
Endothelial-Specific Molecule 1 Inhibition Lessens Productive Angiogenesis and Tumor Metastasis to Overcome Bevacizumab Resistance

Abstract
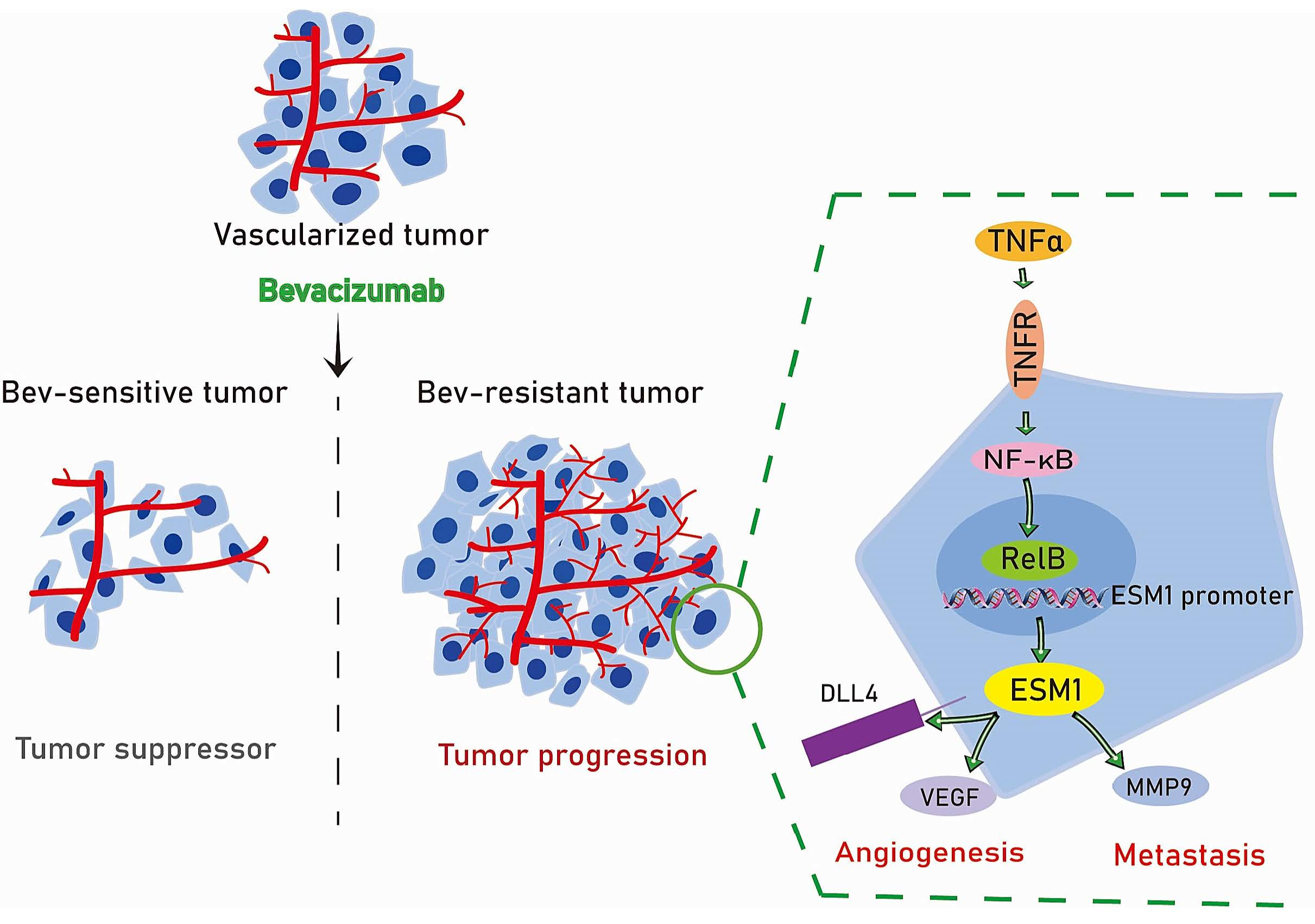
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animal Experiments
2.2. Cell Culture
2.3. Real-Time Polymerase Chain Reaction (PCR) Analysis
2.4. ESM1 Knockdown or Knock-Out Assay
2.5. Immunohistochemistry Staining
2.6. Cell Migration and Invasion
2.7. MTT Assay
2.8. ELISA
2.9. Anti-ESM1 mAbs Preparation
2.10. Statistical Analyses
3. Results
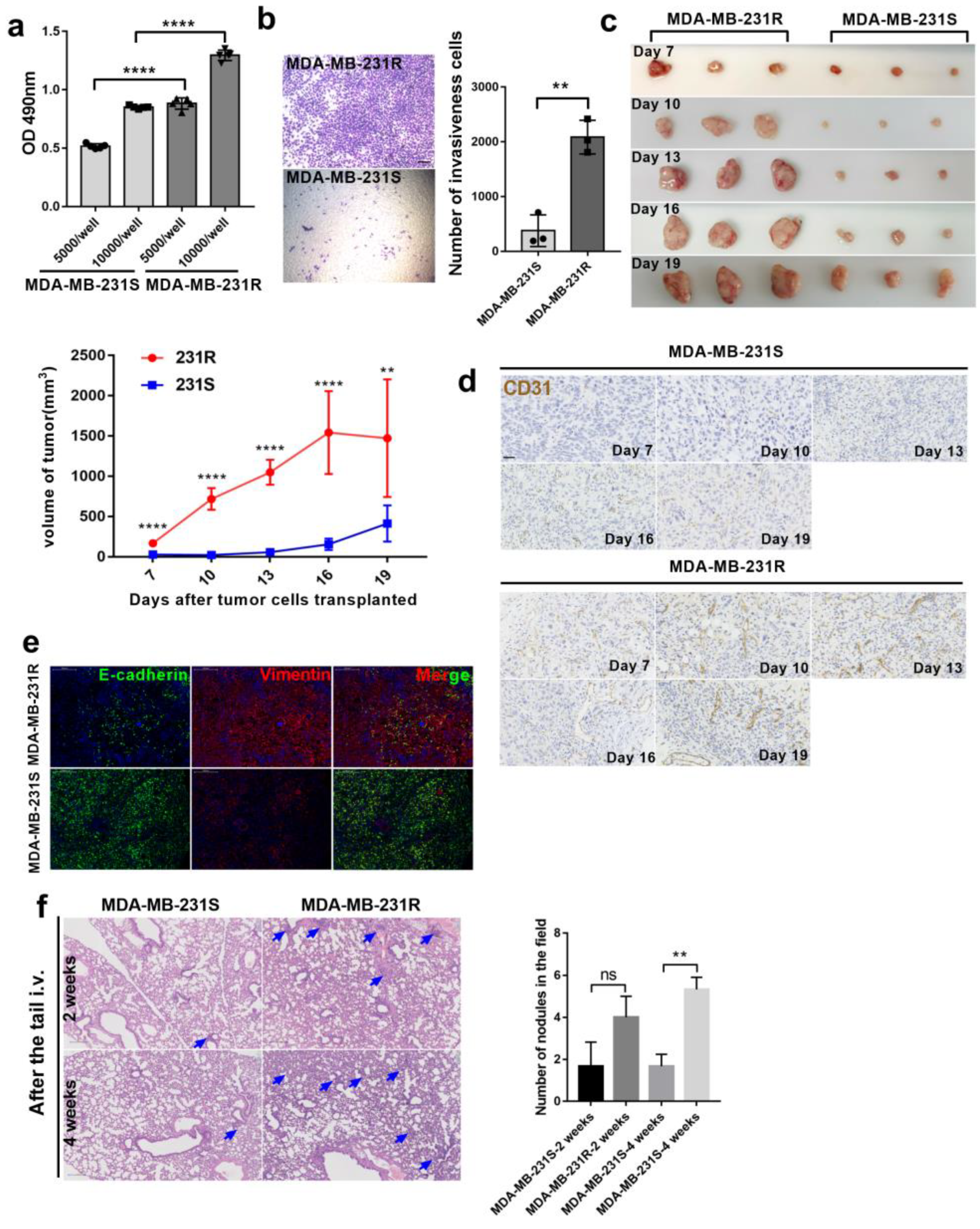
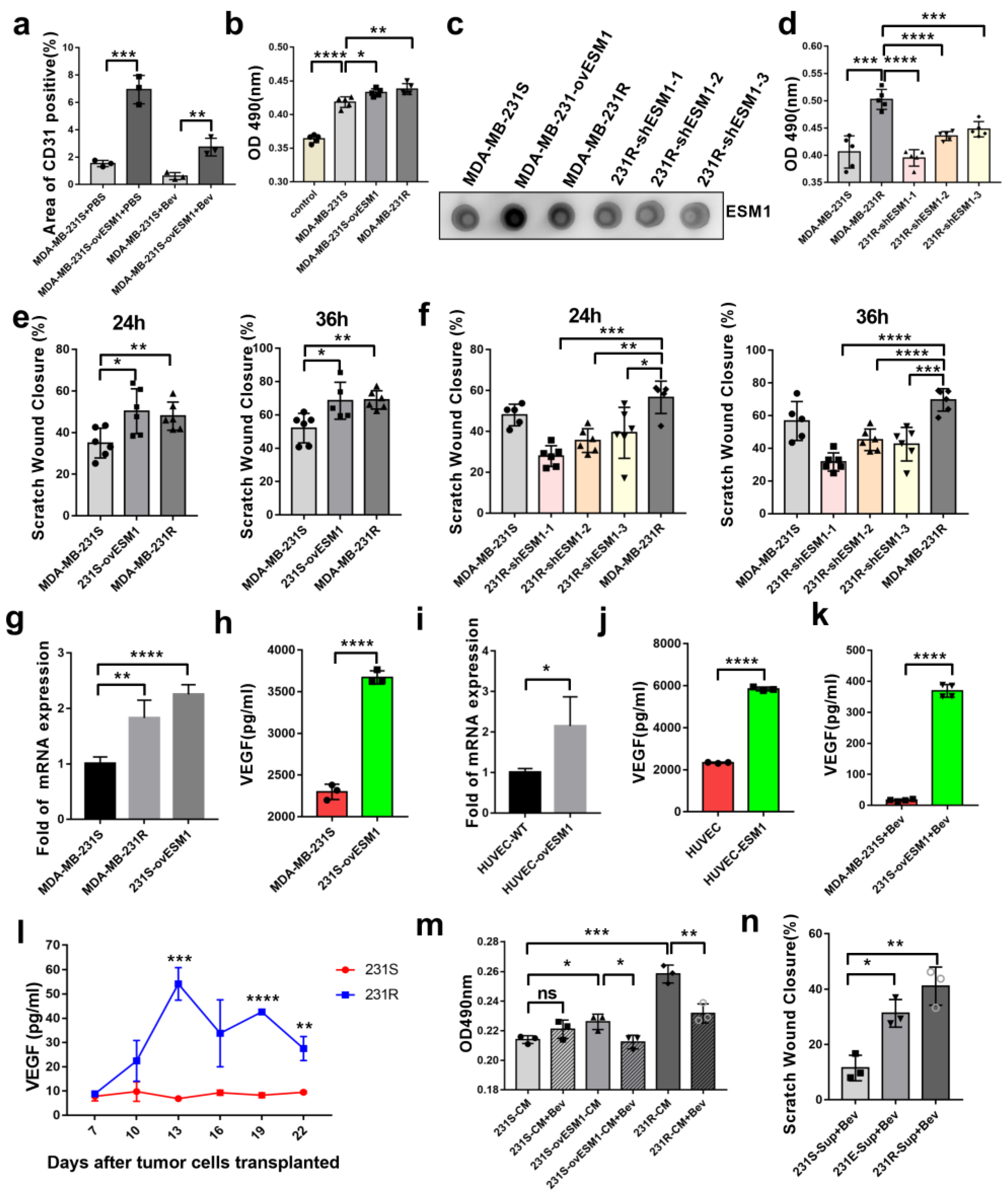
3.1. Bevacizumab-Resistant Tumor Cells Become More Aggressive In Vitro and In Vivo
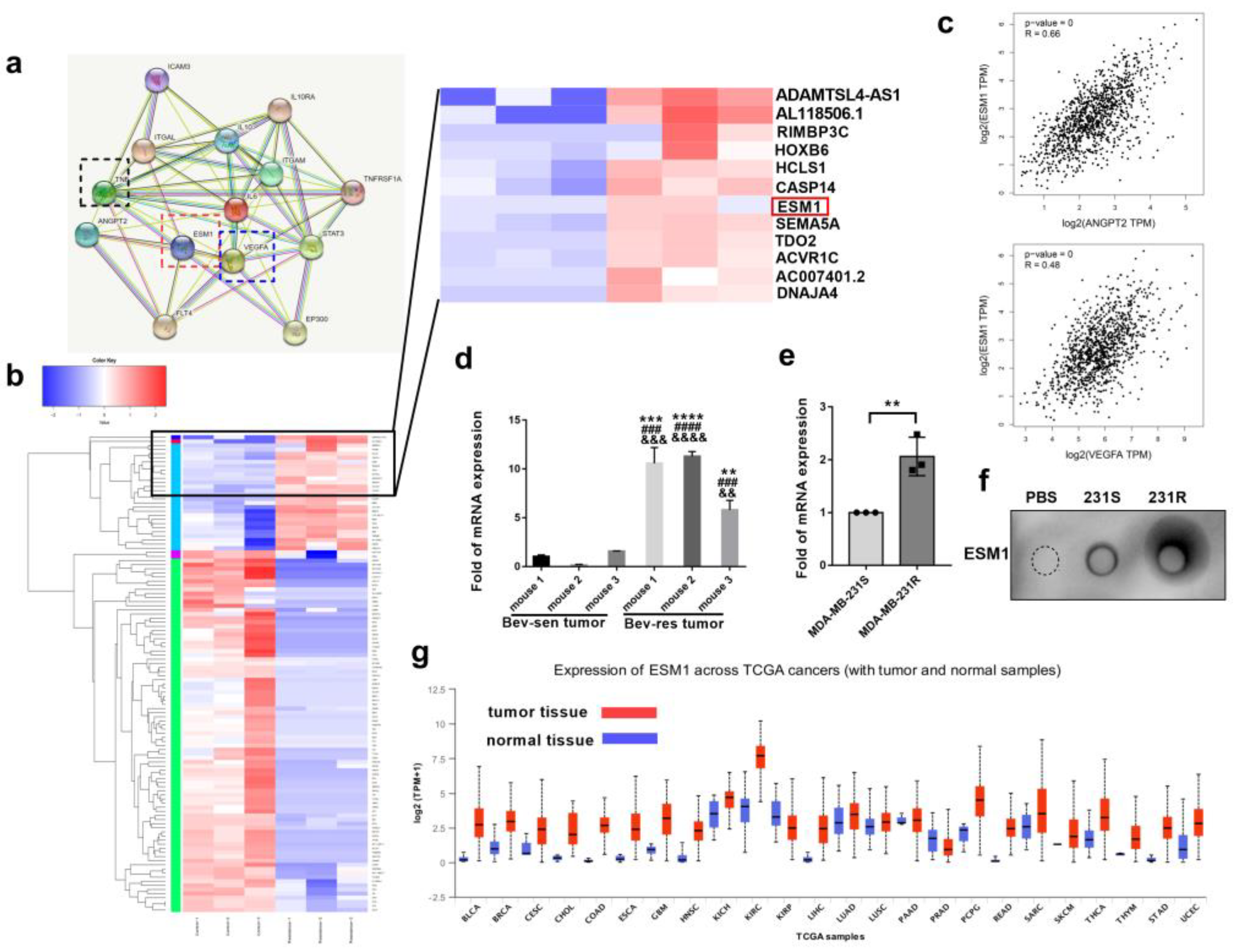
3.2. ESM1 Is Overexpressed in Bevacizumab-Resistant Tumor Cells and Correlates with High Occurrence of Metastasis
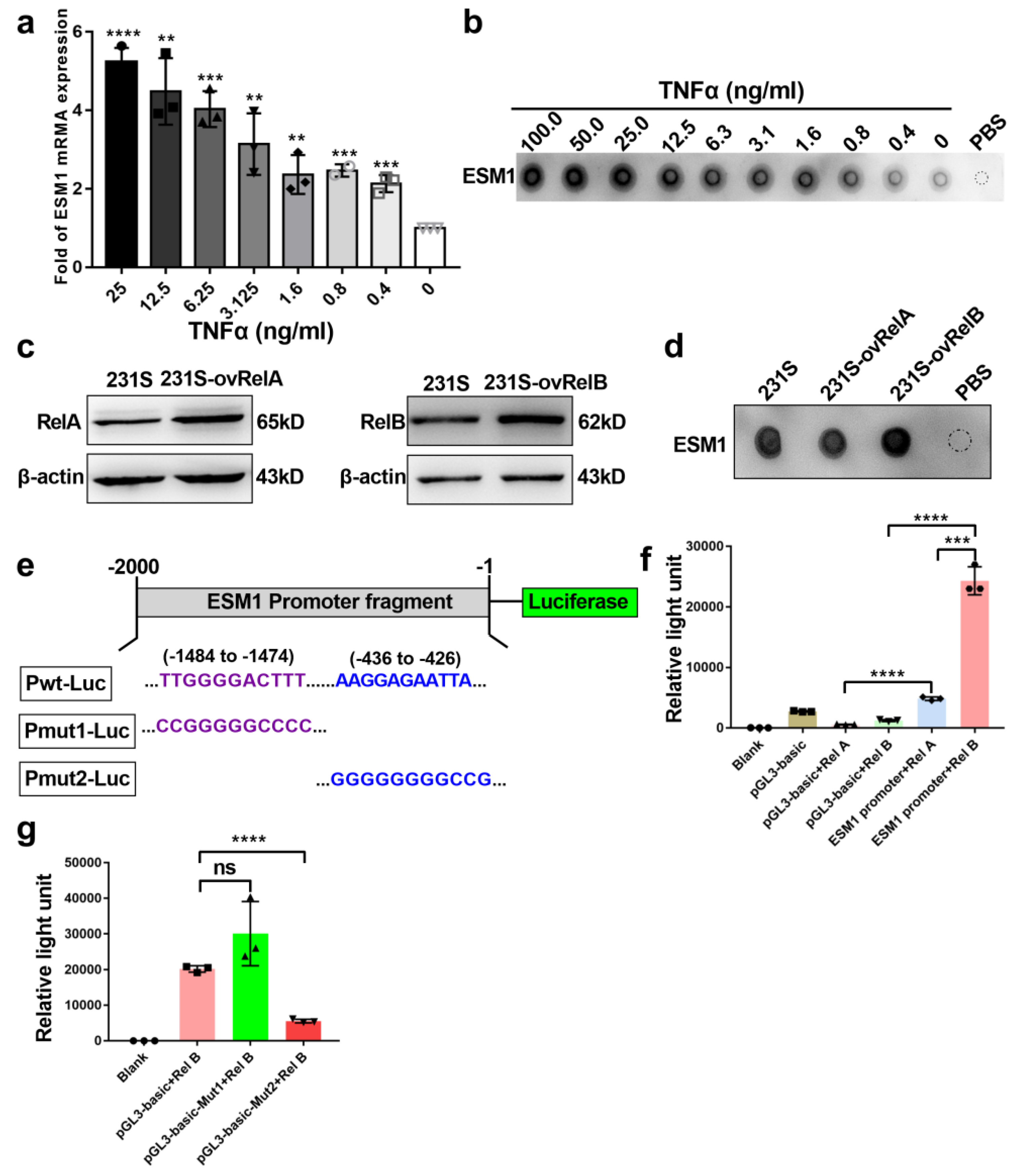
3.3. ESM1 Is Regulated by TNFα- NF-κB- RelB Axis
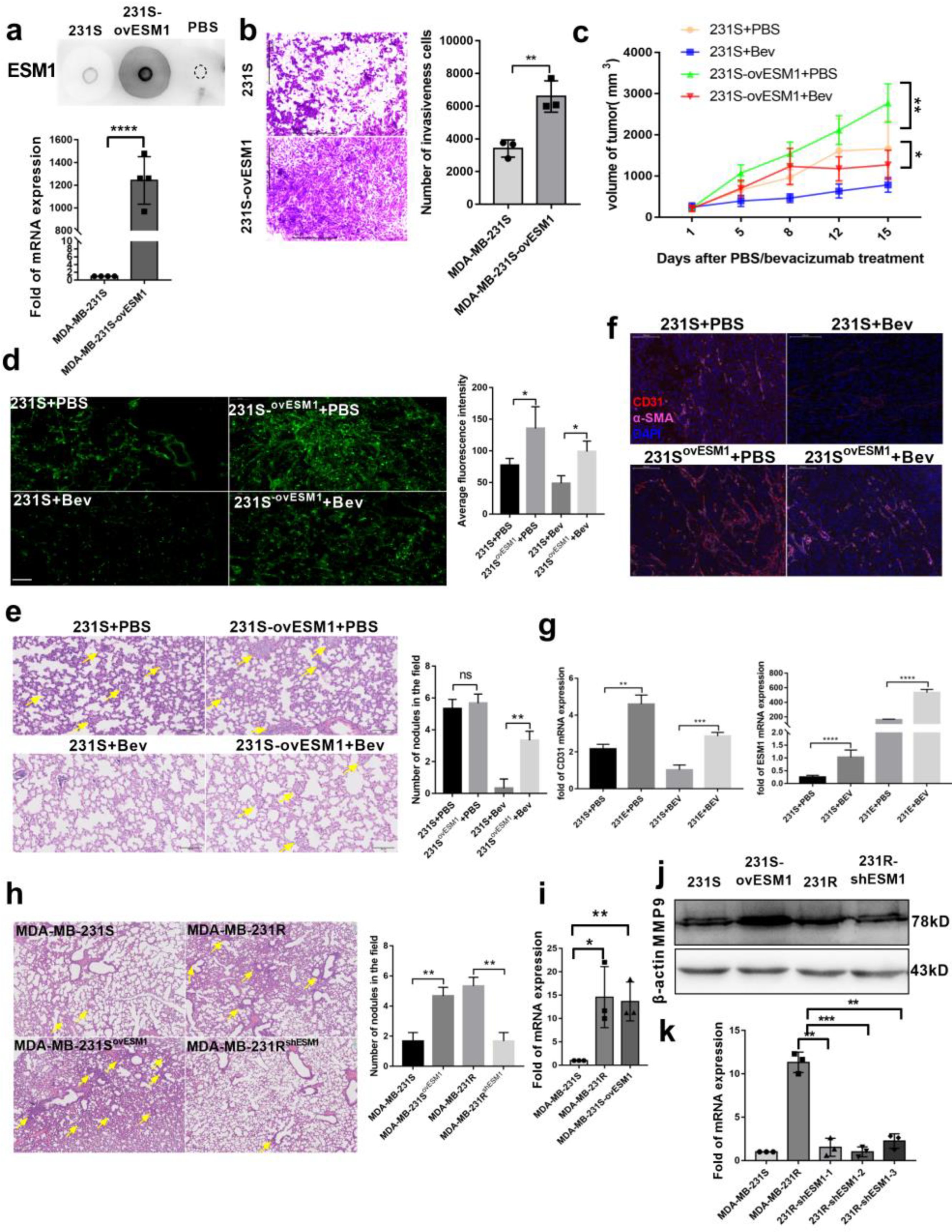
3.4. ESM1 Promoted the Proliferation and Migration of Endothelial Cell In Vitro
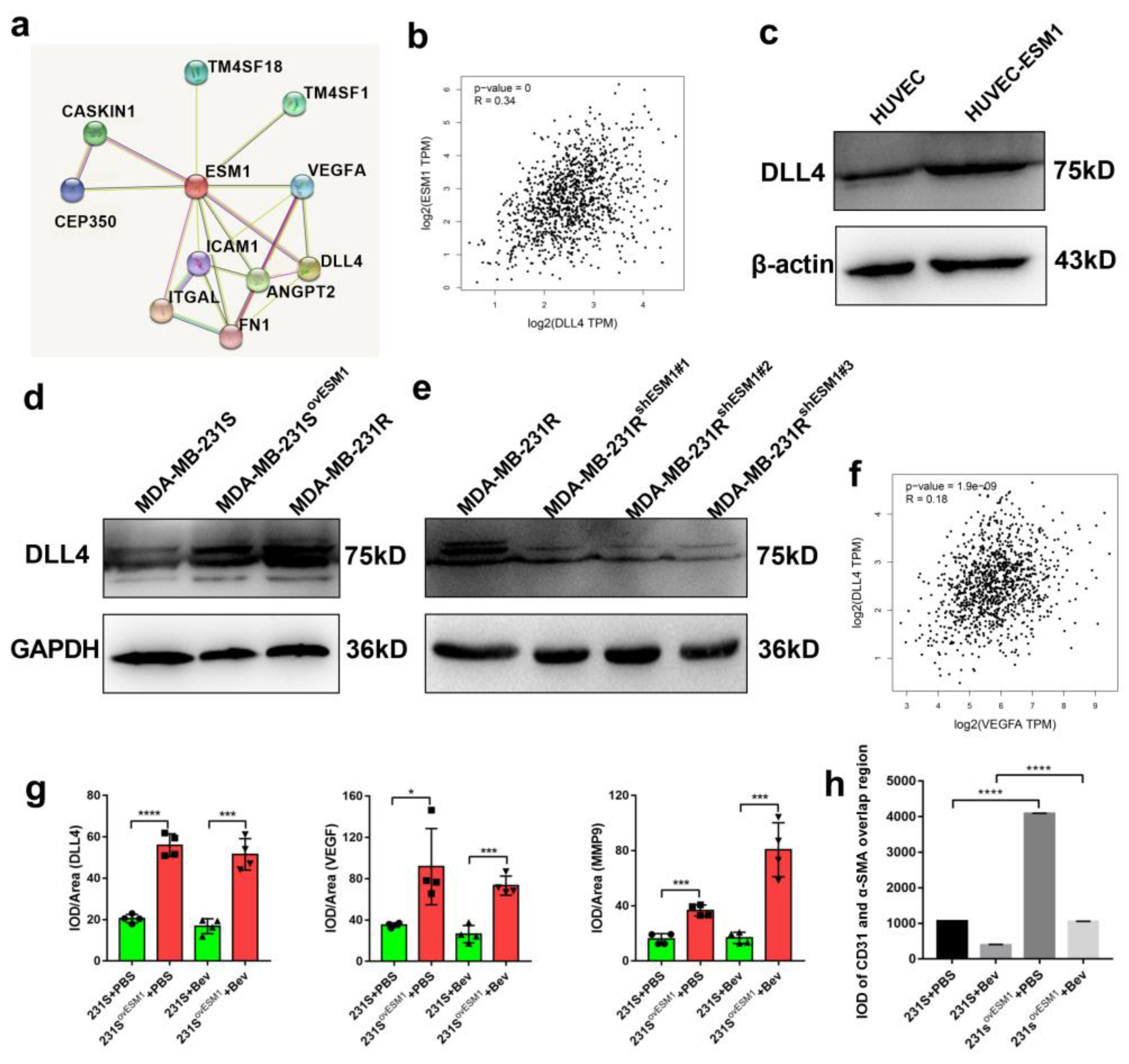
3.5. Effect of ESM1 on In Vivo Productive Angiogenesis, VEGF and DLL4
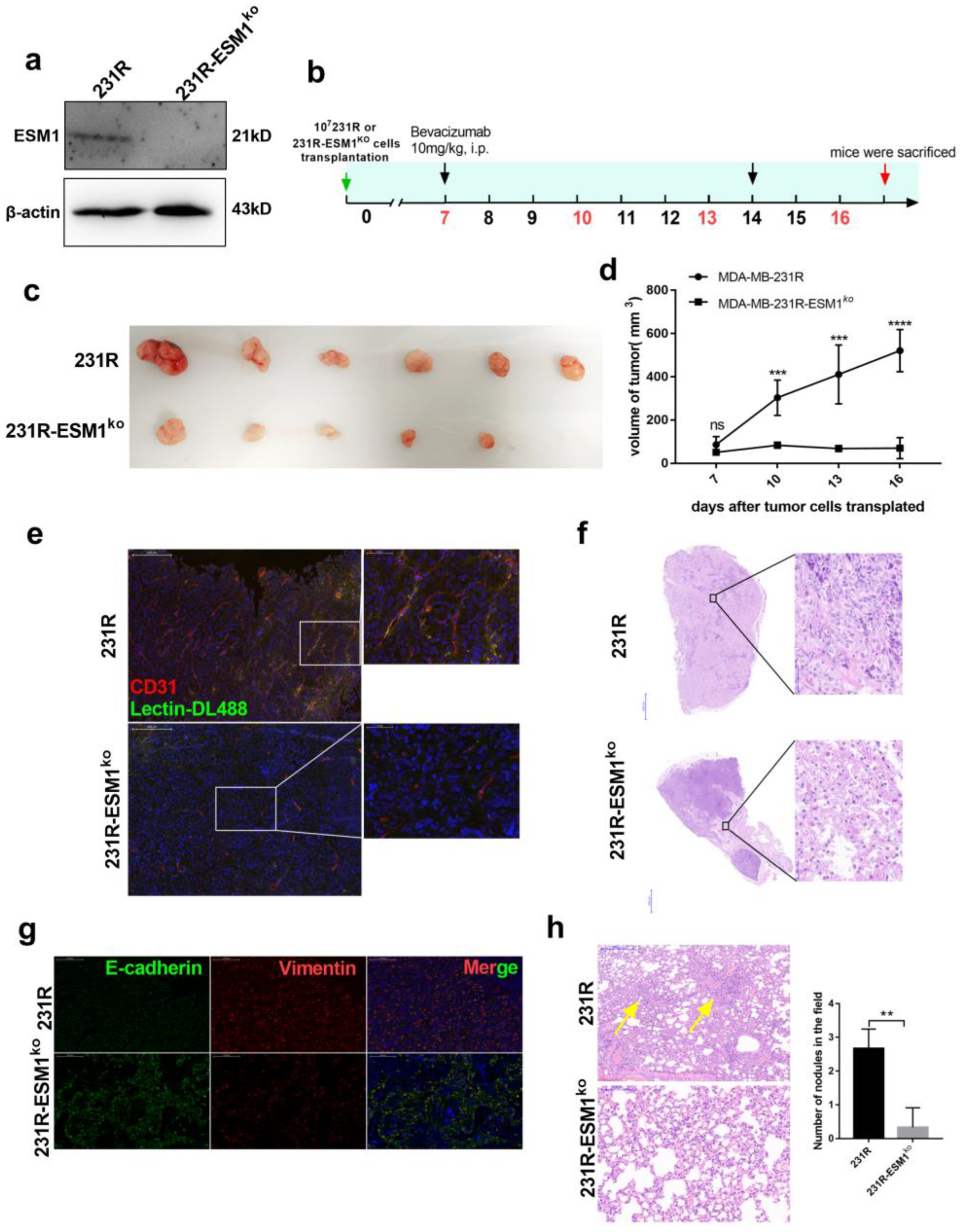
3.6. ESM1 Deletion in Bevacizumab Resistant Cells Inhibits Tumor Growth In Vivo
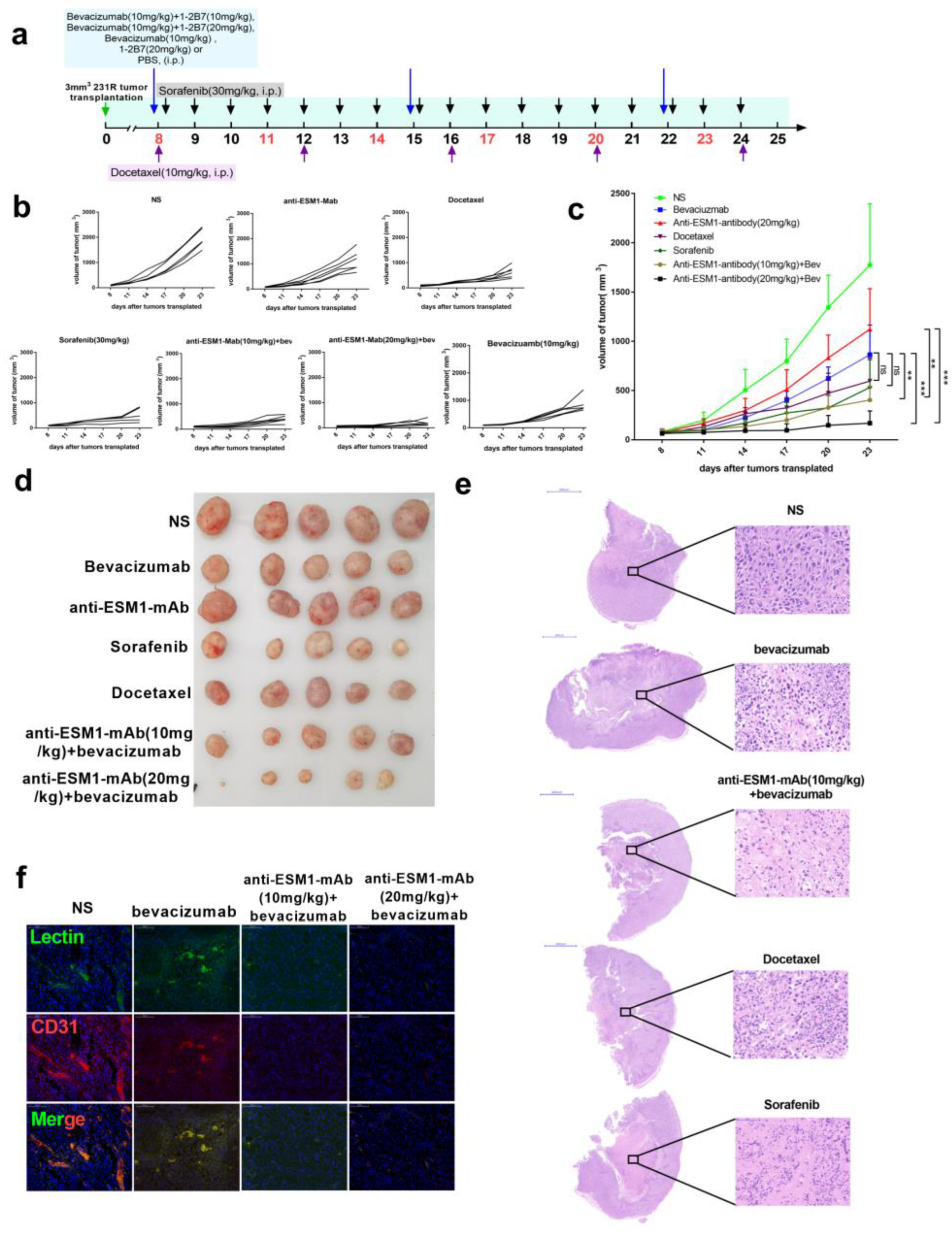
3.7. Targeting ESM1 with an Anti-ESM1 Monoclonal Antibody on Bevacizumab Resistance Increased the Effect of Bevacizumab
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bear, H.D.; Tang, G.; Rastogi, P.; Geyer, C.E., Jr.; Robidoux, A.; Atkins, J.N.; Baez-Diaz, L.; Brufsky, A.M.; Mehta, R.S.; Fehrenbacher, L.; et al. Bevacizumab added to neoadjuvant chemotherapy for breast cancer. N. Engl. J. Med. 2012, 366, 310–320. [Google Scholar] [CrossRef] [Green Version]
- Seto, T.; Higashiyama, M.; Funai, H.; Imamura, F.; Uematsu, K.; Seki, N.; Eguchi, K.; Yamanaka, T.; Ichinose, Y. Prognostic value of expression of vascular endothelial growth factor and its flt-1 and KDR receptors in stage I non-small-cell lung cancer. Lung Cancer 2006, 53, 91–96. [Google Scholar] [CrossRef]
- Goh, B.K.; Yong, W.S. Eighteen-year results in the treatment of early breast carcinoma with mastectomy versus breast conservation therapy. Cancer 2004, 100, 1766–1767. [Google Scholar] [CrossRef]
- Flynn, J.R.; Wang, L.; Gillespie, D.L.; Stoddard, G.J.; Reid, J.K.; Owens, J.; Ellsworth, G.B.; Salzman, K.L.; Kinney, A.Y.; Jensen, R.L. Hypoxia-regulated protein expression, patient characteristics, and preoperative imaging as predictors of survival in adults with glioblastoma multiforme. Cancer 2008, 113, 1032–1042. [Google Scholar] [CrossRef] [PubMed]
- Pamela, K.A.S.; Paley, J.; Gebhard, K.; Mohanraj, D.; Twiggs, L.B.; Carson, F.L.; Ramakrishnan, S. Vascular Endothelial Growth Factor Expression in Early Stage Ovarian Carcinoma. Cancer 1997, 80, 98–106. [Google Scholar]
- Barton, M.K. Bevacizumab in neoadjuvant chemotherapy increases the pathological complete response rate in patients with triple-negative breast cancer. CA Cancer J. Clin. 2014, 64, 155–156. [Google Scholar] [CrossRef] [PubMed]
- Ramezani, S.; Vousooghi, N.; Joghataei, M.T.; Chabok, S.Y. The Role of Kinase Signaling in Resistance to Bevacizumab Therapy for Glioblastoma Multiforme. Cancer Biother. Radiopharm. 2019, 34, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Otani, Y.; Ichikawa, T.; Kurozumi, K.; Inoue, S.; Ishida, J.; Oka, T.; Shimizu, T.; Tomita, Y.; Hattori, Y.; Uneda, A.; et al. Fibroblast growth factor 13 regulates glioma cell invasion and is important for bevacizumab-induced glioma invasion. Oncogene 2017, 37, 777–786. [Google Scholar] [CrossRef] [Green Version]
- Lu, K.V. Mechanisms of evasive resistance to anti-VEGF therapy in glioblastoma. Future Med. 2013, 2, 49–65. [Google Scholar] [CrossRef]
- Hu, Y.L.; DeLay, M.; Jahangiri, A.; Molinaro, A.M.; Rose, S.D.; Carbonell, W.S.; Aghi, M.K. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res. 2012, 72, 1773–1783. [Google Scholar] [CrossRef] [Green Version]
- Horikawa, N.A.; Matsumura, K.; Baba, N.; Hamanishi, T.; Yamaguchi, J.; Murakami, K.; Taki, R.; Ukita, M.; Hosoe, M.; Koshiyama, Y.; et al. Anti-VEGF therapy resistance in ovarian cancer is caused by GM-CSF-induced myeloid-derived suppressor cell recruitment. Br. J. Cancer 2020, 122, 778–788. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Wang, S.; Li, C.; Guo, M.; Xu, Y.; Sun, X.; Yu, J.; Wang, L. The Kinetic Changes of Systemic Inflammatory Factors during Bevacizumab Treatment and Its Prognostic Role in Advanced Non-small Cell Lung Cancer Patients. J. Cancer 2019, 10, 5082–5089. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, M.; Inoue, A.; Ohnishi, T.; Yano, H.; Kanemura, Y.; Kohno, S.; Ohue, S.; Ozaki, S.; Matsumoto, S.; Suehiro, S.; et al. CD44 expression in the tumor periphery predicts the responsiveness to bevacizumab in the treatment of recurrent glioblastoma. Cancer Med. 2021, 10, 2013–2025. [Google Scholar] [CrossRef] [PubMed]
- Gerstner, E.R.; Emblem, K.E.; Yen, Y.F.; Dietrich, J.; Jordan, J.T.; Catana, C.; Wenchin, K.L.; Hooker, J.M.; Duda, D.G.; Rosen, B.R.; et al. Vascular dysfunction promotes regional hypoxia after bevacizumab therapy in recurrent glioblastoma patients. Neurooncol. Adv. 2020, 2, vdaa157. [Google Scholar] [CrossRef]
- Feng, H.; Liu, K.; Shen, X.; Liang, J.; Wang, C.; Qiu, W.; Cheng, X.; Zhao, R. Targeting tumor cell-derived CCL2 as a strategy to overcome Bevacizumab resistance in ETV5(+) colorectal cancer. Cell Death Dis. 2020, 11, 916. [Google Scholar] [CrossRef]
- Liu, Y.; Ji, X.; Kang, N.; Zhou, J.; Liang, X.; Li, J.; Han, T.; Zhao, C.; Yang, T. Tumor necrosis factor alpha inhibition overcomes immunosuppressive M2b macrophage-induced bevacizumab resistance in triple-negative breast cancer. Cell Death Dis. 2020, 11, 993. [Google Scholar] [CrossRef]
- Unterleuthner, D.; Neuhold, P.; Schwarz, K.; Janker, L.; Neuditschko, B.; Nivarthi, H.; Crncec, I.; Kramer, N.; Unger, C.; Hengstschlager, M.; et al. Cancer-associated fibroblast-derived WNT2 increases tumor angiogenesis in colon cancer. Angiogenesis 2020, 23, 159–177. [Google Scholar] [CrossRef] [Green Version]
- Olejarz, W.; Kubiak-Tomaszewska, G.; Chrzanowska, A.; Lorenc, T. Exosomes in Angiogenesis and Anti-angiogenic Therapy in Cancers. Int. J. Mol. Sci. 2020, 21, 5840. [Google Scholar] [CrossRef]
- Li, J.; Liu, X.; Zang, S.; Zhou, J.; Zhang, F.; Sun, B.; Qi, D.; Li, X.; Kong, J.; Jin, D.; et al. Small extracellular vesicle-bound vascular endothelial growth factor secreted by carcinoma-associated fibroblasts promotes angiogenesis in a bevacizumab-resistant manner. Cancer Lett. 2020, 492, 71–83. [Google Scholar] [CrossRef]
- Delehedde, M.; Devenyns, L.; Maurage, C.A.; Vives, R.R. Endocan in cancers: A lesson from a circulating dermatan sulfate proteoglycan. Int. J. Cell Biol. 2013, 2013, 705027. [Google Scholar] [CrossRef] [Green Version]
- Lassalle, P.; Molet, S.; Janin, A.; Heyden, J.V.; Tavernier, J.; Fiers, W.; Devos, R.; Tonnel, A.B. ESM-1 is a novel human endothelial cell-specific molecule expressed in lung and regulated by cytokines. J. Biol. Chem. 1996, 271, 20458–20464. [Google Scholar] [CrossRef] [PubMed]
- Bechard, D.; Meignin, V.; Scherpereel, A.; Oudin, S.; Kervoaze, G.; Bertheau, P.; Janin, A.; Tonnel, A.-B.; Lassalle, P. Characterization of the Secreted Form of Endothelial-Cell-Specific Molecule 1 by Specific Monoclonal Antibodies. J. Vasc. Res. 2000, 37, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.C.; Pan, K.F.; Lee, W.J.; Chang, J.H.; Tan, P.; Gu, C.C.; Chang, W.M.; Yang, S.F.; Hsiao, M.; Hua, K.T.; et al. Circulating Proteoglycan Endocan Mediates EGFR-Driven Progression of Non-Small Cell Lung Cancer. Cancer Res. 2020, 80, 3292–3304. [Google Scholar] [CrossRef] [PubMed]
- Kano, K.; Sakamaki, K.; Oue, N.; Kimura, Y.; Hashimoto, I.; Hara, K.; Maezawa, Y.; Aoyama, T.; Fujikawa, H.; Hiroshima, Y.; et al. Impact of the ESM-1 Gene Expression on Outcomes in Stage II/III Gastric Cancer Patients Who Received Adjuvant S-1 Chemotherapy. In Vivo 2019, 34, 461–467. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Rugira, T.; Ko, Y.S.; Park, S.W.; Yun, S.P.; Kim, H.J. ESM-1 Overexpression is Involved in Increased Tumorigenesis of Radiotherapy-Resistant Breast Cancer Cells. Cancers 2020, 12, 1363. [Google Scholar] [CrossRef] [PubMed]
- Feng, R.; Li, Z.; Wang, X.; Ge, G.; Jia, Y.; Wu, D.; Ji, Y.; Wang, C. Silenced lncRNA SNHG14 restrains the biological behaviors of bladder cancer cells via regulating microRNA-211-3p/ESM1 axis. Cancer Cell Int. 2021, 21, 67. [Google Scholar] [CrossRef]
- Yang, J.; Yang, Q.; Yu, S.; Zhang, X. Endocan: A new marker for cancer and a target for cancer therapy. Biomed. Rep. 2015, 3, 279–283. [Google Scholar] [CrossRef] [Green Version]
- Scherpereel, A.; Gentina, T.; Grigoriu, B.; Sénéchal, S.; Janin, A.; Tsicopoulos, A.; Plénat, F.; Béchard, D.; Tonnel, A.-B.; Lassalle, P. Overexpression of Endocan Induces Tumor Formation. Cancer Res. 2003, 63, 6084–6089. [Google Scholar]
- Kang, Y.H.; Ji, N.Y.; Han, S.R.; Lee, C.I.; Kim, J.W.; Yeom, Y.I.; Kim, Y.H.; Chun, H.K.; Kim, J.W.; Chung, J.W.; et al. ESM-1 regulates cell growth and metastatic process through activation of NF-kappaB in colorectal cancer. Cell. Signal. 2012, 24, 1940–1949. [Google Scholar] [CrossRef]
- del Toro, R.; Prahst, C.; Mathivet, T.; Siegfried, G.; Kaminker, J.S.; Larrivee, B.; Breant, C.; Duarte, A.; Takakura, N.; Fukamizu, A.; et al. Identification and functional analysis of endothelial tip cell-enriched genes. Blood 2010, 116, 4025–4033. [Google Scholar] [CrossRef] [Green Version]
- Thurston, G.; Noguera-Troise, I.; Yancopoulos, G.D. The Delta paradox: DLL4 blockade leads to more tumour vessels but less tumour growth. Nat. Rev. 2007, 7, 327–331. [Google Scholar] [CrossRef]
- Gale, N.W.; Dominguez, M.G.; Noguera, I.; Pan, L.; Hughes, V.; Valenzuela, D.M.; Murphy, A.J.; Adams, N.C.; Lin, H.C.; Holash, J.; et al. Haploinsufficiency of delta-like 4 ligand results in embryonic lethality due to major defects in arterial and vascular development. Proc. Natl. Acad. Sci. USA 2004, 101, 15949–15954. [Google Scholar] [CrossRef] [PubMed]
- Duarte, A.; Hirashima, M.; Benedito, R.; Trindade, A.; Diniz, P.; Bekman, E.; Costa, L.; Henrique, D.; Rossant, J. Dosage-sensitive requirement for mouse Dll4 in artery development. Genes Dev. 2004, 18, 2474–2478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noguera-Troise, C.D.I.; Papadopoulos, N.J.; Coetzee, S.; Boland, P.; Gale, N.W.; Lin, H.C.; Yancopoulos, G.D.; Thurston, G. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Novartis Found Symp. 2007, 283, 106–125. [Google Scholar]
- Mailhos, U.M.C.; Harris, J.L.A.; Bicknell, R.; Ish-Horowicz, D. Delta4, an endothelial specific Notch ligand expressed at sites of physiological and tumor angiogenesis. Differentiation 2001, 69, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Zohny, S.F.; Zamzami, M.A.; Al-Malki, A.L.; Trabulsi, N.H. Highly Expressed DLL4 and JAG1: Their Role in Incidence of Breast Cancer Metastasis. Arch. Med. Res. 2020, 51, 145–152. [Google Scholar] [CrossRef]
- Negri, F.; Bozzetti, C.; Pedrazzi, G.; Azzoni, C.; Bottarelli, L.; Squadrilli, A.; Lagrasta, C.; Tamagnini, I.; Bisagni, A.; Ragazzi, M.; et al. High levels of Notch intracellular cleaved domain are associated with stemness and reduced bevacizumab efficacy in patients with advanced colon cancer. Oncol. Rep. 2019, 42, 2750–2758. [Google Scholar] [CrossRef]
- Yeom, D.H.; Lee, Y.S.; Ryu, I.; Lee, S.; Sung, B.; Lee, H.B.; Kim, D.; Ahn, J.H.; Ha, E.; Choi, Y.S.; et al. ABL001, a Bispecific Antibody Targeting VEGF and DLL4, with Chemotherapy, Synergistically Inhibits Tumor Progression in Xenograft Models. Int. J. Mol. Sci. 2020, 22, 241. [Google Scholar] [CrossRef]
- Gordon, M.S.; Nemunaitis, J.; Barve, M.; Wainberg, Z.A.; Hamilton, E.P.; Ramanathan, R.K.; Sledge, G.W., Jr.; Yue, H.; Morgan-Lappe, S.E.; Blaney, M.; et al. Phase I Open-Label Study Evaluating the Safety, Pharmacokinetics, and Preliminary Efficacy of Dilpacimab in Patients with Advanced Solid Tumors. Mol. Cancer Ther. 2021, 20, 1988–1995. [Google Scholar] [CrossRef]
- Zhang, H.; Shen, Y.-W.; Zhang, L.-J.; Chen, J.-J.; Bian, H.-T.; Gu, W.-J.; Zhang, H.; Chen, H.-Z.; Zhang, W.-D.; Luan, X. Targeting Endothelial Cell-Specific Molecule 1 Protein in Cancer: A Promising Therapeutic Approach. Front. Oncol. 2021, 11, 2002. [Google Scholar] [CrossRef]
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the NFkappaB system. Wiley. Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 227–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parmar, D.; Apte, M. Angiopoietin inhibitors: A review on targeting tumor angiogenesis. Eur. J. Pharmacol. 2021, 899, 174021. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Ku, S.K.; Kim, S.W.; Bae, J.S. Endocan elicits severe vascular inflammatory responses in vitro and in vivo. J. Cell Physiol. 2014, 229, 620–630. [Google Scholar] [CrossRef] [PubMed]
- Lobov, I.; Mikhailova, N. The Role of Dll4/Notch Signaling in Normal and Pathological Ocular Angiogenesis: Dll4 Controls Blood Vessel Sprouting and Vessel Remodeling in Normal and Pathological Conditions. J. Ophthalmol. 2018, 2018, 3565292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, C.; Wu, C.; Xue, R.; Jin, C.; Zheng, C. Suppression of human colon tumor by EERAC through regulating Notch/DLL4/Hes pathway inhibiting angiogenesis in vivo. J. Cancer 2021, 12, 5914–5922. [Google Scholar] [CrossRef]
- Watanabe, M.; Horie, H.; Kurata, Y.; Inoue, Y.; Notsu, T.; Wakimizu, T.; Adachi, M.; Yamamoto, K.; Morikawa, K.; Kuwabara, M.; et al. Esm1 and Stc1 as Angiogenic Factors Responsible for Protective Actions of Adipose-Derived Stem Cell Sheets on Chronic Heart Failure After Rat Myocardial Infarction. Circ. J. 2021, 85, 657–666. [Google Scholar] [CrossRef]
- Rocha, S.F.; Schiller, M.; Jing, D.; Li, H.; Butz, S.; Vestweber, D.; Biljes, D.; Drexler, H.C.; Nieminen-Kelha, M.; Vajkoczy, P.; et al. Esm1 modulates endothelial tip cell behavior and vascular permeability by enhancing VEGF bioavailability. Circ. Res. 2014, 115, 581–590. [Google Scholar] [CrossRef] [Green Version]
- Grigoriu, B.D.; Depontieu, F.; Scherpereel, A.; Gourcerol, D.; Devos, P.; Ouatas, T.; Lafitte, J.-J.; Copin, M.-C.; Tonnel, A.-B.; Lassalle, P. Endocan Expression and Relationship with Survival in Human Non–Small Cell Lung Cancer. Clin. Cancer Res. 2006, 12, 4575–4582. [Google Scholar] [CrossRef] [Green Version]
- Leroy, X.; Aubert, S.; Zini, L.; Franquet, H.; Kervoaze, G.; Villers, A.; Delehedde, M.; Copin, M.-C.; Lassalle, P. Vascular endocan (ESM-1) is markedly overexpressed in clear cell renal cell carcinoma. Histopathology 2010, 56, 180–187. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, N.; Liang, X.; Fan, B.; Zhao, C.; Shen, B.; Ji, X.; Liu, Y. Endothelial-Specific Molecule 1 Inhibition Lessens Productive Angiogenesis and Tumor Metastasis to Overcome Bevacizumab Resistance. Cancers 2022, 14, 5681. https://doi.org/10.3390/cancers14225681
Kang N, Liang X, Fan B, Zhao C, Shen B, Ji X, Liu Y. Endothelial-Specific Molecule 1 Inhibition Lessens Productive Angiogenesis and Tumor Metastasis to Overcome Bevacizumab Resistance. Cancers. 2022; 14(22):5681. https://doi.org/10.3390/cancers14225681
Chicago/Turabian StyleKang, Nannan, Xue Liang, Buxi Fan, Chen Zhao, Beiyu Shen, Xuemei Ji, and Yu Liu. 2022. "Endothelial-Specific Molecule 1 Inhibition Lessens Productive Angiogenesis and Tumor Metastasis to Overcome Bevacizumab Resistance" Cancers 14, no. 22: 5681. https://doi.org/10.3390/cancers14225681