Circulating Tumor DNA—A Novel Biomarker of Tumor Progression and Its Favorable Detection Techniques


Abstract
:Simple Summary
Abstract
1. Introduction
2. Biological Characteristics of ctDNA
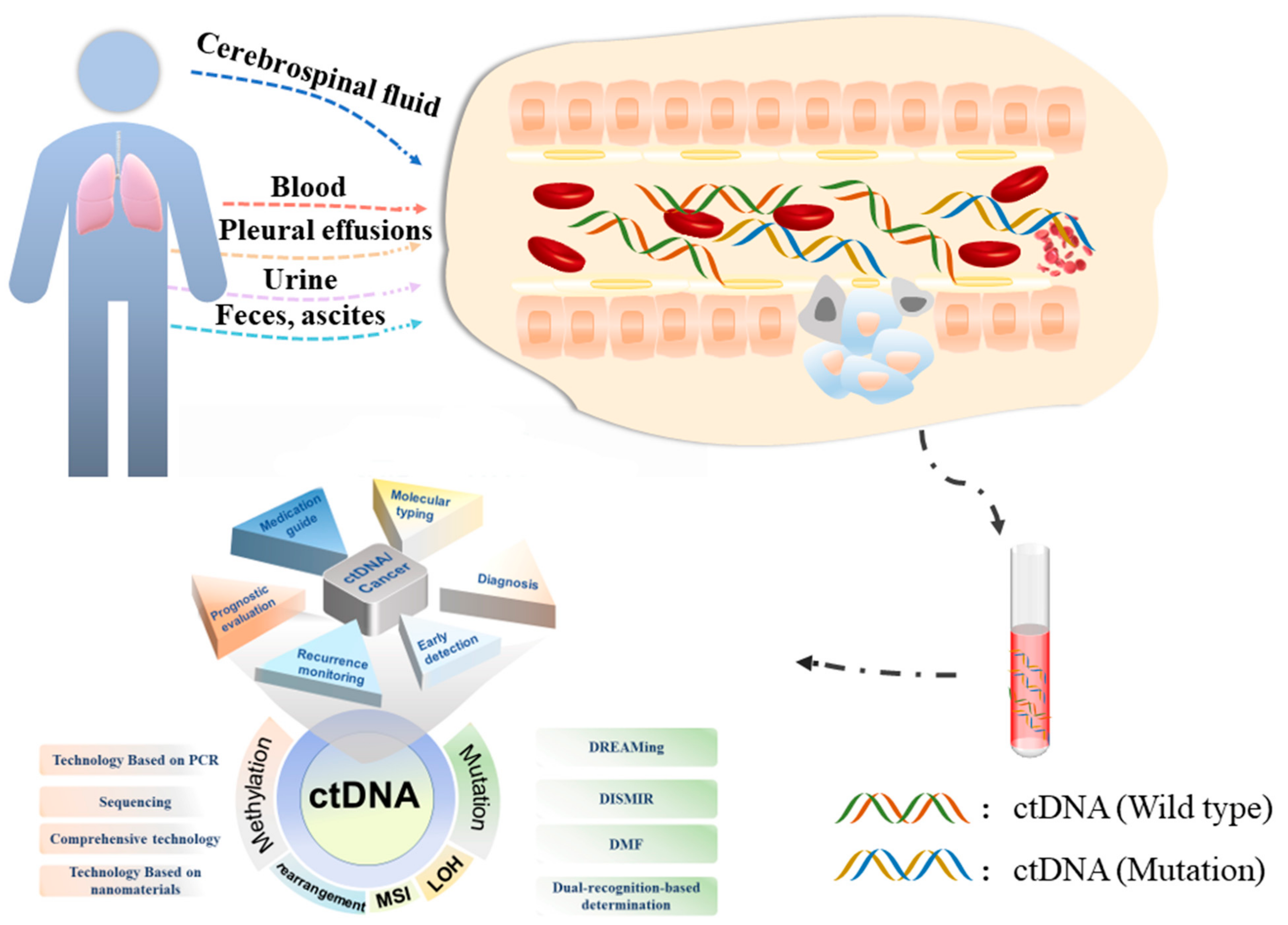
3. ctDNA Preparation
3.1. Blood
3.2. Urine
3.3. Cerebrospinal Fluid
3.4. Feces, Ascites, and Pleural Effusions
4. Detection Technology of ctDNA
4.1. Mutation Detection of ctDNA
4.1.1. Based on PCR
- (1) ARMS-PCR
- (2) COLD-PCR
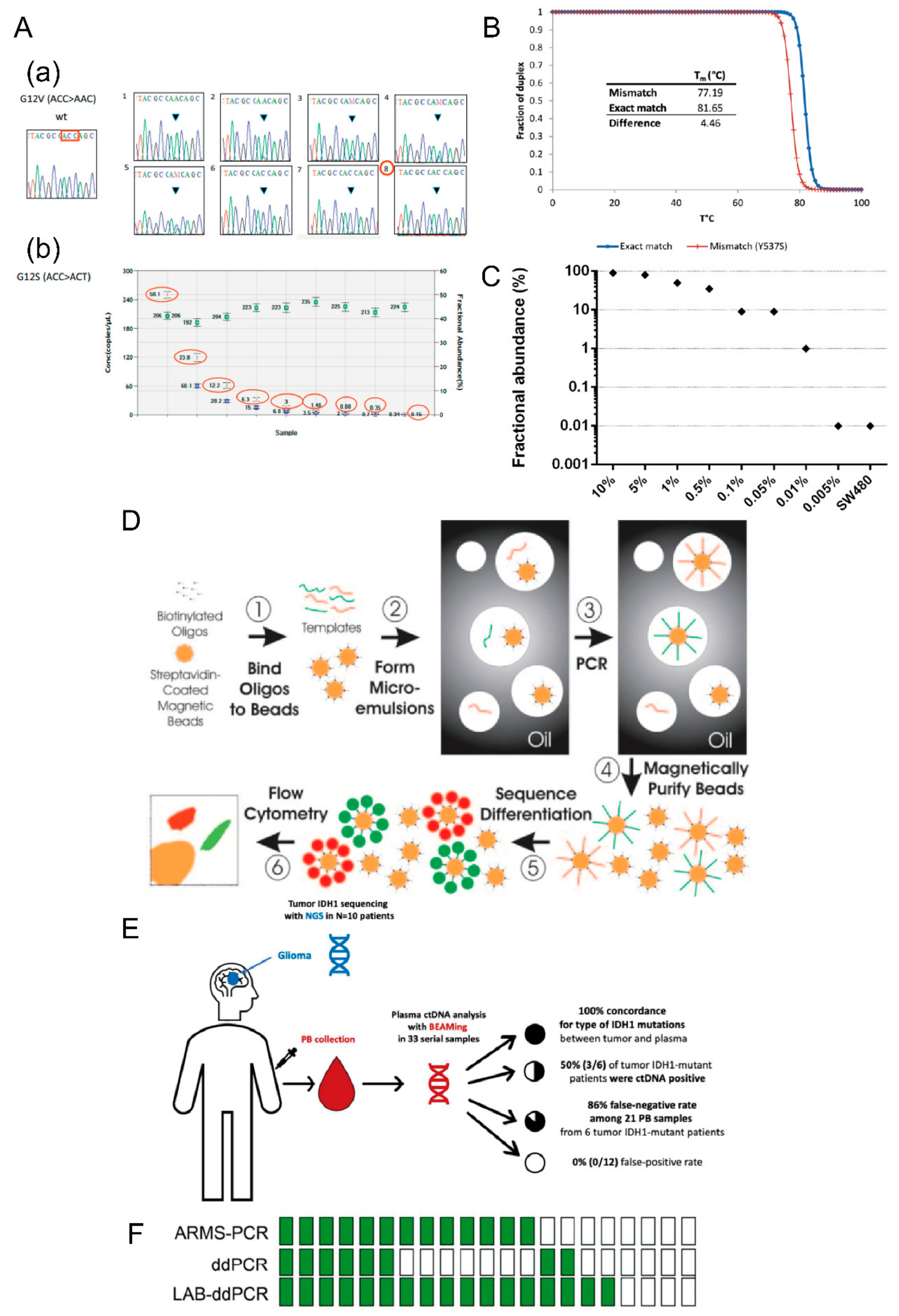
- (3) BEAMing
- (4) ddPCR
4.1.2. Sequencing-Based Detection
4.1.3. Comprehensive Technology
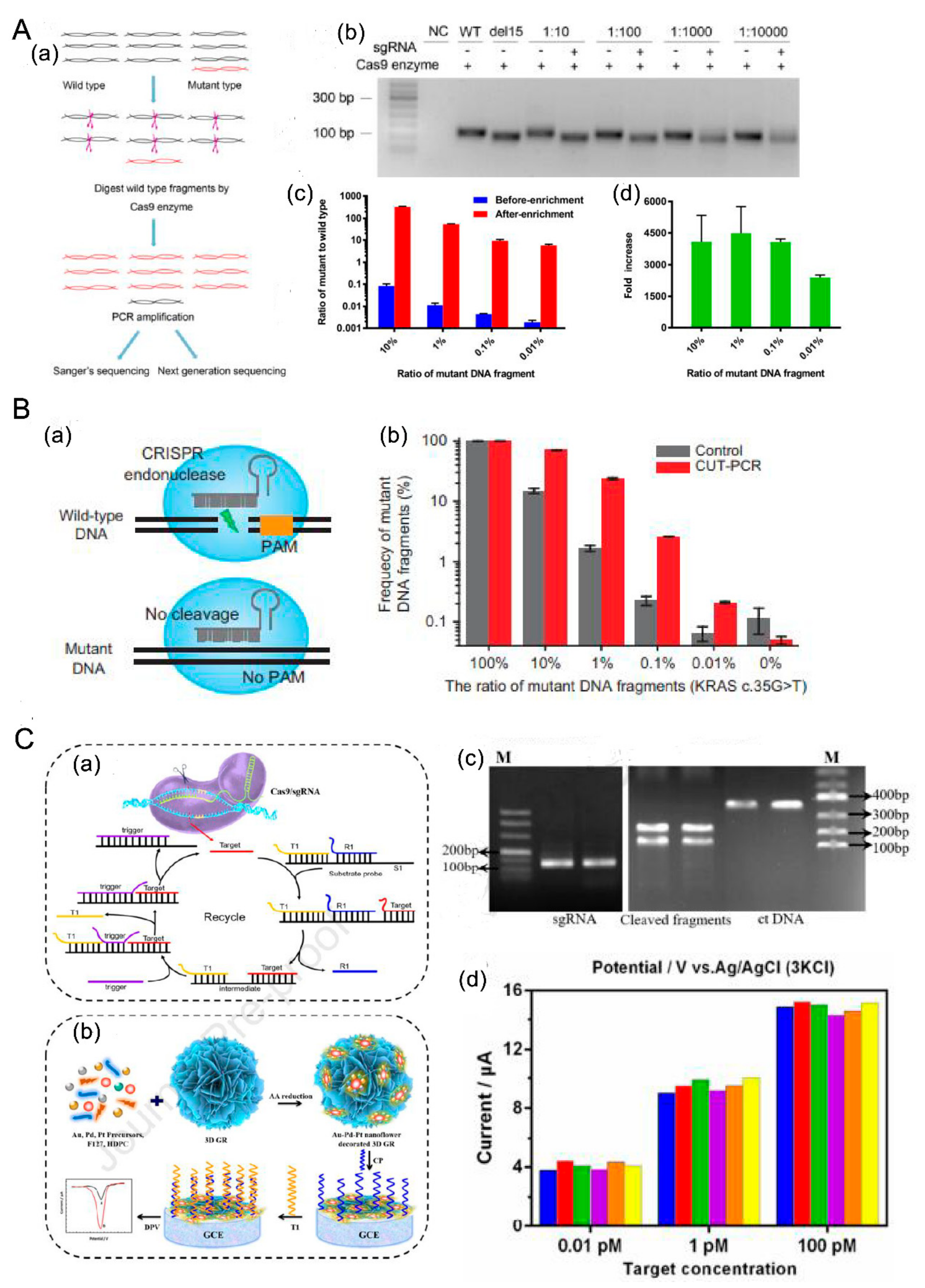
- (1) CRISPR/Cas9 + PCR
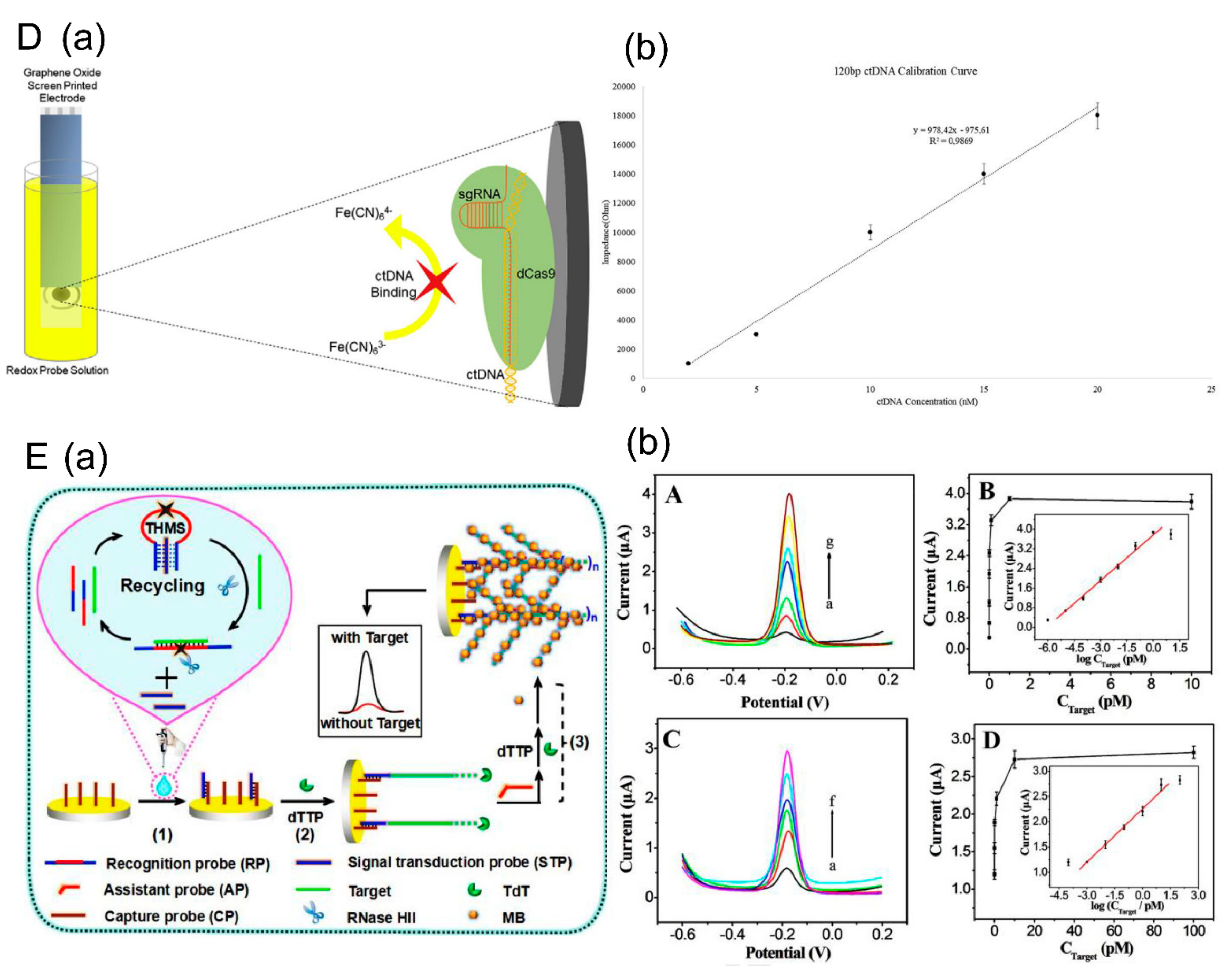
- (2) Based on Functional enzymes
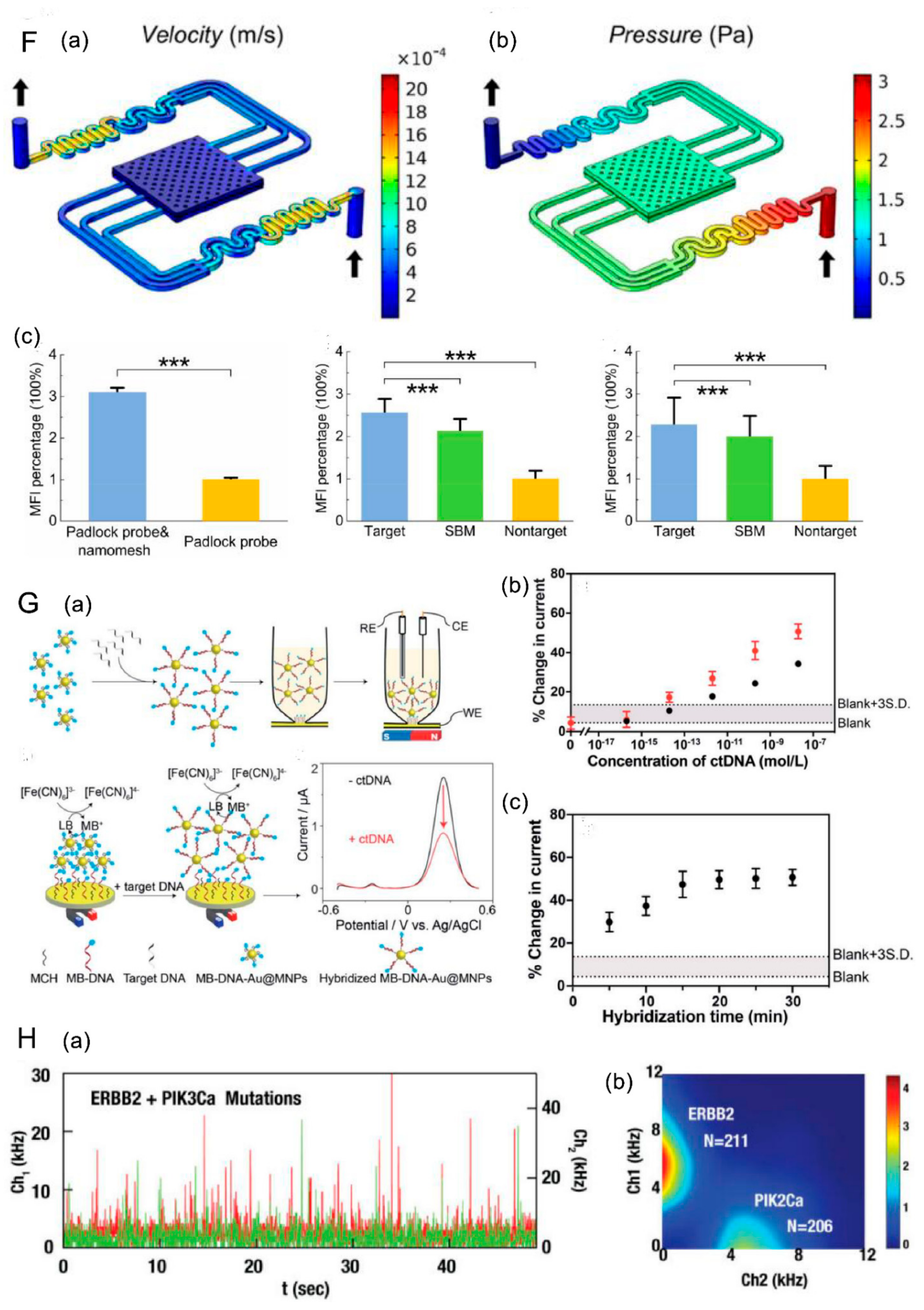
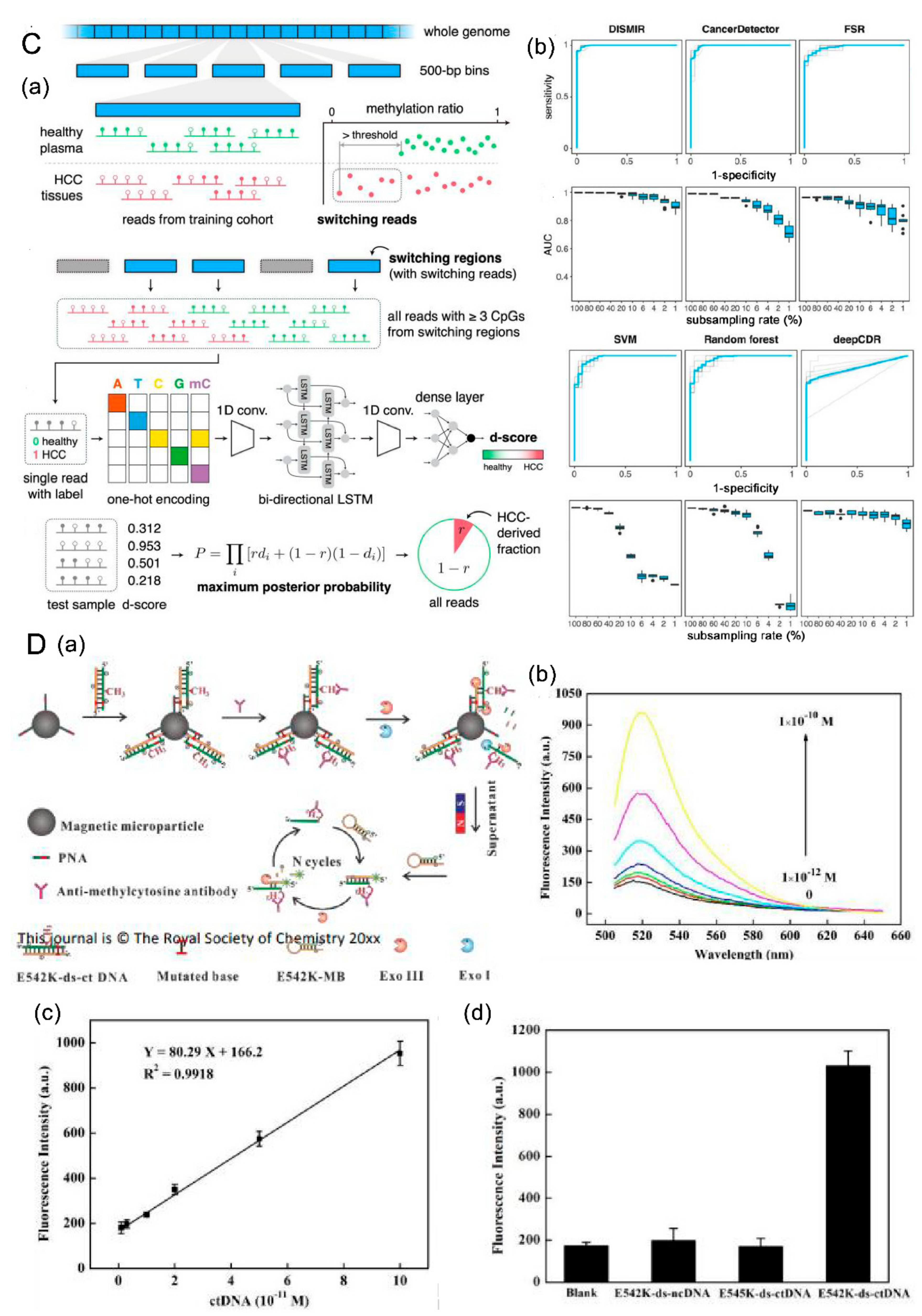
- (3) Detection based on nanomaterials
- (4) Developed Kit
4.2. Methylation Detection of ctDNA
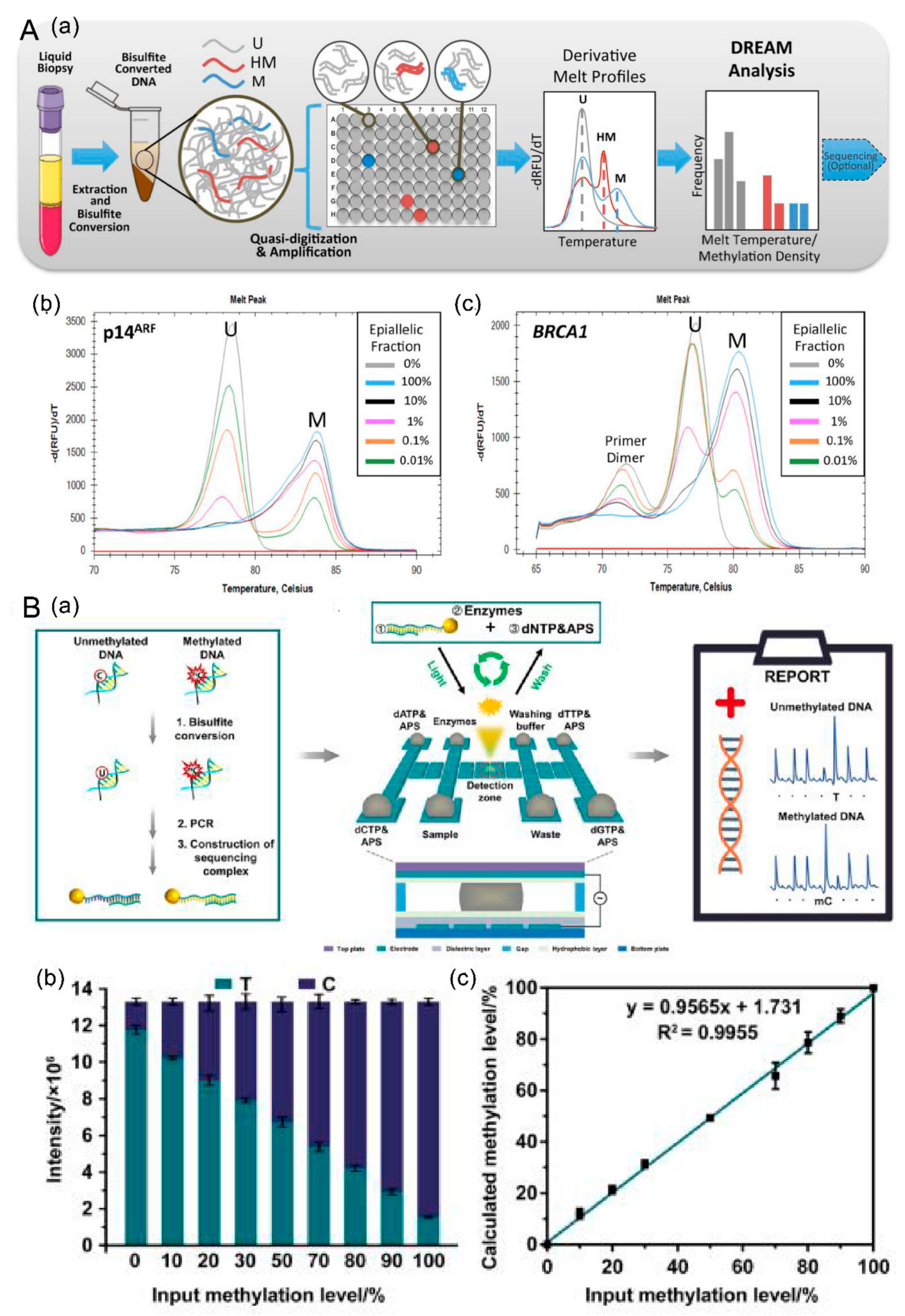
4.2.1. Discrimination of Rare EpiAlleles by Melt (DREAMing)
4.2.2. DISMIR
4.2.3. Detection Based on Digital Microfluidics (dmf)
4.2.4. Dual-Recognition-Based Determination
5. Clinical Application of ctDNA during Cancer Progression
5.1. ctDNA and Early Diagnosis of Tumor
5.2. ctDNA and Prognosis of Tumor
5.3. ctDNA and Tumor Molecular Subtyping Profiles
5.4. ctDNA and Tumor Recurrence Monitoring
5.5. ctDNA and Clinical Medication Guidance
6. Discussion and Perspective
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Kenny, P.A.; Nelson, C.M.; Bissell, M.J. The Ecology of Tumors: By perturbing the microenvironment, wounds and infection may be key to tumor development. Scientist 2006, 20, 30. [Google Scholar] [PubMed]
- Luo, W. Nasopharyngeal Carcinoma Ecology Theory: Cancer as Multidimensional Spatiotemporal “Unity of Ecology and Evolution”. Pathol. Ecosyst. 2022, 2022100226. [Google Scholar] [CrossRef]
- Potosky, A.L.; Miller, B.A.; Albertsen, P.C.; Kramer, B.S. The role of increasing detection in the rising incidence of prostate cancer. JAMA 1995, 273, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Qu, X. Cancer biomarker detection: Recent achievements and challenges. Chem. Soc. Rev. 2015, 44, 2963–2997. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Lee, S.; Yu, H.; Kang, S.H. Ultra-sensitive detection of tumor necrosis factor-alpha on gold nano-patterned protein chip formed via E-beam nanolithography by total internal reflection fluorescence microscopy. J. Nanosci. Nanotechnol. 2010, 10, 3228–3231. [Google Scholar] [CrossRef]
- Vourtsis, A.; Berg, W.A. Breast density implications and supplemental screening. Eur. Radiol. 2019, 29, 1762–1777. [Google Scholar] [CrossRef] [PubMed]
- WHO Guideline for Screening and Treatment of Cervical Pre-Cancer; WHO: Geneva, Switzerland, 2021.
- Helsingen, L.M.; Kalager, M. Colorectal Cancer Screening—Approach, Evidence, and Future Directions. NEJM Evid. 2022, 1. [Google Scholar] [CrossRef]
- Mann, R.M.; Hooley, R.; Barr, R.G.; Moy, L. Novel Approaches to Screening for Breast Cancer. Radiology 2020, 297, 266–285. [Google Scholar] [CrossRef] [PubMed]
- Van Poppel, H.; Albreht, T.; Basu, P.; Hogenhout, R.; Collen, S.; Roobol, M. Serum PSA-based early detection of prostate cancer in Europe and globally: Past, present and future. Nat. Rev. Urol. 2022, 19, 562–572. [Google Scholar] [CrossRef]
- Zinkin, N.T.; Grall, F.; Bhaskar, K.; Otu, H.H.; Spentzos, D.; Kalmowitz, B.; Wells, M.; Guerrero, M.; Asara, J.M.; Libermann, T.A.; et al. Serum proteomics and biomarkers in hepatocellular carcinoma and chronic liver disease. Clin. Cancer Res. 2008, 14, 470–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heitzer, E.; Haque, I.S.; Roberts, C.E.S.; Speicher, M.R. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet. 2019, 20, 71–88. [Google Scholar] [CrossRef]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Xu, J.-W.; Cheng, Y.-G.; Gao, J.-Y.; Hu, S.-Y.; Wang, L.; Zhan, H.-X. Early detection of pancreatic cancer: Where are we now and where are we going? Int. J. Cancer 2017, 141, 231–241. [Google Scholar] [CrossRef] [Green Version]
- Goodman, C.R.; Seagle, B.L.; Friedl, T.W.P.; Rack, B.; Lato, K.; Fink, V.; Cristofanilli, M.; Donnelly, E.D.; Janni, W.; Shahabi, S.; et al. Association of Circulating Tumor Cell Status with Benefit of Radiotherapy and Survival in Early-Stage Breast Cancer. JAMA Oncol. 2018, 4, e180163. [Google Scholar] [CrossRef] [Green Version]
- Olmos, D.; Arkenau, H.T.; Ang, J.E.; Ledaki, I.; Attard, G.; Carden, C.P.; Reid, A.H.; A’Hern, R.; Fong, P.C.; Oomen, N.B.; et al. Circulating tumour cell (CTC) counts as intermediate end points in castration-resistant prostate cancer (CRPC): A single-centre experience. Ann. Oncol. 2009, 20, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.J.; Ewald, A.J. A collective route to metastasis: Seeding by tumor cell clusters. Science 2016, 352, 167–169. [Google Scholar] [CrossRef] [Green Version]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Wang, Y.; Li, C.; Wang, Q.; Sun, B.; Zhang, H.; He, Z.; Sun, J. Cancer-specific calcium nanoregulator suppressing the generation and circulation of circulating tumor cell clusters for enhanced anti-metastasis combinational chemotherapy. Acta Pharm. Sin. B 2021, 11, 3262–3271. [Google Scholar] [CrossRef]
- Larson, M.H.; Pan, W.; Kim, H.J.; Mauntz, R.E.; Stuart, S.M.; Pimentel, M.; Zhou, Y.; Knudsgaard, P.; Demas, V.; Aravanis, A.M.; et al. A comprehensive characterization of the cell-free transcriptome reveals tissue- and subtype-specific biomarkers for cancer detection. Nat. Commun. 2021, 12, 2357. [Google Scholar] [CrossRef]
- Ma, M.; Zhu, H.; Zhang, C.; Sun, X.; Gao, X.; Chen, G. “Liquid biopsy”-ctDNA detection with great potential and challenges. Ann. Transl. Med. 2015, 3, 235. [Google Scholar] [PubMed]
- Chen, W.; Yan, H.; Li, X.; Ge, K.; Wu, J. Circulating tumor DNA detection and its application status in gastric cancer: A narrative review. Transl. Cancer Res. 2021, 10, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Vandekerkhove, G.; Lavoie, J.M.; Annala, M.; Murtha, A.J.; Sundahl, N.; Walz, S.; Sano, T.; Taavitsainen, S.; Ritch, E.; Fazli, L.; et al. Plasma ctDNA is a tumor tissue surrogate and enables clinical-genomic stratification of metastatic bladder cancer. Nat. Commun. 2021, 12, 184. [Google Scholar] [CrossRef]
- Magbanua, M.J.M.; Swigart, L.B.; Wu, H.T.; Hirst, G.L.; Yau, C.; Wolf, D.M.; Tin, A.; Salari, R.; Shchegrova, S.; Pawar, H.; et al. Circulating tumor DNA in neoadjuvant-treated breast cancer reflects response and survival. Ann. Oncol. 2021, 32, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Chen, Z.; Chong, X.; Chen, Y.; Wang, Z.; Yu, R.; Sun, T.; Chen, X.; Shao, Y.; Zhang, X.; et al. Clinical implications of plasma ctDNA features and dynamics in gastric cancer treated with HER2-targeted therapies. Clin. Transl. Med. 2020, 10, e254. [Google Scholar] [CrossRef]
- Mandel, P.M.P. Nuclear acids in human blood plasma. C. R. Seances La Soc. Biol. Ses. Fil. 1948, 3–4, 241–243. [Google Scholar]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F.O.; Hesch, R.D.; Knippers, R. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001, 61, 1659–1665. [Google Scholar]
- Allenson, K.; Castillo, J.; San Lucas, F.A.; Scelo, G.; Kim, D.U.; Bernard, V.; Davis, G.; Kumar, T.; Katz, M.; Overman, M.J.; et al. High prevalence of mutant KRAS in circulating exosome-derived DNA from early-stage pancreatic cancer patients. Ann. Oncol. 2017, 28, 741–747. [Google Scholar] [CrossRef]
- Hause, R.J.; Pritchard, C.C.; Shendure, J.; Salipante, S.J. Classification and characterization of microsatellite instability across 18 cancer types. Nat. Med. 2016, 22, 1342–1350. [Google Scholar] [CrossRef]
- Mayrhofer, M.; De Laere, B.; Whitington, T.; Van Oyen, P.; Ghysel, C.; Ampe, J.; Ost, P.; Demey, W.; Hoekx, L.; Schrijvers, D.; et al. Cell-free DNA profiling of metastatic prostate cancer reveals microsatellite instability, structural rearrangements and clonal hematopoiesis. Genome Med. 2018, 10, 85. [Google Scholar] [CrossRef] [PubMed]
- Kasi, P.M.; Budde, G.; Krainock, M.; Aushev, V.N.; Koyen Malashevich, A.; Malhotra, M.; Olshan, P.; Billings, P.R.; Aleshin, A. Circulating tumor DNA (ctDNA) serial analysis during progression on PD-1 blockade and later CTLA-4 rescue in patients with mismatch repair deficient metastatic colorectal cancer. J. Immunother. Cancer 2022, 10, e003312. [Google Scholar] [CrossRef]
- Han, X.; Zhang, S.; Zhou, D.C.; Wang, D.; He, X.; Yuan, D.; Li, R.; He, J.; Duan, X.; Wendl, M.C.; et al. MSIsensor-ct: Microsatellite instability detection using cfDNA sequencing data. Brief. Bioinform. 2021, 22, bbaa402. [Google Scholar] [CrossRef]
- Steele, C.D.; Abbasi, A.; Islam, S.M.A.; Bowes, A.L.; Khandekar, A.; Haase, K.; Hames-Fathi, S.; Ajayi, D.; Verfaillie, A.; Dhami, P.; et al. Signatures of copy number alterations in human cancer. Nature 2022, 606, 984–991. [Google Scholar] [CrossRef]
- Sokol, E.S.; Feng, Y.X.; Jin, D.X.; Basudan, A.; Lee, A.V.; Atkinson, J.M.; Chen, J.; Stephens, P.J.; Frampton, G.M.; Gupta, P.B.; et al. Loss of function of NF1 is a mechanism of acquired resistance to endocrine therapy in lobular breast cancer. Ann. Oncol. 2019, 30, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Siravegna, G.; Sartore-Bianchi, A.; Mussolin, B.; Cassingena, A.; Amatu, A.; Novara, L.; Buscarino, M.; Corti, G.; Crisafulli, G.; Bartolini, A.; et al. Tracking a CAD-ALK gene rearrangement in urine and blood of a colorectal cancer patient treated with an ALK inhibitor. Ann. Oncol. 2017, 28, 1302–1308. [Google Scholar] [CrossRef]
- Van Loo, K.M.J.; Carvill, G.L.; Becker, A.J.; Conboy, K.; Goldman, A.M.; Kobow, K.; Lopes-Cendes, I.; Reid, C.A.; van Vliet, E.A.; Henshall, D.C. Epigenetic genes and epilepsy—Emerging mechanisms and clinical applications. Nat. Rev. Neurol. 2022, 18, 530–543. [Google Scholar] [CrossRef] [PubMed]
- Azad, T.D.; Chaudhuri, A.A.; Fang, P.; Qiao, Y.; Esfahani, M.S.; Chabon, J.J.; Hamilton, E.G.; Yang, Y.D.; Lovejoy, A.; Newman, A.M.; et al. Circulating Tumor DNA Analysis for Detection of Minimal Residual Disease after Chemoradiotherapy for Localized Esophageal Cancer. Gastroenterology 2020, 158, 494–505e6. [Google Scholar] [CrossRef]
- Ococks, E.; Frankell, A.M.; Soler, N.M.; Grehan, N.; Northrop, A.; Coles, H.; Redmond, A.M.; Devonshire, G.; Weaver, J.M.J.; Hughes, C.; et al. Longitudinal tracking of 97 esophageal adenocarcinomas using liquid biopsy sampling. Ann. Oncol. 2021, 32, 522–532. [Google Scholar] [CrossRef]
- Ricciuti, B.; Jones, G.; Severgnini, M.; Alessi, J.V.; Recondo, G.; Lawrence, M.; Forshew, T.; Lydon, C.; Nishino, M.; Cheng, M.; et al. Early plasma circulating tumor DNA (ctDNA) changes predict response to first-line pembrolizumab-based therapy in non-small cell lung cancer (NSCLC). J. Immunother. Cancer 2021, 9, e001504. [Google Scholar] [CrossRef]
- Luo, H.; Zhao, Q.; Wei, W.; Zheng, L.; Yi, S.; Li, G.; Wang, W.; Sheng, H.; Pu, H.; Mo, H.; et al. Circulating tumor DNA methylation profiles enable early diagnosis, prognosis prediction, and screening for colorectal cancer. Sci. Transl. Med. 2020, 12, eaax7533. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yao, Y.; Xu, Y.; Li, L.; Gong, Y.; Zhang, K.; Zhang, M.; Guan, Y.; Chang, L.; Xia, X.; et al. Pan-cancer circulating tumor DNA detection in over 10,000 Chinese patients. Nat Commun 2021, 12, 11. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Murillas, I.; Chopra, N.; Comino-Mendez, I.; Beaney, M.; Tovey, H.; Cutts, R.J.; Swift, C.; Kriplani, D.; Afentakis, M.; Hrebien, S.; et al. Assessment of Molecular Relapse Detection in Early-Stage Breast Cancer. JAMA Oncol. 2019, 5, 1473–1478. [Google Scholar] [CrossRef]
- Jia, S.; Xie, L.; Li, L.; Qian, Y.; Wang, J.; Wang, S.; Zhang, W.; Qian, B. Values of liquid biopsy in early detection of cancer: Results from meta-analysis. Expert Rev. Mol. Diagn. 2021, 21, 417–427. [Google Scholar] [CrossRef]
- Kang, Q.; Henry, N.L.; Paoletti, C.; Jiang, H.; Vats, P.; Chinnaiyan, A.M.; Hayes, D.F.; Merajver, S.D.; Rae, J.M.; Tewari, M. Comparative analysis of circulating tumor DNA stability In K3EDTA, Streck, and CellSave blood collection tubes. Clin. Biochem. 2016, 49, 1354–1360. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.G.; Moser, T.; Mouliere, F.; Field-Rayner, J.; Eldridge, M.; Riediger, A.L.; Chandrananda, D.; Heider, K.; Wan, J.C.M.; Warren, A.Y.; et al. Comprehensive characterization of cell-free tumor DNA in plasma and urine of patients with renal tumors. Genome Med. 2020, 12, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellini, B.; Pejovic, N.; Feng, W.; Earland, N.; Harris, P.K.; Usmani, A.; Szymanski, J.J.; Qaium, F.; Mudd, J.; Petty, M.; et al. ctDNA MRD Detection and Personalized Oncogenomic Analysis in Oligometastatic Colorectal Cancer from Plasma and Urine. JCO Precis. Oncol. 2021, 5, 378–388. [Google Scholar] [CrossRef]
- Birkenkamp-Demtroder, K.; Christensen, E.; Nordentoft, I.; Knudsen, M.; Taber, A.; Hoyer, S.; Lamy, P.; Agerbaek, M.; Jensen, J.B.; Dyrskjot, L. Monitoring Treatment Response and Metastatic Relapse in Advanced Bladder Cancer by Liquid Biopsy Analysis. Eur. Urol. 2018, 73, 535–540. [Google Scholar] [CrossRef]
- Springer, S.U.; Chen, C.H.; Pena, M.D.C.R.; Li, L.; Douville, C.; Wang, Y.; Cohen, J.D.; Taheri, D.; Silliman, N.; Schaefer, J.; et al. Non-invasive detection of urothelial cancer through the analysis of driver gene mutations and aneuploidy. eLife 2018, 7, e32143. [Google Scholar] [CrossRef]
- Bobillo, S.; Crespo, M.; Escudero, L.; Mayor, R.; Raheja, P.; Carpio, C.; Rubio-Perez, C.; Tazon-Vega, B.; Palacio, C.; Carabia, J.; et al. Cell free circulating tumor DNA in cerebrospinal fluid detects and monitors central nervous system involvement of B-cell lymphomas. Haematologica 2021, 106, 513–521. [Google Scholar] [CrossRef] [Green Version]
- De Mattos-Arruda, L.; Mayor, R.; Ng, C.K.Y.; Weigelt, B.; Martinez-Ricarte, F.; Torrejon, D.; Oliveira, M.; Arias, A.; Raventos, C.; Tang, J.; et al. Cerebrospinal fluid-derived circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nat. Commun. 2015, 6, 8839. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.M.; Shah, R.H.; Pentsova, E.I.; Pourmaleki, M.; Briggs, S.; Distefano, N.; Zheng, Y.; Skakodub, A.; Mehta, S.A.; Campos, C.; et al. Tracking tumour evolution in glioma through liquid biopsies of cerebrospinal fluid. Nature 2019, 565, 654–658. [Google Scholar] [CrossRef]
- ctDNA Profiling of Cerebrospinal Fluid Characterizes Glioma Evolution. Cancer Discov. 2019, 9, 318. [CrossRef]
- Yang, S.; Zhan, X.; Tang, X.; Zhao, S.; Yu, L.; Gao, M.; Luo, D.; Wang, Y.; Chang, K.; Chen, M. A multiplexed circulating tumor DNA detection platform engineered from 3D-coded interlocked DNA rings. Bioact. Mater. 2022, 10, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Han, M.R.; Lee, S.H.; Park, J.Y.; Hong, H.; Ho, J.Y.; Hur, S.Y.; Choi, Y.J. Clinical Implications of Circulating Tumor DNA from Ascites and Serial Plasma in Ovarian Cancer. Cancer Res. Treat. 2020, 52, 779–788. [Google Scholar] [CrossRef] [Green Version]
- Gholamalizadeh, M.; Akbari, M.E.; Doaei, S.; Davoodi, S.H.; Bahar, B.; Tabesh, G.A.; Sadeghi, H.; Razavi Hashemi, M.; Kheyrani, E.; Rastgoo, S.; et al. The Association of Fat-Mass-and Obesity-Associated Gene Polymorphism (rs9939609) with Colorectal Cancer: A Case-Control Study. Front. Oncol. 2021, 11, 732515. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, X.; Wang, L.; Wu, F.; Zhang, X.; Wu, H.; Feng, C.; Liu, M.; Zhang, Y.; Zhang, S. Super-ARMS: A new method for plasma ESR1 mutation detection. Clin. Chim. Acta 2021, 520, 23–28. [Google Scholar] [CrossRef]
- He, C.; Wei, C.; Wen, J.; Chen, S.; Chen, L.; Wu, Y.; Shen, Y.; Bai, H.; Zhang, Y.; Chen, X.; et al. Comprehensive analysis of NGS and ARMS-PCR for detecting EGFR mutations based on 4467 cases of NSCLC patients. J. Cancer Res. Clin. Oncol. 2022, 148, 321–330. [Google Scholar] [CrossRef]
- Jensen, K.; Thakur, S.; Patel, A.; Mendonca-Torres, M.C.; Costello, J.; Gomes-Lima, C.J.; Walter, M.; Wartofsky, L.; Burman, K.D.; Bikas, A.; et al. Detection of BRAFV600E in Liquid Biopsy from Patients with Papillary Thyroid Cancer Is Associated with Tumor Aggressiveness and Response to Therapy. J. Clin. Med. 2020, 9, 2481. [Google Scholar] [CrossRef]
- Lupini, L.; Moretti, A.; Bassi, C.; Schirone, A.; Pedriali, M.; Querzoli, P.; Roncarati, R.; Frassoldati, A.; Negrini, M. High-sensitivity assay for monitoring ESR1 mutations in circulating cell-free DNA of breast cancer patients receiving endocrine therapy. Sci. Rep. 2018, 8, 4371. [Google Scholar] [CrossRef] [Green Version]
- Galbiati, S.; Damin, F.; Burgio, V.; Brisci, A.; Soriani, N.; Belcastro, B.; Di Resta, C.; Gianni, L.; Chiari, M.; Ronzoni, M.; et al. Evaluation of three advanced methodologies, COLD-PCR, microarray and ddPCR, for identifying the mutational status by liquid biopsies in metastatic colorectal cancer patients. Clin. Chim. Acta 2019, 489, 136–143. [Google Scholar] [CrossRef]
- Dressman, D.; Yan, H.; Traverso, G.; Kinzler, K.W.; Vogelstein, B. Transforming single DNA molecules into fluorescent. Proc. Natl. Acad. Sci. USA 2003, 100, 8817–8822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diehl, F.; Li, M.; He, Y.; Kinzler, K.W.; Vogelstein, B.; Dressman, D. BEAMing: Single-molecule PCR on microparticles in water-in-oil emulsions. Nat. Methods 2006, 3, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Kagawa, Y.; Elez, E.; Garcia-Foncillas, J.; Bando, H.; Taniguchi, H.; Vivancos, A.; Akagi, K.; Garcia, A.; Denda, T.; Ros, J.; et al. Combined Analysis of Concordance between Liquid and Tumor Tissue Biopsies for RAS Mutations in Colorectal Cancer with a Single Metastasis Site: The METABEAM Study. Clin. Cancer Res. 2021, 27, 2515–2522. [Google Scholar] [CrossRef]
- Cabezas-Camarero, S.; Garcia-Barberan, V.; Perez-Alfayate, R.; Casado-Farinas, I.; Sloane, H.; Jones, F.S.; Perez-Segura, P. Detection of IDH1 Mutations in Plasma Using BEAMing Technology in Patients with Gliomas. Cancers 2022, 14, 2891. [Google Scholar] [CrossRef]
- Krug, A.K.; Enderle, D.; Karlovich, C.; Priewasser, T.; Bentink, S.; Spiel, A.; Brinkmann, K.; Emenegger, J.; Grimm, D.G.; Castellanos-Rizaldos, E.; et al. Improved EGFR mutation detection using combined exosomal RNA and circulating tumor DNA in NSCLC patient plasma. Ann. Oncol. 2018, 29, 700–706. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; He, Q.; Liang, H.; Cheng, B.; Li, J.; Xiong, S.; Zhao, Y.; Guo, M.; Liu, Z.; He, J.; et al. Diagnostic Accuracy of Droplet Digital PCR and Amplification Refractory Mutation System PCR for Detecting EGFR Mutation in Cell-Free DNA of Lung Cancer: A Meta-Analysis. Front. Oncol. 2020, 10, 290. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Li, J.; Zhao, J.; Ren, P.; Wang, Z.; Wei, B.; Dong, B.; Sun, R.; Wang, X.; Groen, H.J.M.; et al. Developing Ultrasensitive Library-Aliquot-Based Droplet Digital PCR for Detecting T790M in Plasma-Circulating Tumor DNA of Non-small-Cell-Lung-Cancer Patients. Anal. Chem. 2018, 90, 11203–11209. [Google Scholar] [CrossRef]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [Green Version]
- Hasenleithner, S.O.; Speicher, M.R. A clinician’s handbook for using ctDNA throughout the patient journey. Mol. Cancer 2022, 21, 81. [Google Scholar] [CrossRef]
- Wang, L.; Jiang, J.; Li, X.; Li, K.; He, R.; Li, J.; Duan, L.; Luo, W.; Hu, Z.; Luo, D. Improved EGFR mutation detection sensitivity after enrichment by Cas9/sgRNA digestion and PCR amplification. Acta Biochim. Biophys. Sin. 2020, 52, 1316–1324. [Google Scholar] [CrossRef]
- Lee, S.H.; Yu, J.; Hwang, G.H.; Kim, S.; Kim, H.S.; Ye, S.; Kim, K.; Park, J.; Park, D.Y.; Cho, Y.K.; et al. CUT-PCR: CRISPR-mediated, ultrasensitive detection of target DNA using PCR. Oncogene 2017, 36, 6823–6829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Wu, D.; Tu, S.; Yang, C.; Chen, D.; Xu, Y. CRISPR/Cas9 cleavage triggered ESDR for circulating tumor DNA detection based on a 3D graphene/AuPtPd nanoflower biosensor. Biosens. Bioelectron. 2020, 173, 112821. [Google Scholar] [CrossRef] [PubMed]
- Uygun, Z.O.; Yeniay, L.; Gi Rgi, N.S.F. CRISPR-dCas9 powered impedimetric biosensor for label-free detection of circulating tumor DNAs. Anal. Chim. Acta 2020, 1121, 35–41. [Google Scholar] [CrossRef]
- Zhang, W.; Zhao, S.; Xie, Z.; Chen, S.; Huang, Y.; Zhao, Z.; Yi, G. The fluorescence amplification strategy based on 3D DNA walker and CRISPR/Cas12a for the rapid detection of BRAF V600E. Anal. Sci. 2022, 38, 1057–1066. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.F.; Ma, R.N.; Sun, F.; Jia, L.P.; Zhang, W.; Shang, L.; Xue, Q.W.; Jia, W.L.; Wang, H.S. A versatile label-free electrochemical biosensor for circulating tumor DNA based on dual enzyme assisted multiple amplification strategy. Biosens. Bioelectron. 2018, 122, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Patchsung, M.; Jantarug, K.; Pattama, A.; Aphicho, K.; Suraritdechachai, S.; Meesawat, P.; Sappakhaw, K.; Leelahakorn, N.; Ruenkam, T.; Wongsatit, T.; et al. Clinical validation of a Cas13-based assay for the detection of SARS-CoV-2 RNA. Nat. Biomed. Eng. 2020, 4, 1140–1149. [Google Scholar] [CrossRef] [PubMed]
- Broughton, J.P.; Deng, X.; Yu, G.; Fasching, C.L.; Servellita, V.; Singh, J.; Miao, X.; Streithorst, J.A.; Granados, A.; Sotomayor-Gonzalez, A.; et al. CRISPR-Cas12-based detection of SARS-CoV-2. Nat. Biotechnol. 2020, 38, 870–874. [Google Scholar] [CrossRef] [Green Version]
- Arizti-Sanz, J.; Freije, C.A.; Stanton, A.C.; Petros, B.A.; Boehm, C.K.; Siddiqui, S.; Shaw, B.M.; Adams, G.; Kosoko-Thoroddsen, T.F.; Kemball, M.E.; et al. Streamlined inactivation, amplification, and Cas13-based detection of SARS-CoV-2. Nat. Commun. 2020, 11, 5921. [Google Scholar] [CrossRef] [PubMed]
- Casati, B.; Verdi, J.P.; Hempelmann, A.; Kittel, M.; Klaebisch, A.G.; Meister, B.; Welker, S.; Asthana, S.; Di Giorgio, S.; Boskovic, P.; et al. Rapid, adaptable and sensitive Cas13-based COVID-19 diagnostics using ADESSO. Nat. Commun. 2022, 13, 3308. [Google Scholar] [CrossRef]
- Huang, N.; Chen, M.; Chen, S.; Dang, K.; Guo, H.; Wang, X.; Yan, S.; Tian, J.; Liu, Y.; Ye, Q. A Specific Nucleic Acid Microfluidic Capture Device Based on Stable DNA Nanostructure. ACS Appl. Mater. Interfaces 2021, 13, 24487–24492. [Google Scholar] [CrossRef]
- Chen, D.; Wu, Y.; Hoque, S.; Tilley, R.D.; Gooding, J.J. Rapid and ultrasensitive electrochemical detection of circulating tumor DNA by hybridization on the network of gold-coated magnetic nanoparticles. Chem. Sci. 2021, 12, 5196–5201. [Google Scholar] [CrossRef] [PubMed]
- Burck, N.; Gilboa, T.; Gadi, A.; Patkin Nehrer, M.; Schneider, R.J.; Meller, A. Nanopore Identification of Single Nucleotide Mutations in Circulating Tumor DNA by Multiplexed Ligation. Clin. Chem. 2021, 67, 753–762. [Google Scholar] [CrossRef]
- Huang, J.; Wang, L. Cell-Free DNA Methylation Profiling Analysis-Technologies and Bioinformatics. Cancers 2019, 11, 1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhao, S.; Lee, M.; Yin, Y.; Li, J.; Zhou, Y.; Ballester, L.Y.; Esquenazi, Y.; Dashwood, R.H.; Davies, P.J.A.; et al. Reliable tumor detection by whole-genome methylation sequencing of cell-free DNA in cerebrospinal fluid of pediatric medulloblastoma. Sci. Adv. 2020, 6, eabb5427. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Romanel, A.; Conteduca, V.; Casiraghi, N.; Sigouros, M.; Franceschini, G.M.; Orlando, F.; Fedrizzi, T.; Ku, S.Y.; Dann, E.; et al. Circulating tumor DNA profile recognizes transformation to castration-resistant neuroendocrine prostate cancer. J. Clin. Investig. 2020, 130, 1653–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pisanic, T.R., 2nd; Athamanolap, P.; Poh, W.; Chen, C.; Hulbert, A.; Brock, M.V.; Herman, J.G.; Wang, T.H. DREAMing: A simple and ultrasensitive method for assessing intratumor epigenetic heterogeneity directly from liquid biopsies. Nucleic Acids Res. 2015, 43, e154. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wei, L.; Zhang, X.; Zhang, W.; Wang, H.; Zhong, B.; Xie, Z.; Lv, H.; Wang, X. DISMIR: Deep learning-based noninvasive cancer detection by integrating DNA sequence and methylation information of individual cell-free DNA reads. Brief. Bioinform. 2021, 22, bbab250. [Google Scholar] [CrossRef]
- Ruan, Q.; Zou, F.; Wang, Y.; Zhang, Y.; Xu, X.; Lin, X.; Tian, T.; Zhang, H.; Zhou, L.; Zhu, Z.; et al. Sensitive, Rapid, and Automated Detection of DNA Methylation Based on Digital Microfluidics. ACS Appl. Mater. Interfaces 2021, 13, 8042–8048. [Google Scholar] [CrossRef]
- Chen, C.; He, R.; Zhang, Z.; Chen, Y. Dual-recognition-based determination of ctDNA via the clamping function of peptide nucleic acid and terminal protection of small-molecule-linked DNA. Analyst 2020, 145, 7603–7608. [Google Scholar] [CrossRef]
- Leung, M.; Freidin, M.B.; Freydina, D.V.; Von Crease, C.; De Sousa, P.; Barbosa, M.T.; Nicholson, A.G.; Lim, E. Blood-based circulating tumor DNA mutations as a diagnostic and prognostic biomarker for lung cancer. Cancer 2020, 126, 1804–1809. [Google Scholar] [CrossRef]
- Liang, W.; Liu, D.; Li, M.; Wang, W.; Qin, Z.; Zhang, J.; Zhang, Y.; Hu, Y.; Bao, H.; Xiang, Y.; et al. Evaluating the diagnostic accuracy of a ctDNA methylation classifier for incidental lung nodules: Protocol for a prospective, observational, and multicenter clinical trial of 10,560 cases. Transl. Lung Cancer Res. 2020, 9, 2016–2026. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; Ding, X.Q.; Zhu, H.; Wang, R.X.; Pan, X.R.; Tong, J.H. KRAS Mutant Allele Fraction in Circulating Cell-Free DNA Correlates With Clinical Stage in Pancreatic Cancer Patients. Front. Oncol. 2019, 9, 1295. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Yang, W.; Zheng, W.; Chen, X.; Wang, H.; Zhao, L.; Xu, J.; Guo, X. Tumor elastography and its association with cell-free tumor DNA in the plasma of breast tumor patients: A pilot study. Quant. Imaging Med. Surg. 2021, 11, 3518–3534. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef] [Green Version]
- Lennon, A.M.; Buchanan, A.H.; Kinde, I.; Warren, A.; Honushefsky, A.; Cohain, A.T.; Ledbetter, D.H.; Sanfilippo, F.; Sheridan, K.; Rosica, D.; et al. Feasibility of blood testing combined with PET-CT to screen for cancer and guide intervention. Science 2020, 369, eabb9601. [Google Scholar] [CrossRef]
- Chen, X.; Gole, J.; Gore, A.; He, Q.; Lu, M.; Min, J.; Yuan, Z.; Yang, X.; Jiang, Y.; Zhang, T.; et al. Non-invasive early detection of cancer four years before conventional diagnosis using a blood test. Nat. Commun. 2020, 11, 3475. [Google Scholar] [CrossRef]
- Liu, M.C.; Oxnard, G.R.; Klein, E.A.; Swanton, C.; Seiden, M.V.; Consortium, C. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann. Oncol. 2020, 31, 745–759. [Google Scholar] [CrossRef]
- Lam, W.K.J.; Chan, K.C.A.; Lo, Y.M.D. Plasma Epstein-Barr virus DNA as an archetypal circulating tumour DNA marker. J. Pathol. 2019, 247, 641–649. [Google Scholar] [CrossRef] [Green Version]
- Lam, W.K.J.; Jiang, P.; Chan, K.C.A.; Cheng, S.H.; Zhang, H.; Peng, W.; Tse, O.Y.O.; Tong, Y.K.; Gai, W.; Zee, B.C.Y.; et al. Sequencing-based counting and size profiling of plasma Epstein-Barr virus DNA enhance population screening of nasopharyngeal carcinoma. Proc. Natl. Acad. Sci. USA 2018, 115, E5115–E5124. [Google Scholar] [CrossRef] [Green Version]
- Lo, Y.M.; Chan, A.T.; Chan, L.Y.; Leung, S.F.; Lam, C.W.; Huang, D.P.; Johnson, P.J. Molecular prognostication of nasopharyngeal carcinoma by quantitative analysis of circulating Epstein-Barr virus DNA. Cancer Res. 2000, 60, 6878–6881. [Google Scholar]
- Lv, J.; Wu, C.; Li, J.; Chen, F.; He, S.; He, Q.; Zhou, G.; Ma, J.; Sun, Y.; Wei, D.; et al. Improving on-treatment risk stratification of cancer patients with refined response classification and integration of circulating tumor DNA kinetics. BMC Med. 2022, 20, 268. [Google Scholar] [CrossRef] [PubMed]
- Leung, S.-F.; Lo, Y.M.D.; Chan, A.T.C.; To, K.-F.; To, E.; Chan, L.Y.S.; Zee, B.; Huang, D.P.; Johnson, P.J. Disparity of sensitivities in detection of radiation-naïve and postirradiation recurrent nasopharyngeal carcinoma of the undifferentiated type by quantitative analysis of circulating Epstein-Barr virus DNA1,2. Clin. Cancer Res. 2003, 9, 3431–3434. [Google Scholar] [PubMed]
- Sumanasuriya, S.; Seed, G.; Parr, H.; Christova, R.; Pope, L.; Bertan, C.; Bianchini, D.; Rescigno, P.; Figueiredo, I.; Goodall, J.; et al. Elucidating Prostate Cancer Behaviour During Treatment via Low-pass Whole-genome Sequencing of Circulating Tumour DNA. Eur. Urol. 2021, 80, 243–253. [Google Scholar] [CrossRef]
- Fitzpatrick, A.; Iravani, M.; Mills, A.; Childs, L.; Alaguthurai, T.; Clifford, A.; Garcia-Murillas, I.; Van Laere, S.; Dirix, L.; Harries, M.; et al. Assessing CSF ctDNA to Improve Diagnostic Accuracy and Therapeutic Monitoring in Breast Cancer Leptomeningeal Metastasis. Clin. Cancer Res. 2022, 28, 1180–1191. [Google Scholar] [CrossRef] [PubMed]
- Tserpeli, V.; Stergiopoulou, D.; Londra, D.; Giannopoulou, L.; Buderath, P.; Balgkouranidou, I.; Xenidis, N.; Grech, C.; Obermayr, E.; Zeillinger, R.; et al. Prognostic Significance of SLFN11 Methylation in Plasma Cell-Free DNA in Advanced High-Grade Serous Ovarian Cancer. Cancers 2021, 14, 4. [Google Scholar] [CrossRef]
- Lu, Y.; Li, L. The Prognostic Value of Circulating Tumor DNA in Ovarian Cancer: A Meta-Analysis. Technol. Cancer Res. Treat. 2021, 20, 15330338211043784. [Google Scholar] [CrossRef] [PubMed]
- Yi, Z.; Ma, F.; Rong, G.; Liu, B.; Guan, Y.; Li, J.; Sun, X.; Wang, W.; Guan, X.; Mo, H.; et al. The molecular tumor burden index as a response evaluation criterion in breast cancer. Signal. Transduct. Target. Ther. 2021, 6, 251. [Google Scholar] [CrossRef]
- Boons, G.; Vandamme, T.; Marien, L.; Lybaert, W.; Roeyen, G.; Rondou, T.; Papadimitriou, K.; Janssens, K.; Op de Beeck, B.; Simoens, M.; et al. Longitudinal Copy-Number Alteration Analysis in Plasma Cell-Free DNA of Neuroendocrine Neoplasms is a Novel Specific Biomarker for Diagnosis, Prognosis, and Follow-up. Clin. Cancer Res. 2022, 28, 338–349. [Google Scholar] [CrossRef]
- Reichert, Z.R.; Morgan, T.M.; Li, G.; Castellanos, E.; Snow, T.; Dall’Olio, F.G.; Madison, R.W.; Fine, A.D.; Oxnard, G.R.; Graf, R.P.; et al. Prognostic value of plasma circulating tumor DNA fraction across four common cancer types: A real-world outcomes study. Ann Oncol 2022. [Google Scholar] [CrossRef]
- Mo, S.; Dai, W.; Wang, H.; Lan, X.; Ma, C.; Su, Z.; Xiang, W.; Han, L.; Luo, W.; Zhang, L.; et al. Early detection and prognosis prediction for colorectal cancer by circulating tumour DNA methylation haplotypes: A multicentre cohort study. EClinicalMedicine 2023, 55, 101717. [Google Scholar] [CrossRef]
- Goldhirsch, A.; Wood, W.C.; Coates, A.S.; Gelber, R.D.; Thürlimann, B.; Senn, H.J. Strategies for subtypes-dealing with the diversity of breast cancer: Highlights of the St. Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2011. Ann. Oncol. 2011, 22, 1736–1747. [Google Scholar] [PubMed]
- Shi, J.; Wang, Z.; Zhang, J.; Xu, Y.; Xiao, X.; Quan, X.; Bai, Y.; Yang, X.; Ming, Z.; Guo, X.; et al. Genomic Landscape and Tumor Mutational Burden Determination of Circulating Tumor DNA in Over 5,000 Chinese Patients with Lung Cancer. Clin. Cancer Res. 2021, 27, 6184–6196. [Google Scholar] [CrossRef] [PubMed]
- Gay, C.M.; Stewart, C.A.; Park, E.M.; Diao, L.; Groves, S.M.; Heeke, S.; Nabet, B.Y.; Fujimoto, J.; Solis, L.M.; Lu, W.; et al. Patterns of transcription factor programs and immune pathway activation define four major subtypes of SCLC with distinct therapeutic vulnerabilities. Cancer Cell 2021, 39, 346–360.e7. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Brambilla, E.; Faivre-Finn, C.; Sage, J. Small-cell lung cancer. Nat. Rev. Dis. Prim. 2021, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Baine, M.K.; Hsieh, M.-S.; Lai, W.V.; Egger, J.V.; Jungbluth, A.A.; Daneshbod, Y.; Beras, A.; Spencer, R.; Lopardo, J.; Bodd, F.; et al. SCLC Subtypes Defined by ASCL1, NEUROD1, POU2F3, and YAP1: A Comprehensive Immunohistochemical and Histopathologic Characterization. J. Thorac. Oncol. 2020, 15, 1823–1835. [Google Scholar] [CrossRef]
- Chemi, F.; Pearce, S.P.; Clipson, A.; Hill, S.M.; Conway, A.M.; Richardson, S.A.; Kamieniecka, K.; Caeser, R.; White, D.J.; Mohan, S.; et al. cfDNA methylome profiling for detection and subtyping of small cell lung cancers. Nat. Cancer 2022, 3, 1260–1270. [Google Scholar] [CrossRef]
- Vandekerkhove, G.; Struss, W.J.; Annala, M.; Kallio, H.M.L.; Khalaf, D.; Warner, E.W.; Herberts, C.; Ritch, E.; Beja, K.; Loktionova, Y.; et al. Circulating Tumor DNA Abundance and Potential Utility in De Novo Metastatic Prostate Cancer. Eur. Urol. 2019, 75, 667–675. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Zhao, H.; An, K.; Liu, Z.; Hai, L.; Li, R.; Zhou, Y.; Zhao, W.; Jia, Y.; Wu, N.; et al. Whole-genome bisulfite sequencing analysis of circulating tumour DNA for the detection and molecular classification of cancer. Clin. Transl. Med. 2022, 12, e1014. [Google Scholar] [CrossRef] [PubMed]
- Leal, A.; van Grieken, N.C.T.; Palsgrove, D.N.; Phallen, J.; Medina, J.E.; Hruban, C.; Broeckaert, M.A.M.; Anagnostou, V.; Adleff, V.; Bruhm, D.C.; et al. White blood cell and cell-free DNA analyses for detection of residual disease in gastric cancer. Nat. Commun. 2020, 11, 525. [Google Scholar] [CrossRef] [Green Version]
- Qiu, B.; Guo, W.; Zhang, F.; Lv, F.; Ji, Y.; Peng, Y.; Chen, X.; Bao, H.; Xu, Y.; Shao, Y.; et al. Dynamic recurrence risk and adjuvant chemotherapy benefit prediction by ctDNA in resected NSCLC. Nat. Commun. 2021, 12, 6770. [Google Scholar] [CrossRef]
- Xia, Y.; Tang, W.; Qian, X.; Li, X.; Cheng, F.; Wang, K.; Zhang, F.; Zhang, C.; Li, D.; Song, J.; et al. Efficacy and safety of camrelizumab plus apatinib during the perioperative period in resectable hepatocellular carcinoma: A single-arm, open label, phase II clinical trial. J. Immunother. Cancer 2022, 10, e004656. [Google Scholar] [CrossRef] [PubMed]
- Parikh, A.R.; Van Seventer, E.E.; Siravegna, G.; Hartwig, A.V.; Jaimovich, A.; He, Y.; Kanter, K.; Fish, M.G.; Fosbenner, K.D.; Miao, B.; et al. Minimal Residual Disease Detection using a Plasma-only Circulating Tumor DNA Assay in Patients with Colorectal Cancer. Clin. Cancer Res. 2021, 27, 5586–5594. [Google Scholar] [CrossRef] [PubMed]
- Reinert, T.; Henriksen, T.V.; Christensen, E.; Sharma, S.; Salari, R.; Sethi, H.; Knudsen, M.; Nordentoft, I.; Wu, H.T.; Tin, A.S.; et al. Analysis of Plasma Cell-Free DNA by Ultradeep Sequencing in Patients with Stages I to III Colorectal Cancer. JAMA Oncol. 2019, 5, 1124–1131. [Google Scholar] [CrossRef]
- Loupakis, F.; Sharma, S.; Derouazi, M.; Murgioni, S.; Biason, P.; Rizzato, M.D.; Rasola, C.; Renner, D.; Shchegrova, S.; Koyen Malashevich, A.; et al. Detection of Molecular Residual Disease Using Personalized Circulating Tumor DNA Assay in Patients With Colorectal Cancer Undergoing Resection of Metastases. JCO Precis. Oncol. 2021, 5, 1166–1177. [Google Scholar] [CrossRef]
- Polivka, J.; Windrichova, J.; Pesta, M.; Houfkova, K.; Rezackova, H.; Macanova, T.; Vycital, O.; Kucera, R.; Slouka, D.; Topolcan, O. The Level of Preoperative Plasma KRAS Mutations and CEA Predict Survival of Patients Undergoing Surgery for Colorectal Cancer Liver Metastases. Cancers 2020, 12, 2434. [Google Scholar] [CrossRef]
- Tan, L.; Sandhu, S.; Lee, R.J.; Li, J.; Callahan, J.; Ftouni, S.; Dhomen, N.; Middlehurst, P.; Wallace, A.; Raleigh, J.; et al. Prediction and monitoring of relapse in stage III melanoma using circulating tumor DNA. Ann. Oncol. 2019, 30, 804–814. [Google Scholar] [CrossRef]
- Gouda, M.A.; Polivka, J.; Huang, H.J.; Treskova, I.; Pivovarcikova, K.; Fikrle, T.; Woznica, V.; Dustin, D.J.; Call, S.G.; Meric-Bernstam, F.; et al. Ultrasensitive detection of BRAF mutations in circulating tumor DNA of non-metastatic melanoma. ESMO Open 2022, 7, 100357. [Google Scholar] [CrossRef]
- Gale, D.; Heider, K.; Ruiz-Valdepenas, A.; Hackinger, S.; Perry, M.; Marsico, G.; Rundell, V.; Wulff, J.; Sharma, G.; Knock, H.; et al. Residual ctDNA after treatment predicts early relapse in patients with early-stage non-small cell lung cancer. Ann. Oncol. 2022, 33, 500–510. [Google Scholar] [CrossRef]
- Tarazona, N.; Gimeno-Valiente, F.; Gambardella, V.; Zuniga, S.; Rentero-Garrido, P.; Huerta, M.; Rosello, S.; Martinez-Ciarpaglini, C.; Carbonell-Asins, J.A.; Carrasco, F.; et al. Targeted next-generation sequencing of circulating-tumor DNA for tracking minimal residual disease in localized colon cancer. Ann. Oncol. 2019, 30, 1804–1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.S.; Han, Y.; Yun, W.G.; Kwon, W.; Kim, H.; Jeong, H.; Seo, M.S.; Park, Y.; Cho, S.I.; Kim, H.; et al. Parallel Analysis of Pre- and Postoperative Circulating Tumor DNA and Matched Tumor Tissues in Resectable Pancreatic Ductal Adenocarcinoma: A Prospective Cohort Study. Clin. Chem. 2022. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.; Cohen, J.D.; Lahouel, K.; Lo, S.N.; Wang, Y.; Kosmider, S.; Wong, R.; Shapiro, J.; Lee, M.; Harris, S.; et al. Circulating Tumor DNA Analysis Guiding Adjuvant Therapy in Stage II Colon Cancer. N. Engl. J. Med. 2022, 386, 2261–2272. [Google Scholar] [CrossRef] [PubMed]
- Cristofanilli, M.; Rugo, H.S.; Im, S.A.; Slamon, D.J.; Harbeck, N.; Bondarenko, I.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; et al. Overall Survival with Palbociclib and Fulvestrant in Women with HR+/HER2− ABC: Updated Exploratory Analyses of PALOMA-3, a Double-Blind, Phase 3 Randomized Study. Clin. Cancer Res. 2022, 28, 3433–3442. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Wu, J.; Lou, Y.; Shi, Q.; Xu, J.; Zhang, L.; Nie, W.; Qian, J.; Wang, Y.; Zhang, Y.; et al. Blood-based tumour mutation index act as prognostic predictor for immunotherapy and chemotherapy in non-small cell lung cancer patients. Biomark. Res. 2022, 10, 55. [Google Scholar] [CrossRef]
- Sartore-Bianchi, A.; Pietrantonio, F.; Lonardi, S.; Mussolin, B.; Rua, F.; Crisafulli, G.; Bartolini, A.; Fenocchio, E.; Amatu, A.; Manca, P.; et al. Circulating tumor DNA to guide rechallenge with panitumumab in metastatic colorectal cancer: The phase 2 CHRONOS trial. Nat. Med. 2022, 28, 1612–1618. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S.; Velcheti, V.; Mekhail, T.; Yun, C.; Shagan, S.M.; Hu, S.; Chae, Y.K.; Leal, T.A.; Dowell, J.E.; Tsai, M.L.; et al. Blood-based tumor mutational burden as a biomarker for atezolizumab in non-small cell lung cancer: The phase 2 B-F1RST trial. Nat. Med. 2022, 28, 939–945. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Origin of ctDNA | Selective Kit |
|---|---|
| Blood | QIAsymphony DSP Virus/Pathogen Midi Kit |
| MagMAX™ Cell-Free DNA Isolation Kit | |
| DSP Circulating DNA Kit | |
| QIAamp Circulating Nucleic Acid Kit | |
| EliteHealth cfDNA Extraction Kit | |
| Urine | QIAamp Circulating Nucleic Acid Kit |
| Cerebrospinal fluid | QIAamp Circulating Nucleic Acid Kit |
| QIAsymphony DSP Virus/Pathogen Midi Kit | |
| QIAsymphony DSP Circulating DNA Kit | |
| Feces | Fast DNA Stool Mini Kit |
| Ascites | QIAamp Circulating Nucleic Acid Kit |
| Pleural effusions | QIAamp Circulating Nucleic Acid Kit |
| Kit | Target | Cancers | Technology | Specificity | LoD | Company | Country |
|---|---|---|---|---|---|---|---|
| AVENIO ctDNA Expanded Kit | 77 gene panel (including 192Kb, ABL1, AKT1, BRAF, and PIK3CA) | Lung cancer and colorectal cancer | Next-generation sequencing | >99% | 0.10% | Roche | Switzerland |
| AVENIO ctDNA Target Kit | 17 NCCN guideline-aligned genes (81 kb) | Lung cancer and colorectal cancer | Next-generation sequencing | >99% | 0.10% | Roche | Switzerland |
| AVENIO ctDNA Surveillance Kit | 17 NCCN guideline-aligned genes plus 471 frequently mutated, disease-associated regions across 197 genes | Lung cancer and colorectal cancer | Next-generation sequencing | >99% | 0.10% | Roche | Switzerland |
| Super-ARMS® EGFR Mutation Detection Kit | Covers the 42 most frequent EGFR mutations | Non-small cell lung cancer | Real-time PCR | 100% | 0.20% | AmoyDx | China |
| OncoBEAM EGFR Kit V2 (RUO) | 36 EGFR mutations of oncogene exons 18, 19, 20, and 21 (including T790M and C797S) | Non-small cell lung cancer | Highly sensitive BEAMing digital PCR technology | >90% | 0.01% | Sysmex | Japan |
| Guardant360® CDx | Single nucleotide variants (SNVs) and insertions and deletions (indels) in 55 genes; copy number amplifications (CNAs) in 2 genes; fusions in 4 genes | Non-small cell lung cancer | Qualitative next-generation sequencing | 100% | 0.20% | Guardant Health | USA |
| FoundationOne® Liquid CDx | Substitutions and insertions and deletions (indels) in 311 genes, including rearrangements and copy number losses only in BRCA1 and BRCA2 | Non-small cell lung cancer and prostate cancer | Qualitative next-generation sequencing | 100% | 0.40% | Foundation Medicine | USA |
| Guardant Reveal™ | Not published | Early-stage colorectal, breast, and lung cancers | Next-generation sequencing | 100% | 0.01% | Guardant Health | USA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wen, X.; Pu, H.; Liu, Q.; Guo, Z.; Luo, D. Circulating Tumor DNA—A Novel Biomarker of Tumor Progression and Its Favorable Detection Techniques. Cancers 2022, 14, 6025. https://doi.org/10.3390/cancers14246025
Wen X, Pu H, Liu Q, Guo Z, Luo D. Circulating Tumor DNA—A Novel Biomarker of Tumor Progression and Its Favorable Detection Techniques. Cancers. 2022; 14(24):6025. https://doi.org/10.3390/cancers14246025
Chicago/Turabian StyleWen, Xiaosha, Huijie Pu, Quan Liu, Zifen Guo, and Dixian Luo. 2022. "Circulating Tumor DNA—A Novel Biomarker of Tumor Progression and Its Favorable Detection Techniques" Cancers 14, no. 24: 6025. https://doi.org/10.3390/cancers14246025