
Combating Drug Resistance by Exploiting miRNA-200c-Controlled Phase II Detoxification
 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
- Sense: 5′ UAAUACUGCCGGGUAAUGAUGGA 3′;
- Antisense: 5′ UCCAUCAUUACCCGGCAGUAUUA 3′.
- The control siRNA duplex with a scrambled sequence was also obtained from AxoLabs:
- Sense: 5′ AuGuAuuGGccuGuAuuAGdTsdT 3′;
- Antisense: 5′ CuAAuAcAGGCcAAuAcAUdTsdT 3′.
2.2. Cell Culture
2.3. Proteomics Sample Preparation
2.4. Proteomics LC-MS/MS Analysis
2.5. LC-MS/MS Data Analysis
2.6. Generation of 3′UTR GSTM3 Mutations in pISO
2.7. Co-Transfection and Luciferase-Reporter Assay
2.8. RNA-Lysis and Purification
2.9. cDNA Synthesis
2.10. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.11. Protein Lysis and Western Blot
2.12. Analysis of Total Glutathione Using the GSH/GSSG-Glo Assay
2.13. Evaluation of Cell Death Using Propidium Iodide Assay
2.14. Analysis of Long-Term Effects of hsa-miR-200c In Vitro Using the Cellwatcher System
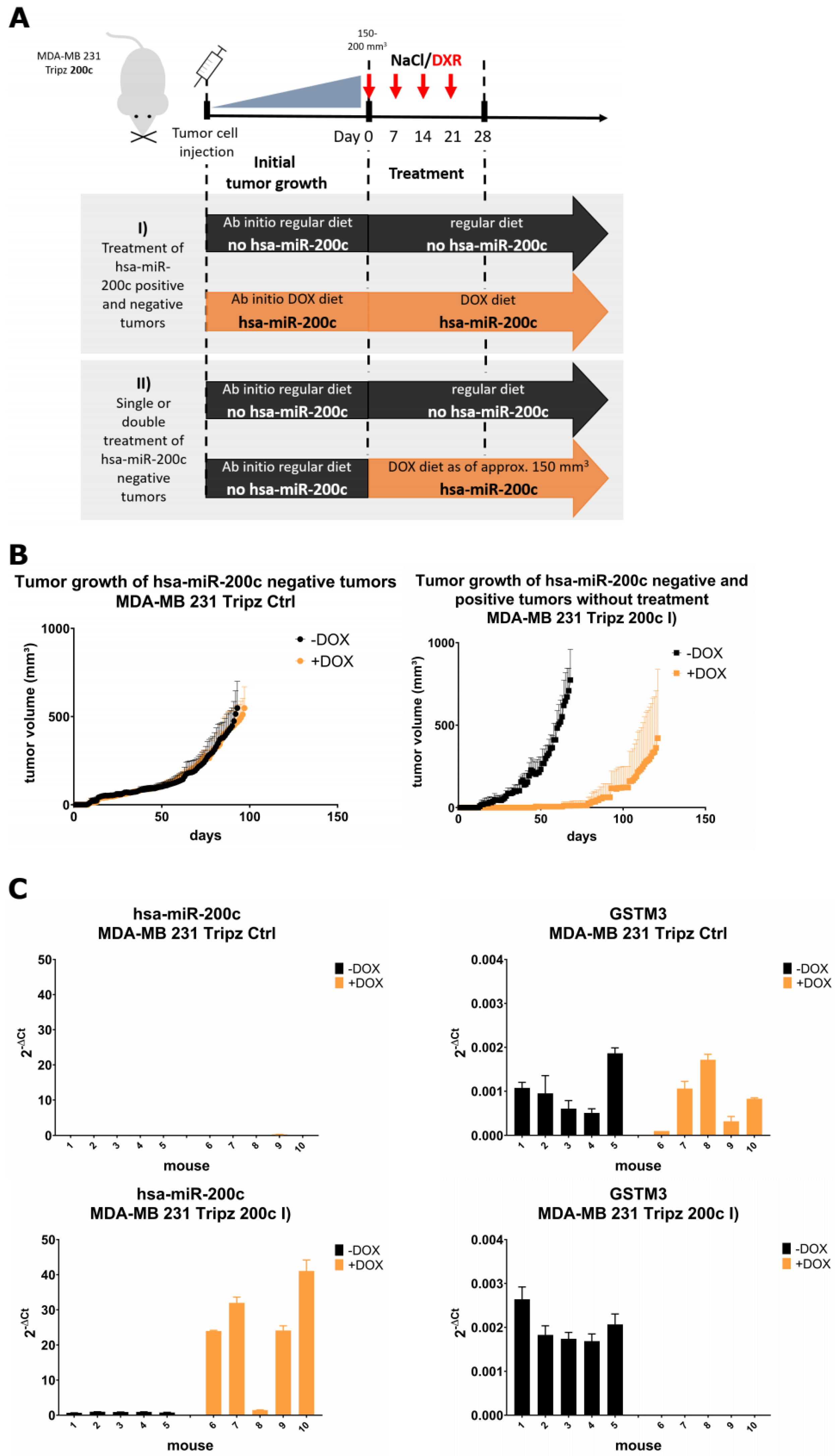
2.15. In Vivo Xenograft Studies of hsa-miR-200c as Genetic Biomarker
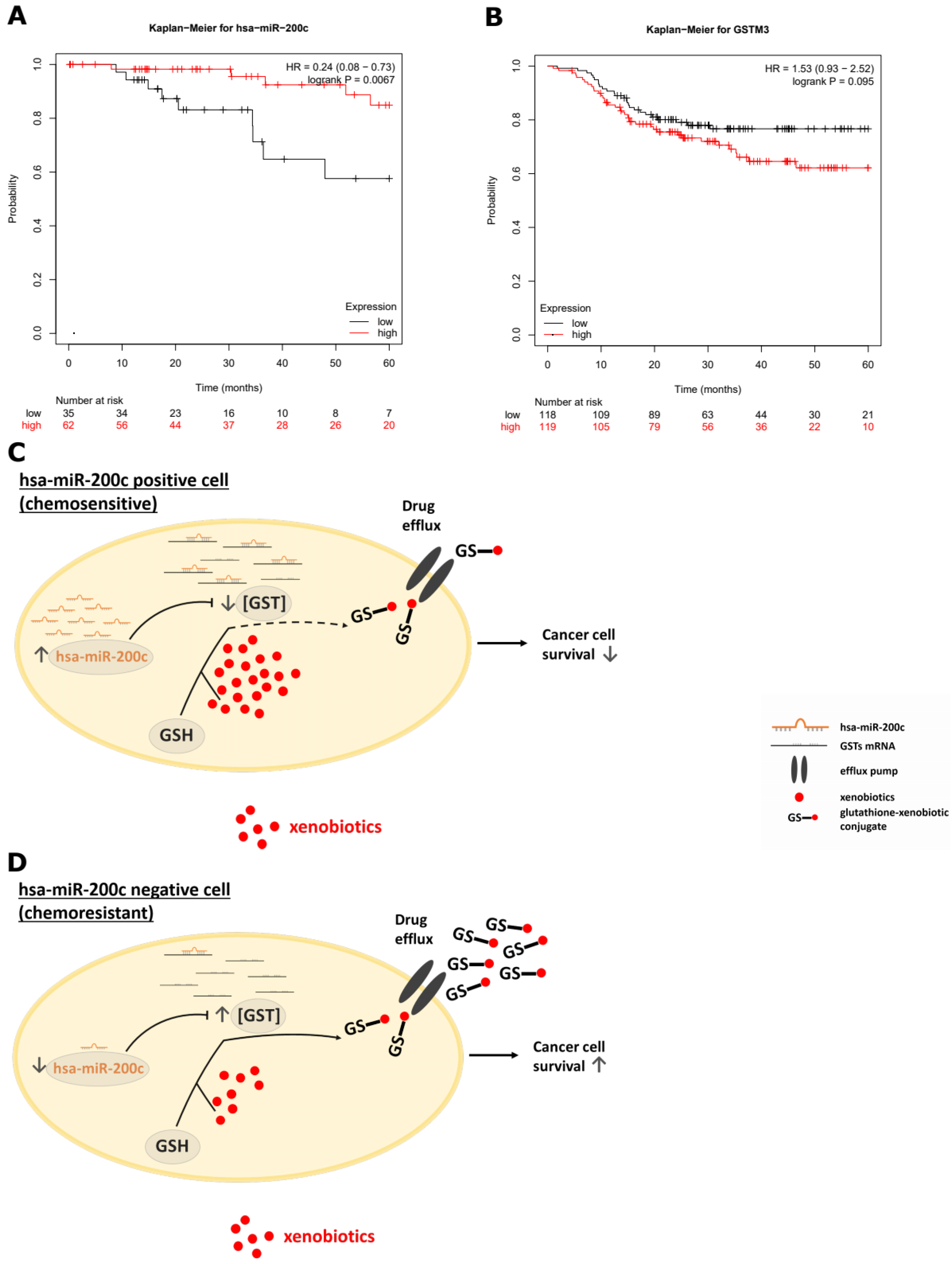
2.16. Clinical Impact Using Kaplan–Meier Plotter
2.17. Software
2.18. Statistical Analysis
3. Results
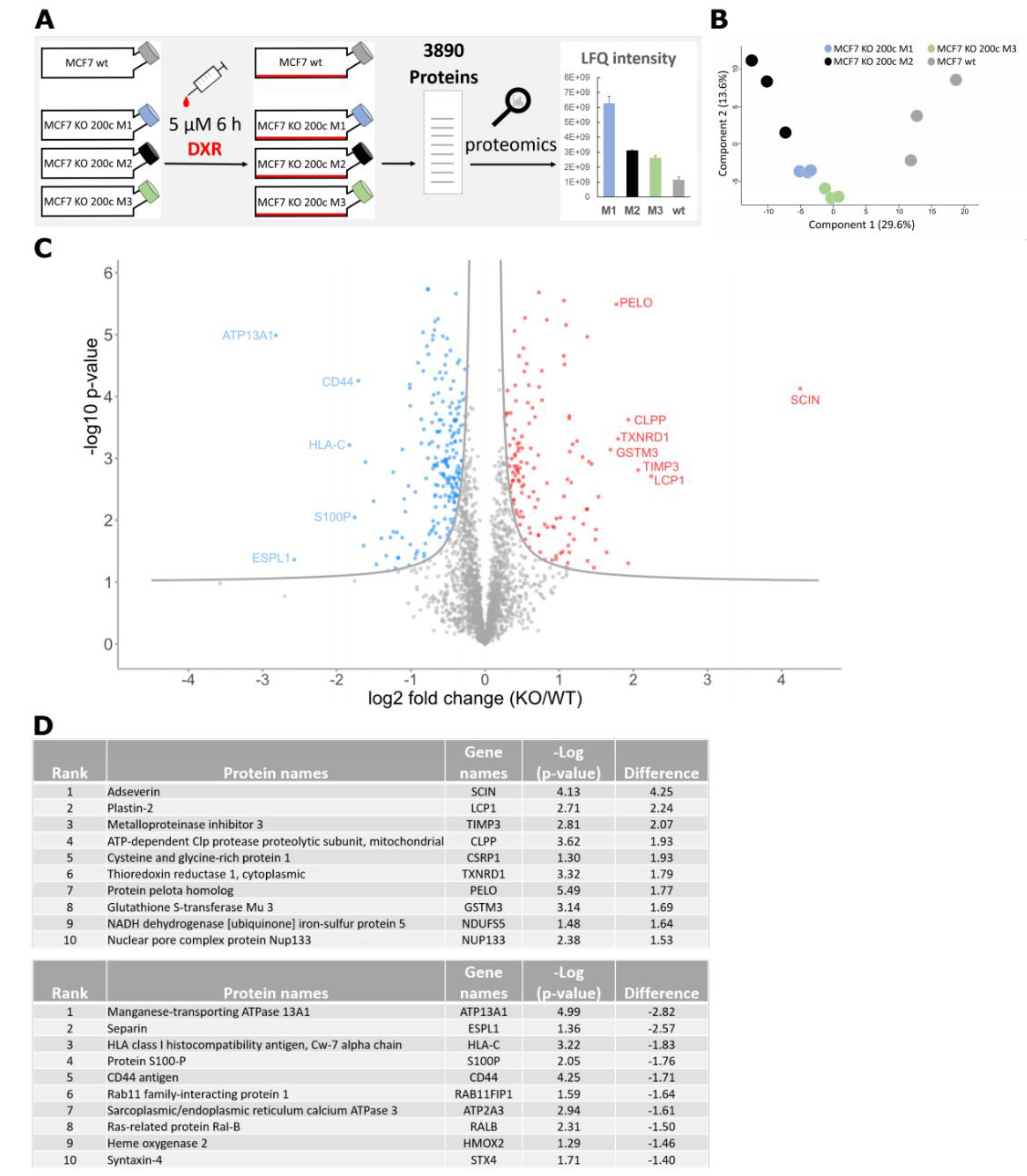
3.1. Proteomic Analysis of a hsa-miR-200c Knockout upon Doxorubicin Treatment Reveals a Higher Abundance of the Glutathione Pathway
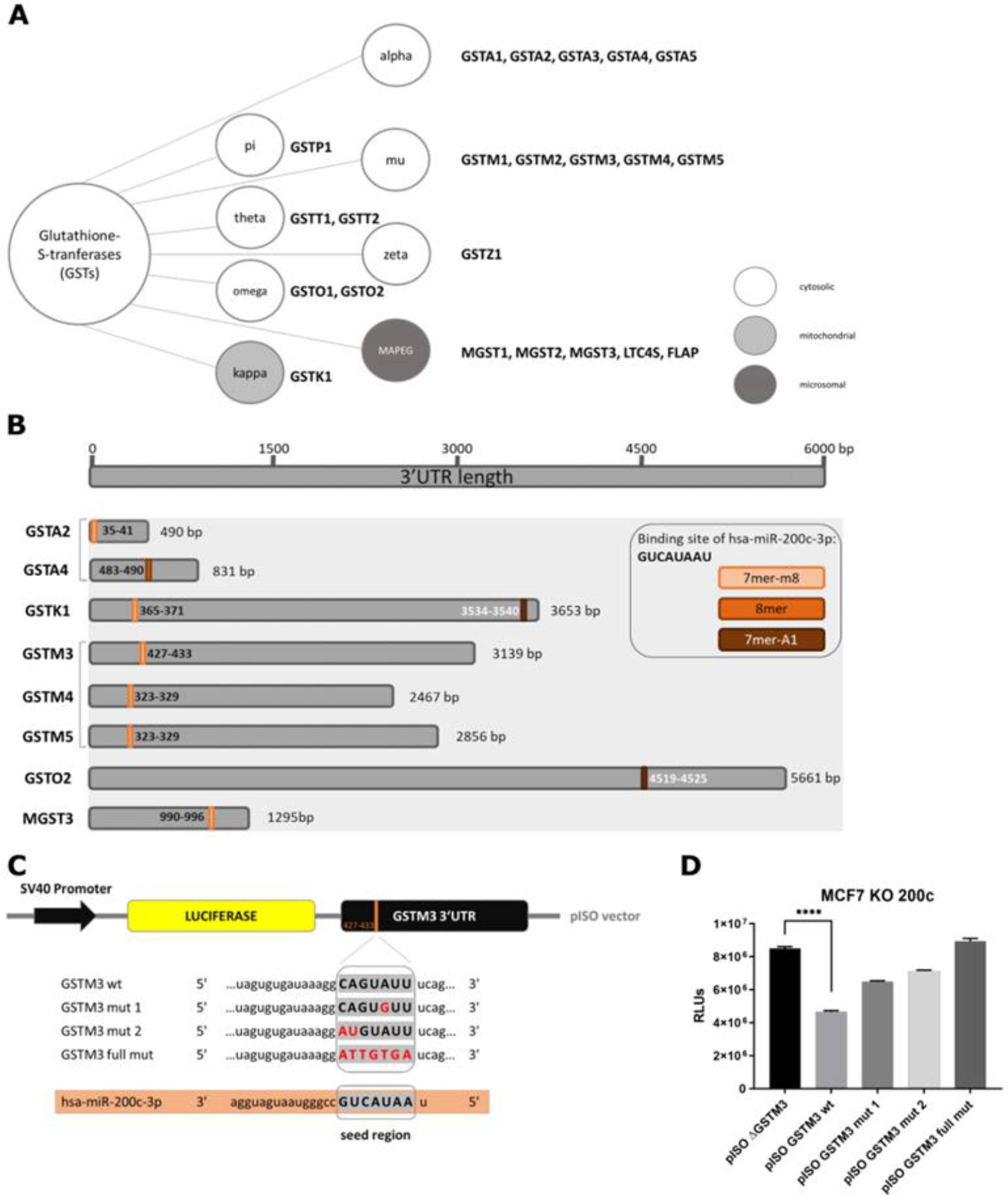
3.2. Glutathione S-Transferase mu 3 Is a Novel Target of hsa-miR-200c-3p
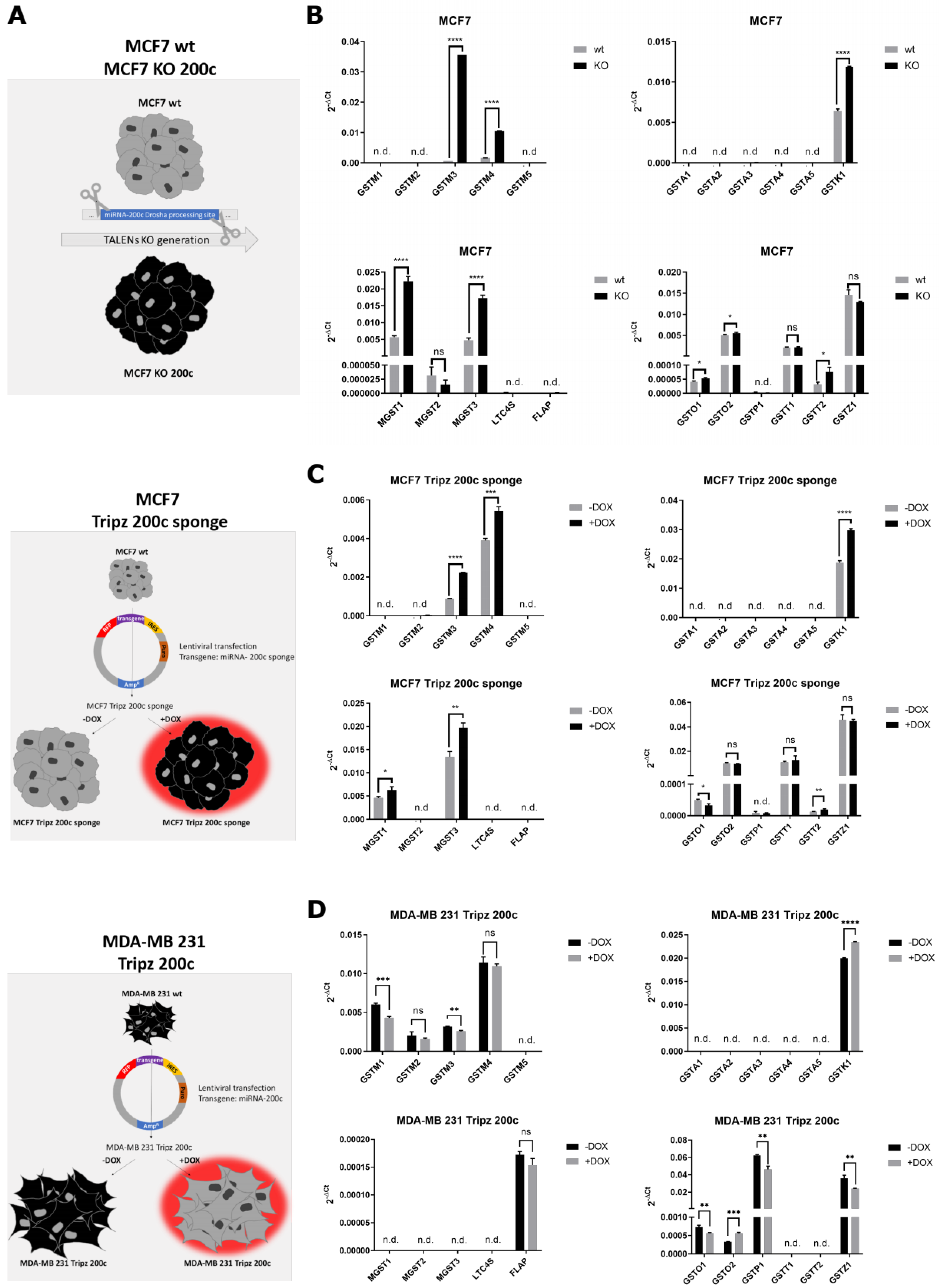
3.3. Hsa-miR-200c Controls the Expression of Additional Glutathione S-Transferases
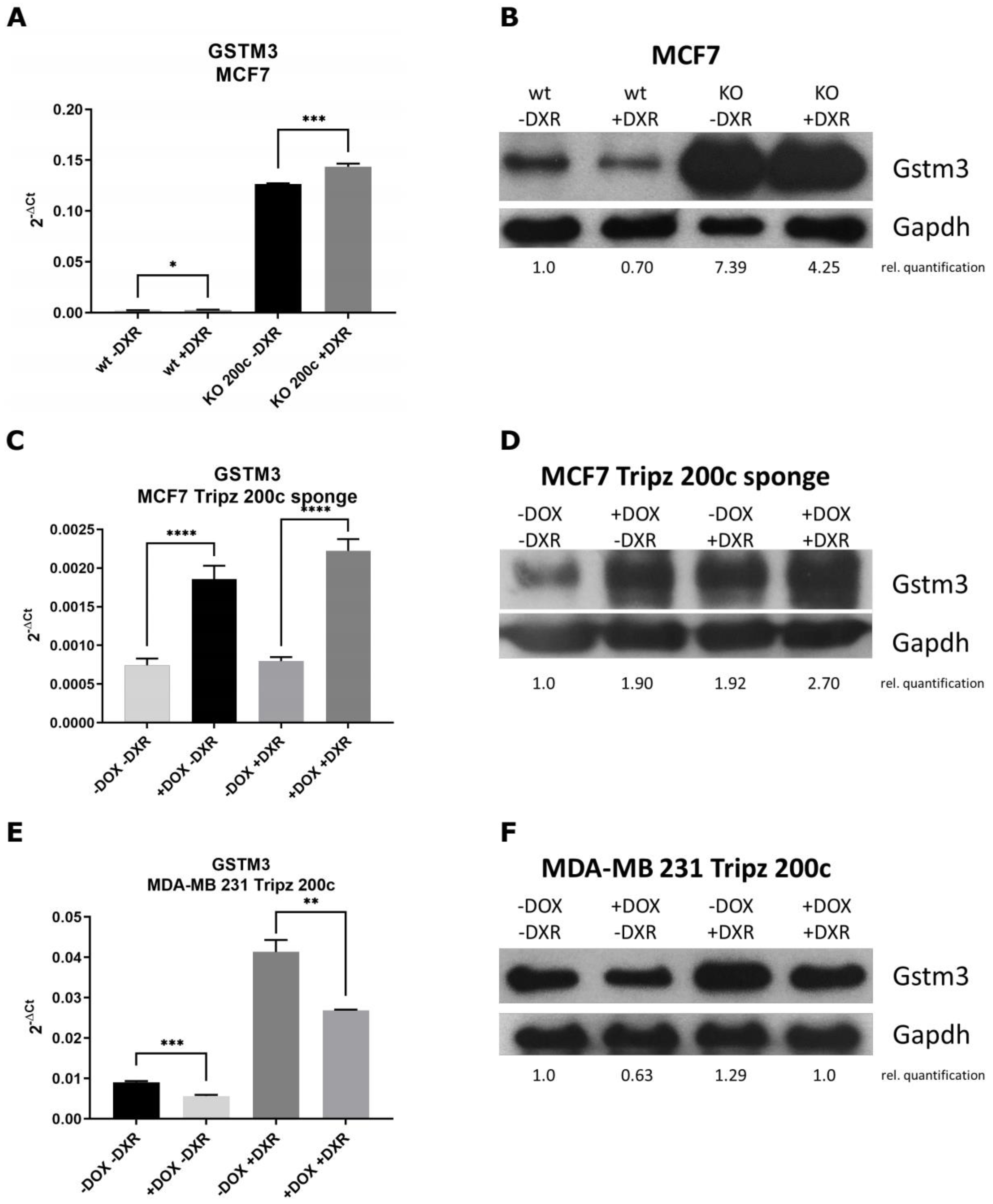
3.4. GSTM3 as Target of the hsa-miR-200c Is Differentially Expressed upon Chemotherapeutic Treatment
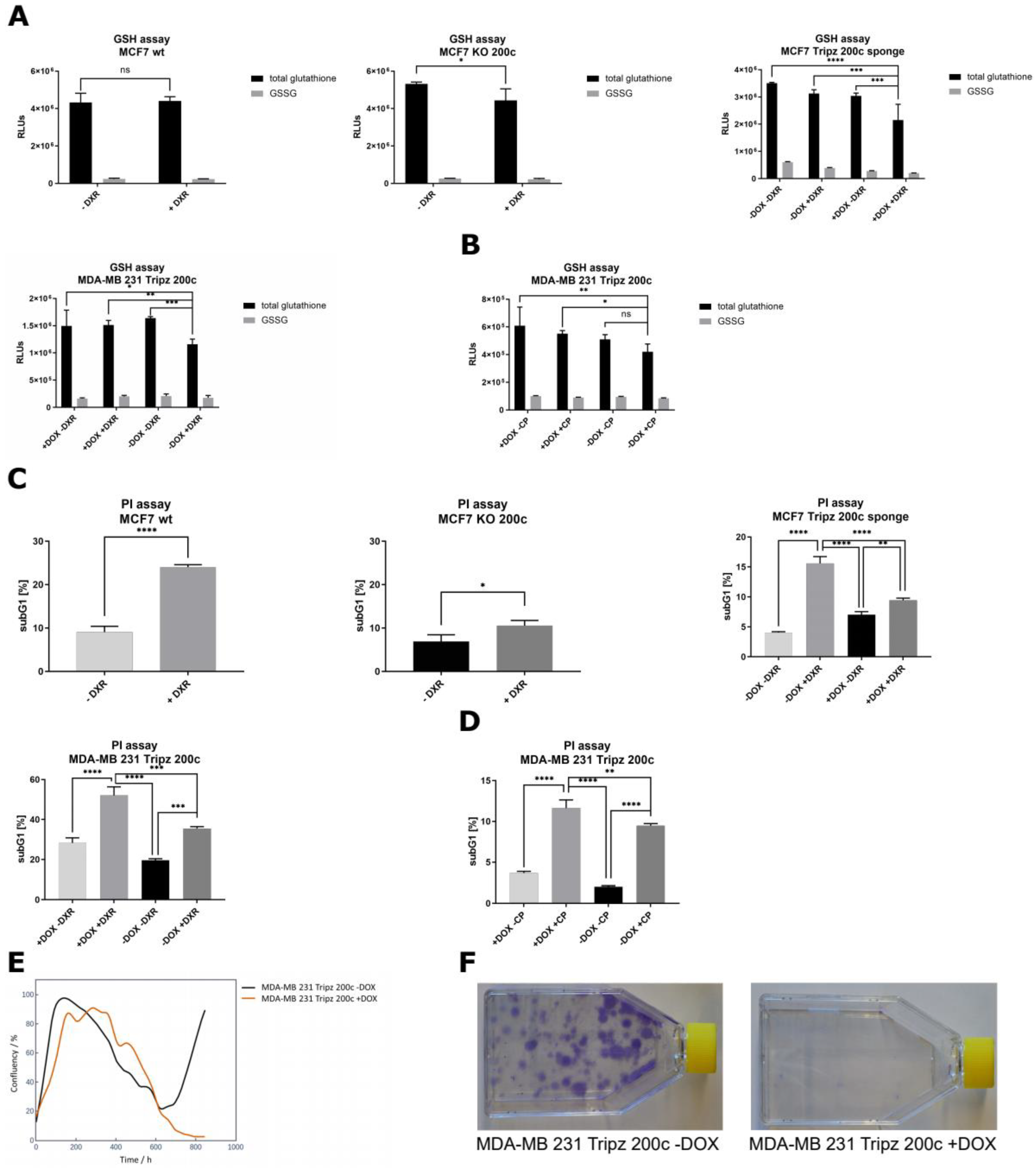
3.5. Hsa-miR-200c Influences the GSH Pool and Mediates Drug Resistance In Vitro
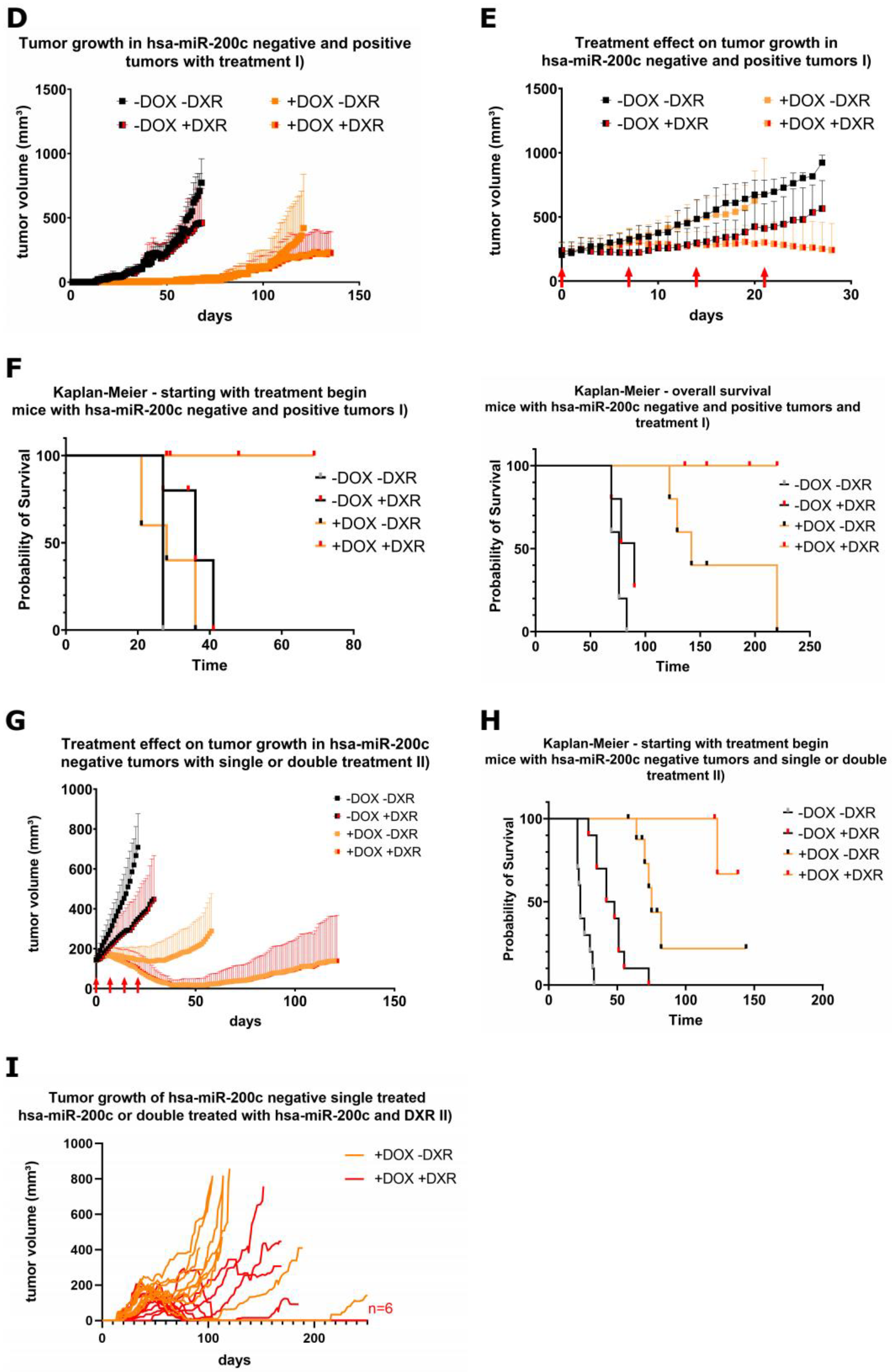
3.6. Xenograft Mouse Models Present Drug Resistance In Vivo upon Modulation of hsa-miR-200c Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Supplementary Material and Methods

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Left Primer | Right Primer | Probe | |
|---|---|---|---|
| GSTA1 | gagaactattgagaggaacaaagagc | tctcctggaggtttctctaagc | 85 |
| GSTA2 | agaaacctccaggagactgcta | tctgccccgtatattggagt | 53 |
| GSTA3 | ctgacattagcctggtggaac | tcagcagagggaagttggag | 19 |
| GSTA4 | cttcctcttgtcctttgtcctc | tgctgccatgatagcttttc | 58 |
| GSTA5 | tgcagaagatttggacaagttaag | ggttgtatttgctggcaatg | 21 |
| GSTP1 | catctccctcatctacaccaacta | aggacctcatggatcagcag | 62 |
| GSTM1 | aggacttcatctcccgcttt | cccagacagccatctttga | 13 |
| GSTM2 | catgacactggggtactgga | tcctcgtagcttgagtctgtgt | 68 |
| GSTM3 | ccaatggctggatgtgaaat | tccaggaggtagggcagat | 85 |
| GSTM4 | tgacctctctgactgggaca | tctgaaggccagagaaccag | 13 |
| GSTM5 | tggacgccttcctaaacttg | aaacaaaagacctcggaggaa | 13 |
| GSTT1 | gtagccatcacggagctgat | gaagaggtcctcccccact | 71 |
| GSTT2 | gctgtttcttgacctggtgtc | tcttgtgctgccctttgac | 28 |
| GSTZ1 | cctgcagaacctgtctgtcc | ccacaagtgatggcgttct | 55 |
| GSTO1 | gcacttttgagctaaggaggaa | caggggattcaggaagtaggt | 12 |
| GSTO2 | gagatgtgggagagaatgcac | gaaggtggtgttctgatactcaag | 8 |
| GSTK1 | tatttggctctgaccggatg | ggtatagggcccatccactt | 62 |
| MGST1 | tcagcatccagttggctttt | aatgggtttaccccagttca | 6 |
| MGST2 | gggtcaccagagtttgagaga | ccttgaagtgacgctgatga | 85 |
| MGST3 | actggtgctgccagctttat | tttcagggtccgtgctgta | 49 |
| LTC4 S | accatgaaggacgaggtagc | tgcagggagaagtaggcttg | 66 |
| FLAP | catcagcgtggtccagaat | caagtgttccggtcctctg | 52 |
| GAPDH | tccactggcgtcttcacc | ggcagagatgatgaccctttt | 45 |
| hsa-miR-200c-3p | gcgtaatactgccgggtaat | PerfeCTa Universal PCR Primer | |
| hsa-miR-191 | gcgcaacggaatcccaaaag | PerfeCTa Universal PCR Primer |
| Proteins | Assay ID |
|---|---|
| ABCB1 | Hs00184500_m1 |
| GAPDH | Hs02758991_g1 |
Appendix B. Quantification of RFP Expression in the Inducible Cell Systems
Appendix C. Proliferation Analysis Using the PHIO Cellwatcher
Appendix D. H&E Staining of In Vivo Tumors
References
- Ma, J.; Dong, C.; Ji, C. MicroRNA and drug resistance. Cancer Gene Ther. 2010, 17, 523–531. [Google Scholar] [CrossRef] [Green Version]
- Lonning, P.E.; Knappskog, S. Mapping genetic alterations causing chemoresistance in cancer: Identifying the roads by tracking the drivers. Oncogene 2013, 32, 5315–5330. [Google Scholar] [CrossRef]
- Si, W.; Shen, J.; Zheng, H.; Fan, W. The role and mechanisms of action of microRNAs in cancer drug resistance. Clin. Epigenet. 2019, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Santos, P.; Almeida, F. Role of Exosomal miRNAs and the Tumor Microenvironment in Drug Resistance. Cells 2020, 9, 1450. [Google Scholar] [CrossRef] [PubMed]
- Konieczkowski, D.J.; Johannessen, C.M.; Garraway, L.A. A Convergence-Based Framework for Cancer Drug Resistance. Cancer Cell 2018, 33, 801–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.; Liu, J.; Zhou, Q.; Song, J.; Zhang, Z.; Li, Z.; Wang, G.; Yuan, W.; Sun, Z. Exosomal Noncoding RNAs and Tumor Drug Resistance. Cancer Res. 2020, 80, 4307–4313. [Google Scholar] [CrossRef] [PubMed]
- Fodale, V.; Pierobon, M.; Liotta, L.; Petricoin, E. Mechanism of Cell Adaptation: When and How Do Cancer Cells Develop Chemoresistance? Cancer J. 2011, 17, 89–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, H.-C. The molecular mechanisms of chemoresistance in cancers. Oncotarget 2017, 8, 59950–59964. [Google Scholar] [CrossRef] [Green Version]
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003, 22, 7369–7375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, W.; Kinsey, M.; Schiffman, J.D.; Lessnick, S.L. Glutathione S-Transferases in Pediatric Cancer. Front. Oncol. 2011, 1, 39. [Google Scholar] [CrossRef]
- Oakley, A. Glutathione transferases: A structural perspective. Drug Metab. Rev. 2011, 43, 138–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Zhang, H.; Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist 2019, 2, 141–160. [Google Scholar] [CrossRef] [Green Version]
- Allocati, N.; Masulli, M.; Di Ilio, C.; Federici, L. Glutathione transferases: Substrates, inihibitors and pro-drugs in cancer and neurodegenerative diseases. Oncogenesis 2018, 7, 8. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, A.; Gupta, S. The multifaceted role of glutathione S-transferases in cancer. Cancer Lett. 2018, 433, 33–42. [Google Scholar] [CrossRef]
- Hayes, J.D.; Flanagan, J.U.; Jowsey, I.R. Glutathione Transferases. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 51–88. [Google Scholar] [CrossRef]
- Sau, A.; Tregno, F.P.; Valentino, F.; Federici, G.; Caccuri, A.M. Glutathione transferases and development of new principles to overcome drug resistance. Arch. Biochem. Biophys. 2010, 500, 116–122. [Google Scholar] [CrossRef]
- Xie, L.; Jing, R.; Qi, J.; Lin, Z.; Ju, S. Drug resistance-related microRNAs in hematological malignancies: Translating basic evidence into therapeutic strategies. Blood Rev. 2015, 29, 33–44. [Google Scholar] [CrossRef]
- Mutlu, M.; Raza, U.; Saatci, Ö.; Eyüpoğlu, E.; Yurdusev, E.; Şahin, Ö. miR-200c: A Versatile Watchdog in Cancer Progression, Emt, and Drug Resistance. J. Mol. Med. 2016, 94, 629–644. [Google Scholar] [CrossRef]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of Microrna Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aurora, E.-K.; Slack, F.J. Oncomirs—Micrornas with a Role in Cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar]
- Ljepoja, B.; García-Roman, J.; Sommer, A.-K.; Fröhlich, T.; Arnold, G.J.; Wagner, E.; Roidl, A. A proteomic analysis of an in vitro knock-out of miR-200c. Sci. Rep. 2018, 8, 6927. [Google Scholar] [CrossRef] [PubMed]
- Kopp, F.; Oak, P.S.; Wagner, E.; Roidl, A. miR-200c Sensitizes Breast Cancer Cells to Doxorubicin Treatment by Decreasing TrkB and Bmi1 Expression. PLoS ONE 2012, 7, e50469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopp, F.; Wagner, E.; Roidl, A. The proto-oncogene KRAS is targeted by miR-200c. Oncotarget 2013, 5, 185–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulrane, L.; McGee, S.F.; Gallagher, W.M.; O’Connor, D.P. miRNA Dysregulation in Breast Cancer. Cancer Res. 2013, 73, 6554–6562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Tetzlaff, M.T.; Cui, R.; Xu, X. miR-200c Inhibits Melanoma Progression and Drug Resistance through Down-Regulation of Bmi-1. Am. J. Pathol. 2012, 181, 1823–1835. [Google Scholar] [CrossRef] [Green Version]
- Kawaguchi, T.; Yan, L.; Qi, Q.; Peng, X.; Gabriel, E.M.; Young, J.; Liu, S.; Takabe, K. Overexpression of Suppressive Micrornas, miR-30 a and miR-200c Are Associated with Improved Survival of Breast Cancer Patients. Sci. Rep. 2017, 7, 15945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, F.; He, H.; Huang, W.; Lin, Y.; Luo, S.; Du, Q.; Duan, R. Decreased expression of MicroRNA-200 family in human breast cancer is associated with lymph node metastasis. Clin. Transl. Oncol. 2015, 18, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Humphries, B.; Yang, C. The Microrna-200 Family: Small Molecules with Novel Roles in Cancer Development, Progression and Therapy. Oncotarget 2015, 6, 6472–6498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Nag, A.; Mandal, C.C. A Comprehensive Review on miR-200c, A Promising Cancer Biomarker with Therapeutic Potential. Curr. Drug Targets 2015, 16, 1381–1403. [Google Scholar] [CrossRef] [PubMed]
- Perdigão-Henriques, R.; Petrocca, F.; Altschuler, G.; Thomas, M.P.; LE, T.N.M.; Tan, S.M.; Hide, W.; A Lieberman, J. miR-200 promotes the mesenchymal to epithelial transition by suppressing multiple members of the Zeb2 and Snail1 transcriptional repressor complexes. Oncogene 2015, 35, 158–172. [Google Scholar] [CrossRef] [PubMed]
- van Staalduinen, J.; Baker, D.; Ten Dijke, P.; van Dam, H. Epithelial–Mesenchymal-Transition-Inducing Transcription Factors: New Targets for Tackling Chemoresistance in Cancer? Oncogene 2018, 37, 6195–6211. [Google Scholar] [CrossRef] [PubMed]
- Cavallari, I.; Ciccarese, F.; Sharova, E.; Urso, L.; Raimondi, V.; Silic-Benussi, M.; D’Agostino, D.M.; Ciminale, V. The miR-200 Family of Micrornas: Fine Tuners of Epithelial-Mesenchymal Transition and Circulating Cancer Biomarkers. Cancers 2021, 13, 5874. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Ginther, C.; Kim, J.; Mosher, N.; Chung, S.; Slamon, D.; Vadgama, J.V. Expression of Wnt3 Activates Wnt/β-Catenin Pathway and Promotes EMT-like Phenotype in Trastuzumab-Resistant HER2-Overexpressing Breast Cancer Cells. Mol. Cancer Res. 2012, 10, 1597–1606. [Google Scholar] [CrossRef] [Green Version]
- Della Corte, C.M.; Bellevicine, C.; Vicidomini, G.; Vitagliano, D.; Malapelle, U.; Accardo, M.; Fabozzi, A.; Fiorelli, A.; Fasano, M.; Papaccio, F.; et al. SMO Gene Amplification and Activation of the Hedgehog Pathway as Novel Mechanisms of Resistance to Anti-Epidermal Growth Factor Receptor Drugs in Human Lung Cancer. Clin. Cancer Res. 2015, 21, 4686–4697. [Google Scholar] [CrossRef] [Green Version]
- Bracken, C.P.; Khew-Goodall, Y.; Goodall, G.J. Network-Based Approaches to Understand the Roles of miR-200 and Other microRNAs in Cancer. Cancer Res. 2015, 75, 2594–2599. [Google Scholar] [CrossRef] [Green Version]
- Sommerova, L.; Ondrouskova, E.; Martisova, A.; Zoumpourlis, V.; Galtsidis, S.; Hrstka, R. Zeb1/miR-200c/Agr2: A New Regulatory Loop Modulating the Epithelial-Mesenchymal Transition in Lung Adenocarcinomas. Cancers 2020, 12, 1614. [Google Scholar] [CrossRef]
- Ljepoja, B.; Schreiber, C.; Gegenfurtner, F.A.; García-Roman, J.; Köhler, B.; Zahler, S.; Rädler, J.O.; Wagner, E.; Roidl, A. Inducible Microrna-200c Decreases Motility of Breast Cancer Cells and Reduces Filamin A. PLoS ONE 2019, 14, e0224314. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved Seed Pairing, Often Flanked by Adenosines, Indicates that Thousands of Human Genes are MicroRNA Targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [Green Version]
- Grimson, A.; Farh, K.K.-H.; Johnston, W.K.; Garrett-Engele, P.; Lim, L.P.; Bartel, D.P. MicroRNA Targeting Specificity in Mammals: Determinants beyond Seed Pairing. Mol. Cell 2007, 27, 91–105. [Google Scholar] [CrossRef] [Green Version]
- Friedman, R.C.; Farh, K.K.-H.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.; Levačić, A.K.; Hörterer, E.; Wilk, U.; Benli-Hoppe, T.; Wang, Y.; Öztürk, Ö.; Luo, J.; Wagner, E. Optimizing Pdna Lipo-Polyplexes: A Balancing Act between Stability and Cargo Release. Biomacromolecules 2021, 22, 1282–1296. [Google Scholar] [CrossRef] [PubMed]
- Sommer, A.-K.; Falcenberg, M.; Ljepoja, B.; Fröhlich, T.; Arnold, G.J.; Wagner, E.; Roidl, A. Downregulation of GRK5 hampers the migration of breast cancer cells. Sci. Rep. 2019, 9, 15548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.-M.; Chen, Y.; Chen, J.; Yang, S.; Gao, F.; Underhill, C.B.; Creswell, K.; Zhang, L. A peptide with three hyaluronan binding motifs inhibits tumor growth and induces apoptosis. Cancer Res. 2003, 63, 5685–5690. [Google Scholar] [PubMed]
- Lánczky, A.; Nagy, Á.; Bottai, G.; Munkácsy, G.; Szabó, A.; Santarpia, L.; Győrffy, B. Mirpower: A Web-Tool to Validate Survival-Associated Mirnas Utilizing Expression Data from 2178 Breast Cancer Patients. Breast. Cancer Res. Treat. 2016, 160, 439–446. [Google Scholar] [CrossRef]
- Győrffy, B. Survival analysis across the entire transcriptome identifies biomarkers with the highest prognostic power in breast cancer. Comput. Struct. Biotechnol. J. 2021, 19, 4101–4109. [Google Scholar] [CrossRef]
- Lánczky, A.; Győrffy, B. Web-Based Survival Analysis Tool Tailored for Medical Research (KMplot): Development and Implementation. J. Med. Internet Res. 2021, 23, e27633. [Google Scholar] [CrossRef]
- Chen, L.; Sun, H.; Wang, C.; Yang, Y.; Zhang, M.; Wong, G. miRNA arm switching identifies novel tumour biomarkers. eBioMedicine 2018, 38, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Ruzza, P.; Calderan, A. Glutathione Transferase (GST)-Activated Prodrugs. Pharmaceutics 2013, 5, 220–231. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Bankhead, A., 3rd; Tian, X.; Tang, J.; Ljungman, M.; Neamati, N. Deletion of Glutathione S-Transferase Omega 1 Activates Type I Interferon Genes and Downregulates Tissue Factor. Cancer Res. 2020, 80, 3692–3705. [Google Scholar] [CrossRef]
- Kalinina, E.V.; Berozov, T.T.; Shtil, A.A.; Chernov, N.N.; Glasunova, V.A.; Novichkova, M.D.; Nurmuradov, N.K. Expression of Genes of Glutathione Transferase Isoforms Gstp1–1, Gsta4–4, and Gstk1–1 in Tumor Cells During the Formation of Drug Resistance to Cisplatin. Bull. Exp. Biol. Med. 2012, 154, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Checa-Rojas, A.; Delgadillo-Silva, L.F.; Velasco-Herrera, M.D.C.; Andrade-Domínguez, A.; Gil, J.; Santillán, O.; Lozano, L.; Toledo-Leyva, A.; Ramírez-Torres, A.; Talamas-Rohana, P.; et al. GSTM3 and GSTP1: Novel players driving tumor progression in cervical cancer. Oncotarget 2018, 9, 21696–21714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louie, S.M.; Grossman, E.A.; Crawford, L.A.; Ding, L.; Camarda, R.; Huffman, T.R.; Miyamoto, D.K.; Goga, A.; Weerapana, E.; Nomura, D.K. GSTP1 Is a Driver of Triple-Negative Breast Cancer Cell Metabolism and Pathogenicity. Cell Chem. Biol. 2016, 23, 567–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.R.; Reindl, K.M. Glutathione S-Transferases in Cancer. Antioxidants 2021, 10, 701. [Google Scholar] [CrossRef] [PubMed]
- El-Aziz, Y.S.A.; Spillane, A.J.; Jansson, P.J.; Sahni, S. Role of ABCB1 in mediating chemoresistance of triple-negative breast cancers. Biosci. Rep. 2021, 41, BSR20204092. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; VandenBoom, T.G., 2nd; Kong, D.; Wang, Z.; Ali, S.; Philip, P.A.; Sarkar, F.H. Up-Regulation of miR-200 and Let-7 by Natural Agents Leads to the Reversal of Epithelial-to-Mesenchymal Transition in Gemcitabine-Resistant Pancreatic Cancer Cells. Cancer Res. 2009, 69, 6704–6712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, W.; Xu, H.; Zhu, D.; Zhi, H.; Wang, T.; Wang, J.; Jiang, B.-H.; Shu, Y.; Liu, P. miR-200bc/429 cluster modulates multidrug resistance of human cancer cell lines by targeting BCL2 and XIAP. Cancer Chemother. Pharmacol. 2011, 69, 723–731. [Google Scholar] [CrossRef]
- Anastasiadou, E.; Messina, E.; Sanavia, T.; Mundo, L.; Farinella, F.; Lazzi, S.; Megiorni, F.; Ceccarelli, S.; Pontecorvi, P.; Marampon, F.; et al. MiR-200c-3p Contrasts PD-L1 Induction by Combinatorial Therapies and Slows Proliferation of Epithelial Ovarian Cancer through Downregulation of β-Catenin and c-Myc. Cells 2021, 10, 519. [Google Scholar] [CrossRef]
- Mansoori, B.; Silvestris, N.; Mohammadi, A.; Khaze, V.; Baghbani, E.; Mokhtarzadeh, A.; Shanehbandi, D.; Derakhshani, A.; Duijf, P.H.G.; Baradaran, B. miR-34a and miR-200c Have an Additive Tumor-Suppressive Effect on Breast Cancer Cells and Patient Prognosis. Genes 2021, 12, 267. [Google Scholar] [CrossRef]
- Song, C.; Liu, L.-Z.; Pei, X.-Q.; Liu, X.; Yang, L.; Ye, F.; Xie, X.; Chen, J.; Tang, H.; Xie, X. miR-200c inhibits breast cancer proliferation by targeting KRAS. Oncotarget 2015, 6, 34968–34978. [Google Scholar] [CrossRef] [Green Version]
- Knezevic, J.; Pfefferle, A.D.; Petrovic, I.; Greene, S.B.; Perou, C.M.; Rosen, J.M. Expression of miR-200c in Claudin-Low Breast Cancer Alters Stem Cell Functionality, Enhances Chemosensitivity and Reduces Metastatic Potential. Oncogene 2015, 34, 5997–6006. [Google Scholar] [CrossRef]
- Jones, R.; Watson, K.; Bruce, A.; Nersesian, S.; Kitz, J.; Moorehead, R. Re-Expression of miR-200c Suppresses Proliferation, Colony Formation and in Vivo Tumor Growth of Murine Claudin-Low Mammary Tumor Cells. Oncotarget 2017, 8, 23727–23749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, K.L.; Jones, R.A.; Bruce, A.; Moorehead, R.A. The miR-200b/200a/429 cluster prevents metastasis and induces dormancy in a murine claudin-low mammary tumor cell line. Exp. Cell Res. 2018, 369, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Schilb, A.L.; Ayat, N.R.; Vaidya, A.M.; Hertz, L.M.; Hall, R.C.; Scheidt, J.H.; Sun, D.; Sun, Z.; Gopalakrishnan, R.; Lu, Z.-R. Efficacy of Targeted ECO/miR-200c Nanoparticles for Modulating Tumor Microenvironment and Treating Triple Negative Breast Cancer as Non-invasively Monitored by MR Molecular Imaging. Pharm. Res. 2021, 38, 1405–1418. [Google Scholar] [CrossRef]
- Carvalho, C.; Santos, R.X.; Cardoso, S.; Correia, S.; Oliveira, P.J.; Santos, M.S.; Moreira, P.I. Doxorubicin: The Good, the Bad and the Ugly Effect. Curr. Med. Chem. 2009, 16, 3267–3285. [Google Scholar] [CrossRef] [PubMed]
- Ghasabi, M.; Majidi, J.; Mansoori, B.; Mohammadi, A.; Shomali, N.; Shirafkan, N.; Baghbani, E.; Kazemi, T.; Baradaran, B. The effect of combined miR-200c replacement and cisplatin on apoptosis induction and inhibition of gastric cancer cell line migration. J. Cell. Physiol. 2019, 234, 22581–22592. [Google Scholar] [CrossRef]
- Vrba, L.; Jensen, T.J.; Garbe, J.C.; Heimark, R.L.; Cress, A.; Dickinson, S.; Stampfer, M.R.; Futscher, B.W. Role for DNA Methylation in the Regulation of miR-200c and miR-141 Expression in Normal and Cancer Cells. PLoS ONE 2010, 5, e8697. [Google Scholar] [CrossRef]
- Tian, Y.; Pan, Q.; Shang, Y.; Zhu, R.; Ye, J.; Liu, Y.; Zhong, X.; Li, S.; He, Y.; Chen, L.; et al. Microrna-200 (miR-200) Cluster Regulation by Achaete Scute-Like 2 (Ascl2): Impact on the Epithelial-Mesenchymal Transition in Colon Cancer Cells. J. Biol. Chem. 2014, 289, 36101–36115. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhou, Y.; Cheng, H.; Tian, J.; Yang, S. Roles of a Tmpo-As1/Microrna-200c/Tmeff2 Cerna Network in the Malignant Behaviors and 5-Fu Resistance of Ovarian Cancer Cells. Exp. Mol. Pathol. 2020, 115, 104481. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, F.; Xiang, Z.; Huang, T.; Zhou, W.B. LncRNA XIST promotes chemoresistance of breast cancer cells to doxorubicin by sponging miR-200c-3p to upregulate ANLN. Clin. Exp. Pharmacol. Physiol. 2020, 47, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Cai, Y.; Wang, C.; Huang, M.; Chen, J. LncRNA GAS5 regulates the proliferation, migration, invasion and apoptosis of brain glioma cells through targeting GSTM3 expression. The effect of LncRNA GAS5 on glioma cells. J. Neuro-Oncol. 2019, 143, 525–536. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Stewart, S.A.; Dykxhoorn, D.M.; Palliser, D.; Mizuno, H.; Yu, E.Y.; An, D.S.; Sabatini, D.M.; Chen, I.S.; Hahn, W.C.; Sharp, P.A.; et al. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 2003, 9, 493–501. [Google Scholar] [CrossRef] [Green Version]
- Marfels, C.; Hoehn, M.; Wagner, E.; Günther, M. Characterization of in vivo chemoresistant human hepatocellular carcinoma cells with transendothelial differentiation capacities. BMC Cancer 2013, 13, 176. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Köhler, B.; Dubovik, S.; Hörterer, E.; Wilk, U.; Stöckl, J.B.; Tekarslan-Sahin, H.; Ljepoja, B.; Paulitschke, P.; Fröhlich, T.; Wagner, E.; et al. Combating Drug Resistance by Exploiting miRNA-200c-Controlled Phase II Detoxification. Cancers 2022, 14, 5554. https://doi.org/10.3390/cancers14225554
Köhler B, Dubovik S, Hörterer E, Wilk U, Stöckl JB, Tekarslan-Sahin H, Ljepoja B, Paulitschke P, Fröhlich T, Wagner E, et al. Combating Drug Resistance by Exploiting miRNA-200c-Controlled Phase II Detoxification. Cancers. 2022; 14(22):5554. https://doi.org/10.3390/cancers14225554
Chicago/Turabian StyleKöhler, Bianca, Sviatlana Dubovik, Elisa Hörterer, Ulrich Wilk, Jan Bernd Stöckl, Hande Tekarslan-Sahin, Bojan Ljepoja, Philipp Paulitschke, Thomas Fröhlich, Ernst Wagner, and et al. 2022. "Combating Drug Resistance by Exploiting miRNA-200c-Controlled Phase II Detoxification" Cancers 14, no. 22: 5554. https://doi.org/10.3390/cancers14225554