
Targeting Protein Kinase C for Cancer Therapy


Abstract
:Simple Summary
Abstract
1. Introduction
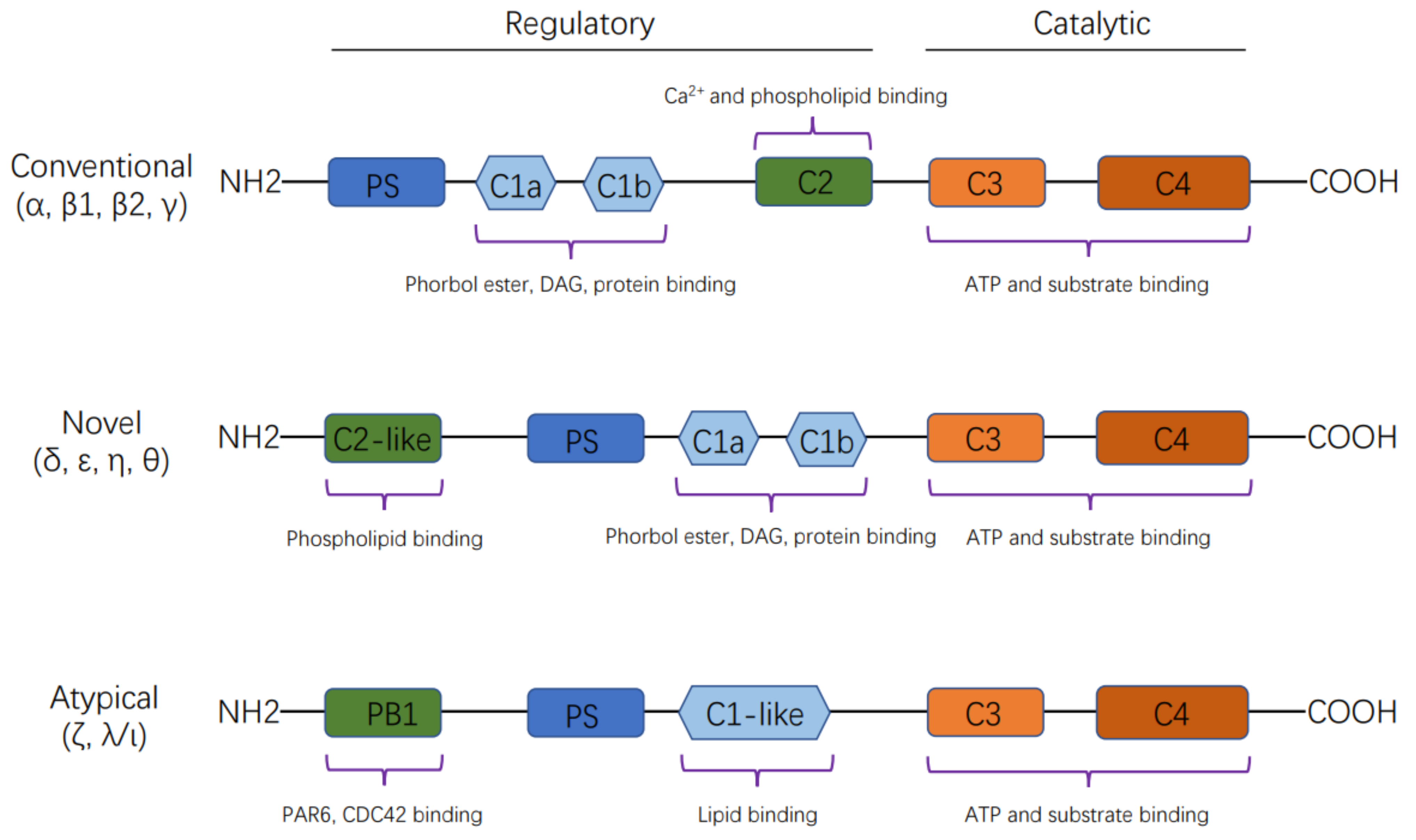
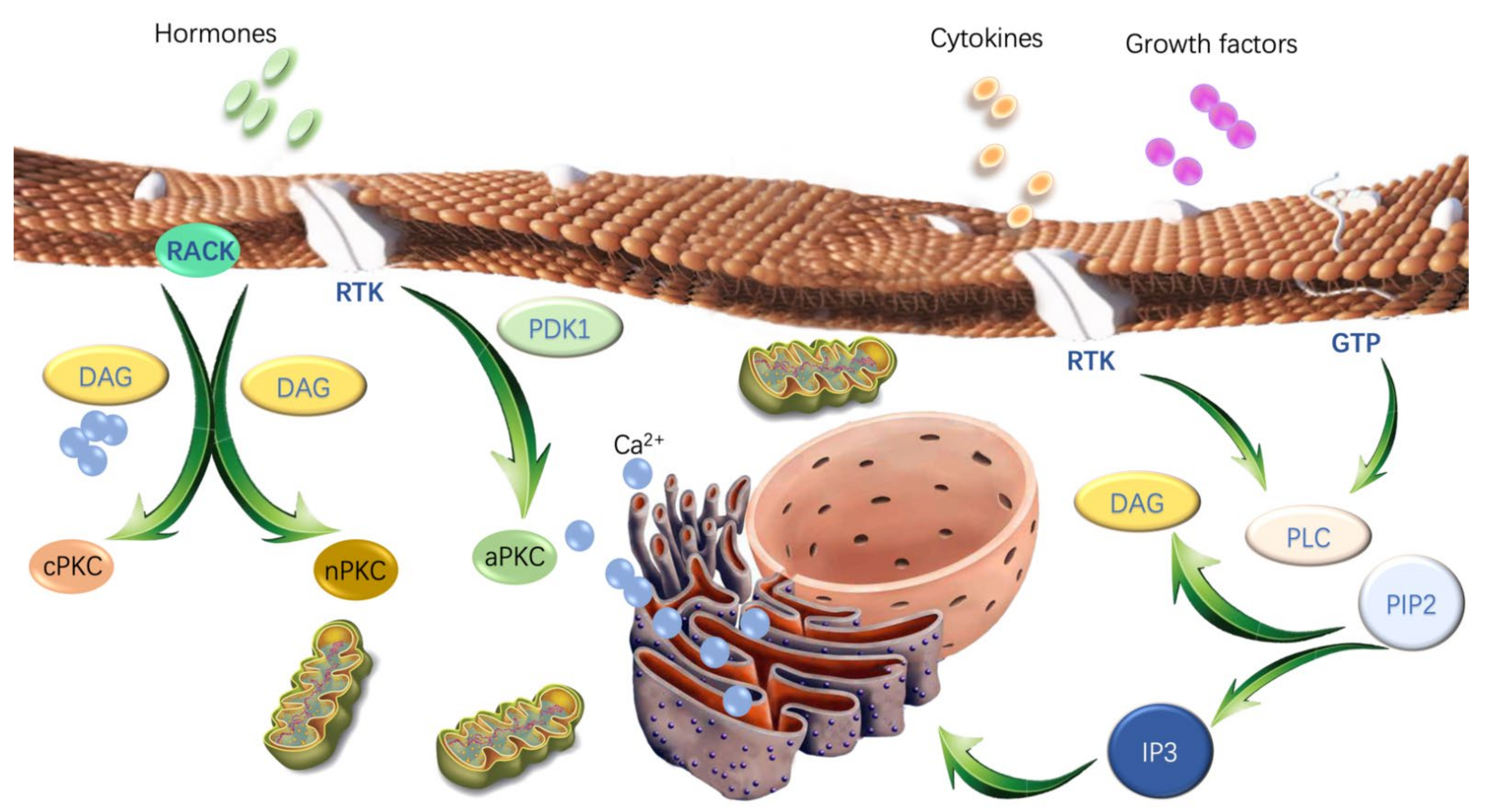
2. The Structural Features and Activation of PKC Isoforms
3. PKCs in Cancer
3.1. Differential Expression and Mutation of PKCs in Cancers
3.2. The Role of PKC in Anti-cancer Treatment-Induced Cell Death
3.2.1. PKCs and Apoptosis
3.2.2. PKCs and Autophagy
3.2.3. PKCs and Necrosis
3.2.4. PKCs and Other Cell Death Modalities
3.3. PKCs and Epithelial-Mesenchymal Transition (EMT)
3.4. PKCs and Tumor Microenvironment (TME)
3.5. PKCs and Tumor Metabolism
4. PKC Inhibitors for Cancer Therapy
4.1. ATP-Competitive Molecule Inhibitors
4.2. Phorbol Esters and Other Derivatives
4.3. Lipid Analogs
4.4. Others
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Prescott, S.L.; Irvine, J.; Dunstan, J.A.; Hii, C.; Ferrante, A. Protein kinase Czeta: A novel protective neonatal T-cell marker that can be upregulated by allergy prevention strategies. J. Allergy Clin. Immunol. 2007, 120, 200–206. [Google Scholar] [CrossRef]
- Anderson, K.; Fitzgerald, M.; Dupont, M.; Wang, T.; Paz, N.; Dorsch, M.; Healy, A.; Xu, Y.; Ocain, T.; Schopf, L.; et al. Mice deficient in PKC theta demonstrate impaired in vivo T cell activation and protection from T cell-mediated inflammatory diseases. Autoimmunity 2006, 39, 469–478. [Google Scholar] [CrossRef]
- Geraldes, P.; King, G.L. Activation of Protein Kinase C Isoforms and Its Impact on Diabetic Complications. Circ. Res. 2010, 106, 1319–1331. [Google Scholar] [CrossRef] [Green Version]
- Zanin-Zhorov, A.; Dustin, M.L.; Blazar, B.R. PKC-theta function at the immunological synapse: Prospects for therapeutic targeting. Trends Immunol. 2011, 32, 358–363. [Google Scholar] [CrossRef] [Green Version]
- Altman, A.; Kong, K.-F. Protein kinase C inhibitors for immune disorders. Drug Discov. Today 2014, 19, 1217–1221. [Google Scholar] [CrossRef] [Green Version]
- Isakov, N. Protein kinase C (PKC) isoforms in cancer, tumor promotion and tumor suppression. Semin. Cancer Biol. 2018, 48, 36–52. [Google Scholar] [CrossRef] [PubMed]
- Takai, Y.; Kishimoto, A.; Inoue, M.; Nishizuka, Y. Studies on a cyclic nucleotide-independent protein kinase and its proenzyme in mammalian tissues. I. Purification and characterization of an active enzyme from bovine cerebellum. J. Biol. Chem. 1977, 252, 7603–7609. [Google Scholar] [CrossRef]
- Newton, A.C. Protein Kinase C: Structure, Function, and Regulation. J. Biol. Chem. 1995, 270, 28495–28498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, R.; Benedetti, L.G.; Abera, M.B.; Wang, H.; Abba, M.; Kazanietz, M.G. Protein kinase C and cancer: What we know and what we do not. Oncogene 2014, 33, 5225–5237. [Google Scholar] [CrossRef] [Green Version]
- Newton, A.C. Regulation of the ABC kinases by phosphorylation: Protein kinase C as a paradigm. Biochem. J. 2003, 370, 361–371. [Google Scholar] [CrossRef]
- Newton, A.C. Protein Kinase C: Structural and Spatial Regulation by Phosphorylation, Cofactors, and Macromolecular Interactions. Chem. Rev. 2001, 101, 2353–2364. [Google Scholar] [CrossRef]
- Newton, A.C. Protein kinase C as a tumor suppressor. Semin. Cancer Biol. 2017, 48, 18–26. [Google Scholar] [CrossRef]
- Gould, C.M.; Newton, A.C. The life and death of protein kinase C. Curr. Drug Targets 2008, 9, 614–625. [Google Scholar] [CrossRef]
- Balendran, A.; Hare, G.R.; Kieloch, A.; Williams, M.R.; Alessi, D.R. Further evidence that 3-phosphoinositide-dependent protein kinase-1 (PDK1) is required for the stability and phosphorylation of protein kinase C (PKC) isoforms. FEBS Lett. 2000, 484, 217–223. [Google Scholar] [CrossRef]
- Parker, P.J. Protein Kinase C Phosphorylation: An Introduction. Methods Mol. Biol. 2003, 233, 159–162. [Google Scholar]
- Mochly-Rosen, D. Localization of protein kinases by anchoring proteins: A theme in signal transduction. Science 1995, 268, 247–251. [Google Scholar] [CrossRef]
- Merida, I.; Arranz-Nicolas, J.; Rodriguez-Rodriguez, C.; Avila-Flores, A. Diacylglycerol kinase control of protein kinase C. Biochem. J. 2019, 476, 1205–1219. [Google Scholar] [CrossRef]
- Lu, Z.; Liu, D.; Hornia, A.; Devonish, W.; Pagano, M.; Foster, D.A. Activation of protein kinase C triggers its ubiquitination and degradation. Mol Cell Biol 1998, 18, 839–845. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-W.; Smith, L.; Pettit, G.R.; Vinitsky, A.; Smith, J.B. Ubiquitination of Protein Kinase C-α and Degradation by the Proteasome. J. Biol. Chem. 1996, 271, 20973–20976. [Google Scholar] [CrossRef] [Green Version]
- Jiao, J.; Garg, V.; Yang, B.; Elton, T.S.; Hu, K. Protein kinase C-epsilon induces caveolin-dependent internalization of vascular adenosine 5’-triphosphate-sensitive K+ channels. Hypertension 2008, 52, 499–506. [Google Scholar] [CrossRef] [Green Version]
- Koren, R.; Langzam, L.; Paz, A.; Livne, P.M.; Gal, R.; Sampson, S.R. Protein kinase C (PKC) isoenzymes immunohistochemistry in lymph node revealing solution-fixed, paraffin-embedded bladder tumors. Appl. Immunohistochem. Mol. Morphol. 2000, 8, 166–171. [Google Scholar] [CrossRef]
- Tsai, J.H.; Hsieh, Y.S.; Kuo, S.J.; Chen, S.T.; Yu, S.Y.; Huang, C.Y.; Chang, A.C.; Wang, Y.W.; Tsai, M.T.; Liu, J.Y. Alteration in the expression of protein kinase C isoforms in human hepatocellular carcinoma. Cancer Lett. 2000, 161, 171–175. [Google Scholar] [CrossRef]
- Lin, Y.M.; Su, C.C.; Su, W.W.; Hwang, J.M.; Hsu, H.H.; Tsai, C.H.; Wang, Y.C.; Tsai, F.J.; Huang, C.Y.; Liu, J.Y.; et al. Expression of protein kinase C isoforms in cancerous breast tissue and adjacent normal breast tissue. Chin. J. Physiol. 2012, 55, 55–61. [Google Scholar]
- Pan, Q.; Bao, L.W.; Kleer, C.G.; Sabel, M.S.; Griffith, K.A.; Teknos, T.N.; Merajver, S.D. Protein kinase C epsilon is a predictive biomarker of aggressive breast cancer and a validated target for RNA interference anticancer therapy. Cancer Res 2005, 65, 8366–8371. [Google Scholar] [CrossRef] [Green Version]
- Koren, R.; Ben Meir, D.; Langzam, L.; Dekel, Y.; Konichezky, M.; Baniel, J.; Livne, P.M.; Gal, R.; Sampson, S.R. Expression of protein kinase C isoenzymes in benign hyperplasia and carcinoma of prostate. Oncol. Rep. 2004, 11, 321–326. [Google Scholar] [CrossRef]
- Antal, C.E.; Hudson, A.M.; Kang, E.; Zanca, C.; Wirth, C.; Stephenson, N.L.; Trotter, E.W.; Gallegos, L.L.; Miller, C.J.; Furnari, F.B.; et al. Cancer-associated protein kinase C mutations reveal kinase’s role as tumor suppressor. Cell 2015, 160, 489–502. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Prevostel, C.; Alvaro, V.; de Boisvilliers, F.; Martin, A.; Jaffiol, C.; Joubert, D. The natural protein kinase C alpha mutant is present in human thyroid neoplasms. Oncogene 1995, 11, 669–674. [Google Scholar]
- Vallentin, A.; Lo, T.C.; Joubert, D. A single point mutation in the V3 region affects protein kinase Calpha targeting and accumulation at cell-cell contacts. Mol. Cell. Biol. 2001, 21, 3351–3363. [Google Scholar] [CrossRef] [Green Version]
- Gould, C.M.; Kannan, N.; Taylor, S.S.; Newton, A.C. The chaperones Hsp90 and Cdc37 mediate the maturation and stabilization of protein kinase C through a conserved PXXP motif in the C-terminal tail. J. Biol. Chem. 2009, 284, 4921–4935. [Google Scholar] [CrossRef] [Green Version]
- Callender, J.A.; Yang, Y.; Lorden, G.; Stephenson, N.L.; Jones, A.C.; Brognard, J.; Newton, A.C. Protein kinase Calpha gain-of-function variant in Alzheimer’s disease displays enhanced catalysis by a mechanism that evades down-regulation. Proc. Natl. Acad. Sci. USA 2018, 115, E5497–E5505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridge, J.A.; Liu, X.Q.; Sumegi, J.; Nelson, M.; Reyes, C.; Bruch, L.A.; Rosenblum, M.; Puccioni, M.J.; Bowdino, B.S.; McComb, R.D. Identification of a novel, recurrent SLC44A1-PRKCA fusion in papillary glioneuronal tumor. Brain Pathol. 2013, 23, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Panagopoulos, I.; Gorunova, L.; Bjerkehagen, B.; Lobmaier, I.; Heim, S. LAMTOR1-PRKCD and NUMA1-SFMBT1 fusion genes identified by RNA sequencing in aneurysmal benign fibrous histiocytoma with t(3;11)(p21;q13). Cancer Genet. 2015, 208, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Haughian, J.M.; Jackson, T.A.; Koterwas, D.M.; Bradford, A.P. Endometrial cancer cell survival and apoptosis is regulated by protein kinase C alpha and delta. Endocr. Relat. Cancer 2006, 13, 1251–1267. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, T. Positive and negative regulation of radiation-induced apoptosis by protein kinase C. J. Radiat. Res. 2008, 49, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Shu, C.W.; Cheng, N.L.; Chang, W.M.; Tseng, T.L.; Lai, Y.K. Transactivation of hsp70-1/2 in geldanamycin-treated human non-small cell lung cancer H460 cells: Involvement of intracellular calcium and protein kinase C. J. Cell Biochem. 2005, 94, 1199–1209. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Q.; Hu, W.; Evers, B.M. Regulation of phorbol ester-mediated TRAF1 induction in human colon cancer cells through a PKC/RAF/ERK/NF-kappaB-dependent pathway. Oncogene 2004, 23, 1885–1895. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Dean, N.M.; Glazer, R.I. Induction of p53-dependent, insulin-like growth factor-binding protein-3-mediated apoptosis in glioblastoma multiforme cells by a protein kinase Calpha antisense oligonucleotide. Mol. Pharmacol. 1999, 55, 396–402. [Google Scholar] [CrossRef]
- Ding, L.; Wang, H.; Lang, W.; Xiao, L. Protein kinase C-epsilon promotes survival of lung cancer cells by suppressing apoptosis through dysregulation of the mitochondrial caspase pathway. J. Biol. Chem. 2002, 277, 35305–35313. [Google Scholar] [CrossRef] [Green Version]
- Martin, P.M.; Hussaini, I.M. PKC eta as a therapeutic target in glioblastoma multiforme. Expert Opin. Ther. Targets 2005, 9, 299–313. [Google Scholar] [CrossRef]
- Li, N.; Zhang, W. Protein kinase C beta inhibits autophagy and sensitizes cervical cancer Hela cells to cisplatin. Biosci. Rep. 2017, 37, BSR20160445. [Google Scholar] [CrossRef] [Green Version]
- Humphries, M.J.; Limesand, K.H.; Schneider, J.C.; Nakayama, K.I.; Anderson, S.M.; Reyland, M.E. Suppression of apoptosis in the protein kinase Cdelta null mouse in vivo. J. Biol. Chem. 2006, 281, 9728–9737. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, K.; Wang, H.G.; Miki, Y.; Kufe, D. Protein kinase Cdelta is responsible for constitutive and DNA damage-induced phosphorylation of Rad9. EMBO J. 2003, 22, 1431–1441. [Google Scholar] [CrossRef] [Green Version]
- Mazumder, S.; De, R.; Debsharma, S.; Bindu, S.; Maity, P.; Sarkar, S.; Saha, S.J.; Siddiqui, A.A.; Banerjee, C.; Nag, S.; et al. Indomethacin impairs mitochondrial dynamics by activating the PKCzeta-p38-DRP1 pathway and inducing apoptosis in gastric cancer and normal mucosal cells. J. Biol. Chem. 2019, 294, 8238–8258. [Google Scholar] [CrossRef]
- Datta, R.; Kojima, H.; Yoshida, K.; Kufe, D. Caspase-3-mediated cleavage of protein kinase C theta in induction of apoptosis. J. Biol. Chem. 1997, 272, 20317–20320. [Google Scholar] [CrossRef] [Green Version]
- Manicassamy, S.; Sun, Z. The critical role of protein kinase C-theta in Fas/Fas ligand-mediated apoptosis. J. Immunol. 2007, 178, 312–319. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.Y.; Kim, M.J.; Kang, C.M.; Bae, S.; Cho, C.K.; Soh, J.W.; Kim, J.H.; Kang, S.; Chung, H.Y.; Lee, Y.S.; et al. Activation of Bak and Bax through c-abl-protein kinase Cdelta-p38 MAPK signaling in response to ionizing radiation in human non-small cell lung cancer cells. J. Biol. Chem. 2006, 281, 7049–7059. [Google Scholar] [CrossRef] [Green Version]
- Zhan, B.; Kong, C.; Zhang, Z.; Dong, X.; Zhang, N. Inhibition of PKCalpha reduces the ability of migration of kidney cancer cells but has no impact on cell apoptosis. Exp. Ther. Med. 2017, 13, 2473–2479. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, T.; Yukawa, O.; Azuma, C.; Ohyama, H.; Wang, B.; Kojima, S.; Hayata, I.; Hama-Inaba, H. Involvement of protein kinase C-related anti-apoptosis signaling in radiation-induced apoptosis in murine thymic lymphoma(3SBH5) cells. Radiat. Res. 2004, 161, 528–534. [Google Scholar] [CrossRef]
- Umemori, Y.; Kuribayashi, K.; Nirasawa, S.; Kondoh, T.; Tanaka, M.; Kobayashi, D.; Watanabe, N. Protein kinase C zeta regulates survivin expression and inhibits apoptosis in colon cancer. Int. J. Oncol. 2014, 45, 1043–1050. [Google Scholar] [CrossRef]
- Byerly, J.H.; Port, E.R.; Irie, H.Y. PRKCQ inhibition enhances chemosensitivity of triple-negative breast cancer by regulating Bim. Breast Cancer Res. 2020, 22, 72. [Google Scholar] [CrossRef]
- Basu, A. Regulation of Autophagy by Protein Kinase C-epsilon in Breast Cancer Cells. Int. J. Mol. Sci. 2020, 21, 4247. [Google Scholar] [CrossRef]
- Zhou, N.; Wei, Z.; Qi, Z.; Chen, L. Abscisic Acid-Induced Autophagy Selectively via MAPK/JNK Signalling Pathway in Glioblastoma. Cell. Mol. Neurobiol. 2021, 41, 813–826. [Google Scholar] [CrossRef]
- Xu, Z.; Han, X.; Ou, D.; Liu, T.; Li, Z.; Jiang, G.; Liu, J.; Zhang, J. Targeting PI3K/AKT/mTOR-mediated autophagy for tumor therapy. Appl. Microbiol. Biotechnol. 2020, 104, 575–587. [Google Scholar] [CrossRef]
- Kudo, Y.; Sugimoto, M.; Arias, E.; Kasashima, H.; Cordes, T.; Linares, J.F.; Duran, A.; Nakanishi, Y.; Nakanishi, N.; L’Hermitte, A.; et al. PKClambda/iota Loss Induces Autophagy, Oxidative Phosphorylation, and NRF2 to Promote Liver Cancer Progression. Cancer Cell 2020, 38, 247–262.e11. [Google Scholar] [CrossRef]
- Tan, S.H.; Shui, G.; Zhou, J.; Li, J.J.; Bay, B.H.; Wenk, M.R.; Shen, H.M. Induction of autophagy by palmitic acid via protein kinase C-mediated signaling pathway independent of mTOR (mammalian target of rapamycin). J. Biol. Chem. 2012, 287, 14364–14376. [Google Scholar] [CrossRef] [Green Version]
- Deng, Q.; Jiang, L.; Mao, L.; Song, X.H.; He, C.Q.; Li, X.L.; Zhang, Z.H.; Zeng, H.C.; Chen, J.X.; Long, D.X. The role of protein kinase C alpha in tri-ortho-cresyl phosphate-induced autophagy in human neuroblastoma SK-N-SH cells. J. Appl. Toxicol. 2020, 40, 1480–1490. [Google Scholar] [CrossRef]
- Wang, F.; Xu, C.; Reece, E.A.; Li, X.; Wu, Y.; Harman, C.; Yu, J.; Dong, D.; Wang, C.; Yang, P.; et al. Protein kinase C-alpha suppresses autophagy and induces neural tube defects via miR-129-2 in diabetic pregnancy. Nat. Commun. 2017, 8, 15182. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Liu, C.; Jia, L. The roles of PKCs in regulating autophagy. J. Cancer Res. Clin. Oncol. 2018, 144, 2303–2311. [Google Scholar] [CrossRef]
- Kumar, D.; Shankar, S.; Srivastava, R.K. Rottlerin-induced autophagy leads to the apoptosis in breast cancer stem cells: Molecular mechanisms. Mol. Cancer 2013, 12, 171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saberi, B.; Shinohara, M.; Ybanez, M.D.; Hanawa, N.; Gaarde, W.A.; Kaplowitz, N.; Han, D. Regulation of H2O2-induced necrosis by PKC and AMP-activated kinase signaling in primary cultured hepatocytes. Am. J. Physiol. Cell Physiol. 2008, 295, C50–C63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baek, M.W.; Cho, H.S.; Kim, S.H.; Kim, W.J.; Jung, J.Y. Ascorbic Acid Induces Necrosis in Human Laryngeal Squamous Cell Carcinoma via ROS, PKC, and Calcium Signaling. J. Cell. Physiol. 2017, 232, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Han, S.I.; Lee, S.Y.; Youk, H.S.; Moon, J.Y.; Duong, H.Q.; Park, M.J.; Joo, Y.M.; Park, H.G.; Kim, Y.J.; et al. Protein kinase C-ERK1/2 signal pathway switches glucose depletion-induced necrosis to apoptosis by regulating superoxide dismutases and suppressing reactive oxygen species production in A549 lung cancer cells. J. Cell. Physiol. 2007, 211, 371–385. [Google Scholar] [CrossRef]
- Calcinotto, A.; Kohli, J.; Zagato, E.; Pellegrini, L.; Demaria, M.; Alimonti, A. Cellular Senescence: Aging, Cancer, and Injury. Physiol. Rev. 2019, 99, 1047–1078. [Google Scholar] [CrossRef]
- Park, J.W.; Lee, Y.H.; Bae, Y.S. Protein kinase C downregulation induces senescence via FoxO3a inhibition in HCT116 and HEK293 cells. Biochem. Biophys. Res. Commun. 2017, 493, 1548–1554. [Google Scholar] [CrossRef]
- Paget, J.A.; Restall, I.J.; Daneshmand, M.; Mersereau, J.A.; Simard, M.A.; Parolin, D.A.; Lavictoire, S.J.; Amin, M.S.; Islam, S.; Lorimer, I.A. Repression of cancer cell senescence by PKCiota. Oncogene 2012, 31, 3584–3596. [Google Scholar] [CrossRef] [Green Version]
- Zurgil, U.; Ben-Ari, A.; Atias, K.; Isakov, N.; Apte, R.; Livneh, E. PKCeta promotes senescence induced by oxidative stress and chemotherapy. Cell Death Dis. 2014, 5, e1531. [Google Scholar] [CrossRef] [Green Version]
- Basu, A.; Pal, D.; Blaydes, R. Differential effects of protein kinase C-eta on apoptosis versus senescence. Cell. Signal. 2019, 55, 1–7. [Google Scholar] [CrossRef]
- Dachert, J.; Ehrenfeld, V.; Habermann, K.; Dolgikh, N.; Fulda, S. Targeting ferroptosis in rhabdomyosarcoma cells. Int. J. Cancer 2020, 146, 510–520. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Savagner, P. The epithelial-mesenchymal transition (EMT) phenomenon. Ann. Oncol. 2010, 21, vii89–vii92. [Google Scholar] [CrossRef]
- Singh, R.; Lei, P.; Andreadis, S.T. PKC-delta binds to E-cadherin and mediates EGF-induced cell scattering. Exp. Cell Res. 2009, 315, 2899–2913. [Google Scholar] [CrossRef]
- Wang, J.; Wu, J.; Hong, J.; Chen, R.; Xu, K.; Niu, W.; Peng, C.; Liu, E.; Wang, J.; Liu, S.; et al. PKC promotes the migration of colon cancer cells by regulating the internalization and recycling of integrin alphavbeta6. Cancer Lett. 2011, 311, 38–47. [Google Scholar] [CrossRef]
- Kim, S.; Chun, S.Y.; Kwon, Y.S.; Nam, K.S. Crosstalk between Wnt signaling and Phorbol ester-mediated PKC signaling in MCF-7 human breast cancer cells. Biomed. Pharmacother. 2016, 77, 114–119. [Google Scholar] [CrossRef]
- Lin, C.W.; Shen, S.C.; Chien, C.C.; Yang, L.Y.; Shia, L.T.; Chen, Y.C. 12-O-tetradecanoylphorbol-13-acetate-induced invasion/migration of glioblastoma cells through activating PKCalpha/ERK/NF-kappaB-dependent MMP-9 expression. J. Cell Physiol. 2010, 225, 472–481. [Google Scholar] [CrossRef]
- Gunaratne, A.; Thai, B.L.; di Guglielmo, G.M. Atypical protein kinase C phosphorylates Par6 and facilitates transforming growth factor beta-induced epithelial-to-mesenchymal transition. Mol. Cell Biol. 2013, 33, 874–886. [Google Scholar] [CrossRef] [Green Version]
- Jain, K.; Basu, A. Protein Kinase C-epsilon Promotes EMT in Breast Cancer. Breast Cancer 2014, 8, 61–67. [Google Scholar]
- Qi, H.; Sun, B.; Zhao, X.; Du, J.; Gu, Q.; Liu, Y.; Cheng, R.; Dong, X. Wnt5a promotes vasculogenic mimicry and epithelial-mesenchymal transition via protein kinase Calpha in epithelial ovarian cancer. Oncol. Rep. 2014, 32, 771–779. [Google Scholar] [CrossRef] [Green Version]
- Qian, Y.; Yao, W.; Yang, T.; Yang, Y.; Liu, Y.; Shen, Q.; Zhang, J.; Qi, W.; Wang, J. aPKC-iota/P-Sp1/Snail signaling induces epithelial-mesenchymal transition and immunosuppression in cholangiocarcinoma. Hepatology 2017, 66, 1165–1182. [Google Scholar] [CrossRef] [Green Version]
- Tian, L.; Deng, Z.; Xu, L.; Yang, T.; Yao, W.; Ji, L.; Lu, Y.; Zhang, J.; Liu, Y.; Wang, J. Downregulation of ASPP2 promotes gallbladder cancer metastasis and macrophage recruitment via aPKC-iota/GLI1 pathway. Cell Death Dis. 2018, 9, 1115. [Google Scholar] [CrossRef] [PubMed]
- Pfeifhofer, C.; Gruber, T.; Letschka, T.; Thuille, N.; Lutz-Nicoladoni, C.; Hermann-Kleiter, N.; Braun, U.; Leitges, M.; Baier, G. Defective IgG2a/2b class switching in PKC alpha-/- mice. J. Immunol. 2006, 176, 6004–6011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thuille, N.; Gruber, T.; Bock, G.; Leitges, M.; Baier, G. Protein kinase C beta is dispensable for TCR-signaling. Mol. Immunol. 2004, 41, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.A.; Pitarresi, J.R.; Sharma, N.; Palettas, M.; Cuitino, M.C.; Sizemore, S.T.; Yu, L.; Sanderlin, A.; Rosol, T.J.; Mehta, K.D.; et al. Protein kinase C Beta in the tumor microenvironment promotes mammary tumorigenesis. Front. Oncol. 2014, 4, 87. [Google Scholar] [CrossRef] [Green Version]
- Martini, S.; Pozzi, G.; Carubbi, C.; Masselli, E.; Galli, D.; Di Nuzzo, S.; Banchini, A.; Gobbi, G.; Vitale, M.; Mirandola, P. PKCepsilon promotes human Th17 differentiation: Implications in the pathophysiology of psoriasis. Eur. J. Immunol. 2018, 48, 644–654. [Google Scholar] [CrossRef]
- Brezar, V.; Tu, W.J.; Seddiki, N. PKC-Theta in Regulatory and Effector T-cell Functions. Front. Immunol. 2015, 6, 530. [Google Scholar] [CrossRef] [Green Version]
- Kong, K.F.; Fu, G.; Zhang, Y.; Yokosuka, T.; Casas, J.; Canonigo-Balancio, A.J.; Becart, S.; Kim, G.; Yates, J.R., 3rd; Kronenberg, M.; et al. Protein kinase C-eta controls CTLA-4-mediated regulatory T cell function. Nat. Immunol. 2014, 15, 465–472. [Google Scholar] [CrossRef] [Green Version]
- Martin, J.D.; Fukumura, D.; Duda, D.G.; Boucher, Y.; Jain, R.K. Reengineering the Tumor Microenvironment to Alleviate Hypoxia and Overcome Cancer Heterogeneity. Cold Spring Harb. Perspect Med. 2016, 6, a027094. [Google Scholar] [CrossRef] [Green Version]
- Sledge, G.W., Jr.; Gokmen-Polar, Y. Protein kinase C-beta as a therapeutic target in breast cancer. Semin. Oncol. 2006, 33, S15–S18. [Google Scholar] [CrossRef]
- Kim, J.; Choi, Y.L.; Vallentin, A.; Hunrichs, B.S.; Hellerstein, M.K.; Peehl, D.M.; Mochly-Rosen, D. Centrosomal PKCbetaII and pericentrin are critical for human prostate cancer growth and angiogenesis. Cancer Res. 2008, 68, 6831–6839. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; He, S.; Wang, M.; Zhou, L.; Zhang, Z.; Feng, X.; Yu, Y.; Ma, J.; Dai, C.; Zhang, S.; et al. The Caspase-3/PKCdelta/Akt/VEGF-A Signaling Pathway Mediates Tumor Repopulation during Radiotherapy. Clin. Cancer Res. 2019, 25, 3732–3743. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Cheng, T. Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010, 29, 313–324. [Google Scholar] [CrossRef] [Green Version]
- Tandon, M.; Othman, A.H.; Winogradzki, M.; Pratap, J. Bone metastatic breast cancer cells display downregulation of PKC-zeta with enhanced glutamine metabolism. Gene 2021, 775, 145419. [Google Scholar] [CrossRef]
- Ma, L.; Tao, Y.; Duran, A.; Llado, V.; Galvez, A.; Barger, J.F.; Castilla, E.A.; Chen, J.; Yajima, T.; Porollo, A.; et al. Control of nutrient stress-induced metabolic reprogramming by PKCzeta in tumorigenesis. Cell 2013, 152, 599–611. [Google Scholar] [CrossRef] [Green Version]
- Ma, D.; Wang, P.; Fang, Q.; Yu, Z.; Zhou, Z.; He, Z.; Wei, D.; Yu, K.; Lu, T.; Zhang, Y.; et al. Low-dose staurosporine selectively reverses BCR-ABL-independent IM resistance through PKC-alpha-mediated G2/M phase arrest in chronic myeloid leukaemia. Artif. Cells Nanomed. Biotechnol. 2018, 46, S208–S216. [Google Scholar] [CrossRef]
- Ge, X.; Chen, J.; Li, L.; Ding, P.; Wang, Q.; Zhang, W.; Li, L.; Lv, X.; Zhou, D.; Jiang, Z.; et al. Midostaurin potentiates rituximab antitumor activity in Burkitt’s lymphoma by inducing apoptosis. Cell Death Dis. 2018, 10, 8. [Google Scholar] [CrossRef]
- Koh, J.; Kubota, T.; Migita, T.; Abe, S.; Hashimoto, M.; Hosoda, Y.; Kitajima, M. UCN-01 (7-hydroxystaurosporine) inhibits the growth of human breast cancer xenografts through disruption of signal transduction. Breast Cancer 2002, 9, 50–54. [Google Scholar] [CrossRef]
- Edelman, M.J.; Bauer, K.S., Jr.; Wu, S.; Smith, R.; Bisacia, S.; Dancey, J. Phase I and pharmacokinetic study of 7-hydroxystaurosporine and carboplatin in advanced solid tumors. Clin. Cancer Res. 2007, 13, 2667–2674. [Google Scholar] [CrossRef] [Green Version]
- Kortmansky, J.; Shah, M.A.; Kaubisch, A.; Weyerbacher, A.; Yi, S.; Tong, W.; Sowers, R.; Gonen, M.; O’Reilly, E.; Kemeny, N.; et al. Phase I trial of the cyclin-dependent kinase inhibitor and protein kinase C inhibitor 7-hydroxystaurosporine in combination with Fluorouracil in patients with advanced solid tumors. J. Clin. Oncol. 2005, 23, 1875–1884. [Google Scholar] [CrossRef]
- Wu, X.; Li, J.; Zhu, M.; Fletcher, J.A.; Hodi, F.S. Protein kinase C inhibitor AEB071 targets ocular melanoma harboring GNAQ mutations via effects on the PKC/Erk1/2 and PKC/NF-kappaB pathways. Mol. Cancer Ther. 2012, 11, 1905–1914. [Google Scholar] [CrossRef] [Green Version]
- Giovannetti, E.; Honeywell, R.; Hanauske, A.R.; Tekle, C.; Kuenen, B.; Sigmond, J.; Giaccone, G.; Peters, G.J. Pharmacological aspects of the enzastaurin-pemetrexed combination in non-small cell lung cancer (NSCLC). Curr. Drug Targets 2010, 11, 12–28. [Google Scholar] [CrossRef]
- Griner, E.M.; Kazanietz, M.G. Protein kinase C and other diacylglycerol effectors in cancer. Nat. Rev. Cancer 2007, 7, 281–294. [Google Scholar] [CrossRef]
- Oskoueian, E.; Abdullah, N.; Ahmad, S. Phorbol esters from Jatropha meal triggered apoptosis, activated PKC-delta, caspase-3 proteins and down-regulated the proto-oncogenes in MCF-7 and HeLa cancer cell lines. Molecules 2012, 17, 10816–10830. [Google Scholar] [CrossRef] [Green Version]
- Newton, A.C. Protein kinase C: Perfectly balanced. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 208–230. [Google Scholar] [CrossRef]
- Marshall, J.L.; Bangalore, N.; El-Ashry, D.; Fuxman, Y.; Johnson, M.; Norris, B.; Oberst, M.; Ness, E.; Wojtowicz-Praga, S.; Bhargava, P.; et al. Phase I study of prolonged infusion Bryostatin-1 in patients with advanced malignancies. Cancer Biol. Ther. 2002, 1, 409–416. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Bermejo, M.L.; Leskow, F.C.; Fujii, T.; Wang, Q.; Blumberg, P.M.; Ohba, M.; Kuroki, T.; Han, K.C.; Lee, J.; Marquez, V.E.; et al. Diacylglycerol (DAG)-lactones, a new class of protein kinase C (PKC) agonists, induce apoptosis in LNCaP prostate cancer cells by selective activation of PKCalpha. J. Biol. Chem. 2002, 277, 645–655. [Google Scholar] [CrossRef] [Green Version]
- Hamada, M.; Sumi, T.; Iwai, S.; Nakazawa, M.; Yura, Y. Induction of endonuclease G-mediated apopotosis in human oral squamous cell carcinoma cells by protein kinase C inhibitor safingol. Apoptosis 2006, 11, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Dickson, M.A.; Carvajal, R.D.; Merrill, A.H., Jr.; Gonen, M.; Cane, L.M.; Schwartz, G.K. A phase I clinical trial of safingol in combination with cisplatin in advanced solid tumors. Clin. Cancer Res. 2011, 17, 2484–2492. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, J.; O’Connor, P.M.; Jackman, J.; Schubert, C.; Ueberall, F.; Kohn, K.W.; Grunicke, H. The protein kinase C inhibitor ilmofosine (BM 41 440) arrests cells in G2 phase and suppresses CDC2 kinase activation through a mechanism different from that of DNA damaging agents. Biochem. Biophys. Res. Commun. 1994, 199, 937–943. [Google Scholar] [CrossRef]
- Hanauske, A.R.; Degen, D.; Marshall, M.H.; Hilsenbeck, S.G.; McPhillips, J.J.; Von Hoff, D.D. Preclinical activity of ilmofosine against human tumor colony forming units in vitro. Anticancer Drugs 1992, 3, 43–46. [Google Scholar] [CrossRef]
- Giantonio, B.J.; Derry, C.; McAleer, C.; McPhillips, J.J.; O’Dwyer, P.J. Phase I and pharmacokinetic study of the cytotoxic ether lipid ilmofosine administered by weekly two-hour infusion in patients with advanced solid tumors. Clin. Cancer Res. 2004, 10, 1282–1288. [Google Scholar] [CrossRef] [Green Version]
- Woolley, P.V.; Schultz, C.J.; Rodriguez, G.I.; Gams, R.A.; Rowe, K.W., Jr.; Dadey, M.L.; Von Hoff, D.D.; McPhillips, J.J. A phase II trial of ilmofosine in non-small cell bronchogenic carcinoma. Investig. New Drugs 1996, 14, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Leighl, N.B.; Dent, S.; Clemons, M.; Vandenberg, T.A.; Tozer, R.; Warr, D.G.; Crump, R.M.; Hedley, D.; Pond, G.R.; Dancey, J.E.; et al. A Phase 2 study of perifosine in advanced or metastatic breast cancer. Breast Cancer Res. Treat. 2008, 108, 87–92. [Google Scholar] [CrossRef]
- Karagul, M.I.; Aktas, S.; Yilmaz, S.N.; Yetkin, D.; Celikcan, H.D.; Cevik, O.S. Perifosine and vitamin D combination induces apoptotic and non-apoptotic cell death in endometrial cancer cells. EXCLI J. 2020, 19, 532–546. [Google Scholar] [PubMed]
- Knowling, M.; Blackstein, M.; Tozer, R.; Bramwell, V.; Dancey, J.; Dore, N.; Matthews, S.; Eisenhauer, E. A phase II study of perifosine (D-21226) in patients with previously untreated metastatic or locally advanced soft tissue sarcoma: A National Cancer Institute of Canada Clinical Trials Group trial. Investig. New Drugs 2006, 24, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Regala, R.P.; Thompson, E.A.; Fields, A.P. Atypical protein kinase C iota expression and aurothiomalate sensitivity in human lung cancer cells. Cancer Res. 2008, 68, 5888–5895. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.Q.; Yang, Y.; Wang, J.M.; Du, G.S.; Shen, Q.; Liu, Y.; Zhang, J.; Hu, J.L.; Zhu, P.; Qi, W.P.; et al. The aPKCiota blocking agent ATM negatively regulates EMT and invasion of hepatocellular carcinoma. Cell Death Dis. 2014, 5, e1129. [Google Scholar] [CrossRef] [Green Version]
- Trani, M.; Sorrentino, A.; Busch, C.; Landstrom, M. Pro-apoptotic effect of aurothiomalate in prostate cancer cells. Cell Cycle 2009, 8, 306–313. [Google Scholar] [CrossRef]
- Castanotto, D.; Stein, C.A. Antisense oligonucleotides in cancer. Curr. Opin. Oncol. 2014, 26, 584–589. [Google Scholar] [CrossRef]
- Roychowdhury, D.; Lahn, M. Antisense therapy directed to protein kinase C-alpha (Affinitak, LY900003/ISIS 3521): Potential role in breast cancer. Semin Oncol. 2003, 30, 30–33. [Google Scholar] [CrossRef]
- Rao, S.; Watkins, D.; Cunningham, D.; Dunlop, D.; Johnson, P.; Selby, P.; Hancock, B.W.; Fegan, C.; Culligan, D.; Schey, S.; et al. Phase II study of ISIS 3521, an antisense oligodeoxynucleotide to protein kinase C alpha, in patients with previously treated low-grade non-Hodgkin’s lymphoma. Ann Oncol 2004, 15, 1413–1418. [Google Scholar] [CrossRef]
- Marshall, J.L.; Eisenberg, S.G.; Johnson, M.D.; Hanfelt, J.; Dorr, F.A.; El-Ashry, D.; Oberst, M.; Fuxman, Y.; Holmlund, J.; Malik, S. A phase II trial of ISIS 3521 in patients with metastatic colorectal cancer. Clin Colorectal Cancer 2004, 4, 268–274. [Google Scholar] [CrossRef]
- Cripps, M.C.; Figueredo, A.T.; Oza, A.M.; Taylor, M.J.; Fields, A.L.; Holmlund, J.T.; McIntosh, L.W.; Geary, R.S.; Eisenhauer, E.A. Phase II randomized study of ISIS 3521 and ISIS 5132 in patients with locally advanced or metastatic colorectal cancer: A National Cancer Institute of Canada clinical trials group study. Clin Cancer Res 2002, 8, 2188–2192. [Google Scholar]
- Newton, A.C.; Brognard, J. Reversing the Paradigm: Protein Kinase C as a Tumor Suppressor. Trends Pharmacol. Sci. 2017, 38, 438–447. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Structure | Name | Specificity | Inhibition Mechanism |
|---|---|---|---|
 | Staurosporine | Pan-PKCs | competitive interfere with the ATP binding site |
 | Midostaurin (PKC412, CGP41251, N-benzoylstaurosporine) | cPKCs, nPKCs | competitive interfere with the ATP binding site |
 | 7-Hydroxystaurosporine (UCN-01) | cPKCs, nPKCs | competitive interfere with the ATP binding site |
 | Sotrastaurin (AEB071) | PKCα, β, θ, δ | competitive interfere with the ATP binding site |
 | Enzastaurin (LY317615) | PKCβ | competitive interfere with the ATP binding site |
 | Bryostatin 1 | PKCα, PKCε, and PKCη | competitive interfere with the phorbol ester binding site |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, S.; Li, Q.; Huang, Q.; Cheng, J. Targeting Protein Kinase C for Cancer Therapy. Cancers 2022, 14, 1104. https://doi.org/10.3390/cancers14051104
He S, Li Q, Huang Q, Cheng J. Targeting Protein Kinase C for Cancer Therapy. Cancers. 2022; 14(5):1104. https://doi.org/10.3390/cancers14051104
Chicago/Turabian StyleHe, Sijia, Qi Li, Qian Huang, and Jin Cheng. 2022. "Targeting Protein Kinase C for Cancer Therapy" Cancers 14, no. 5: 1104. https://doi.org/10.3390/cancers14051104