
Targeting HIF-2α in the Tumor Microenvironment: Redefining the Role of HIF-2α for Solid Cancer Therapy

 , , , , and
, , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Role of HIF-2α in Development
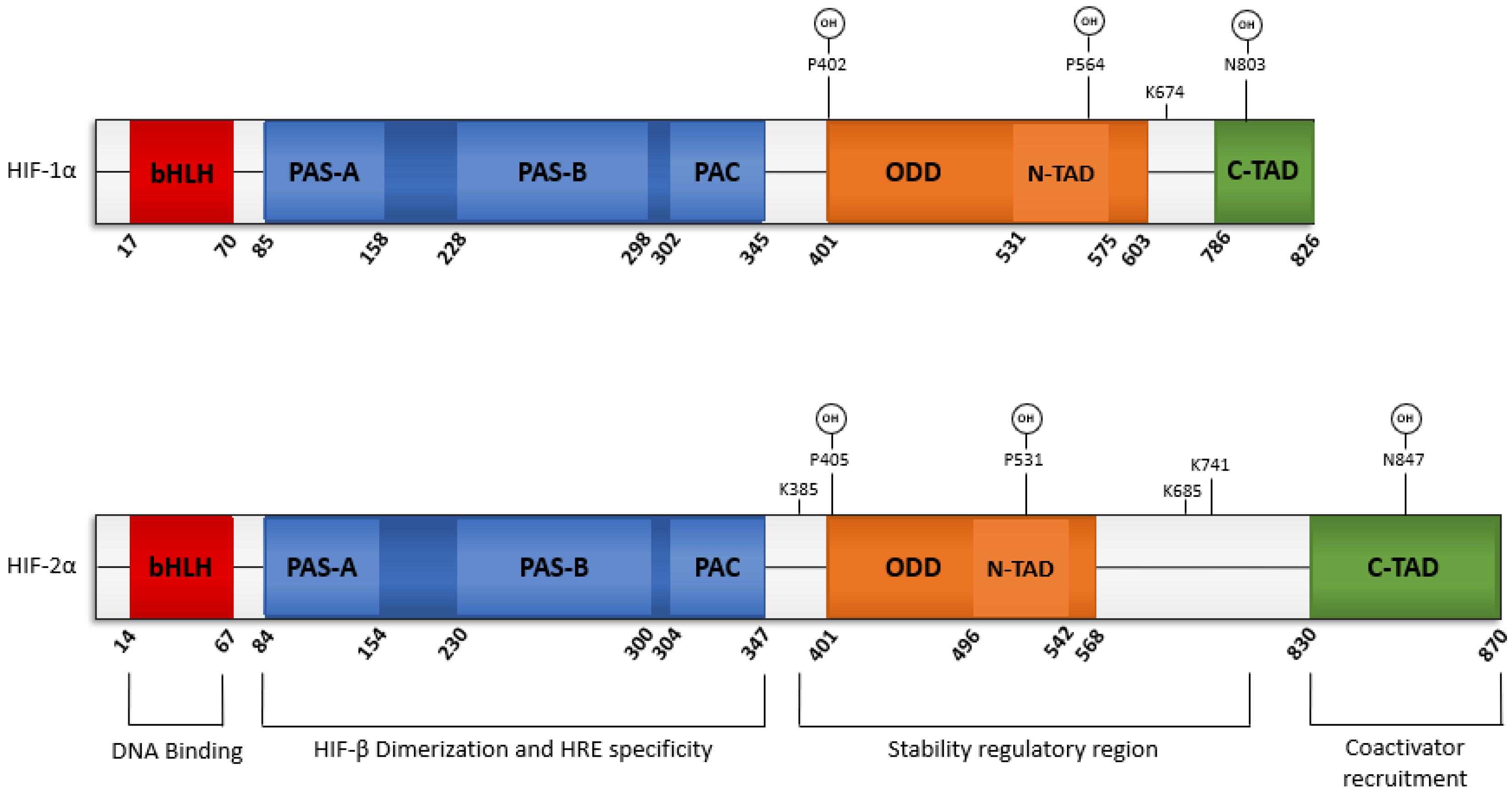
2.1. Structure of EPAS1
2.2. Genetic Variations of EPAS1
2.3. HIF-2α Mediates Hypoxia-Induced Translation
2.4. Translational Regulation of HIF-2α
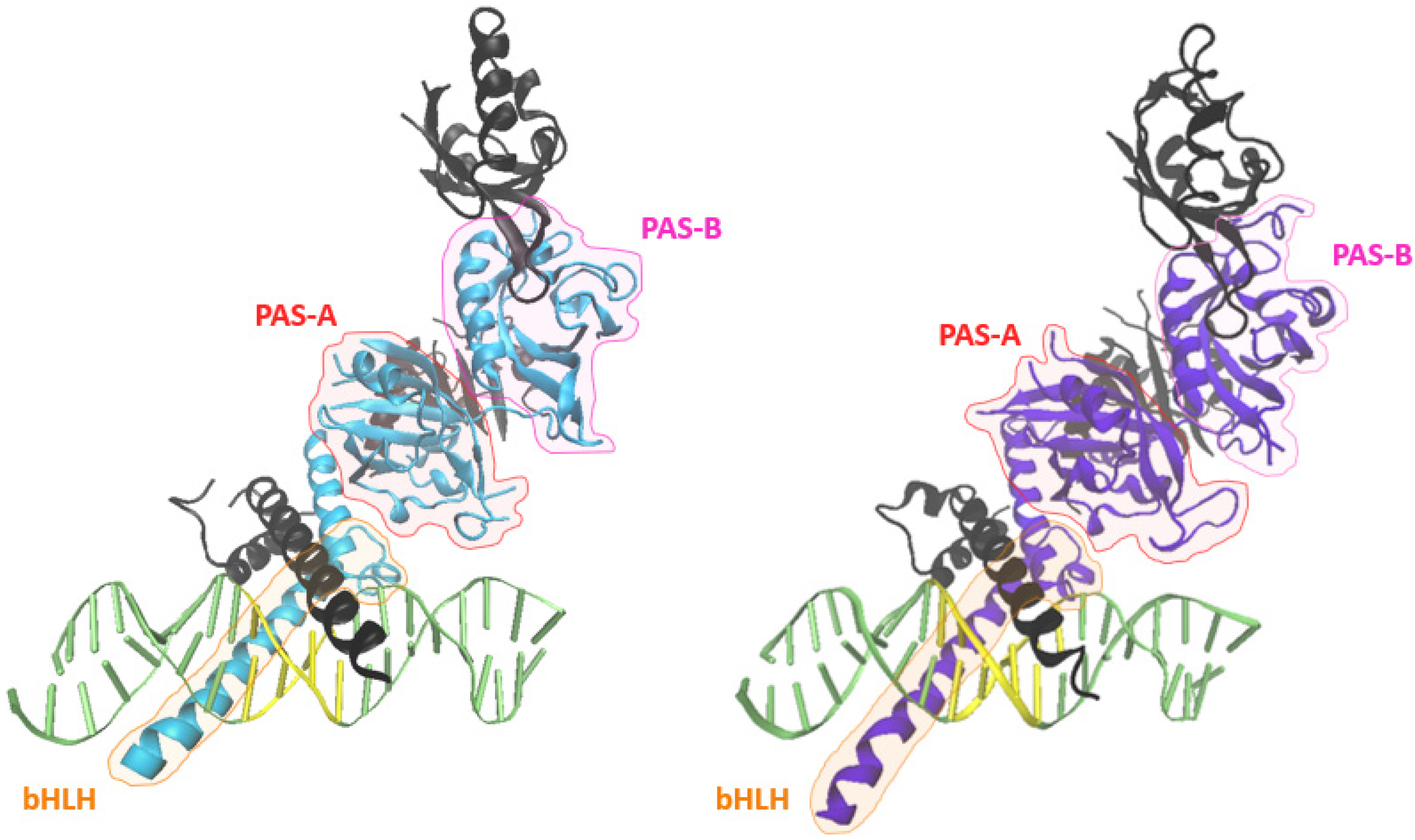
2.5. Structure of HIF-2α
2.6. PTMs Regulate HIF-2α Stability
2.7. Spatiotemporal Dynamics of HIF-α
2.8. PTMs Regulate the HIF Switch
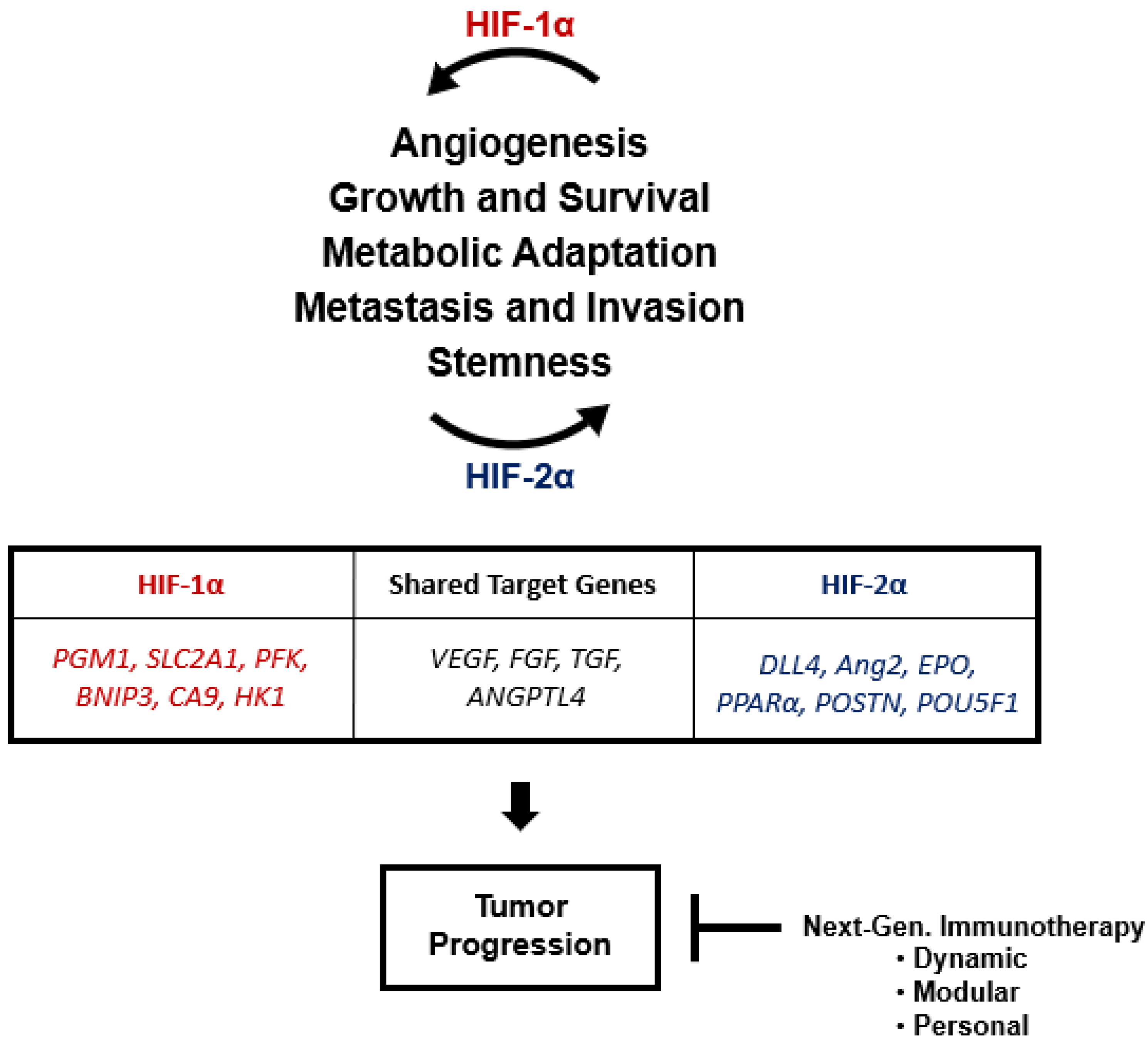
3. Role of HIF-2α in Tumor Progression
3.1. The Hypoxic TME
3.2. EPAS1 Mutations and Cancer
3.3. Stroma
3.4. Metastasis
3.5. Angiogenesis
3.6. Stemness
4. Hypoxia and Cancer Therapy
5. The HIF-α Debate
6. Future Perspectives
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Shao, C.; Yang, F.; Miao, S.; Liu, W.; Wang, C.; Shu, Y.; Shen, H. Role of hypoxia-induced exosomes in tumor biology. Mol. Cancer 2018, 17, 120. [Google Scholar] [CrossRef] [PubMed]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Investig. 2013, 123, 3664–3671. [Google Scholar] [CrossRef] [Green Version]
- Walsh, J.C.; Lebedev, A.; Aten, E.; Madsen, K.; Marciano, L.; Kolb, H.C. The Clinical Importance of Assessing Tumor Hypoxia: Relationship of Tumor Hypoxia to Prognosis and Therapeutic Opportunities. Antioxidants Redox Signal. 2014, 21, 1516–1554. [Google Scholar] [CrossRef]
- Jing, X.; Yang, F.; Shao, C.; Wei, K.; Xie, M.; Shen, H.; Shu, Y. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol. Cancer 2019, 18, 157. [Google Scholar] [CrossRef] [Green Version]
- Gilkes, D.M. (Ed.) Hypoxia and Cancer Metastasis; Springer: Cham, Switzerland, 2019. [Google Scholar] [CrossRef]
- Liao, D.; Johnson, R.S. Hypoxia: A key regulator of angiogenesis in cancer. Cancer Metastasis Rev. 2007, 26, 281–290. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Zhang, S.; Gong, Z.; Li, X.; Cao, K.; Deng, H.; He, Y.; et al. The role of microenvironment in tumor angiogenesis. J. Exp. Clin. Cancer Res. 2020, 39, 204. [Google Scholar] [CrossRef]
- Rankin, E.; Nam, J.-M.; Giaccia, A.J. Hypoxia: Signaling the Metastatic Cascade. Trends Cancer 2016, 2, 295–304. [Google Scholar] [CrossRef] [Green Version]
- Vaupel, P. The Role of Hypoxia-Induced Factors in Tumor Progression. Oncologist 2004, 9 (Suppl. S5), 10–17. [Google Scholar] [CrossRef]
- Yang, G.; Shi, R.; Zhang, Q. Hypoxia and Oxygen-Sensing Signaling in Gene Regulation and Cancer Progression. Int. J. Mol. Sci. 2020, 21, 8162. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, K.; Song, G.; Ouyang, G. Role of hypoxia in the hallmarks of human cancer. J. Cell. Biochem. 2009, 107, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, N.R.; Semenza, G.L. Oxygen Sensing and Homeostasis. Physiology 2015, 30, 340–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L.; Agani, F.; Iyer, N.; Kotch, L.; Laughner, E.; Leung, S.; Yu, A. Regulation of cardiovascular development and physiology by hypoxia-inducible factor 1. Ann. N. Y. Acad. Sci. 1999, 874, 262–268. [Google Scholar] [CrossRef]
- Koh, M.Y.; Lemos, R.; Liu, X.; Powis, G. The Hypoxia-Associated Factor Switches Cells from HIF-1α- to HIF-2α-Dependent Signaling Promoting Stem Cell Characteristics, Aggressive Tumor Growth and Invasion. Cancer Res. 2011, 71, 4015–4027. [Google Scholar] [CrossRef] [Green Version]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1α and HIF2α: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2011, 12, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Hypoxia-inducible nuclear factors bind to an enhancer element located 3′ to the human erythropoietin gene Hypoxia-inducible nuclear factors bind to an enhancer element located 3′ to the human erythropoietin gene. Proc. Natl. Acad. Sci. USA 1991, 88, 5680–5684. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L.; Wang, G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell. Biol. 1992, 12, 5447–5454. [Google Scholar] [CrossRef] [Green Version]
- Wiener, C.M.; Bootha, G.; Semenza, G.L. In Vivo Expression of mRNAs Encoding Hypoxia-Inducible Factor 1. Biochem. Biophys. Res. Commun. 1996, 225, 485–488. [Google Scholar] [CrossRef]
- Semenza, G.L.; Agani, F.; Booth, G.; Forsythe, J.; Iyer, N.; Jiang, B.-H.; Leung, S.; Roe, R.; Wiener, C.; Yu, A. Structural and functional analysis of hypoxia-inducible factor 1. Kidney Int. 1997, 51, 553–555. [Google Scholar] [CrossRef] [Green Version]
- Weidemann, A.; Johnson, R.S. Biology of HIF-1α. Cell Death Differ. 2008, 15, 621–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 2009, 29, 625–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flamme, I.; Frölich, T.; Risau, W. Molecular mechanisms of vasculogenesis and embryonic angiogenesis. J. Cell. Physiol. 1997, 173, 206–210. [Google Scholar] [CrossRef]
- Jain, S.; Maltepe, E.; Lu, M.M.; Simon, C.; Bradfield, C. Expression of ARNT, ARNT2, HIF1α, HIF2α and Ah receptor mRNAs in the developing mouse. Mech. Dev. 1998, 73, 117–123. [Google Scholar] [CrossRef]
- Wiesener, M.S.; Jürgensen, J.S.; Rosenberger, C.; Scholze, C.K.; Hörstrup, J.H.; Warnecke, C.; Mandriota, S.J.; Bechmann, I.; Frei, U.A.; Pugh, C.W.; et al. Widespread, hypoxia-inducible expression of HIF-2α in distinct cell populations of different organs. FASEB J. 2002, 17, 271–273. [Google Scholar] [CrossRef] [Green Version]
- Smythies, J.; Sun, M.; Masson, N.; Salama, R.; Simpson, P.D.; Murray, E.; Neumann, V.; Cockman, M.; Choudhry, H.; Ratcliffe, P.J.; et al. Inherent DNA-binding specificities of the HIF-1α and HIF-2α transcription factors in chromatin. EMBO Rep. 2018, 20, e46401. [Google Scholar] [CrossRef]
- Schödel, J.; Oikonomopoulos, S.; Ragoussis, J.; Pugh, C.W.; Ratcliffe, P.J.; Mole, D.R. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood 2011, 117, e207–e217. [Google Scholar] [CrossRef] [Green Version]
- Graham, A.M.; Presnell, J.S. Hypoxia Inducible Factor (HIF) transcription factor family expansion, diversification, divergence and selection in eukaryotes. PLoS ONE 2017, 12, e0179545. [Google Scholar] [CrossRef] [Green Version]
- Chaudhuri, P.K.; Low, B.C.; Lim, C.T. Mechanobiology of Tumor Growth. Chem. Rev. 2018, 118, 6499–6515. [Google Scholar] [CrossRef]
- Graham, K.; Unger, E. Overcoming tumor hypoxia as a barrier to radiotherapy, chemotherapy and immunotherapy in cancer treatment. Int. J. Nanomed. 2018, 13, 6049–6058. [Google Scholar] [CrossRef] [Green Version]
- Arora, S.; Balasubramaniam, S.; Zhang, H.; Berman, T.; Narayan, P.; Suzman, D.; Bloomquist, E.; Tang, S.; Gong, Y.; Sridhara, R.; et al. FDA Approval Summary: Olaparib Monotherapy or in Combination with Bevacizumab for the Maintenance Treatment of Patients with Advanced Ovarian Cancer. Oncologist 2020, 26, e164–e172. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.-M.; Hwang, Y.-C.; Liu, I.-J.; Lee, C.-C.; Tsai, H.-Z.; Li, H.-J.; Wu, H.-C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1. [Google Scholar] [CrossRef] [PubMed]
- Fallah, J.; Rini, B.I. HIF Inhibitors: Status of Current Clinical Development. Curr. Oncol. Rep. 2019, 21, 6. [Google Scholar] [CrossRef] [PubMed]
- Rapisarda, A.; Zalek, J.; Hollingshead, M.; Braunschweig, T.; Uranchimeg, B.; Bonomi, C.A.; Borgel, S.D.; Carter, J.P.; Hewitt, S.M.; Shoemaker, R.H.; et al. Schedule-dependent Inhibition of Hypoxia-inducible Factor-1 Protein Accumulation, Angiogenesis, and Tumor Growth by Topotecan in U251-HRE Glioblastoma Xenografts. Cancer Res. 2004, 64, 6845–6848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapisarda, A.; Uranchimeg, B.; Scudiero, D.A.; Selby, M.; Sausville, E.A.; Shoemaker, R.H.; Melillo, G. Identification of small molecule inhibitors of hypoxia-inducible factor 1 transcriptional activation pathway. Cancer Res. 2002, 62, 4316–4324. [Google Scholar] [PubMed]
- Kummar, S.; Raffeld, M.; Juwara, L.; Horneffer, Y.; Strassberger, A.; Allen, D.; Steinberg, S.M.; Rapisarda, A.; Spencer, S.D.; Figg, W.D.; et al. Multihistology, Target-Driven Pilot Trial of Oral Topotecan as an Inhibitor of Hypoxia-Inducible Factor-1α in Advanced Solid Tumors. Clin. Cancer Res. 2011, 17, 5123–5131. [Google Scholar] [CrossRef] [Green Version]
- Ema, M.; Taya, S.; Yokotani, N.; Sogawa, K.; Matsuda, Y.; Fujii-Kuriyama, Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1α regulates the VEGF expression and is potentially involved in lung and vascular development. Proc. Natl. Acad. Sci. USA 1997, 94, 4273–4278. [Google Scholar] [CrossRef] [Green Version]
- Flamme, I.; Fröhlich, T.; von Reutern, M.; Kappel, A.; Damert, A.; Risau, W. HRF, a putative basic helix-loop-helix-PAS-domain transcription factor is closely related to hypoxia-inducible factor-1α and developmentally expressed in blood vessels. Mech. Dev. 1997, 63, 51–60. [Google Scholar] [CrossRef]
- Hogenesch, J.B.; Chan, W.K.; Jackiw, V.H.; Brown, R.C.; Gu, Y.-Z.; Pray-Grant, M.; Perdew, G.H.; Bradfield, C.A. Characterization of a Subset of the Basic-Helix-Loop-Helix-PAS Superfamily That Interacts with Components of the Dioxin Signaling Pathway. J. Biol. Chem. 1997, 272, 8581–8593. [Google Scholar] [CrossRef] [Green Version]
- Tian, H.; McKnight, S.L.; Russell, D.W. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997, 11, 72–82. [Google Scholar] [CrossRef] [Green Version]
- Kotch, L.E.; Iyer, N.V.; Laughner, E.; Semenza, G.L. Defective Vascularization of HIF-1α-Null Embryos Is Not Associated with VEGF Deficiency but with Mesenchymal Cell Death. Dev. Biol. 1999, 209, 254–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, N.V.; Kotch, L.E.; Agani, F.; Leung, S.W.; Laughner, E.; Wenger, R.H.; Gassmann, M.; Gearhart, J.D.; Lawler, A.M.; Yu, A.Y.; et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1α. Genes Dev. 1998, 12, 149–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Compernolle, V.; Brusselmans, K.; Acker, T.; Hoet, P.; Tjwa, M.; Beck, H.; Plaisance, S.; Dor, Y.; Keshet, E.; Lupu, F.; et al. Loss of HIF-2α and inhibition of VEGF impair fetal lung maturation, whereas treatment with VEGF prevents fatal respiratory distress in premature mice. Nat. Med. 2002, 8, 702–710. [Google Scholar] [CrossRef]
- Tian, H.; Hammer, R.E.; Matsumoto, A.M.; Russell, D.W.; McKnight, S.L. The hypoxia-responsive transcription factor EPAS1 is essential for catecholamine homeostasis and protection against heart failure during embryonic development. Genes Dev. 1998, 12, 3320–3324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, J.; Zhang, L.; Drysdale, L.; Fong, G.-H. The transcription factor EPAS-1/hypoxia-inducible factor 2alpha plays an important role in vascular remodeling. Proc. Natl. Acad. Sci. USA 2000, 97, 8386–8391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scortegagna, M.; Ding, K.; Oktay, Y.; Gaur, A.; Thurmond, F.; Yan, L.-J.; Marck, B.T.; Matsumoto, A.M.; Shelton, J.M.; Richardson, J.; et al. Multiple organ pathology, metabolic abnormalities and impaired homeostasis of reactive oxygen species in Epas1−/− mice. Nat. Genet. 2003, 35, 331–340. [Google Scholar] [CrossRef]
- Mastrogiannaki, M.; Matak, P.; Keith, B.; Simon, M.C.; Vaulont, S.; Peyssonnaux, C. HIF-2α, but not HIF-1α, promotes iron absorption in mice. J. Clin. Investig. 2009, 119, 1159–1166. [Google Scholar] [CrossRef]
- Shah, Y.M.; Matsubara, T.; Ito, S.; Yim, S.-H.; Gonzalez, F.J. Intestinal Hypoxia-Inducible Transcription Factors Are Essential for Iron Absorption following Iron Deficiency. Cell Metab. 2009, 9, 152–164. [Google Scholar] [CrossRef] [Green Version]
- Morita, M.; Ohneda, O.; Yamashita, T.; Takahashi, S.; Suzuki, N.; Nakajima, O.; Kawauchi, S.; Ema, M.; Shibahara, S.; Udono, T.; et al. HLF/HIF-2α is a key factor in retinopathy of prematurity in association with erythropoietin. EMBO J. 2003, 22, 1134–1146. [Google Scholar] [CrossRef] [Green Version]
- Gruber, M.; Hu, C.; Johnson, R.S.; Brown, E.J.; Keith, B.; Simon, M.C. Acute postnatal ablation of Hif-2α results in anemia. Proc. Natl. Acad. Sci. USA 2007, 104, 2301–2306. [Google Scholar] [CrossRef] [Green Version]
- Rankin, E.; Biju, M.P.; Liu, Q.; Unger, T.L.; Rha, J.; Johnson, R.; Simon, M.C.; Keith, B.; Haase, V.H. Hypoxia-inducible factor–2 (HIF-2) regulates hepatic erythropoietin in vivo. J. Clin. Investig. 2007, 117, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Scortegagna, M.; Ding, K.; Zhang, Q.; Oktay, Y.; Bennett, M.J.; Shelton, J.M.; Richardson, J.A.; Moe, O.; Garcia, J.A. HIF-2α regulates murine hematopoietic development in an erythropoietin-dependent manner. Blood 2005, 105, 3133–3140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.-H.; Huang, X.-W.; Qun, L.; Li, Y.-N.; Wang, Y.; Liu, C.; Ma, Y.; Liu, Q.-M.; Sun, K.; Qian, F.; et al. Two functional loci in the promoter of EPAS1 gene involved in high-altitude adaptation of Tibetans. Sci. Rep. 2014, 4, 7465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, H.-C.; Lin, P.-Y.; Chung, T.-C.; Chao, C.-M.; Lai, L.-C.; Tsai, M.-H.; Chuang, E.Y. DBCAT: Database of CpG Islands and Analytical Tools for Identifying Comprehensive Methylation Profiles in Cancer Cells. J. Computat. Biol. 2011, 18, 1013–1017. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-M.; Lee, J.; Noh, K.-M.; Choi, W.-Y.; Jeon, S.; Oh, G.T.; Kim-Ha, J.; Jin, Y.; Cho, S.-W.; Kim, Y.-J. Intragenic CpG islands play important roles in bivalent chromatin assembly of developmental genes. Proc. Natl. Acad. Sci. USA 2017, 114, E1885–E1894. [Google Scholar] [CrossRef] [Green Version]
- Bird, A.; Taggart, M.; Frommer, M.; Miller, O.J.; Macleod, D. A fraction of the mouse genome that is derived from islands of nonmethylated, CpG-rich DNA. Cell 1985, 40, 91–99. [Google Scholar] [CrossRef]
- Esteller, M. CpG island hypermethylation and tumor suppressor genes: A booming present, a brighter future. Oncogene 2002, 21, 5427–5440. [Google Scholar] [CrossRef] [Green Version]
- Long, H.K.; King, H.W.; Patient, R.K.; Odom, D.T.; Klose, R.J. Protection of CpG islands from DNA methylation is DNA-encoded and evolutionarily conserved. Nucleic Acids Res. 2016, 44, 6693–6706. [Google Scholar] [CrossRef] [Green Version]
- Rawłuszko-Wieczorek, A.A.; Horbacka, K.; Krokowicz, P.; Misztal, M.; Jagodziński, P.P. Prognostic Potential of DNA Methylation and Transcript Levels of HIF1A and EPAS1 in Colorectal Cancer. Mol. Cancer Res. 2014, 12, 1112–1127. [Google Scholar] [CrossRef] [Green Version]
- Krinner, S.; Heitzer, A.P.; Diermeier, S.; Obermeier, I.; Längst, G.; Wagner, R. CpG domains downstream of TSSs promote high levels of gene expression. Nucleic Acids Res. 2014, 42, 3551–3564. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Kline, D.D.; Dick, T.E.; Prabhakar, N.R. Chronic Intermittent Hypdxia Enhances Carotid Body Chemoreceptor Response to Low Oxygen. Front. Modeling Control. Breath. 2001, 499, 33–38. [Google Scholar] [CrossRef]
- Xu, S.; Li, S.; Yang, Y.; Tan, J.; Lou, H.; Jin, W.; Yang, L.; Pan, X.; Wang, J.; Shen, Y.; et al. A Genome-Wide Search for Signals of High-Altitude Adaptation in Tibetans. Mol. Biol. Evol. 2010, 28, 1003–1011. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.-S.; Wang, S.; Li, Y.; Jhala, Y.; Thakur, M.; Otecko, N.; Si, J.-F.; Chen, H.-M.; Shapiro, B.; Nielsen, R.; et al. Ancient Hybridization with an Unknown Population Facilitated High-Altitude Adaptation of Canids. Mol. Biol. Evol. 2020, 37, 2616–2629. [Google Scholar] [CrossRef]
- Werhahn, G.; Liu, Y.; Meng, Y.; Cheng, C.; Lu, Z.; Atzeni, L.; Deng, Z.; Kun, S.; Shao, X.; Lu, Q.; et al. Himalayan wolf distribution and admixture based on multiple genetic markers. J. Biogeogr. 2020, 47, 1272–1285. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Jiang, C.; Luo, Y.; Liu, F.; Gao, Y. An EPAS1 Haplotype Is Associated with High Altitude Polycythemia in Male Han Chinese at the Qinghai-Tibetan Plateau. Wilderness Environ. Med. 2014, 25, 392–400. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Yang, Z.; Zhang, H.; Cui, C.; Qi, X.; Luo, X.; Tao, X.; Wu, T.; Ouzhuluobu, B.; Danzengduojie, C.; et al. Genetic Variations in Tibetan Populations and High-Altitude Adaptation at the Himalayas. Mol. Biol. Evol. 2010, 28, 1075–1081. [Google Scholar] [CrossRef] [Green Version]
- Kafri, M.; Metzl-Raz, E.; Jona, G.; Barkai, N. The Cost of Protein Production. Cell Rep. 2015, 14, 22–31. [Google Scholar] [CrossRef] [Green Version]
- Hochachka, P.W.; Buck, L.T.; Doll, C.J.; Land, S. Unifying theory of hypoxia tolerance: Molecular/metabolic defense and rescue mechanisms for surviving oxygen lack. Proc. Natl. Acad. Sci. USA 1996, 93, 9493–9498. [Google Scholar] [CrossRef] [Green Version]
- Chee, N.T.; Lohse, I.; Brothers, S.P. mRNA-to-protein translation in hypoxia. Mol. Cancer 2019, 18, 49. [Google Scholar] [CrossRef]
- Uniacke, J.; Holterman, C.E.; Lachance, G.; Franovic, A.; Jacob, M.D.; Fabian, M.R.; Payette, J.; Holcik, M.; Pause, A.; Lee, S. An oxygen-regulated switch in the protein synthesis machinery. Nature 2012, 486, 126–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.-C.; Hsu, M.; Tarn, W.-Y. Cell stress modulates the function of splicing regulatory protein RBM4 in translation control. Proc. Natl. Acad. Sci. USA 2007, 104, 2235–2240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, M.; Ler, L.W.; Fabian, M.R.; Siddiqui, N.; Mullin, M.; Henderson, V.C.; Alain, T.; Fonseca, B.D.; Karashchuk, G.; Bennett, C.F.; et al. A Novel 4EHP-GIGYF2 Translational Repressor Complex Is Essential for Mammalian Development. Mol. Cell. Biol. 2012, 32, 3585–3593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, J.D.; Wang, M.; Audas, T.E.; Kwon, D.; Carlsson, S.K.; Timpano, S.; Evagelou, S.L.; Brothers, S.; Gonzalgo, M.L.; Krieger, J.R.; et al. Systemic Reprogramming of Translation Efficiencies on Oxygen Stimulus. Cell Rep. 2016, 14, 1293–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanova, I.G.; Park, C.V.; Kenneth, N.S. Translating the Hypoxic Response—the Role of HIF Protein Translation in the Cellular Response to Low Oxygen. Cells 2019, 8, 114. [Google Scholar] [CrossRef] [Green Version]
- Zimmer, M.; Ebert, B.L.; Neil, C.; Brenner, K.; Papaioannou, I.; Melas, A.; Tolliday, N.; Lamb, J.; Pantopoulos, K.; Golub, T.; et al. Small-Molecule Inhibitors of HIF-2a Translation Link Its 5′UTR Iron-Responsive Element to Oxygen Sensing. Mol. Cell 2008, 32, 838–848. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, M.; Galy, B.; Muckenthaler, M.U.; Hentze, M.W. Iron-regulatory proteins limit hypoxia-inducible factor-2α expression in iron deficiency. Nat. Struct. Mol. Biol. 2007, 14, 420–426. [Google Scholar] [CrossRef]
- Mathew, L.K.; Lee, S.S.; Skuli, N.; Rao, S.; Keith, B.; Nathanson, K.L.; Lal, P.; Simon, M.C. Restricted Expression of miR-30c-2-3p and miR-30a-3p in Clear Cell Renal Cell Carcinomas Enhances HIF2α Activity. Cancer Discov. 2013, 4, 53–60. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Pu, J.; Qi, T.; Qi, M.; Yang, C.; Li, S.; Huang, K.; Zheng, L.; Tong, Q. MicroRNA-145 inhibits the growth, invasion, metastasis and angiogenesis of neuroblastoma cells through targeting hypoxia-inducible factor 2 alpha. Oncogene 2012, 33, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Crews, S.T. Control of cell lineage-specific development and transcription by bHLH-PAS proteins. Genes Dev. 1998, 12, 607–620. [Google Scholar] [CrossRef] [Green Version]
- Kolonko, M.; Greb-Markiewicz, B. bHLH-PAS Proteins: Their Structure and Intrinsic Disorder. Int. J. Mol. Sci. 2019, 20, 3653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kewley, R.J.; Whitelaw, M.L.; Chapman-Smith, A. The mammalian basic helix–loop–helix/PAS family of transcriptional regulators. Int. J. Biochem. Cell Biol. 2004, 36, 189–204. [Google Scholar] [CrossRef]
- Wenger, R.H.; Stiehl, D.P.; Camenisch, G. Integration of Oxygen Signaling at the Consensus HRE. Sci. STKE 2005, 2005, re12. [Google Scholar] [CrossRef] [Green Version]
- Petrella, B.L.; Lohi, J.; Brinckerhoff, C. Identification of membrane type-1 matrix metalloproteinase as a target of hypoxia-inducible factor-2α in von Hippel–Lindau renal cell carcinoma. Oncogene 2004, 24, 1043–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.L.; Jiang, B.-H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.E.; Arany, Z.; Livingston, D.M.; Bunn, H.F. Activation of Hypoxia-inducible Transcription Factor Depends Primarily upon Redox-sensitive Stabilization of Its α Subunit. J. Biol. Chem. 1996, 271, 32253–32259. [Google Scholar] [CrossRef] [Green Version]
- Wenger, R.H.; Rolfs, A.; Spielmann, P.; Zimmermann, D.R.; Gassmann, M. Mouse Hypoxia-Inducible Factor-1α Is Encoded by Two Different mRNA Isoforms: Expression from a Tissue-Specific and a Housekeeping-Type Promoter. Blood 1998, 91, 3471–3480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Q.; Cong, X.; Yun, Z. Differential Hypoxic Regulation of Hypoxia-Inducible Factors 1α and 2α. Mol. Cancer Res. 2011, 9, 757–765. [Google Scholar] [CrossRef] [Green Version]
- Moroz, E.; Carlin, S.; Dyomina, K.; Burke, S.; Thaler, H.T.; Blasberg, R.; Serganova, I. Real-Time Imaging of HIF-1α Stabilization and Degradation. PLoS ONE 2009, 4, e5077. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, Z.; Yang, C.; Ryska, A.; Ji, Y.; Hou, Y.; Graybill, S.D.; Bullova, P.; Lubensky, I.; Klöppel, G.; Pacak, K. HIF2A gain-of-function mutations detected in duodenal gangliocytic paraganglioma. Endocr. Relat. Cancer 2016, 23, L13–L16. [Google Scholar] [CrossRef] [Green Version]
- Lando, D.; Peet, D.J.; Whelan, D.A.; Gorman, J.J.; Whitelaw, M.L. Asparagine Hydroxylation of the HIF Transactivation Domain: A Hypoxic Switch. Science 2002, 295, 858–861. [Google Scholar] [CrossRef] [PubMed]
- Appelhoff, R.J.; Tian, Y.-M.; Raval, R.R.; Turley, H.; Harris, A.L.; Pugh, C.W.; Ratcliffe, P.J.; Gleadle, J. Differential Function of the Prolyl Hydroxylases PHD1, PHD2, and PHD3 in the Regulation of Hypoxia-inducible Factor. J. Biol. Chem. 2004, 279, 38458–38465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, D.; Li, L.-S.; Heaton-Johnson, K.J.; Arsenault, P.; Master, S.R.; Lee, F.S. Prolyl Hydroxylase Domain Protein 2 (PHD2) Binds a Pro-Xaa-Leu-Glu Motif, Linking It to the Heat Shock Protein 90 Pathway. J. Biol. Chem. 2013, 288, 9662–9674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagnall, J.; Leedale, J.; Taylor, S.E.; Spiller, D.G.; White, M.R.H.; Sharkey, K.J.; Bearon, R.N.; Sée, V. Tight Control of Hypoxia-inducible Factor-α Transient Dynamics Is Essential for Cell Survival in Hypoxia. J. Biol. Chem. 2014, 289, 5549–5564. [Google Scholar] [CrossRef] [Green Version]
- Takeda, K.; Ho, V.C.; Takeda, H.; Duan, L.-J.; Nagy, A.; Fong, G.-H. Placental but Not Heart Defects Are Associated with Elevated Hypoxia-Inducible Factor α Levels in Mice Lacking Prolyl Hydroxylase Domain Protein 2. Mol. Cell. Biol. 2006, 26, 8336–8346. [Google Scholar] [CrossRef] [Green Version]
- Ohh, M.; Park, C.W.; Ivan, M.; Hoffman, M.A.; Kim, T.-Y.; Huang, E.; Pavletich, N.; Chau, V.; Kaelin, W.G. Ubiquitination of hypoxia-inducible factor requires direct binding to the β-domain of the von Hippel–Lindau protein. Nat. Cell Biol. 2000, 2, 423–427. [Google Scholar] [CrossRef]
- Koh, M.Y.; Powis, G. Passing the baton: The HIF switch. Trends Biochem. Sci. 2012, 37, 364–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ema, M.; Hirota, K.; Mimura, J.; Abe, H.; Yodoi, J.; Sogawa, K.; Poellinger, L.; Fujii-Kuriyama, Y. Molecular mechanisms of transcription activation by HLF and HIF1alpha in response to hypoxia: Their stabilization and redox signal-induced interaction with CBP/p300. EMBO J. 1999, 18, 1905–1914. [Google Scholar] [CrossRef] [Green Version]
- Koivunen, P.; Hirsilä, M.; Günzler, V.; Kivirikko, K.I.; Myllyharju, J. Catalytic Properties of the Asparaginyl Hydroxylase (FIH) in the Oxygen Sensing Pathway Are Distinct from Those of Its Prolyl 4-Hydroxylases. J. Biol. Chem. 2004, 279, 9899–9904. [Google Scholar] [CrossRef] [Green Version]
- Arany, Z.; Huang, E.; Eckner, R.; Bhattacharya, S.; Jiang, C.; Goldberg, M.A.; Bunn, H.F.; Livingston, D.M. An essential role for p300/CBP in the cellular response to hypoxia. Proc. Natl. Acad. Sci. USA 1996, 93, 12969–12973. [Google Scholar] [CrossRef] [Green Version]
- Kallio, P.J.; Okamoto, K.; O’Brien, S.; Carrero, P.; Makino, Y.; Tanaka, H.; Poellinger, L. Signal transduction in hypoxic cells: Inducible nuclear translocation and recruitment of the CBP/p300 coactivator by the hypoxia-inducible factor-1alpha. EMBO J. 1998, 17, 6573–6586. [Google Scholar] [CrossRef] [PubMed]
- Bouthelier, A.; Meléndez-Rodríguez, F.; Urrutia, A.A.; Aragonés, J. Differential Contribution of N- and C-Terminal Regions of HIF1α and HIF2α to Their Target Gene Selectivity. Int. J. Mol. Sci. 2020, 21, 9401. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.-J.; Sataur, A.; Wang, L.; Chen, H.; Simon, M.C. The N-terminal transactivation domain confers target gene specificity of hypoxia-inducible factors HIF-1alpha and HIF-2alpha. Mol. Biol. Cell 2007, 18, 4528–4542. [Google Scholar] [CrossRef] [Green Version]
- Mengelbier, L.H.; Fredlund, E.; Löfstedt, T.; Noguera, R.; Navarro, S.; Nilsson, H.; Pietras, A.; Vallon-Christersson, J.; Borg, Å.; Gradin, K.; et al. Recruitment of HIF-1α and HIF-2α to common target genes is differentially regulated in neuroblastoma: HIF-2α promotes an aggressive phenotype. Cancer Cell 2006, 10, 413–423. [Google Scholar] [CrossRef] [Green Version]
- Holmquist, L.; Jögi, A.; Påhlman, S. Phenotypic persistence after reoxygenation of hypoxic neuroblastoma cells. Int. J. Cancer 2005, 116, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Bartoszewski, R.; Moszyńska, A.; Serocki, M.; Cabaj, A.; Polten, A.; Ochocka, R.; Dell’Italia, L.; Bartoszewska, S.; Króliczewski, J.; Dąbrowski, M.; et al. Primary endothelial Cell–Specific regulation of Hypoxia-Inducible factor (HIF)-1 and HIF-2 and their target gene expression profiles during hypoxia. FASEB J. 2019, 33, 7929–7941. [Google Scholar] [CrossRef] [PubMed]
- Uchida, T.; Rossignol, F.; Matthay, M.A.; Mounier, R.; Couette, S.; Clottes, E.; Clerici, C. Prolonged Hypoxia Differentially Regulates Hypoxia-inducible Factor (HIF)-1α and HIF-2α Expression in Lung Epithelial Cells. J. Biol. Chem. 2004, 279, 14871–14878. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, H.; Jögi, A.; Beckman, S.; Harris, A.L.; Poellinger, L.; Påhlman, S. HIF-2α expression in human fetal paraganglia and neuroblastoma: Relation to sympathetic differentiation, glucose deficiency, and hypoxia. Exp. Cell Res. 2005, 303, 447–456. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD-Visual Molecular Dynamics. J. Molec. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Schulz, K.; Milke, L.; Rübsamen, D.; Menrad, H.; Schmid, T.; Brüne, B. HIF-1α protein is upregulated in HIF-2α depleted cells via enhanced translation. FEBS Lett. 2012, 586, 1652–1657. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.-J.; Wang, L.-Y.; Chodosh, L.A.; Keith, B.; Simon, M.C. Differential Roles of Hypoxia-Inducible Factor 1α (HIF-1α) and HIF-2α in Hypoxic Gene Regulation. Mol. Cell. Biol. 2003, 23, 9361–9374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, S.E.; Bagnall, J.; Mason, D.; Levy, R.; Fernig, D.G.; See, V. Differential sub-nuclear distribution of hypoxia-inducible factors (HIF)-1 and -2 alpha impacts on their stability and mobility. Open Biol. 2016, 6, 160195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Downes, N.; Laham-Karam, N.; Kaikkonen, M.U.; Ylä-Herttuala, S. Differential but Complementary HIF1α and HIF2α Transcriptional Regulation. Mol. Ther. 2018, 26, 1735–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raval, R.R.; Lau, K.W.; Tran, M.G.B.; Sowter, H.M.; Mandriota, S.J.; Li, J.-L.; Pugh, C.W.; Maxwell, P.H.; Harris, A.L.; Ratcliffe, P.J. Contrasting Properties of Hypoxia-Inducible Factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-Associated Renal Cell Carcinoma. Mol. Cell. Biol. 2005, 25, 5675–5686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, N.; O’dea, E.L.; Doedens, A.; Kim, J.-W.; Weidemann, A.; Stockmann, C.; Asagiri, M.; Simon, M.C.; Hoffmann, A.; Johnson, R.S. Differential activation and antagonistic function of HIF-a isoforms in macrophages are essential for NO homeo-stasis. Genes Dev. 2010, 24, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Skuli, N.; Majmundar, A.J.; Krock, B.L.; Mesquita, R.C.; Mathew, L.K.; Quinn, Z.L.; Runge, A.; Liu, L.; Kim, M.N.; Liang, J.; et al. Endothelial HIF-2α regulates murine pathological angiogenesis and revascularization processes. J. Clin. Investig. 2012, 122, 1427–1443. [Google Scholar] [CrossRef]
- Bayer, C.; Vaupel, P. Acute versus chronic hypoxia in tumors. Strahlenther. Onkol. 2012, 188, 616–627. [Google Scholar] [CrossRef]
- Saxena, K.; Jolly, M.K. Acute vs. Chronic vs. Cyclic Hypoxia: Their Differential Dynamics, Molecular Mechanisms, and Effects on Tumor Progression. Biomolecules 2019, 9, 339. [Google Scholar] [CrossRef] [Green Version]
- Seton-Rogers, S. Hypoxia: HIF switch. Nat. Cancer 2011, 11, 391. [Google Scholar] [CrossRef]
- Zheng, F.; Chen, J.; Zhang, X.; Wang, Z.; Chen, J.; Lin, X.; Huang, H.; Fu, W.; Liang, J.; Wu, W.; et al. The HIF-1α antisense long non-coding RNA drives a positive feedback loop of HIF-1α mediated transactivation and glycolysis. Nat. Commun. 2021, 12, 1341. [Google Scholar] [CrossRef]
- Haronikova, L.; Olivares-Illana, V.; Wang, L.; Karakostis, K.; Chen, S.; Fåhraeus, R. The p53 mRNA: An integral part of the cellular stress response. Nucleic Acids Res. 2019, 47, 3257–3271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.-H.; Lee, Y.-M.; Chun, Y.-S.; Chen, J.; Kim, J.-E.; Park, J.-W. Sirtuin 1 Modulates Cellular Responses to Hypoxia by Deacetylating Hypoxia-Inducible Factor 1α. Mol. Cell 2010, 38, 864–878. [Google Scholar] [CrossRef]
- Chen, R.; Xu, M.; Hogg, R.T.; Li, J.; Little, B.; Gerard, R.D.; Garcia, J.A. The Acetylase/Deacetylase Couple CREB-binding Protein/Sirtuin 1 Controls Hypoxia-inducible Factor 2 Signaling. J. Biol. Chem. 2012, 287, 30800–30811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gradin, K.; McGuire, J.; Wenger, R.H.; Kvietikova, I.; Fhitelaw, M.L.; Toftgård, R.; Tora, L.; Gassmann, M.; Poellinger, L. Functional interference between hypoxia and dioxin signal transduction pathways: Competition for recruitment of the Arnt transcription factor. Mol. Cell. Biol. 1996, 16, 5221–5231. [Google Scholar] [CrossRef] [Green Version]
- Minet, E.; Mottet, D.; Michel, G.; Roland, I.; Raes, M.; Remacle, J.; Michiels, C. Hypoxia-induced activation of HIF-1: Role of HIF-1α-Hsp90 interaction. FEBS Lett. 1999, 460, 251–256. [Google Scholar] [CrossRef]
- Katschinski, D.M.; Le, L.; Schindler, S.G.; Thomas, T.; Voss, A.K.; Wenger, R.H. Interaction of the PAS B Domain with HSP90 Accelerates Hypoxia-Inducible Factor-1α Stabilization. Cell. Physiol. Biochem. 2004, 14, 351–360. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Zhong, J.; Chang, R.; Hu, H.; Pandey, A.; Semenza, G.L. Hsp70 and CHIP Selectively Mediate Ubiquitination and Degradation of Hypoxia-inducible Factor (HIF)-1α but Not HIF-2α. J. Biol. Chem. 2010, 285, 3651–3663. [Google Scholar] [CrossRef] [Green Version]
- Koh, M.Y.; Spivak-Kroizman, T.R.; Powis, G. HIF-1 regulation: Not so easy come, easy go. Trends Biochem. Sci. 2008, 33, 526–534. [Google Scholar] [CrossRef]
- Dioum, E.M.; Chen, R.; Alexander, M.S.; Zhang, Q.; Hogg, R.T.; Gerard, R.D.; Garcia, J.A. Regulation of Hypoxia-Inducible Factor 2α Signaling by the Stress-Responsive Deacetylase Sirtuin 1. Science 2009, 324, 1289–1293. [Google Scholar] [CrossRef]
- Cavadas, M.; Mesnieres, M.; Crifo, B.; Manresa, M.C.; Selfridge, A.C.; Scholz, C.C.; Cummins, E.; Cheong, A.; Taylor, C. REST mediates resolution of HIF-dependent gene expression in prolonged hypoxia. Sci. Rep. 2015, 5, 17851. [Google Scholar] [CrossRef] [Green Version]
- Cavadas, M.; Mesnieres, M.; Crifo, B.; Manresa, M.C.; Selfridge, A.C.; Keogh, C.E.; Fabian, Z.; Scholz, C.C.; Nolan, K.A.; Rocha, L.M.A.; et al. REST is a hypoxia-responsive transcriptional repressor. Sci. Rep. 2016, 6, 31355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, R.; SenBanerjee, S.; Lin, Z.; Mir, S.; Hamik, A.; Wang, P.; Mukherjee, P.; Mukhopadhyay, D.; Jain, M.K. Inhibition of Vascular Permeability Factor/Vascular Endothelial Growth Factor-mediated Angiogenesis by the Kruppel-like Factor KLF2. J. Biol. Chem. 2005, 280, 28848–28851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartoszewski, R.; Serocki, M.; Janaszak-Jasiecka, A.; Bartoszewska, S.; Kochan-Jamrozy, K.; Piotrowski, A.; Króliczewski, J.; Collawn, J.F. miR-200b downregulates Kruppel Like Factor 2 (KLF2) during acute hypoxia in human endothelial cells. Eur. J. Cell Biol. 2017, 96, 758–766. [Google Scholar] [CrossRef] [PubMed]
- Kawanami, D.; Mahabeleshwar, G.H.; Lin, Z.; Atkins, G.B.; Hamik, A.; Haldar, S.M.; Maemura, K.; LaManna, J.C.; Jain, M.K. Kruppel-like Factor 2 Inhibits Hypoxia-inducible Factor 1α Expression and Function in the Endothelium. J. Biol. Chem. 2009, 284, 20522–20530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartoszewska, S.; Rochan, K.; Piotrowski, A.; Kamysz, W.; Ochocka, R.J.; Collawn, J.F.; Bartoszewski, R. The hypoxia-inducible miR-429 regulates hypoxia-inducible factor-1α expression in human endothelial cells through a negative feedback loop. FASEB J. 2014, 29, 1467–1479. [Google Scholar] [CrossRef] [Green Version]
- Carroll, V.; Ashcroft, M. Regulation of Angiogenic Factors by HDM2 in Renal Cell Carcinoma. Cancer Res. 2008, 68, 545–552. [Google Scholar] [CrossRef] [Green Version]
- To, K.; Sedelnikova, O.; Samons, M.; Bonner, W.M.; Huang, L.E. The phosphorylation status of PAS-B distinguishes HIF-1α from HIF-2α in NBS1 repression. EMBO J. 2006, 25, 4784–4794. [Google Scholar] [CrossRef] [Green Version]
- Pangou, E.; Befani, C.; Mylonis, I.; Samiotaki, M.; Panayotou, G.; Simos, G.; Liakos, P. HIF-2α phosphorylation by CK1δ promotes erythropoietin secretion in liver cancer cells under hypoxia. J. Cell Sci. 2016, 129, 4213–4226. [Google Scholar] [CrossRef] [Green Version]
- Truffi, M.; Sorrentino, L.; Corsi, F. Fibroblasts in the Tumor Microenvironment. In Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2020; Volume 1234, pp. 15–29. ISBN 9783030371838. [Google Scholar]
- Frangioni, J.V. New Technologies for Human Cancer Imaging. J. Clin. Oncol. 2008, 26, 4012–4021. [Google Scholar] [CrossRef]
- Liu, D. Cancer biomarkers for targeted therapy. Biomark. Res. 2019, 7, 25–27. [Google Scholar] [CrossRef]
- Bader, S.B.; Dewhirst, M.W.; Hammond, E.M. Cyclic Hypoxia: An Update on Its Characteristics, Methods to Measure It and Biological Implications in Cancer. Cancers 2020, 13, 23. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-H.; Xu, L.-Y.; Wu, Y.-P.; Ke, Z.-B.; Huang, P.; Lin, F.; Li, X.-D.; Xue, X.-Y.; Wei, Y.; Zheng, Q.-S.; et al. Tumor volume: A new prognostic factor of oncological outcome of localized clear cell renal cell carcinoma. BMC Cancer 2021, 21, 79. [Google Scholar] [CrossRef] [PubMed]
- Grimes, D.R.; Kelly, C.; Bloch, K.; Partridge, M. A method for estimating the oxygen consumption rate in multicellular tumour spheroids. J. R. Soc. Interface 2014, 11, 20131124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimes, D.R.; Kannan, P.; Warren, D.R.; Markelc, B.; Bates, R.; Muschel, R.; Partridge, M. Correction to ‘Estimating oxygen distribution from vasculature in three-dimensional tumour tissue’. J. R. Soc. Interface 2016, 13, 20160362. [Google Scholar] [CrossRef] [PubMed]
- Al Tameemi, W.; Dale, T.P.; Al-Jumaily, R.M.K.; Forsyth, N.R. Hypoxia-Modified Cancer Cell Metabolism. Front. Cell Dev. Biol. 2019, 7, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michiels, C.; Tellier, C.; Feron, O. Cycling hypoxia: A key feature of the tumor microenvironment. Biochim. Biophys. Acta 2016, 1866, 76–86. [Google Scholar] [CrossRef]
- Bredholt, G.; Mannelqvist, M.; Stefansson, I.M.; Birkeland, E.; Bø, T.H.; Oyan, A.M.; Trovik, J.; Kalland, K.-H.; Jonassen, I.; Salvesen, H.B.; et al. Tumor necrosis is an important hallmark of aggressive endometrial cancer and associates with hypoxia, angiogenesis and inflammation responses. Oncotarget 2015, 6, 39676–39691. [Google Scholar] [CrossRef] [Green Version]
- Edwards, J.G.; Swinson, D.E.; Jones, J.L.; Muller, S.; Waller, D.A.; O’Byrne, K.J. Tumor Necrosis Correlates With Angiogenesis and Is a Predictor of Poor Prognosis in Malignant Mesothelioma. Chest 2003, 124, 1916–1923. [Google Scholar] [CrossRef] [Green Version]
- Lam, J.S.; Shvarts, O.; Said, J.W.; Pantuck, A.J.; Seligson, D.B.; Aldridge, M.E.; Bui, M.H.T.; Liu, X.; Horvath, S.; Figlin, R.A.; et al. Clinicopathologic and molecular correlations of necrosis in the primary tumor of patients with renal cell carcinoma. Cancer 2005, 103, 2517–2525. [Google Scholar] [CrossRef]
- Liu, Z.-G.; Jiao, D. Necroptosis, tumor necrosis and tumorigenesis. Cell Stress 2020, 4, 1–8. [Google Scholar] [CrossRef]
- Pressley, M.; Gallaher, J.A.; Brown, J.S.; Tomaszewski, M.R.; Borad, P.; Damaghi, M.; Gillies, R.J.; Whelan, C.J. Cycling hypoxia selects for constitutive HIF stabilization. Sci. Rep. 2021, 11, 5777. [Google Scholar] [CrossRef] [PubMed]
- Roig, E.M.; Yaromina, A.; Houben, R.; Groot, A.J.; Dubois, L.; Vooijs, M. Prognostic Role of Hypoxia-Inducible Factor-2α Tumor Cell Expression in Cancer Patients: A Meta-Analysis. Front. Oncol. 2018, 8, 224. [Google Scholar] [CrossRef] [PubMed]
- Gkagkalidis, K.; Kampantais, S.; Dimitriadis, G.; Gourvas, V.; Kapoukranidou, D.; Mironidou-Tzouveleki, M. Expression of HIF-2a in clear-cell renal cell carcinoma independently predicts overall survival. Med. Mol. Morphol. 2020, 53, 229–237. [Google Scholar] [CrossRef]
- Biswas, S.; Troy, H.; Leek, R.; Chung, Y.-L.; Li, J.-L.; Raval, R.R.; Turley, H.; Gatter, K.; Pezzella, F.; Griffiths, J.R.; et al. Effects of HIF-1 and HIF2 on Growth and Metabolism of Clear-Cell Renal Cell Carcinoma 786-0 Xenografts. J. Oncol. 2010, 2010, 757908. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Han, F.; Fan, Y.; Lian, B.; Xiao, J.; Sun, W.; Han, D.; Kou, H.; Li, C.; Wu, B. Notch3 is involved in the proliferation of renal cancer cells via regulation of cell cycle progression and HIF-2α. Oncol. Lett. 2020, 20, 379. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Yang, Q.; Wang, D.; Luo, F.; Liu, X.; Xue, J.; Yang, P.; Xu, H.; Lu, J.; Zhang, A.; et al. MicroRNA-191, regulated by HIF-2α, is involved in EMT and acquisition of a stem cell-like phenotype in arsenite-transformed human liver epithelial cells. Toxicol. Vitr. 2017, 48, 128–136. [Google Scholar] [CrossRef]
- Jarman, E.J.; Ward, C.; Turnbull, A.K.; Martinez-Perez, C.; Meehan, J.; Xintaropoulou, C.; Sims, A.H.; Langdon, S.P. HER2 regulates HIF-2α and drives an increased hypoxic response in breast cancer. Breast Cancer Res. 2019, 21, 10. [Google Scholar] [CrossRef]
- Yang, H.; Geng, Y.; Wang, P.; Zhou, Y.; Yang, H.; Huo, Y.; Zhang, H.; Li, Y.; He, H.; Tian, X.; et al. Extracellular ATP promotes breast cancer invasion and epithelial-mesenchymal transition via hypoxia-inducible factor 2α signaling. Cancer Sci. 2019, 110, 2456–2470. [Google Scholar] [CrossRef]
- Hao, T.; Li, C.X.; Ding, X.Y.; Xing, X.J. MicroRNA-363-3p/p21(Cip1/Waf1) axis is regulated by HIF-2α in mediating stemness of melanoma cells. Neoplasma 2019, 66, 427–436. [Google Scholar] [CrossRef]
- Renfrow, J.J.; Soike, M.H.; West, J.L.; Ramkissoon, S.H.; Metheny-Barlow, L.; Mott, R.T.; Kittel, C.A.; D’Agostino, R.B., Jr.; Tatter, S.B.; Laxton, A.W.; et al. Attenuating hypoxia driven malignant behavior in glioblastoma with a novel hypoxia-inducible factor 2 alpha inhibitor. Sci. Rep. 2020, 10, 15195. [Google Scholar] [CrossRef]
- Seidel, S.; Garvalov, B.K.; Wirta, V.; Von Stechow, L.; Schänzer, A.; Meletis, K.; Wolter, M.; Sommerlad, D.; Henze, A.-T.; Nistér, M.; et al. A hypoxic niche regulates glioblastoma stem cells through hypoxia inducible factor 2α. Brain 2010, 133, 983–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhen, Q.; Zhang, Y.; Gao, L.; Wang, R.; Chu, W.; Zhao, X.; Li, Z.; Li, H.; Zhang, B.; Lv, B.; et al. EPAS1 promotes peritoneal carcinomatosis of non-small-cell lung cancer by enhancing mesothelial–mesenchymal transition. Strahlenther. Onkol. 2020, 197, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Li, Y.; Ren, H.; Zhou, H.; Ning, Q.; Chen, X.; Hu, T.; Yang, L. PDK4 promotes tumorigenesis and cisplatin resistance in lung adenocarcinoma via transcriptional regulation of EPAS1. Cancer Chemother. Pharmacol. 2020, 87, 207–215. [Google Scholar] [CrossRef]
- Cao, M.; You, A.; Cui, W.; Zhang, S.; Guo, Z.; Chen, L.; Zhu, X.; Zhang, W.; Guo, H.; Deng, D.; et al. Cross talk between oxidative stress and hypoxia via thioredoxin and HIF-2α drives metastasis of hepatocellular carcinoma. FASEB J. 2020, 34, 5892–5905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saint-Martin, A.; Martínez-Ríos, J.; Castañeda-Patlán, M.C.; Sarabia-Sánchez, M.A.; Tejeda-Muñoz, N.; Chinney-Herrera, A.; Soldevila, G.; Benelli, R.; Santoyo-Ramos, P.; Poggi, A.; et al. Functional Interaction of Hypoxia-Inducible Factor 2-Alpha and Autophagy Mediates Drug Resistance in Colon Cancer Cells. Cancers 2019, 11, 755. [Google Scholar] [CrossRef] [Green Version]
- Das, B.; Pal, B.; Bhuyan, R.; Li, H.; Sarma, A.; Gayan, S.; Talukdar, J.; Sandhya, S.; Bhuyan, S.; Gogoi, G.; et al. MYC Regulates the HIF2α Stemness Pathway via Nanog and Sox2 to Maintain Self-Renewal in Cancer Stem Cells versus Non-Stem Cancer Cells. Cancer Res. 2019, 79, 4015–4025. [Google Scholar] [CrossRef]
- Peng, S.; Zhang, J.; Tan, X.; Huang, Y.; Xu, J.; Silk, N.; Zhang, D.; Liu, Q.; Jiang, J. The VHL/HIF Axis in the Development and Treatment of Pheochromocytoma/Paraganglioma. Front. Endocrinol. 2020, 11, 586857. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2017, 46, D1062–D1067. [Google Scholar] [CrossRef] [Green Version]
- Toledo, R.A. New HIF2α inhibitors: Potential implications as therapeutics for advanced pheochromocytomas and paragangliomas. Endocr.-Relat. Cancer 2017, 24, C9–C19. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Fan, D.; Adah, D.; Wu, Z.; Liu, R.; Yan, Q.; Zhang, Y.; Du, Z.; Wang, D.; Li, Y.; et al. CRISPR/Cas9-mediated hypoxia inducible factor-1α knockout enhances the antitumor effect of transarterial embolization in hepatocellular carcinoma. Oncol. Rep. 2018, 40, 2547–2557. [Google Scholar] [CrossRef]
- Lorenzo, F.R.; Yang, C.; Fui, M.N.T.; Vankayalapati, H.; Zhuang, Z.; Huynh, T.; Grossmann, M.; Pacak, K.; Prchal, J.T. A novel EPAS1/HIF2A germline mutation in a congenital polycythemia with paraganglioma. Klin. Wochenschr. 2012, 91, 507–512. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, Z.; Yang, C.; Lorenzo, F.; Merino, M.; Fojo, T.; Kebebew, E.; Popovic, V.; Stratakis, C.A.; Prchal, J.T.; Pacak, K. SomaticHIF2AGain-of-Function Mutations in Paraganglioma with Polycythemia. N. Engl. J. Med. 2012, 367, 922–930. [Google Scholar] [CrossRef] [Green Version]
- Percy, M.J.; Furlow, P.W.; Lucas, G.S.; Li, X.; Lappin, T.R.; Frances McMullin, M.; Lee, F.S. A Gain-of-Function Mutation in the HIF2A Gene in Familial Erythrocytosis. N. Engl. J. Med. 2008, 358, 162–168. [Google Scholar] [CrossRef] [Green Version]
- Percy, M.J.; Beer, P.A.; Campbell, G.; Dekker, A.W.; Green, A.R.; Oscier, D.; Rainey, M.G.; Van Wijk, R.; Wood, M.; Lappin, T.R.J.; et al. Novel exon 12 mutations in the HIF2A gene associated with erythrocytosis. Blood 2008, 111, 5400–5402. [Google Scholar] [CrossRef] [PubMed]
- Islam, F.; Gopalan, V.; Law, S.; Lam, A.K.; Pillai, S. Molecular Deregulation of EPAS1 in the Pathogenesis of Esophageal Squamous Cell Carcinoma. Front. Oncol. 2020, 10, 1534. [Google Scholar] [CrossRef] [PubMed]
- Putra, A.C.; Eguchi, H.; Lee, K.L.; Yamane, Y.; Gustine, E.; Isobe, T.; Nishiyama, M.; Hiyama, K.; Poellinger, L.; Tanimoto, K. The A Allele at rs13419896 of EPAS1 Is Associated with Enhanced Expression and Poor Prognosis for Non-Small Cell Lung Cancer. PLoS ONE 2015, 10, e0134496. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, O.; Mole, D.R. HIF Pathways in Clear Cell Renal Cancer. In Biomarkers and Bioanalysis Overview; Nunes, A.C.F., Ed.; IntechOpen: London, UK, 2021. [Google Scholar] [CrossRef]
- Biswas, S.; Roy, S.; Banerjee, J.; Hussain, S.-R.A.; Khanna, S.; Meenakshisundaram, G.; Kuppusamy, P.; Friedman, A.; Sen, C.K. Hypoxia inducible microRNA 210 attenuates keratinocyte proliferation and impairs closure in a murine model of ischemic wounds. Proc. Natl. Acad. Sci. USA 2010, 107, 6976–6981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoefflin, R.; Harlander, S.; Schäfer, S.; Metzger, P.; Kuo, F.; Schönenberger, D.; Adlesic, M.; Peighambari, A.; Seidel, P.; Chen, C.-Y.; et al. HIF-1α and HIF-2α differently regulate tumour development and inflammation of clear cell renal cell carcinoma in mice. Nat. Commun. 2020, 11, 4111. [Google Scholar] [CrossRef] [PubMed]
- Casazza, A.; Laoui, D.; Wenes, M.; Rizzolio, S.; Bassani, N.; Mambretti, M.; Deschoemaeker, S.; Van Ginderachter, J.A.; Tamagnone, L.; Mazzone, M. Impeding Macrophage Entry into Hypoxic Tumor Areas by Sema3A/Nrp1 Signaling Blockade Inhibits Angiogenesis and Restores Antitumor Immunity. Cancer Cell 2013, 24, 695–709. [Google Scholar] [CrossRef] [Green Version]
- Vickman, R.E.; Faget, D.V.; Beachy, P.; Beebe, D.; Bhowmick, N.A.; Cukierman, E.; Deng, W.-M.; Granneman, J.G.; Hildesheim, J.; Kalluri, R.; et al. Deconstructing tumor heterogeneity: The stromal perspective. Oncotarget 2020, 11, 3621–3632. [Google Scholar] [CrossRef]
- Labani-Motlagh, A.; Ashja-Mahdavi, M.; Loskog, A. The Tumor Microenvironment: A Milieu Hindering and Obstructing Antitumor Immune Responses. Front. Immunol. 2020, 11, 940. [Google Scholar] [CrossRef] [PubMed]
- Abdulla, O.A.; Nagarkatti, P.S.; Nagarkatti, M. Regulation of macrophages in tumor microenvironment by microRNA in T cell lymphoma-bearing mice. J. Immunol. 2020, 204, 164.18. [Google Scholar]
- Talks, K.L.; Turley, H.; Gatter, K.C.; Maxwell, P.; Pugh, C.; Ratcliffe, P.; Harris, A.L. The Expression and Distribution of the Hypoxia-Inducible Factors HIF-1α and HIF-2α in Normal Human Tissues, Cancers, and Tumor-Associated Macrophages. Am. J. Pathol. 2000, 157, 411–421. [Google Scholar] [CrossRef]
- Leek, R.D.; Talks, K.L.; Pezzella, F.; Turley, H.; Campo, L.; Brown, N.S.; Bicknell, R.; Taylor, M.; Gatter, K.C.; Harris, A.L. Relation of Hypoxia-inducible Factor-2α (HIF-2α) Expression in Tumor-infiltrative Macrophages to Tumor Angiogenesis and the Oxidative Thymidine Phosphorylase Pathway in Human Breast Cancer. Cancer Res. 2002, 62, 1326–1329. [Google Scholar] [PubMed]
- Dehn, S.; Deberge, M.; Yeap, X.-Y.; Yvan-Charvet, L.; Fang, D.; Eltzschig, H.K.; Miller, S.D.; Thorp, E.B. HIF-2α in Resting Macrophages Tempers Mitochondrial Reactive Oxygen Species to Selectively Repress MARCO-Dependent Phagocytosis. J. Immunol. 2016, 197, 3639–3649. [Google Scholar] [CrossRef] [Green Version]
- Skuli, N.; Liu, L.; Runge, A.; Wang, T.; Yuan, L.; Patel, S.; Iruela-Arispe, L.; Simon, M.C.; Keith, B. Endothelial deletion of hypoxia-inducible factor–2α (HIF-2α) alters vascular function and tumor angiogenesis. Blood 2009, 114, 469–477. [Google Scholar] [CrossRef] [Green Version]
- Mazzone, M.; Dettori, D.; de Oliveira, R.L.; Loges, S.; Schmidt, T.; Jonckx, B.; Tian, Y.-M.; Lanahan, A.A.; Pollard, P.; de Almodovar, C.R.; et al. Heterozygous Deficiency of PHD2 Restores Tumor Oxygenation and Inhibits Metastasis via Endothelial Normalization. Cell 2009, 136, 839–851. [Google Scholar] [CrossRef] [Green Version]
- Carlsson, L.M.; Jonsson, J.; Edlund, T.; Marklund, S.L. Mice lacking extracellular superoxide dismutase are more sensitive to hyperoxia. Proc. Natl. Acad. Sci. USA 1995, 92, 6264–6268. [Google Scholar] [CrossRef] [Green Version]
- O’Leary, B.R.; Fath, M.; Bellizzi, A.; Hrabe, J.; Button, A.M.; Allen, B.; Case, A.; Altekruse, S.; Wagner, B.A.; Buettner, G.; et al. Loss of SOD3 (EcSOD) Expression Promotes an Aggressive Phenotype in Human Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2015, 21, 1741–1751. [Google Scholar] [CrossRef] [Green Version]
- Teoh-Fitzgerald, M.L.; Fitzgerald, M.P.; Jensen, T.J.; Futscher, B.W.; Domann, F.E. Genetic and Epigenetic Inactivation of Extracellular Superoxide Dismutase Promotes an Invasive Phenotype in Human Lung Cancer by Disrupting ECM Homeostasis. Mol. Cancer Res. 2011, 10, 40–51. [Google Scholar] [CrossRef] [Green Version]
- Mira, E.; Carmona-Rodríguez, L.; Pérez-Villamil, B.; Casas, J.; Fernández-Aceñero, M.J.; Martinez-Rey, D.; Martín-González, P.; Heras-Murillo, I.; Cabezas, M.P.; Tardáguila, M.; et al. SOD3 improves the tumor response to chemotherapy by stabilizing endothelial HIF-2α. Nat. Commun. 2018, 9, 575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmona-Rodríguez, L.; Martínez-Rey, D.; Mira, E.; Mañes, S. SOD3 boosts T cell infiltration by normalizing the tumor endothelium and inducing laminin-α4. OncoImmunology 2020, 9, 1794163. [Google Scholar] [CrossRef] [PubMed]
- Pietrobon, V.; Marincola, F.M. Hypoxia and the phenomenon of immune exclusion. J. Transl. Med. 2021, 19, 9. [Google Scholar] [CrossRef] [PubMed]
- Hsu, T.-S.; Lin, Y.-L.; Wang, Y.-A.; Mo, S.-T.; Chi, P.-Y.; Lai, A.C.-Y.; Pan, H.-Y.; Chang, Y.-J.; Lai, M.-Z. HIF-2α is indispensable for regulatory T cell function. Nat. Commun. 2020, 11, 5005. [Google Scholar] [CrossRef] [PubMed]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal. Transduct. Target. Ther. 2020, 5, 28. [Google Scholar] [CrossRef]
- Paget, S. The distribution of secondary growths in cancer of the breast. Cancer Metastasis Rev. 1989, 2, 98–101. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Kang, Y. Hypoxia and Hypoxia-Inducible Factors: Master Regulators of Metastasis. Clin. Cancer Res. 2010, 16, 5928–5935. [Google Scholar] [CrossRef] [Green Version]
- Steunou, A.-L.; Ducoux-Petit, M.; Lazar, I.; Monsarrat, B.; Erard, M.; Muller, C.; Clottes, E.; Schiltz, O.; Nieto, L. Identification of the Hypoxia-inducible Factor 2α Nuclear Interactome in Melanoma Cells Reveals Master Proteins Involved in Melanoma Development. Mol. Cell. Proteom. 2013, 12, 736–748. [Google Scholar] [CrossRef] [Green Version]
- Saikolappan, S.; Kumar, B.; Shishodia, G.; Koul, S.; Koul, H.K. Reactive oxygen species and cancer: A complex interaction. Cancer Lett. 2019, 452, 132–143. [Google Scholar] [CrossRef]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.-F.; Yang, X.-L.; Zhao, Y.; Tian, Q.; Chen, M.-T.; Zhao, Y.-Y.; Jin, W. Loss of TMEM126A promotes extracellular matrix remodeling, epithelial-to-mesenchymal transition, and breast cancer metastasis by regulating mitochondrial retrograde signaling. Cancer Lett. 2018, 440–441, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Ju, M.K.; Jeon, H.M.; Lee, Y.J.; Kim, C.H.; Park, H.G.; Han, S.I.; Kang, H.S. Reactive oxygen species induce epithelial-mesenchymal transition, glycolytic switch, and mitochondrial repression through the Dlx-2/Snail signaling pathways in MCF-7 cells. Mol. Med. Rep. 2019, 20, 2339–2346. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Li, W.; Zhou, Y.; Zhang, Y.; Huang, S.; Xu, X.; Li, Z.; Guo, Q. The overexpression and nuclear translocation of Trx-1 during hypoxia confers on HepG2 cells resistance to DDP, and GL-V9 reverses the resistance by suppressing the Trx-1/Ref-1 axis. Free Radic. Biol. Med. 2015, 82, 29–41. [Google Scholar] [CrossRef]
- Branco-Price, C.; Zhang, N.; Schnelle, M.; Evans, C.; Katschinski, D.M.; Liao, D.; Ellies, L.; Johnson, R.S. Endothelial Cell HIF-1α and HIF-2α Differentially Regulate Metastatic Success. Cancer Cell 2012, 21, 52–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flier, J.S.; Underhill, L.H.; Dvorak, H.F. Tumors: Wounds That Do Not Heal. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [CrossRef]
- Dvorak, H.F. Tumors: Wounds That Do Not Heal—Redux. Cancer Immunol. Res. 2015, 3, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, T.; Ohneda, K.; Nagano, M.; Miyoshi, C.; Kaneko, N.; Miwa, Y.; Yamamoto, M.; Ohneda, O.; Fujii-Kuriyama, Y. Hypoxia-inducible Transcription Factor-2α in Endothelial Cells Regulates Tumor Neovascularization through Activation of Ephrin A1. J. Biol. Chem. 2008, 283, 18926–18936. [Google Scholar] [CrossRef] [Green Version]
- Coulthard, M.G.; Morgan, M.; Woodruff, T.M.; Arumugam, T.V.; Taylor, S.M.; Carpenter, T.C.; Lackmann, M.; Boyd, A.W. Eph/Ephrin Signaling in Injury and Inflammation. Am. J. Pathol. 2012, 181, 1493–1503. [Google Scholar] [CrossRef] [Green Version]
- Larson, J.; Schomberg, S.; Schroeder, W.; Carpenter, T.C. Endothelial EphA receptor stimulation increases lung vascular permeability. Am. J. Physiol. Cell. Mol. Physiol. 2008, 295, L431–L439. [Google Scholar] [CrossRef]
- Dixit, V.M.; Green, S.; Sarma, V.; Holzman, L.B.; Wolf, F.W.; O’Rourke, K.; Ward, P.; Prochownik, E.V.; Marks, R.M. Tumor necrosis factor-alpha induction of novel gene products in human endothelial cells including a macrophage-specific chemotaxin. J. Biol. Chem. 1990, 265, 2973–2978. [Google Scholar] [CrossRef]
- Ritsu, M.; Kanno, E.; Tanno, H.; Imai, Y.; Maruyama, R.; Tachi, M.; Kawakami, K.; Ishii, K. Critical role of tumor necrosis factor-α in the early process of wound healing in skin. J. Dermatol. Dermatol. Surg. 2017, 21, 14–19. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Zhao, X.-P.; Song, K.; Shang, Z.-J. Ephrin-A1 Is Upregulated by Hypoxia in Cancer Cells and Promotes Angiogenesis of HUVECs through a Coordinated Cross-Talk with eNOS. PLoS ONE 2013, 8, e74464. [Google Scholar] [CrossRef] [Green Version]
- Chan, B.; Sukhatme, V.P. Receptor tyrosine kinase EphA2 mediates thrombin-induced upregulation of ICAM-1 in endothelial cells in vitro. Thromb. Res. 2009, 123, 745–752. [Google Scholar] [CrossRef] [Green Version]
- Biao-Xue, R.; Xi-Guang, C.; Shuan-Ying, Y.; Wei, L.; Zong-Juan, M. EphA2-dependent molecular targeting therapy for malignant tumors. Curr. Cancer Drug Targets 2011, 11, 1082–1097. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Zhou, N.; Zhao, W.-D.; Liu, D.-X.; Liang, Y.; Fang, W.-G.; Li, B.; Chen, Y.-H. Inactivation of EphA2 promotes tight junction formation and impairs angiogenesis in brain endothelial cells. Microvasc. Res. 2011, 82, 113–121. [Google Scholar] [CrossRef]
- Cheng, N.; Brantley, D.M.; Liu, H.; Lin, Q.; Enriquez, M.; Gale, N.; Yancopoulos, G.; Cerretti, D.P.; Daniel, T.; Chen, J. Blockade of EphA receptor tyrosine kinase activation inhibits vascular endothelial cell growth factor-induced angiogenesis. Mol. Cancer Res. 2002, 1, 2–11. [Google Scholar]
- De Andrés, J.L.; Griñán-Lisón, C.; Jiménez, G.; Marchal, J.A. Cancer stem cell secretome in the tumor microenvironment: A key point for an effective personalized cancer treatment. J. Hematol. Oncol. 2020, 13, 136. [Google Scholar] [CrossRef]
- Aponte, P.M.; Caicedo, A. Stemness in Cancer: Stem Cells, Cancer Stem Cells, and Their Microenvironment. Stem Cells Int. 2017, 2017, 5619472. [Google Scholar] [CrossRef]
- Morizane, R.; Fujii, S.; Monkawa, T.; Hiratsuka, K.; Yamaguchi, S.; Homma, K.; Itoh, H. miR-363 induces transdifferentiation of human kidney tubular cells to mesenchymal phenotype. Clin. Exp. Nephrol. 2015, 20, 394–401. [Google Scholar] [CrossRef]
- Covello, K.L.; Kehler, J.; Yu, H.; Gordan, J.D.; Arsham, A.M.; Hu, C.-J.; Labosky, P.A.; Simon, M.C.; Keith, B. HIF-2α regulates Oct-4: Effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 2006, 20, 557–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, X.; Zhao, Y.; Li, X.; Tao, Z.; Hou, M.; Ma, H. Overexpression of HIF-2a-Dependent NEAT1 Promotes the Progression of Non-Small Cell Lung Cancer through miR-1013p/SOX9/Wnt/β-Catenin Signal Pathway. Cell. Physiol. Biochem. 2019, 52, 368–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, H.; Wang, S.; Wu, J.; Yang, S.; Gray, S.; Ng, C.S.; Du, J.; Underwood, M.J.; Li, M.-Y.; Chen, G.G. EGFR-AS1/HIF2A regulates the expression of FOXP3 to impact the cancer stemness of smoking-related non-small cell lung cancer. Ther. Adv. Med. Oncol. 2019, 11, 1758835919855228. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.-J.; Wang, Y.; Yuan, W.-D.; Yuan, J.-Q.; Yuan, K. HIF-2α not HIF-1α overexpression confers poor prognosis in non–small cell lung cancer. Tumor Biol. 2017, 39, 1010428317709637. [Google Scholar] [CrossRef] [Green Version]
- Koukourakis, M.I.; Giatromanolaki, A.; Skarlatos, J.; Corti, L.; Blandamura, S.; Piazza, M.; Gatter, K.C.; Harris, A.L. Hypoxia inducible factor (HIF-1a and HIF-2a) expression in early esophageal cancer and response to photodynamic therapy and radi-otherapy. Cancer Res. 2001, 61, 1830–1832. [Google Scholar] [PubMed]
- Sørensen, B.S.; Horsman, M.R. Tumor Hypoxia: Impact on Radiation Therapy and Molecular Pathways. Front. Oncol. 2020, 10, 562. [Google Scholar] [CrossRef] [PubMed]
- Grimes, D.R.; Partridge, M. A mechanistic investigation of the oxygen fixation hypothesis and oxygen enhancement ratio. Biomed. Phys. Eng. Express 2015, 1, 045209. [Google Scholar] [CrossRef]
- Carnero, A.; Lleonart, M. The hypoxic microenvironment: A determinant of cancer stem cell evolution. Insid. Cell 2015, 1, 96–105. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, L.; Li, X.-F. Targeting Hypoxia: Hypoxia-Activated Prodrugs in Cancer Therapy. Front. Oncol. 2021, 11, 700407. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, L.; Li, X.-F. The Hypoxia-Activated Prodrug TH-302: Exploiting Hypoxia in Cancer Therapy. Front. Pharmacol. 2021, 12, 524. [Google Scholar] [CrossRef]
- van der Wiel, A.M.; Jackson-Patel, V.; Niemans, R.; Yaromina, A.; Liu, E.; Marcus, D.; Mowday, A.M.; Lieuwes, N.G.; Biemans, R.; Lin, X.; et al. Selectively Targeting Tumor Hypoxia with the Hypoxia-Activated Prodrug CP-506. Mol. Cancer Ther. 2021, 20, 2372–2383. [Google Scholar] [CrossRef] [PubMed]
- McKeage, M.J.; Gu, Y.; Wilson, W.R.; Hill, A.; Amies, K.; Melink, T.J.; Jameson, M.B. A phase I trial of PR-104, a pre-prodrug of the bioreductive prodrug PR-104A, given weekly to solid tumour patients. BMC Cancer 2011, 11, 432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laubach, J.P.; Liu, C.-J.; Raje, N.S.; Yee, A.J.; Armand, P.; Schlossman, R.L.; Rosenblatt, J.; Hedlund, J.; Martin, M.; Reynolds, C.H.; et al. A Phase I/II Study of Evofosfamide, A Hypoxia-activated Prodrug with or without Bortezomib in Subjects with Relapsed/Refractory Multiple Myeloma. Clin. Cancer Res. 2018, 25, 478–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindblom, E.K.; Ureba, A.; Dasu, A.; Wersäll, P.; Even, A.J.G.; Van Elmpt, W.; Lambin, P.; Toma-Dasu, I. Impact of SBRT fractionation in hypoxia dose painting—Accounting for heterogeneous and dynamic tumor oxygenation. Med. Phys. 2019, 46, 2512–2521. [Google Scholar] [CrossRef]
- Zhang, R.; Feng, L.; Dong, Z.; Wang, L.; Liang, C.; Chen, J.; Ma, Q.; Chen, Q.; Wang, Y.; Liu, Z. Glucose & oxygen exhausting liposomes for combined cancer starvation and hypoxia-activated therapy. Biomaterials 2018, 162, 123–131. [Google Scholar] [CrossRef]
- Shirai, Y.; Chow, C.; Kambe, G.; Suwa, T.; Kobayashi, M.; Takahashi, I.; Harada, H.; Nam, J.-M. An Overview of the Recent Development of Anticancer Agents Targeting the HIF-1 Transcription Factor. Cancers 2021, 13, 2813. [Google Scholar] [CrossRef]
- Ma, Z.; Wang, L.Z.; Cheng, J.-T.; Lam, W.S.T.; Ma, X.; Xiang, X.; Wong, A.L.-A.; Goh, B.C.; Gong, Q.; Sethi, G.; et al. Targeting Hypoxia-Inducible Factor-1-Mediated Metastasis for Cancer Therapy. Antioxidants Redox Signal. 2021, 34, 1484–1497. [Google Scholar] [CrossRef]
- Ma, Z.; Xiang, X.; Li, S.; Xie, P.; Gong, Q.; Goh, B.-C.; Wang, L. Targeting hypoxia-inducible factor-1, for cancer treatment: Recent advances in developing small-molecule inhibitors from natural compounds. Semin. Cancer Biol. 2020, in press. [Google Scholar] [CrossRef]
- Kong, X.; Lin, Z.; Liang, D.; Fath, D.; Sang, N.; Caro, J. Histone Deacetylase Inhibitors Induce VHL and Ubiquitin-Independent Proteasomal Degradation of Hypoxia-Inducible Factor 1α. Mol. Cell. Biol. 2006, 26, 2019–2028. [Google Scholar] [CrossRef] [Green Version]
- Albadari, N.; Deng, S.; Li, W. The transcriptional factors HIF-1 and HIF-2 and their novel inhibitors in cancer therapy. Expert Opin. Drug Discov. 2019, 14, 667–682. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal. Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef] [Green Version]
- Leopoldo, M.; Nardulli, P.; Contino, M.; Leonetti, F.; Luurtsema, G.; Colabufo, N.A. An updated patent review on P-glycoprotein inhibitors (2011–2018). Expert Opin. Ther. Pat. 2019, 29, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Courtney, K.D.; Ma, Y.; de Leon, A.D.; Christie, A.; Xie, Z.; Woolford, L.; Singla, N.; Joyce, A.; Hill, H.; Madhuranthakam, A.J.; et al. HIF-2 Complex Dissociation, Target Inhibition, and Acquired Resistance with PT2385, a First-in-Class HIF-2 Inhibitor, in Patients with Clear Cell Renal Cell Carcinoma. Clin. Cancer Res. 2020, 26, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Hill, H.; Christie, A.; Kim, M.S.; Holloman, E.; Pavia-Jimenez, A.; Homayoun, F.; Ma, Y.; Patel, N.; Yell, P.; et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 2016, 539, 112–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, M.; Li, Y.; Lu, Y.; Wang, M.; Li, Y.; Wang, C.; Li, Q.; Zhao, H. The regulation of immune checkpoints by the hypoxic tumor microenvironment. PeerJ 2021, 9, e11306. [Google Scholar] [CrossRef]
- Lequeux, A.; Noman, M.Z.; Xiao, M.; Sauvage, D.; Van Moer, K.; Viry, E.; Bocci, I.; Hasmim, M.; Bosseler, M.; Berchem, G.; et al. Impact of hypoxic tumor microenvironment and tumor cell plasticity on the expression of immune checkpoints. Cancer Lett. 2019, 458, 13–20. [Google Scholar] [CrossRef]
- Samanta, D.; Park, Y.; Ni, X.; Li, H.; Zahnow, C.A.; Gabrielson, E.; Pan, F.; Semenza, G.L. Chemotherapy induces enrichment of CD47+/CD73+/PDL1+ immune evasive triple-negative breast cancer cells. Proc. Natl. Acad. Sci. USA 2018, 115, E1239–E1248. [Google Scholar] [CrossRef] [Green Version]
- Palazon, A.; Martínez-Forero, I.; Teijeira, A.; Morales-Kastresana, A.; Alfaro, C.; Sanmamed, M.F.; Perez-Gracia, J.L.; Penuelas, I.; Hervas-Stubbs, S.; Rouzaut, A.; et al. The HIF-1α Hypoxia Response in Tumor-Infiltrating T Lymphocytes Induces Functional CD137 (4-1BB) for Immunotherapy. Cancer Discov. 2012, 2, 608–623. [Google Scholar] [CrossRef] [Green Version]
- Zaini, M.N.; Patel, S.A.; Syafruddin, S.E.; Rodrigues, P.; Vanharanta, S. Endogenous HIF2A reporter systems for high-throughput functional screening. Sci. Rep. 2018, 8, 12063. [Google Scholar] [CrossRef]
- Nakazawa, M.S.; Eisinger-Mathason, T.S.K.; Sadri, N.; Ochocki, J.D.; Gade, T.P.F.; Amin, R.K.; Simon, M.C. Epigenetic re-expression of HIF-2α suppresses soft tissue sarcoma growth. Nat. Commun. 2016, 7, 10539. [Google Scholar] [CrossRef] [Green Version]
- Mazumdar, J.; Hickey, M.M.; Pant, D.K.; Durham, A.C.; Sweet-Cordero, A.; Vachani, A.; Jacks, T.; Chodosh, L.A.; Kissil, J.L.; Simon, M.C.; et al. HIF-2 deletion promotes Kras-driven lung tumor development. Proc. Natl. Acad. Sci. USA 2010, 107, 14182–14187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acker, T.; Diez-Juan, A.; Aragones, J.; Tjwa, M.; Brusselmans, K.; Moons, L.; Fukumura, D.; Moreno-Murciano, M.P.; Herbert, J.-M.; Burger, A.; et al. Genetic evidence for a tumor suppressor role of HIF-2α. Cancer Cell 2005, 8, 131–141. [Google Scholar] [CrossRef] [Green Version]
- Westerlund, I.; Shi, Y.; Toskas, K.; Fell, S.M.; Li, S.; Surova, O.; Södersten, E.; Kogner, P.; Nyman, U.; Schlisio, S.; et al. Combined epigenetic and differentiation-based treatment inhibits neuroblastoma tumor growth and links HIF2α to tumor suppression. Proc. Natl. Acad. Sci. USA 2017, 114, E6137–E6146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiavarina, B.; Whitaker-Menezes, D.; Migneco, G.; Martinez-Outschoorn, U.E.; Pavlides, S.; Howell, A.; Tanowitz, H.B.; Casimiro, M.C.; Wang, C.; Pestell, R.G.; et al. HIF1-alpha functions as a tumor promoter in cancer-associated fibroblasts, and as a tumor suppressor in breast cancer cells. Cell Cycle 2010, 9, 3534–3551. [Google Scholar] [CrossRef] [Green Version]
- Velasco-Hernandez, T.; Hyrenius-Wittsten, A.; Rehn, M.; Bryder, D.; Cammenga, J. HIF-1α can act as a tumor suppressor gene in murine acute myeloid leukemia. Blood 2014, 124, 3597–3607. [Google Scholar] [CrossRef] [Green Version]
- Maranchie, J.; Vasselli, J.R.; Riss, J.; Bonifacino, J.S.; Linehan, W.; Klausner, R.D. The contribution of VHL substrate binding and HIF1-α to the phenotype of VHL loss in renal cell carcinoma. Cancer Cell 2002, 1, 247–255. [Google Scholar] [CrossRef] [Green Version]
- Shen, C.; Beroukhim, R.; Schumacher, S.E.; Zhou, J.; Chang, M.; Signoretti, S.; Kaelin, W.G., Jr. Genetic and Functional Studies Implicate HIF1α as a 14q Kidney Cancer Suppressor Gene. Cancer Discov. 2011, 1, 222–235. [Google Scholar] [CrossRef] [Green Version]
- Imtiyaz, H.Z.; Williams, E.P.; Hickey, M.M.; Patel, S.A.; Durham, A.C.; Yuan, L.-J.; Hammond, R.; Gimotty, P.A.; Keith, B.; Simon, M.C. Hypoxia-inducible factor 2α regulates macrophage function in mouse models of acute and tumor inflammation. J. Clin. Investig. 2010, 120, 2699–2714. [Google Scholar] [CrossRef] [Green Version]
- He, D.; Wang, D.; Lu, P.; Yang, N.; Xue, Z.; Zhu, X.; Zhang, P.; Fan, G. Single-cell RNA sequencing reveals heterogeneous tumor and immune cell populations in early-stage lung adenocarcinomas harboring EGFR mutations. Oncogene 2020, 40, 355–368. [Google Scholar] [CrossRef]
- Susen, R.M.; Bauer, R.; Olesch, C.; Fuhrmann, D.; Fink, A.F.; Dehne, N.; Jain, A.; Ebersberger, I.; Schmid, T.; Brüne, B. Macrophage HIF-2α regulates tumor-suppressive Spint1 in the tumor microenvironment. Mol. Carcinog. 2019, 58, 2127–2138. [Google Scholar] [CrossRef]
- Dang, C.V. MYC on the Path to Cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Sun, X.-X.; Qian, D.Z.; Dai, M.-S. Molecular Crosstalk Between MYC and HIF in Cancer. Front. Cell Dev. Biol. 2020, 8, 590576. [Google Scholar] [CrossRef] [PubMed]
- Koshiji, M.; To, K.; Hammer, S.; Kumamoto, K.; Harris, A.; Modrich, P.; Huang, L.E. HIF-1α Induces Genetic Instability by Transcriptionally Downregulating MutSα Expression. Mol. Cell 2005, 17, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gao, P.; Fukuda, R.; Kumar, G.; Krishnamachary, B.; Zeller, K.I.; Dang, C.; Semenza, G.L. HIF-1 Inhibits Mitochondrial Biogenesis and Cellular Respiration in VHL-Deficient Renal Cell Carcinoma by Repression of C-MYC Activity. Cancer Cell 2007, 11, 407–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, W.J.; Qiu, B.; Nakazawa, M.S.; Qing, G.; Simon, M.C. MYC Degradation under Low O2Tension Promotes Survival by Evading Hypoxia-Induced Cell Death. Mol. Cell. Biol. 2013, 33, 3494–3504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordan, J.D.; Lal, P.; Dondeti, V.R.; Letrero, R.; Parekh, K.N.; Oquendo, C.E.; Greenberg, R.A.; Flaherty, K.T.; Rathmell, W.K.; Keith, B.; et al. HIF-α Effects on c-Myc Distinguish Two Subtypes of Sporadic VHL-Deficient Clear Cell Renal Carcinoma. Cancer Cell 2008, 14, 435–446. [Google Scholar] [CrossRef] [Green Version]
- Florczyk, U.; Czauderna, S.; Stachurska, A.; Tertil, M.; Nowak, W.; Kozakowska, M.; Poellinger, L.; Jozkowicz, A.; Loboda, A.; Dulak, J. Opposite effects of HIF-1α and HIF-2α on the regulation of IL-8 expression in endothelial cells. Free Radic. Biol. Med. 2011, 51, 1882–1892. [Google Scholar] [CrossRef] [Green Version]
- Qing, G.; Skuli, N.; Mayes, P.A.; Pawel, B.; Martinez, D.; Maris, J.M.; Simon, M.C. Combinatorial Regulation of Neuroblastoma Tumor Progression by N-Myc and Hypoxia Inducible Factor HIF-1α. Cancer Res. 2010, 70, 10351–10361. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-W.; Gao, P.; Liu, Y.-C.; Semenza, G.L.; Dang, C.V. Hypoxia-Inducible Factor 1 and Dysregulated c-Myc Cooperatively Induce Vascular Endothelial Growth Factor and Metabolic Switches Hexokinase 2 and Pyruvate Dehydrogenase Kinase 1. Mol. Cell. Biol. 2007, 27, 7381–7393. [Google Scholar] [CrossRef] [Green Version]
- Blagosklonny, M.; Gudkov, A.; Bharti, S.K.; Mironchik, Y.; Wildes, F.; Penet, M.-F.; Goggins, E.; Krishnamachary, B.; Bhujwalla, Z.M. Introducing, OncoTarget. Oncotarget 2010, 1, 2. [Google Scholar] [CrossRef]
- Shih, H.J.; Chang, H.F.; Chen, C.L.; Torng, P.-L. Differential expression of hypoxia-inducible factors related to the invasiveness of epithelial ovarian cancer. Sci. Rep. 2021, 11, 22925. [Google Scholar] [CrossRef] [PubMed]
- Bertout, J.A.; Majmundar, A.J.; Gordan, J.D.; Lam, J.C.; Ditsworth, D.; Keith, B.; Brown, E.J.; Nathanson, K.; Simon, M.C. HIF2 inhibition promotes p53 pathway activity, tumor cell death, and radiation responses. Proc. Natl. Acad. Sci. USA 2009, 106, 14391–14396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juillerat, A.; Marechal, A.; Filhol, J.M.; Valogne, Y.; Valton, J.; Duclert, A.; Duchateau, P.; Poirot, L. An oxygen sensitive self-decision making engineered CAR T-cell. Sci. Rep. 2017, 7, srep39833. [Google Scholar] [CrossRef] [PubMed]
- Kosti, P.; Opzoomer, J.W.; Larios-Martinez, K.I.; Henley-Smith, R.; Scudamore, C.L.; Okesola, M.; Taher, M.Y.; Davies, D.M.; Muliaditan, T.; Larcombe-Young, D.; et al. Hypoxia-sensing CAR T cells provide safety and efficacy in treating solid tumors. Cell Rep. Med. 2021, 2, 100227. [Google Scholar] [CrossRef]
- Liao, Q.; He, H.; Mao, Y.; Ding, X.; Zhang, X.; Xu, J. Engineering T cells with hypoxia-inducible chimeric antigen receptor (HiCAR) for selective tumor killing. Biomark. Res. 2020, 8, 56. [Google Scholar] [CrossRef]
- Li, X.; Wu, Y.; Zhang, R.; Bai, W.; Ye, T.; Wang, S. Oxygen-Based Nanocarriers to Modulate Tumor Hypoxia for Ameliorated Anti-Tumor Therapy: Fabrications, Properties, and Future Directions. Front. Mol. Biosci. 2021, 8, 683519. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Prognosis | Comparison Made to HIF-1α | Evidence of HIF-1α Involvement | Method(s) | Reference |
|---|---|---|---|---|---|
| Neuroblastoma | Advanced clinical stage | No | N/A | Western blot, RT-PCR | [80] |
| Angiogenesis, aggressive phenotype, growth | Yes | Transiently expressed | Western blot, qPCR | [105] | |
| Aggressive phenotype, metastasis | Yes | Transiently expressed | Western blot, RT-qPCR | [106] | |
| Aggressive phenotype | Yes | Transiently expressed | Western blot, qPCR | [109] | |
| Stemness | Yes | Transiently expressed | Western blot, RT-qPCR | [89] | |
| Clear cell renal carcinoma | Poor overall survival | Yes | Lower Fuhrman grade | Immunohistochemistry | [155] |
| Oxidative phenotype | Yes | Basal expression, decreased growth | Immunohistochemistry | [156] | |
| Cell cycle progression | No | N/A | Western blot | [157] | |
| Arsenite- transformed liver cancer | Epithelial-mesenchymal transition, stemness | No | N/A | Western blot | [158] |
| Breast cancer | Worse disease-specific survival (HER2+) | Yes | Independent normal expression | Western blot, RT-PCR, immunohistochemistry | [159] |
| Epithelial-mesenchymal transition, invasion | Yes | Independent normal expression | Western blot, qPCR | [160] | |
| Melanoma | Stemness | Yes | Independent overexpression | Western blot, siRNA, | [161] |
| Glioblastoma | Increasing grade, mortality | No | N/A | Immunohistochemistry | [162] |
| Stemness | Yes | Independent overexpression | Western blot, immunochemistry | [163] | |
| Non-small-cell lung cancer | Mesothelial- mesenchymal transition | No | N/A | Western blot, shRNA | [164] |
| Lung adenocarcinoma | Growth, resistance | No | N/A | qt-PCR, shRNA | [165] |
| Hepatocellular carcinoma | Metastasis | Yes | Transiently expressed | Western blot, shRNA | [166] |
| Colon cancer | Resistance | Yes | Co-expressed | Western blot, siRNA | [167] |
| Cancer Stem Cells | Stemness, self-renewal | No | N/A | qPCR, siRNA, ELISA | [168] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davis, L.; Recktenwald, M.; Hutt, E.; Fuller, S.; Briggs, M.; Goel, A.; Daringer, N. Targeting HIF-2α in the Tumor Microenvironment: Redefining the Role of HIF-2α for Solid Cancer Therapy. Cancers 2022, 14, 1259. https://doi.org/10.3390/cancers14051259
Davis L, Recktenwald M, Hutt E, Fuller S, Briggs M, Goel A, Daringer N. Targeting HIF-2α in the Tumor Microenvironment: Redefining the Role of HIF-2α for Solid Cancer Therapy. Cancers. 2022; 14(5):1259. https://doi.org/10.3390/cancers14051259
Chicago/Turabian StyleDavis, Leah, Matthias Recktenwald, Evan Hutt, Schuyler Fuller, Madison Briggs, Arnav Goel, and Nichole Daringer. 2022. "Targeting HIF-2α in the Tumor Microenvironment: Redefining the Role of HIF-2α for Solid Cancer Therapy" Cancers 14, no. 5: 1259. https://doi.org/10.3390/cancers14051259