [Google Scholar] [CrossRef]
- Hirayama, K.; Yasutake, A.; Inoue, M. Effect of sex hormones on the fate of methylmercury and on glutathione metabolism in mice. Biochem. Pharmacol. 1987, 36, 1919–1924. [Google Scholar] [CrossRef]
- Ma, Y.; Kawabata, T.; Hamazaki, S.; Ogino, T.; Okada, S. Sex differences in oxidative damage in ddY mouse kidney treated with a renal carcinogen, iron nitrilotriacetate. Carcinogenesis 1998, 19, 1983–1988. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.C.; Reisman, S.A.; Klaassen, C.D. Tissue distribution, hormonal regulation, ontogeny, diurnal expression, and induction of mouse cystine transporters Slc3a1 and Slc7a9. Free Radic. Res. 2020, 54, 525–534. [Google Scholar] [CrossRef]
- Xu, F.; Guan, Y.; Xue, L.; Zhang, P.; Li, M.; Gao, M.; Chong, T. The roles of ferroptosis regulatory gene SLC7A11 in renal cell carcinoma: A multi-omics study. Cancer Med. 2021, 10, 9078–9096. [Google Scholar] [CrossRef]
- Sato, H.; Shiiya, A.; Kimata, M.; Maebara, K.; Tamba, M.; Sakakura, Y.; Makino, N.; Sugiyama, F.; Yagami, K.; Moriguchi, T.; et al. Redox imbalance in cystine/glutamate transporter-deficient mice. J. Biol. Chem. 2005, 280, 37423–37429. [Google Scholar] [CrossRef] [Green Version]
- Harris, A.N.; Castro, R.A.; Lee, H.W.; Verlander, J.W.; Weiner, I.D. Role of the renal androgen receptor in sex differences in ammonia metabolism. Am. J. Physiol. Renal Physiol. 2021, 321, F629–F644. [Google Scholar] [CrossRef]
- Tarade, D.; Ohh, M. The HIF and other quandaries in VHL disease. Oncogene 2018, 37, 139–147. [Google Scholar] [CrossRef]
- Kikuchi, Y.; Kobayashi, E.; Nishizawa, M.; Hamazaki, S.; Okada, S.; Hino, O. Cloning of the rat homologue of the von Hippel-Lindau tumor suppressor gene and its non-somatic mutation in rat renal cell carcinomas. Jpn. J. Cancer Res 1995, 86, 905–909. [Google Scholar] [CrossRef]
- Toyokuni, S.; Okada, K.; Kondo, S.; Nishioka, H.; Tanaka, T.; Nishiyama, Y.; Hino, O.; Hiai, H. Development of high-grade renal cell carcinomas in rats independently of somatic mutations in the Tsc2 and VHL tumor suppressor genes. Jpn. J. Cancer Res. 1998, 89, 814–820. [Google Scholar] [CrossRef]
- Nishiyama, Y.; Suwa, H.; Okamoto, K.; Fukumoto, M.; Hiai, H.; Toyokuni, S. Low incidence of point mutations in H-, K- and N-ras oncogenes and p53 tumor suppressor gene in renal cell carcinoma and peritoneal mesothelioma of Wistar rats induced by ferric nitrilotriacetate. Jpn. J. Cancer Res. 1995, 86, 1150–1158. [Google Scholar] [CrossRef]
- Akiyama, T.; Hamazaki, S.; Okada, S. Absence of ras mutations and low incidence of p53 mutations in renal cell carcinomas induced by ferric nitrilotriacetate. Jpn. J. Cancer Res. 1995, 86, 1143–1149. [Google Scholar] [CrossRef]
- Harlander, S.; Schonenberger, D.; Toussaint, N.C.; Prummer, M.; Catalano, A.; Brandt, L.; Moch, H.; Wild, P.J.; Frew, I.J. Combined mutation in Vhl, Trp53 and Rb1 causes clear cell renal cell carcinoma in mice. Nat. Med. 2017, 23, 869–877. [Google Scholar] [CrossRef] [Green Version]
- Albers, J.; Rajski, M.; Schonenberger, D.; Harlander, S.; Schraml, P.; von Teichman, A.; Georgiev, S.; Wild, P.J.; Moch, H.; Krek, W.; et al. Combined mutation of Vhl and Trp53 causes renal cysts and tumours in mice. EMBO Mol. Med. 2013, 5, 949–964. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.F.; Cohn, S.; Christie, A.; McKenzie, T.; Wolff, N.; Do, Q.N.; Madhuranthakam, A.J.; Pedrosa, I.; Wang, T.; Dey, A.; et al. Modeling Renal Cell Carcinoma in Mice: Bap1 and Pbrm1 Inactivation Drive Tumor Grade. Cancer Discov. 2017, 7, 900–917. [Google Scholar] [CrossRef] [Green Version]
- Choueiri, T.K.; Bauer, T.M.; Papadopoulos, K.P.; Plimack, E.R.; Merchan, J.R.; McDermott, D.F.; Michaelson, M.D.; Appleman, L.J.; Thamake, S.; Perini, R.F.; et al. Inhibition of hypoxia-inducible factor-2alpha in renal cell carcinoma with belzutifan: A phase 1 trial and biomarker analysis. Nat. Med. 2021, 27, 802–805. [Google Scholar] [CrossRef]
- Henske, E.P.; Cornejo, K.M.; Wu, C.L. Renal Cell Carcinoma in Tuberous Sclerosis Complex. Genes 2021, 12, 1585. [Google Scholar] [CrossRef]
- Satake, N.; Urakami, S.; Hirayama, Y.; Izumi, K.; Hino, O. Biallelic mutations of the Tsc2 gene in chemically induced rat renal cell carcinoma. Int. J. Cancer. 1998, 77, 895–900. [Google Scholar] [CrossRef]
- Hino, O.; Kobayashi, E.; Hirayama, Y.; Kobayashi, T.; Kubo, Y.; Tsuchiya, H.; Kikuchi, Y.; Mitani, H. Molecular genetic basis of renal carcinogenesis in the Eker rat model of tuberous sclerosis (Tsc2). Mol. Carcinog. 1995, 14, 23–27. [Google Scholar] [CrossRef]
- Knapek, D.F.; Serrano, M.; Beach, D.; Trono, D.; Walker, C.L. Association of rat p15INK4B/p16INK4 deletions with monosomy 5 in kidney epithelial cell lines but not primary renal tumors. Cancer Res. 1995, 55, 1607–1612. [Google Scholar]
- Tanaka, T.; Iwasa, Y.; Kondo, S.; Hiai, H.; Toyokuni, S. High incidence of allelic loss on chromosome 5 and inactivation of p15INK4B and p16INK4A tumor suppressor genes in oxystress-induced renal cell carcinoma of rats. Oncogene 1999, 18, 3793–3797. [Google Scholar] [CrossRef] [Green Version]
- Hiroyasu, M.; Ozeki, M.; Kohda, H.; Echizenya, M.; Tanaka, T.; Hiai, H.; Toyokuni, S. Specific allelic loss of p16 (INK4A) tumor suppressor gene after weeks of iron-mediated oxidative damage during rat renal carcinogenesis. Am. J. Pathol. 2002, 160, 419–424. [Google Scholar] [CrossRef] [Green Version]
- Jafri, M.; Wake, N.C.; Ascher, D.B.; Pires, D.E.; Gentle, D.; Morris, M.R.; Rattenberry, E.; Simpson, M.A.; Trembath, R.C.; Weber, A.; et al. Germline Mutations in the CDKN2B Tumor Suppressor Gene Predispose to Renal Cell Carcinoma. Cancer Discov. 2015, 5, 723–729. [Google Scholar] [CrossRef] [Green Version]
- Deguchi, J.; Kawabata, T.; Kondo, A.; Okada, S. Transforming growth factor-alpha expression of renal proximal tubules in Wistar rats treated with ferric and aluminum nitrilotriacetate. Jpn. J. Cancer Res. 1993, 84, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Akatsuka, S.; Li, G.H.; Mori, K.; Takahashi, T.; Toyokuni, S. Ferroptosis resistance determines high susceptibility of murine A/J strain to iron-induced renal carcinogenesis. Cancer Sci. 2021, 113, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Akatsuka, S.; Li, G.H.; Toyokuni, S. Superiority of rat over murine model for studies on the evolution of cancer genome. Free Radic. Res. 2018, 52, 1323–1327. [Google Scholar] [CrossRef] [PubMed]
- Kanki, K.; Umemura, T.; Kitamura, Y.; Ishii, Y.; Kuroiwa, Y.; Kodama, Y.; Itoh, K.; Yamamoto, M.; Nishikawa, A.; Hirose, M. A possible role of nrf2 in prevention of renal oxidative damage by ferric nitrilotriacetate. Toxicol. Pathol. 2008, 36, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Aleksunes, L.M.; Goedken, M.J.; Chen, C.; Reisman, S.A.; Manautou, J.E.; Klaassen, C.D. Coordinated induction of Nrf2 target genes protects against iron nitrilotriacetate (FeNTA)-induced nephrotoxicity. Toxicol. Appl. Pharmacol. 2008, 231, 364–373. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, M.; Sharma, S.D.; Okazaki, Y.; Fujisawa, M.; Okada, S. Dietary supplementation of curcumin enhances antioxidant and phase II metabolizing enzymes in ddY male mice: Possible role in protection against chemical carcinogenesis and toxicity. Pharmacol. Toxicol. 2003, 92, 33–38. [Google Scholar] [CrossRef]
- Okazaki, Y.; Iqbal, M.; Okada, S. Suppressive effects of dietary curcumin on the increased activity of renal ornithine decarboxylase in mice treated with a renal carcinogen, ferric nitrilotriacetate. Biochim. Biophys. Acta 2005, 1740, 357–366. [Google Scholar] [CrossRef] [Green Version]
- Okada, S.; Hamazaki, S.; Ebina, Y.; Li, J.L.; Midorikawa, O. Nephrotoxicity and its prevention by vitamin E in ferric nitrilotriacetate-promoted lipid peroxidation. Biochim. Biophys. Acta 1987, 922, 28–33. [Google Scholar] [CrossRef]
- Zhang, D.; Okada, S.; Yu, Y.; Zheng, P.; Yamaguchi, R.; Kasai, H. Vitamin E inhibits apoptosis, DNA modification, and cancer incidence induced by iron-mediated peroxidation in Wistar rat kidney. Cancer Res. 1997, 57, 2410–2414. [Google Scholar]
- Mizote, A.; Hida, A.I.; Hosako, M.; Fujisawa, M.; Kamekawa, M.; Okada, S. Effects of phlebotomy on the growth of ferric nitrilotriacetate-induced renal cell carcinoma. Acta Med. Okayama 2002, 56, 199–204. [Google Scholar] [CrossRef]
- Shirase, T.; Mori, K.; Okazaki, Y.; Itoh, K.; Yamamoto, M.; Tabuchi, M.; Kishi, F.; Jiang, L.; Akatsuka, S.; Nakao, K.; et al. Suppression of SLC11A2 expression is essential to maintain duodenal integrity during dietary iron overload. Am. J. Pathol. 2010, 177, 677–685. [Google Scholar] [CrossRef]
- Tong, W.H.; Sourbier, C.; Kovtunovych, G.; Jeong, S.Y.; Vira, M.; Ghosh, M.; Romero, V.V.; Sougrat, R.; Vaulont, S.; Viollet, B.; et al. The glycolytic shift in fumarate-hydratase-deficient kidney cancer lowers AMPK levels, increases anabolic propensities and lowers cellular iron levels. Cancer Cell 2011, 20, 315–327. [Google Scholar] [CrossRef] [Green Version]
- Mastrogiannaki, M.; Matak, P.; Keith, B.; Simon, M.C.; Vaulont, S.; Peyssonnaux, C. HIF-2alpha, but not HIF-1alpha, promotes iron absorption in mice. J. Clin. Investig. 2009, 119, 1159–1166. [Google Scholar] [CrossRef]
- Hirota, K. HIF-alpha Prolyl Hydroxylase Inhibitors and Their Implications for Biomedicine: A Comprehensive Review. Biomedicines 2021, 9, 468. [Google Scholar] [CrossRef]
- Shawki, A.; Anthony, S.R.; Nose, Y.; Engevik, M.A.; Niespodzany, E.J.; Barrientos, T.; Ohrvik, H.; Worrell, R.T.; Thiele, D.J.; Mackenzie, B. Intestinal DMT1 is critical for iron absorption in the mouse but is not required for the absorption of copper or manganese. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G635–G647. [Google Scholar] [CrossRef] [Green Version]
- Pacak, K.; Jochmanova, I.; Prodanov, T.; Yang, C.; Merino, M.J.; Fojo, T.; Prchal, J.T.; Tischler, A.S.; Lechan, R.M.; Zhuang, Z. New syndrome of paraganglioma and somatostatinoma associated with polycythemia. J. Clin. Oncol. 2013, 31, 1690–1698. [Google Scholar] [CrossRef] [Green Version]
- Rosenblum, J.S.; Wang, H.; Dmitriev, P.M.; Cappadona, A.J.; Mastorakos, P.; Xu, C.; Jha, A.; Edwards, N.; Donahue, D.R.; Munasinghe, J.; et al. Developmental vascular malformations in EPAS1 gain-of-function syndrome. JCI Insight 2021, 6, e144368. [Google Scholar] [CrossRef]
- Saxena, N.; Maio, N.; Crooks, D.R.; Ricketts, C.J.; Yang, Y.; Wei, M.H.; Fan, T.W.; Lane, A.N.; Sourbier, C.; Singh, A.; et al. SDHB-Deficient Cancers: The Role of Mutations That Impair Iron Sulfur Cluster Delivery. J. Natl. Cancer Inst. 2016, 108, djv287. [Google Scholar] [CrossRef] [Green Version]
- Yakirevich, E.; Ali, S.M.; Mega, A.; McMahon, C.; Brodsky, A.S.; Ross, J.S.; Allen, J.; Elvin, J.A.; Safran, H.; Resnick, M.B. A Novel SDHA-deficient Renal Cell Carcinoma Revealed by Comprehensive Genomic Profiling. Am. J. Surg. Pathol. 2015, 39, 858–863. [Google Scholar] [CrossRef]
- Kamai, T.; Higashi, S.; Murakami, S.; Arai, K.; Namatame, T.; Kijima, T.; Abe, H.; Jamiyan, T.; Ishida, K.; Shirataki, H.; et al. Single nucleotide variants of succinate dehydrogenase A gene in renal cell carcinoma. Cancer Sci. 2021, 112, 3375–3387. [Google Scholar] [CrossRef]
- Linehan, W.M.; Schmidt, L.S.; Crooks, D.R.; Wei, D.; Srinivasan, R.; Lang, M.; Ricketts, C.J. The Metabolic Basis of Kidney Cancer. Cancer Discov. 2019, 9, 1006–1021. [Google Scholar] [CrossRef] [Green Version]
- Nath, K.A.; Hebbel, R.P. Sickle cell disease: Renal manifestations and mechanisms. Nat. Rev. Nephrol. 2015, 11, 161–171. [Google Scholar] [CrossRef] [Green Version]
- Msaouel, P.; Tannir, N.M.; Walker, C.L. A Model Linking Sickle Cell Hemoglobinopathies and SMARCB1 Loss in Renal Medullary Carcinoma. Clin. Cancer Res. 2018, 24, 2044–2049. [Google Scholar] [CrossRef] [Green Version]
- Msaouel, P.; Malouf, G.G.; Su, X.; Yao, H.; Tripathi, D.N.; Soeung, M.; Gao, J.; Rao, P.; Coarfa, C.; Creighton, C.J.; et al. Comprehensive Molecular Characterization Identifies Distinct Genomic and Immune Hallmarks of Renal Medullary Carcinoma. Cancer Cell 2020, 37, 720–734. [Google Scholar] [CrossRef]
- Schmidt, L.S.; Linehan, W.M. Genetic predisposition to kidney cancer. Semin. Oncol. 2016, 43, 566–574. [Google Scholar] [CrossRef] [Green Version]
- Alaghehbandan, R.; Trpkov, K.; Tretiakova, M.; Luis, A.S.; Rogala, J.D.; Hes, O. Comprehensive Review of Numerical Chromosomal Aberrations in Chromophobe Renal Cell Carcinoma Including Its Variant Morphologies. Adv. Anat. Pathol. 2021, 28, 8–20. [Google Scholar] [CrossRef]
- Lang, M.; Vocke, C.D.; Ricketts, C.J.; Metwalli, A.R.; Ball, M.W.; Schmidt, L.S.; Linehan, W.M. Clinical and Molecular Characterization of Microphthalmia-associated Transcription Factor (MITF)-related Renal Cell Carcinoma. Urology 2021, 149, 89–97. [Google Scholar] [CrossRef]
- Gallan, A.J.; Parilla, M.; Segal, J.; Ritterhouse, L.; Antic, T. BAP1-Mutated Clear Cell Renal Cell Carcinoma. Am. J. Clin. Pathol. 2021, 155, 718–728. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Hamazaki, S.; Toyokuni, S.; Midorikawa, O. Induction of mesothelioma by intraperitoneal injections of ferric saccharate in male Wistar rats. Br. J. Cancer 1989, 60, 708–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Q.; Akatsuka, S.; Yamashita, Y.; Ohara, H.; Nagai, H.; Okazaki, Y.; Takahashi, T.; Toyokuni, S. Homozygous deletion of CDKN2A/2B is a hallmark of iron-induced high-grade rat mesothelioma. Lab. Investig. 2010, 90, 360–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minami, D.; Takigawa, N.; Kato, Y.; Kudo, K.; Isozaki, H.; Hashida, S.; Harada, D.; Ochi, N.; Fujii, M.; Kubo, T.; et al. Downregulation of TBXAS1 in an iron-induced malignant mesothelioma model. Cancer Sci. 2015, 106, 1296–1302. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; Conrad, M. The Metabolic Underpinnings of Ferroptosis. Cell Metab. 2020, 32, 920–937. [Google Scholar] [CrossRef]
- Nagai, H.; Okazaki, Y.; Chew, S.H.; Misawa, N.; Yasui, H.; Toyokuni, S. Deferasirox induces mesenchymal-epithelial transition in crocidolite-induced mesothelial carcinogenesis in rats. Cancer Prev. Res. 2013, 6, 1222–1230. [Google Scholar] [CrossRef] [Green Version]
- Ohara, Y.; Chew, S.H.; Shibata, T.; Okazaki, Y.; Yamashita, K.; Toyokuni, S. Phlebotomy as a preventive measure for crocidolite-induced mesothelioma in male rats. Cancer Sci. 2018, 109, 330–339. [Google Scholar] [CrossRef] [Green Version]
- Kato, J.; Miyanishi, K.; Kobune, M.; Nakamura, T.; Takada, K.; Takimoto, R.; Kawano, Y.; Takahashi, S.; Takahashi, M.; Sato, Y.; et al. Long-term phlebotomy with low-iron diet therapy lowers risk of development of hepatocellular carcinoma from chronic hepatitis C. J. Gastroenterol. 2007, 42, 830–836. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Hassannia, B.; Vandenabeele, P.; Vanden Berghe, T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef]
- Miyazawa, T. Lipid hydroperoxides in nutrition, health, and diseases. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2021, 97, 161–196. [Google Scholar] [CrossRef]
- Ratan, R.R. The Chemical Biology of Ferroptosis in the Central Nervous System. Cell Chem. Biol. 2020, 27, 479–498. [Google Scholar] [CrossRef]
- Ward, R.J.; Dexter, D.T.; Martin-Bastida, A.; Crichton, R.R. Is Chelation Therapy a Potential Treatment for Parkinson’s Disease? Int. J. Mol. Sci. 2021, 22, 3338. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Bayir, H.; Anthonymuthu, T.S.; Tyurina, Y.Y.; Patel, S.J.; Amoscato, A.A.; Lamade, A.M.; Yang, Q.; Vladimirov, G.K.; Philpott, C.C.; Kagan, V.E. Achieving Life through Death: Redox Biology of Lipid Peroxidation in Ferroptosis. Cell Chem. Biol. 2020, 27, 387–408. [Google Scholar] [CrossRef]
- Ran, Q.; Van Remmen, H.; Gu, M.; Qi, W.; Roberts, L.J., 2nd; Prolla, T.; Richardson, A. Embryonic fibroblasts from Gpx4+/− mice: A novel model for studying the role of membrane peroxidation in biological processes. Free Radic. Biol. Med. 2003, 35, 1101–1109. [Google Scholar] [CrossRef]
- Thomas, J.P.; Maiorino, M.; Ursini, F.; Girotti, A.W. Protective action of phospholipid hydroperoxide glutathione peroxidase against membrane-damaging lipid peroxidation. In situ reduction of phospholipid and cholesterol hydroperoxides. J. Biol. Chem. 1990, 265, 454–461. [Google Scholar] [CrossRef]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of Mitochondria in Ferroptosis. Mol. Cell 2019, 73, 354–363. [Google Scholar] [CrossRef] [Green Version]
- Shimada, K.; Hayano, M.; Pagano, N.C.; Stockwell, B.R. Cell-Line Selectivity Improves the Predictive Power of Pharmacogenomic Analyses and Helps Identify NADPH as Biomarker for Ferroptosis Sensitivity. Cell Chem. Biol. 2016, 23, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [Green Version]
- Belavgeni, A.; Meyer, C.; Stumpf, J.; Hugo, C.; Linkermann, A. Ferroptosis and Necroptosis in the Kidney. Cell Chem. Biol. 2020, 27, 448–462. [Google Scholar] [CrossRef]
- Linkermann, A.; Skouta, R.; Himmerkus, N.; Mulay, S.R.; Dewitz, C.; De Zen, F.; Prokai, A.; Zuchtriegel, G.; Krombach, F.; Welz, P.S.; et al. Synchronized renal tubular cell death involves ferroptosis. Proc. Natl. Acad. Sci. USA 2014, 111, 16836–16841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonnus, W.; Meyer, C.; Steinebach, C.; Belavgeni, A.; von Massenhausen, A.; Gonzalez, N.Z.; Maremonti, F.; Gembardt, F.; Himmerkus, N.; Latk, M.; et al. Dysfunction of the key ferroptosis-surveilling systems hypersensitizes mice to tubular necrosis during acute kidney injury. Nat. Commun. 2021, 12, 4402. [Google Scholar] [CrossRef] [PubMed]
- Miess, H.; Dankworth, B.; Gouw, A.M.; Rosenfeldt, M.; Schmitz, W.; Jiang, M.; Saunders, B.; Howell, M.; Downward, J.; Felsher, D.W.; et al. The glutathione redox system is essential to prevent ferroptosis caused by impaired lipid metabolism in clear cell renal cell carcinoma. Oncogene 2018, 37, 5435–5450. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Sato, M.; Mishima, E.; Sato, H.; Proneth, B.; Conrad, M. Sorafenib fails to trigger ferroptosis across a wide range of cancer cell lines. Cell Death Dis. 2021, 12, 698. [Google Scholar] [CrossRef] [PubMed]
- Toyokuni, S.; Luo, X.P.; Tanaka, T.; Uchida, K.; Hiai, H.; Lehotay, D.C. Induction of a wide range of C(2–12) aldehydes and C(7–12) acyloins in the kidney of Wistar rats after treatment with a renal carcinogen, ferric nitrilotriacetate. Free Radic. Biol. Med. 1997, 22, 1019–1027. [Google Scholar] [CrossRef]
- Deiana, M.; Aruoma, O.I.; Rosa, A.; Crobu, V.; Casu, V.; Piga, R.; Dessi, M.A. The effect of ferric-nitrilotriacetic acid on the profile of polyunsaturated fatty acids in the kidney and liver of rats. Toxicol. Lett. 2001, 123, 125–133. [Google Scholar] [CrossRef]
- Yasuda, S.; Watanabe, S.; Kobayashi, T.; Hata, N.; Misawa, Y.; Utsumi, H.; Okuyama, H. Dietary docosahexaenoic acid enhances ferric nitrilotriacetate-induced oxidative damage in mice but not when additional alpha-tocopherol is supplemented. Free Radic. Res. 1999, 30, 199–205. [Google Scholar] [CrossRef]
- Okazaki, Y.; Iqbal, M.; Kawakami, N.; Yamamoto, Y.; Toyokuni, S.; Okada, S. A beverage containing fermented black soybean ameliorates ferric nitrilotriacetate-induced renal oxidative damage in rats. J. Clin. Biochem. Nutr. 2010, 47, 198–207. [Google Scholar] [CrossRef] [Green Version]
- Ran, Q.; Liang, H.; Ikeno, Y.; Qi, W.; Prolla, T.A.; Roberts, L.J., 2nd; Wolf, N.; Van Remmen, H.; Richardson, A. Reduction in glutathione peroxidase 4 increases life span through increased sensitivity to apoptosis. J. Gerontol. A Biol. Sci. Med. Sci. 2007, 62, 932–942. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; La Vecchia, C.; Negri, E.; DesMeules, M.; Mery, L.; Canadian Cancer Registries Epidemiology Research, G. Dietary vitamin C, E, and carotenoid intake and risk of renal cell carcinoma. Cancer Causes Control 2009, 20, 1451–1458. [Google Scholar] [CrossRef]
- Liao, Z.; Fang, Z.; Gou, S.; Luo, Y.; Liu, Y.; He, Z.; Li, X.; Peng, Y.; Fu, Z.; Li, D.; et al. The role of diet in renal cell carcinoma incidence: An umbrella review of meta-analyses of observational studies. BMC Med. 2022, 20, 39. [Google Scholar] [CrossRef]
Figure 1.
The mechanism of Fe-NTA-induced oxidative injury in the renal proximal tubules of rodents. Approximately 95–99% of all amino acids are reabsorbed in the proximal convoluted tubule and proximal straight tubule; thus, with very few exceptions, individual amino acids are transported by more than one transporter to provide backup capacity for absorption [34]. The intracellular GSH is exported from the cytoplasm to the lumen through GSH exporters, such as multidrug resistance-associated protein (Mrp) 1–5, cystic fibrosis transmembrane regulator (Cftr), organic anion transporting polypeptide (Oatp) 1 and 2, ATP-binding cassette superfamily G2 (Abcg2), or gap junction hemi-channels [27]. GSH is degraded by membrane-associated enzymes, such as γ-glutamyl transpeptidase (γ-Gtp) [8,27]. The Cys-Gly can be degraded by membrane-bound dipeptidases (Dpep) [8,27] or imported to the cytoplasm directly by amino acid transporters [27,34]. Among the constituents of GSH, Gly is transported by many amino acids transporters [34], while Cys reduces Fe(III)-NTA to yield Fe(II)-NTA and cystine (CssC), triggering iron-dependent oxidative injury and carcinogenesis in the renal proximal tubules [8]. The CssC is transported into the cytoplasm via the related b0 amino acid transporter/b0,+ amino acid transporter (rBAT/b0,+AT), which is also called the Slc3a1/Slc7a9 complex, with anionic amino acids (Lys and Arg) and ornithine [34,35] or rBAT with Slc7a13, also known as aspartate/glutamate transporter 1 (Agt1), with cationic amino acids (Asp and Glu) [36]. In physiological conditions, more than 90% of CssC is reabsorbed via rBAT/b0,+AT, and the residual CssC is reabsorbed by rBAT/AGT1 in the renal proximal tubules [36].
Figure 1.
The mechanism of Fe-NTA-induced oxidative injury in the renal proximal tubules of rodents. Approximately 95–99% of all amino acids are reabsorbed in the proximal convoluted tubule and proximal straight tubule; thus, with very few exceptions, individual amino acids are transported by more than one transporter to provide backup capacity for absorption [34]. The intracellular GSH is exported from the cytoplasm to the lumen through GSH exporters, such as multidrug resistance-associated protein (Mrp) 1–5, cystic fibrosis transmembrane regulator (Cftr), organic anion transporting polypeptide (Oatp) 1 and 2, ATP-binding cassette superfamily G2 (Abcg2), or gap junction hemi-channels [27]. GSH is degraded by membrane-associated enzymes, such as γ-glutamyl transpeptidase (γ-Gtp) [8,27]. The Cys-Gly can be degraded by membrane-bound dipeptidases (Dpep) [8,27] or imported to the cytoplasm directly by amino acid transporters [27,34]. Among the constituents of GSH, Gly is transported by many amino acids transporters [34], while Cys reduces Fe(III)-NTA to yield Fe(II)-NTA and cystine (CssC), triggering iron-dependent oxidative injury and carcinogenesis in the renal proximal tubules [8]. The CssC is transported into the cytoplasm via the related b0 amino acid transporter/b0,+ amino acid transporter (rBAT/b0,+AT), which is also called the Slc3a1/Slc7a9 complex, with anionic amino acids (Lys and Arg) and ornithine [34,35] or rBAT with Slc7a13, also known as aspartate/glutamate transporter 1 (Agt1), with cationic amino acids (Asp and Glu) [36]. In physiological conditions, more than 90% of CssC is reabsorbed via rBAT/b0,+AT, and the residual CssC is reabsorbed by rBAT/AGT1 in the renal proximal tubules [36].

Figure 2.
The dysregulated signaling pathways in RCC. RCC is caused by several signaling pathways, such as HIF, MET kinase, mTOR, TCA cycle, cell cycle regulation, and chromatin modification and regulation. Brown tags represent a gene that causes RCC via germline or somatic alterations, such as VHL (von Hippel–Lindau), BAP1 (BRCA1-associated protein 1), PBRM1 (polybromo 1), MET, PTEN (phosphatase and tensin homolog), TSC1/2 (tuberous sclerosis 1/2), FLCN (folliculin), MiTF (microphthalmia-associated transcription factor), SDHB/C/D (succinate dehydrogenase B/C/D), FH (fumarate hydratase), and CDKN2B (cyclin-dependent kinase inhibitor 2B). Orange tags represent a gene that is frequently mutated in sporadic RCCs, such as SDHA, TFEB (transcriptional factor EB), TFE3 (transcriptional factor E3), SMARCB1, and CDKN2A. The blue arrow indicates the activation pathway, and the red line indicates signal transduction suppression. Acronyms: PHDs (prolyl hydroxylases), HGF (hepatocyte growth factor), EPO (erythropoietin), AMPK (AMP-activated protein kinase), FNIP1/2 (folliculin-interacting protein 1/2), RHEB (ras-homolog expressed in brain), VEGF (vascular endothelial growth factor), PDGF (platelet-derived growth factor), TETs (ten-eleven translocations), and KDMs (lysine-specific demethylases).
Figure 2.
The dysregulated signaling pathways in RCC. RCC is caused by several signaling pathways, such as HIF, MET kinase, mTOR, TCA cycle, cell cycle regulation, and chromatin modification and regulation. Brown tags represent a gene that causes RCC via germline or somatic alterations, such as VHL (von Hippel–Lindau), BAP1 (BRCA1-associated protein 1), PBRM1 (polybromo 1), MET, PTEN (phosphatase and tensin homolog), TSC1/2 (tuberous sclerosis 1/2), FLCN (folliculin), MiTF (microphthalmia-associated transcription factor), SDHB/C/D (succinate dehydrogenase B/C/D), FH (fumarate hydratase), and CDKN2B (cyclin-dependent kinase inhibitor 2B). Orange tags represent a gene that is frequently mutated in sporadic RCCs, such as SDHA, TFEB (transcriptional factor EB), TFE3 (transcriptional factor E3), SMARCB1, and CDKN2A. The blue arrow indicates the activation pathway, and the red line indicates signal transduction suppression. Acronyms: PHDs (prolyl hydroxylases), HGF (hepatocyte growth factor), EPO (erythropoietin), AMPK (AMP-activated protein kinase), FNIP1/2 (folliculin-interacting protein 1/2), RHEB (ras-homolog expressed in brain), VEGF (vascular endothelial growth factor), PDGF (platelet-derived growth factor), TETs (ten-eleven translocations), and KDMs (lysine-specific demethylases).

Figure 3.
Iron-regulated HIF-α expression. (A) HIF-α is hydroxylated by PHDs, which are suppressed by molecular oxygen depletion or iron chelation. The hydroxylations at the proline in HIF-α induce recognition by the VHL complex, which initiates ubiquitination-dependent proteasomal degradation. Competitive inhibitors of α-ketoglutarate are prescribed for renal anemia to inhibit PHDs, and, in turn, stabilize HIF-α [75]. (B) HIF2α, which is translationally suppressed by IRE/IRPs as FPN (ferroportin), FTH (ferritin heavy chain), and FTL (ferritin light chain), transcribes DMT1 and FPN [74]. The increased cytoplasmic iron may activate PHDs, while IRE/IRP-dependent translational inhibition is decreased.
Figure 3.
Iron-regulated HIF-α expression. (A) HIF-α is hydroxylated by PHDs, which are suppressed by molecular oxygen depletion or iron chelation. The hydroxylations at the proline in HIF-α induce recognition by the VHL complex, which initiates ubiquitination-dependent proteasomal degradation. Competitive inhibitors of α-ketoglutarate are prescribed for renal anemia to inhibit PHDs, and, in turn, stabilize HIF-α [75]. (B) HIF2α, which is translationally suppressed by IRE/IRPs as FPN (ferroportin), FTH (ferritin heavy chain), and FTL (ferritin light chain), transcribes DMT1 and FPN [74]. The increased cytoplasmic iron may activate PHDs, while IRE/IRP-dependent translational inhibition is decreased.

Figure 4.
Ferroptotic signaling pathway. (A) Among phospholipids (PLs), PE-esterified AA or AdA are predominantly located in the inner leaflet of the plasma membrane or nonbilayer arrangements in contrast to the high PC confinement on the outer membrane or ordered bilayer organization of PC-PUFAs [103]. The lipid hydroperoxides (PL-PUFA-OOH or PE-AA-OOH) are detoxified to alcohol (PL-PUFA-OH or PE-AA-OH) by GPX4. On the other hand, PL-PUFA-OOH and PE-AA-OOH yield iron-mediated peroxyl (R-OO•) or alkoxyl (R-O•) radicals; finally, reactive electrophiles transduce lipid death signals. These ferroptotic signals are attenuated by suppressing lipid peroxidation with vitamin E or iron chelation with DFO while being enhanced by lipoxygenases (LOXs) or acyl-CoA synthetase long chain family member 4 (ACSL4) or lysophosphatidylcholine acyltransferase 3 (LPCAT3), which mediate PE esterification with PUFAs in addition to PC and phosphatidylserine (PS). The ferroptosis inhibitors are shown in red, while the ferroptosis inducers are shown in blue. (B) The compounds that trigger glutaminolysis or that inhibit the cystine/GSH/GPX4 axis, FSP1/CoQ10/α-Toc (ferroptosis suppressor protein 1/coenzymeQ10) axis, GCH1/BH4/DHFR (guanosine triphosphate cyclohydrolase 1/tetrahydrobiopterin/dihydrofolate reductase) axis, or NADPH synthesis are categorized as ferroptosis inducers that may initiate tumor cell death, while PUFA metabolism suppressors or iron chelators are designated as ferroptosis inhibitors that may confer the prevention of ischemic injury, neurodegeneration, or pancreatic β-cell loss. In addition to these beneficial roles of ferroptosis, the protection of tumor-initiating cells is also speculated.
Figure 4.
Ferroptotic signaling pathway. (A) Among phospholipids (PLs), PE-esterified AA or AdA are predominantly located in the inner leaflet of the plasma membrane or nonbilayer arrangements in contrast to the high PC confinement on the outer membrane or ordered bilayer organization of PC-PUFAs [103]. The lipid hydroperoxides (PL-PUFA-OOH or PE-AA-OOH) are detoxified to alcohol (PL-PUFA-OH or PE-AA-OH) by GPX4. On the other hand, PL-PUFA-OOH and PE-AA-OOH yield iron-mediated peroxyl (R-OO•) or alkoxyl (R-O•) radicals; finally, reactive electrophiles transduce lipid death signals. These ferroptotic signals are attenuated by suppressing lipid peroxidation with vitamin E or iron chelation with DFO while being enhanced by lipoxygenases (LOXs) or acyl-CoA synthetase long chain family member 4 (ACSL4) or lysophosphatidylcholine acyltransferase 3 (LPCAT3), which mediate PE esterification with PUFAs in addition to PC and phosphatidylserine (PS). The ferroptosis inhibitors are shown in red, while the ferroptosis inducers are shown in blue. (B) The compounds that trigger glutaminolysis or that inhibit the cystine/GSH/GPX4 axis, FSP1/CoQ10/α-Toc (ferroptosis suppressor protein 1/coenzymeQ10) axis, GCH1/BH4/DHFR (guanosine triphosphate cyclohydrolase 1/tetrahydrobiopterin/dihydrofolate reductase) axis, or NADPH synthesis are categorized as ferroptosis inducers that may initiate tumor cell death, while PUFA metabolism suppressors or iron chelators are designated as ferroptosis inhibitors that may confer the prevention of ischemic injury, neurodegeneration, or pancreatic β-cell loss. In addition to these beneficial roles of ferroptosis, the protection of tumor-initiating cells is also speculated.


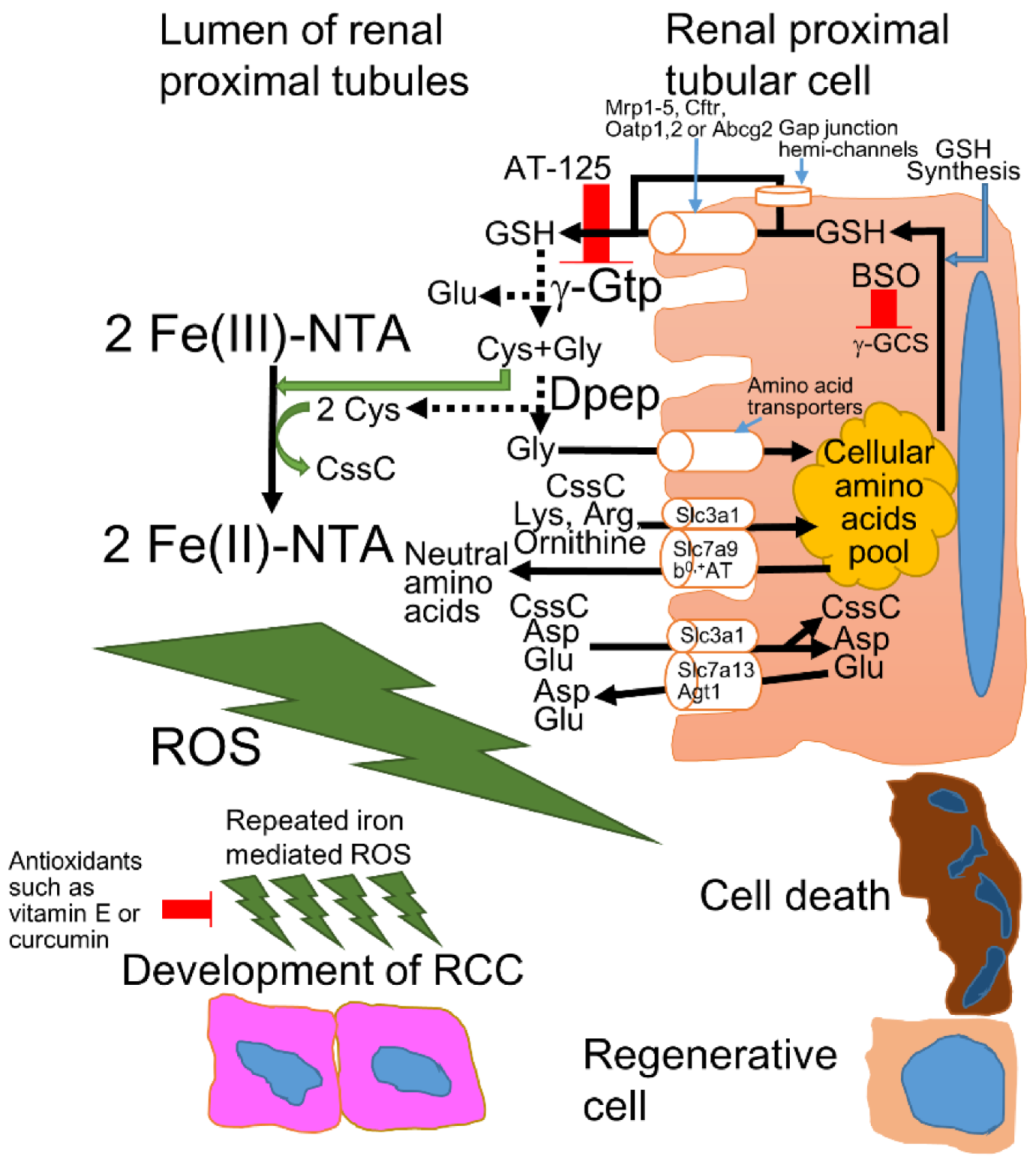{kind=link}
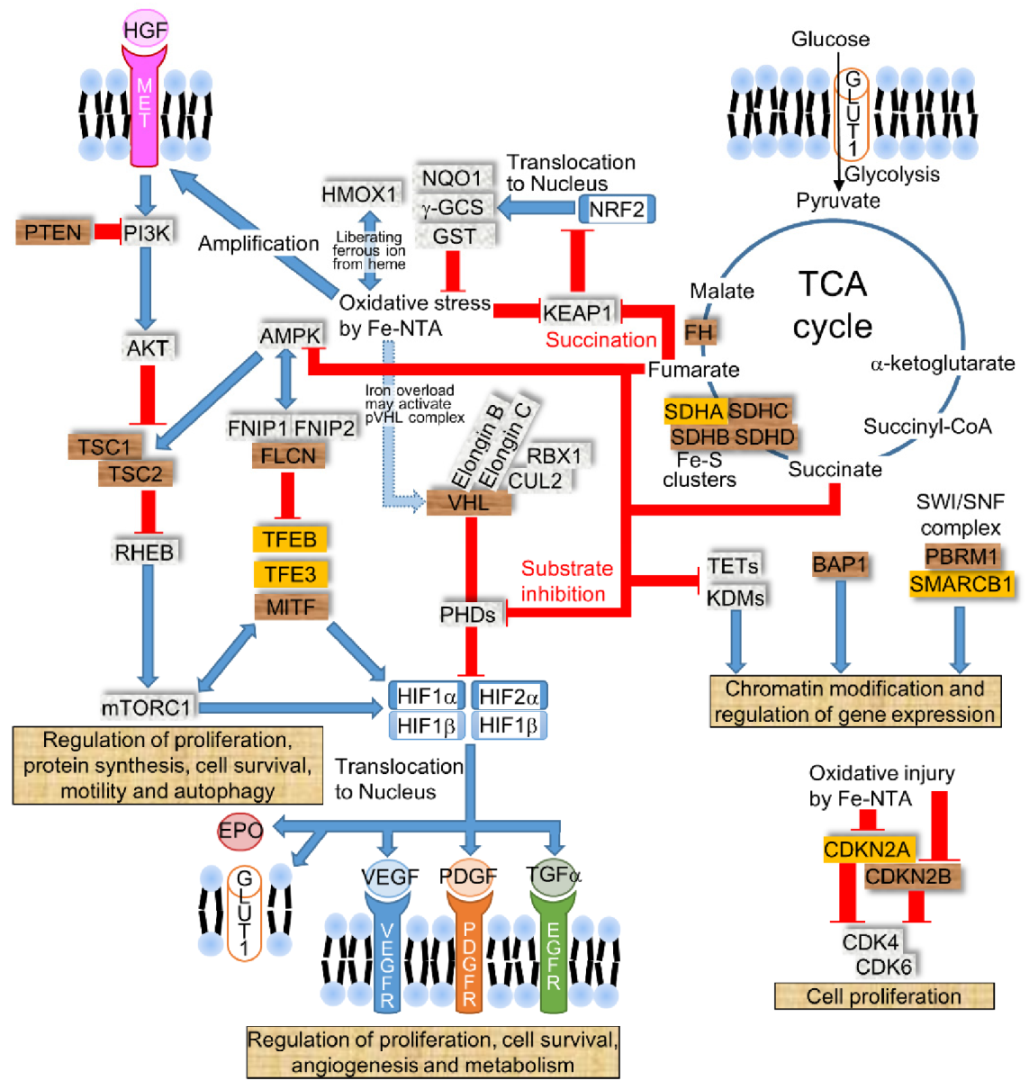{kind=link}
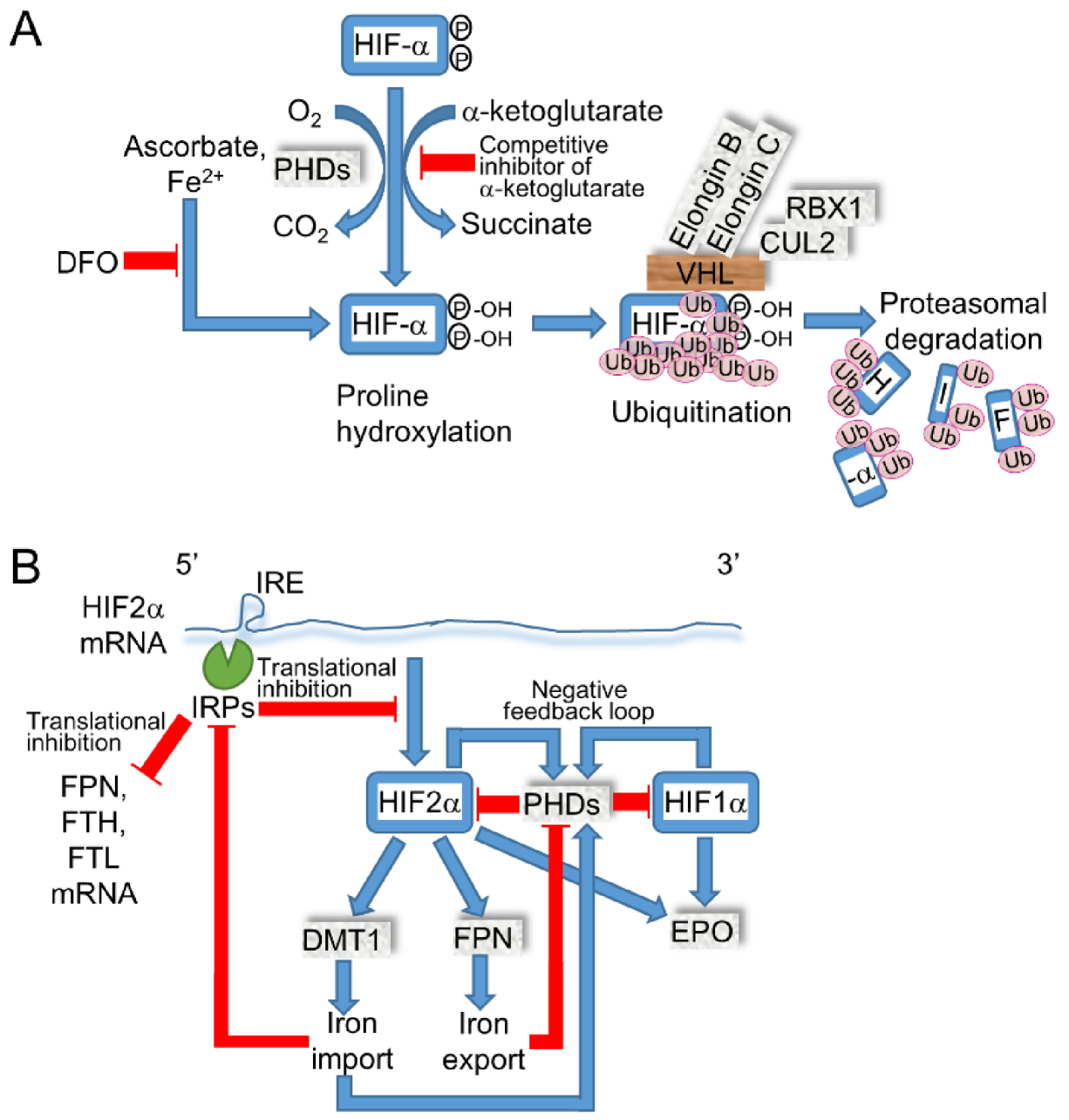{kind=link}
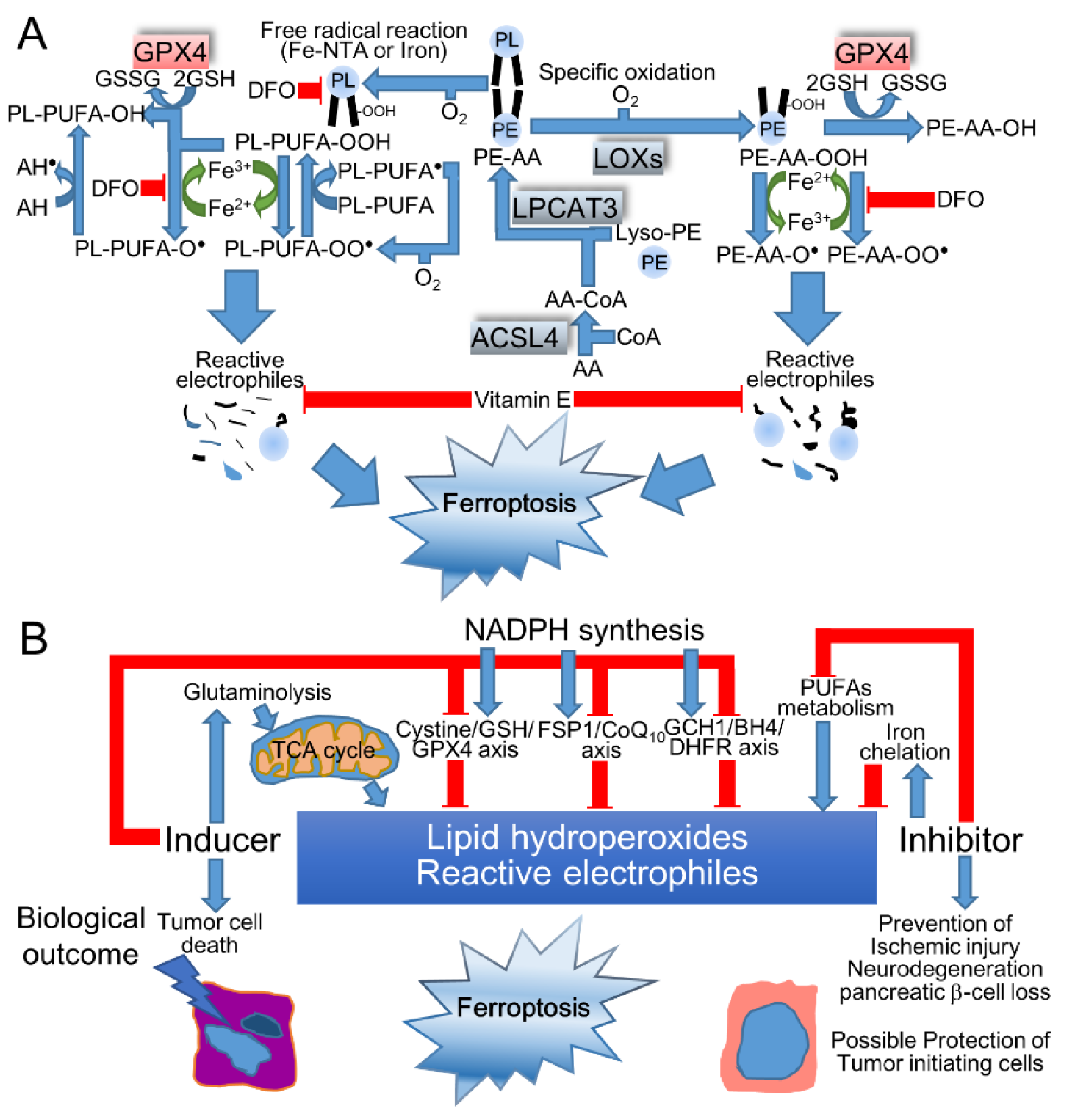{kind=link}
Table 1.
Inherited renal cancer syndromes with predisposing genes: the chromosomal location, predominant histopathology, and activated signaling pathway.
Table 1.
Inherited renal cancer syndromes with predisposing genes: the chromosomal location, predominant histopathology, and activated signaling pathway.
| Syndrome | Predisposing Gene | Chromosomal Location | Predominant Histopathology | Activated Signaling Pathway |
|---|---|---|---|---|
| von Hippel–Lindau (VHL) disease | VHL | 3p25 | Clear cell | HIF pathway |
| Hereditary papillary renal cell carcinoma | MET | 7q31 | Papillary (type 1) | MET kinase pathway |
| Birt–Hogg–Dubé syndrome | FLCN (Folliculin) | 17p11 | Chromophobe, hybrid oncocytic, and clear cell | mTOR pathway |
| Hereditary leiomyomatosis and renal cell carcinoma (HLRCC) | FH (Fumarate hydratase) | 1q43 | Papillary (type 2) | HIF pathway, NRF2 pathway, and TCA cycle pathway |
| Succinate dehydrogenase-deficient renal cell carcinoma (SDH-deficient RCC) | SDHB SDHC SDHD | 1p36 1q23 11q23 | Oncocytic is most common and clear cell is less frequent | HIF pathway and TCA cycle pathway |
| Tuberous sclerosis complex (TSC) | TSC1 TSC2 | 9q34 16p13 | Heterogeneous; chromophobe, oncocytic, and cystic | mTOR pathway |
| Cowden syndrome | PTEN | 10q23 | Chromophobe | PI3/AKT/mTOR pathway |
| Inherited RCC | CDKN2B | 9q21 | Clear cell | Cell cycle regulation |
| BAP1 tumor predisposition syndrome | BAP1 (BRCA1-associated Protein 1) | 3p21 | # Clear cell or eosinophilic, high-grade nuclei | Dysregulated BAP1 transcriptional activity, including metabolic reprogramming |
| MiTF-associated cancer syndrome | MiTF (Microphthalmia-associated transcription factor) | 3p14 | ## Papillary (type 1) and/or clear cell or demonstrate TFE3-positive Xp11 translocation RCC | Dysregulated MiTF transcriptional activity, including mTOR pathway |
# In sporadic BAP1-mutated RCC, its histopathology sometimes overlaps with Xp11 translocation RCC, which is characterized by papillary and epithelioid growth [89]. ## Due to the small number of reports, the histological subtype of RCC that is associated with the MiTF mutation has shown variable characteristics [88].
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Okazaki, Y. The Role of Ferric Nitrilotriacetate in Renal Carcinogenesis and Cell Death: From Animal Models to Clinical Implications. Cancers 2022, 14, 1495. https://doi.org/10.3390/cancers14061495
AMA Style
Okazaki Y. The Role of Ferric Nitrilotriacetate in Renal Carcinogenesis and Cell Death: From Animal Models to Clinical Implications. Cancers. 2022; 14(6):1495. https://doi.org/10.3390/cancers14061495
Chicago/Turabian StyleOkazaki, Yasumasa. 2022. "The Role of Ferric Nitrilotriacetate in Renal Carcinogenesis and Cell Death: From Animal Models to Clinical Implications" Cancers 14, no. 6: 1495. https://doi.org/10.3390/cancers14061495
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.