
Compound C Inhibits Ovarian Cancer Progression via PI3K-AKT-mTOR-NFκB Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Reagents and Chemicals
2.3. Antibodies
2.4. Primary OvCa Cell Isolation and Maintenance
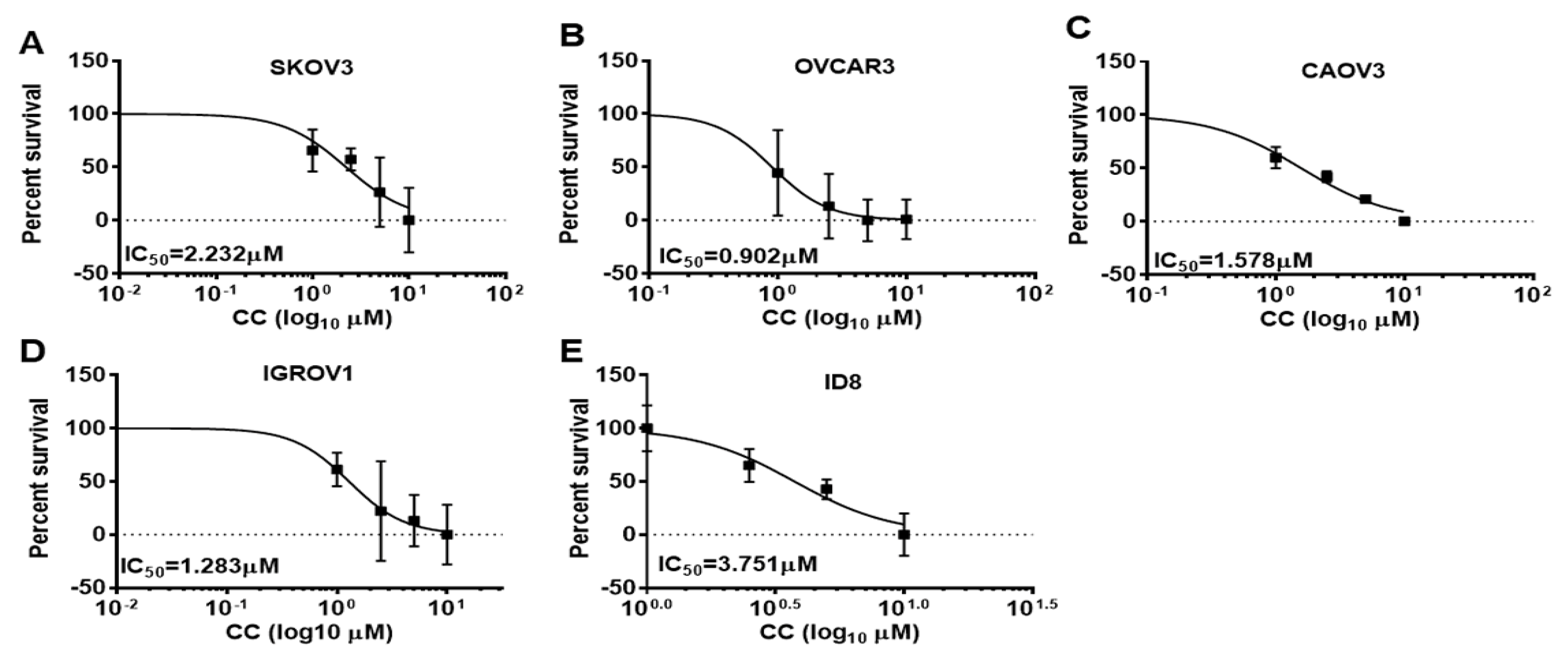
2.5. Proliferation Assays
2.6. Colony Survival Assay
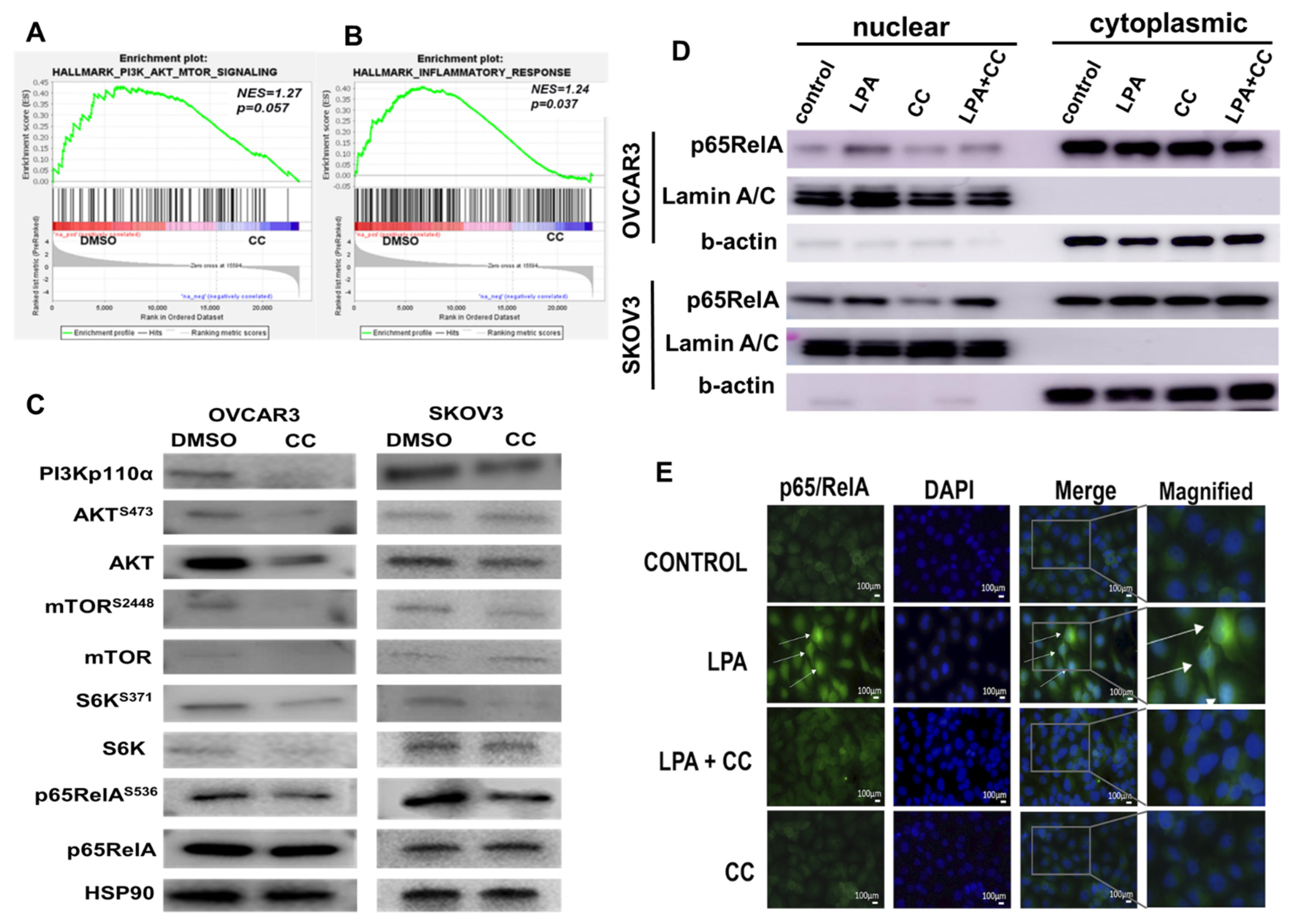
2.7. Gene Set Enrichment Analysis (GSEA)
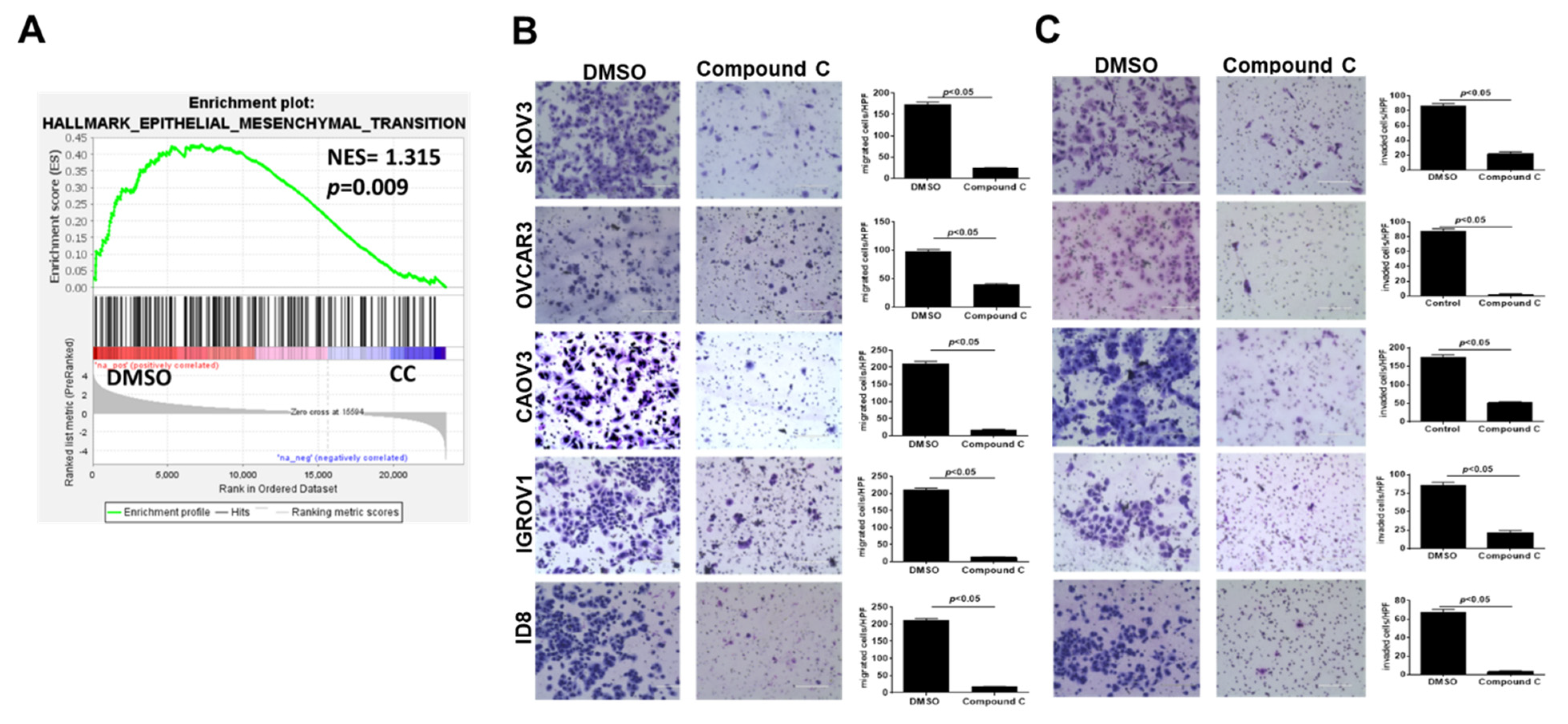
2.8. Migration, Invasion, and Chemotaxis Assays
2.9. Co-Culture Assays
2.10. Subcellular Fractionation
2.11. Western Blots (WB)
2.12. Immunofluorescence
2.13. Plasmids, Lentiviral Packaging, and Viral Transduction
2.14. In Vitro Adhesion Assays
2.15. In Vivo OvCa Cell Homing/Adhesion Assays
2.16. RNA Extraction and Real-Time Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
2.17. Measurement of Mitochondrial Mass MitoTracker Staining
2.18. In Vivo Tumor Xenografts
2.19. Immunohistochemistry (IHC)
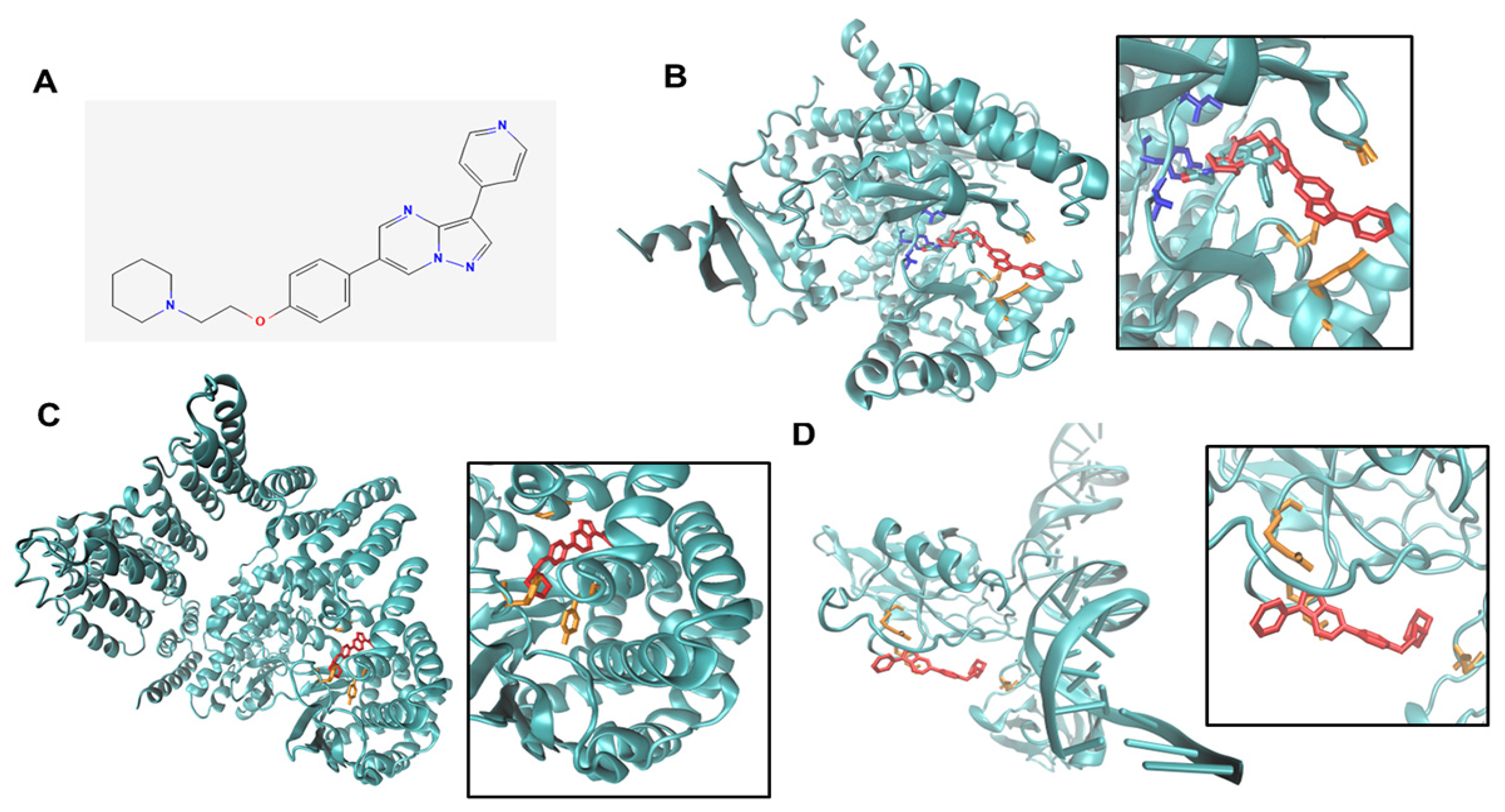
2.20. Docking
2.21. Statistical Analysis
3. Results
3.1. Compound C Inhibits OvCa Proliferation and Clonogenic Survival
3.2. Compound C Inhibits OvCa Cell Migration and Invasiveness
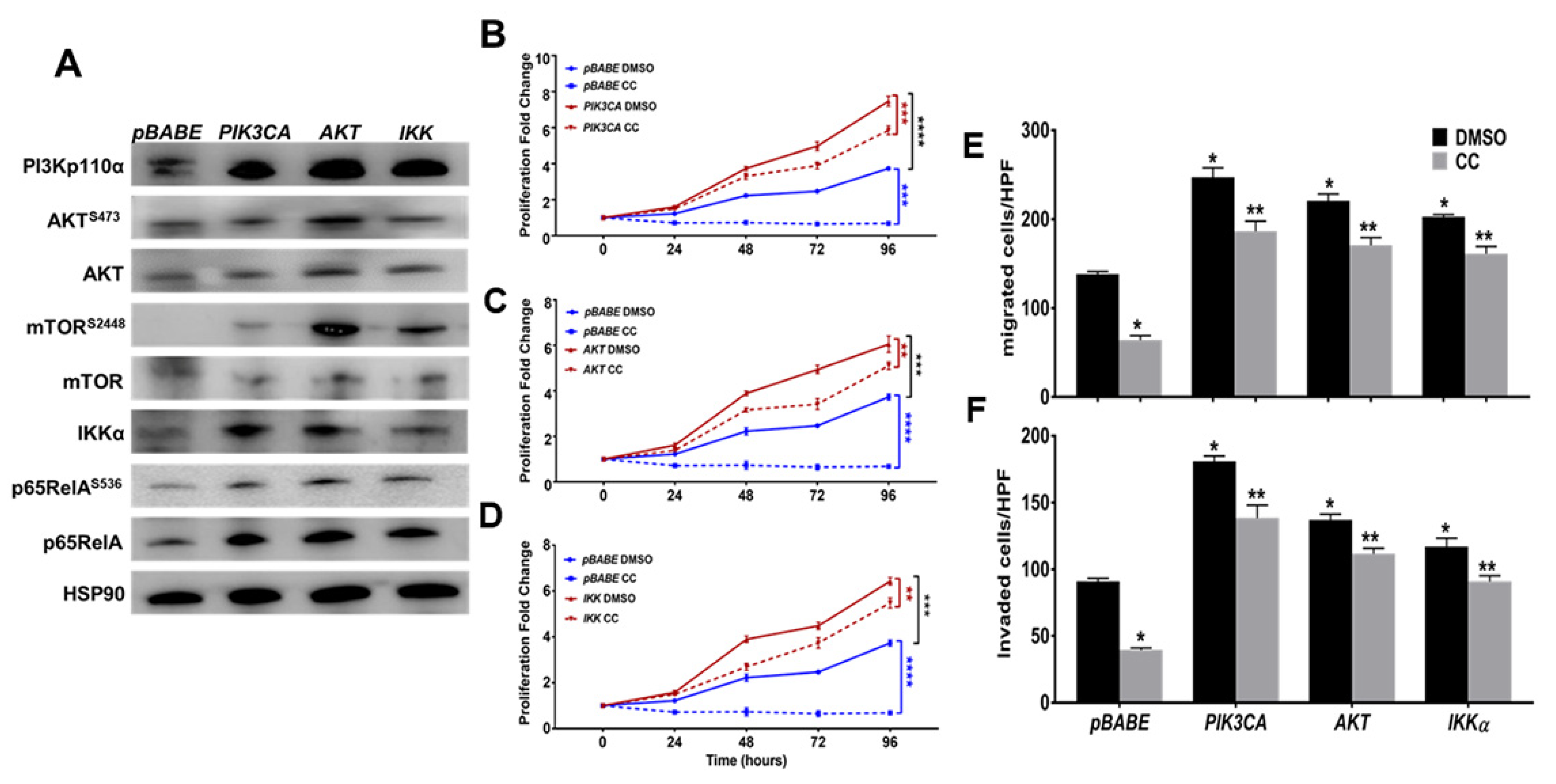
3.3. Compound C Inhibits PI3K-AKT-mTOR and NFκB in OvCa
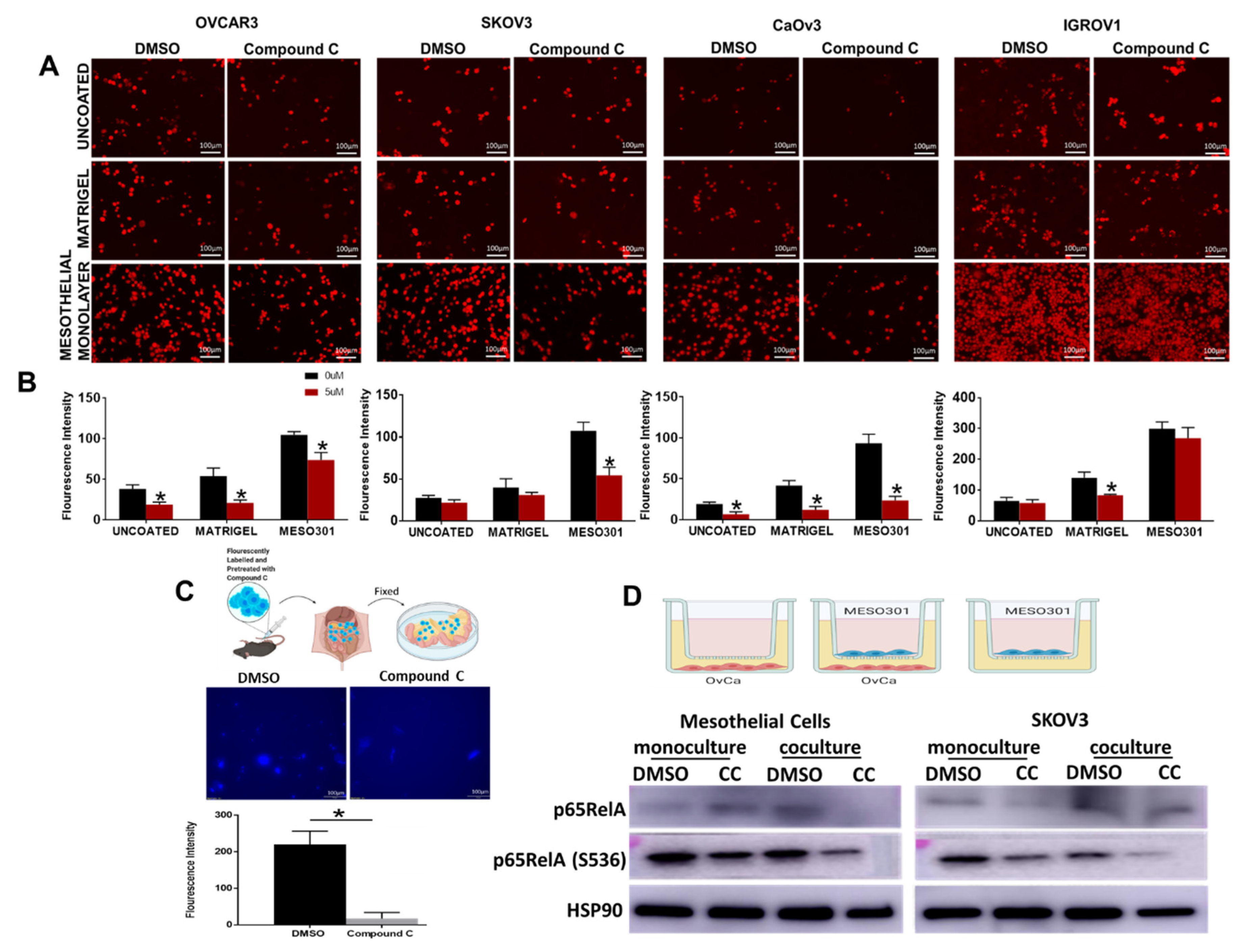
3.4. Compound C Inhibits OvCa Cells-Mesothelial Interactions In Vitro and In Vivo
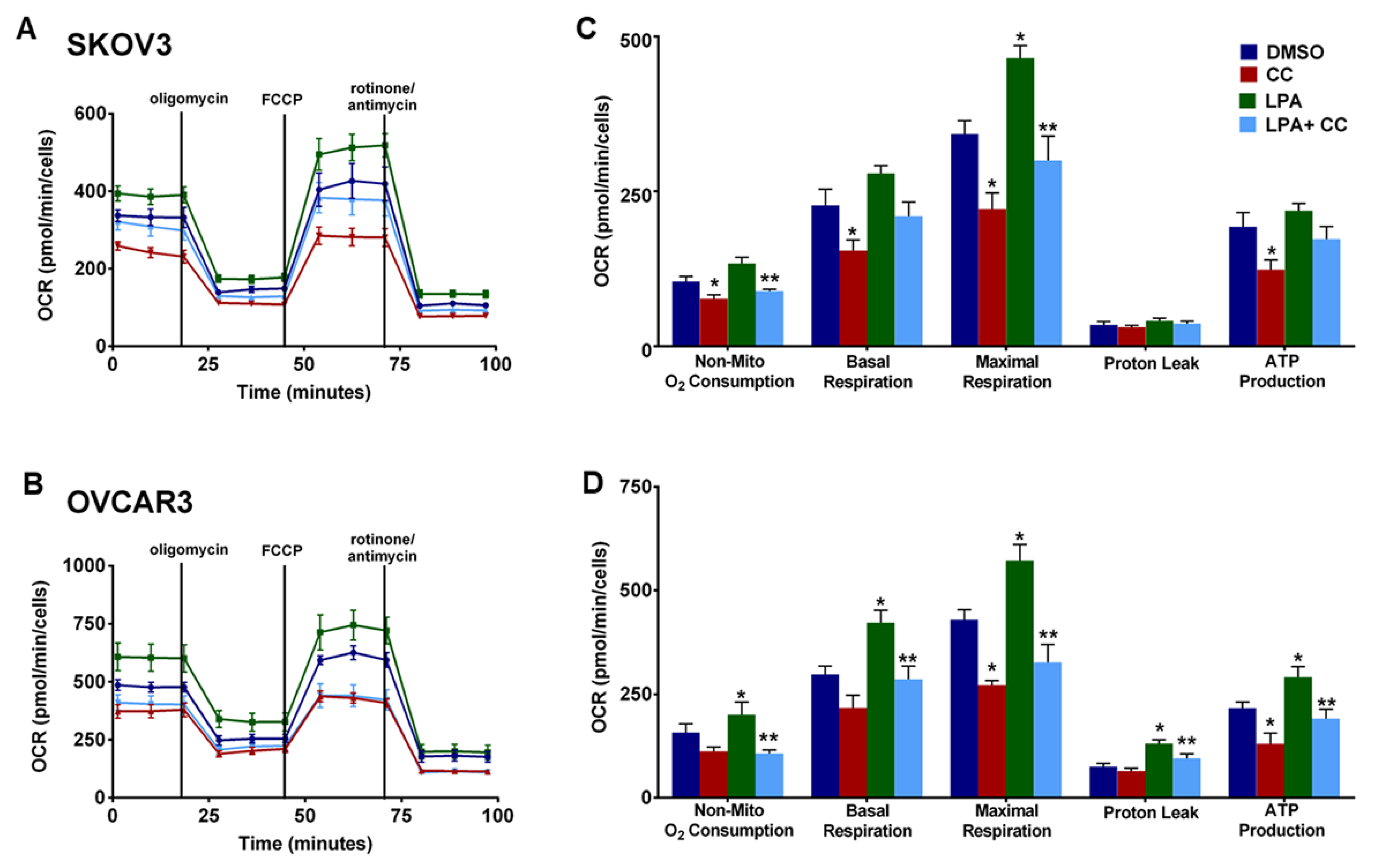
3.5. Compound C Inhibits OvCa Cell-Macrophage Crosstalk
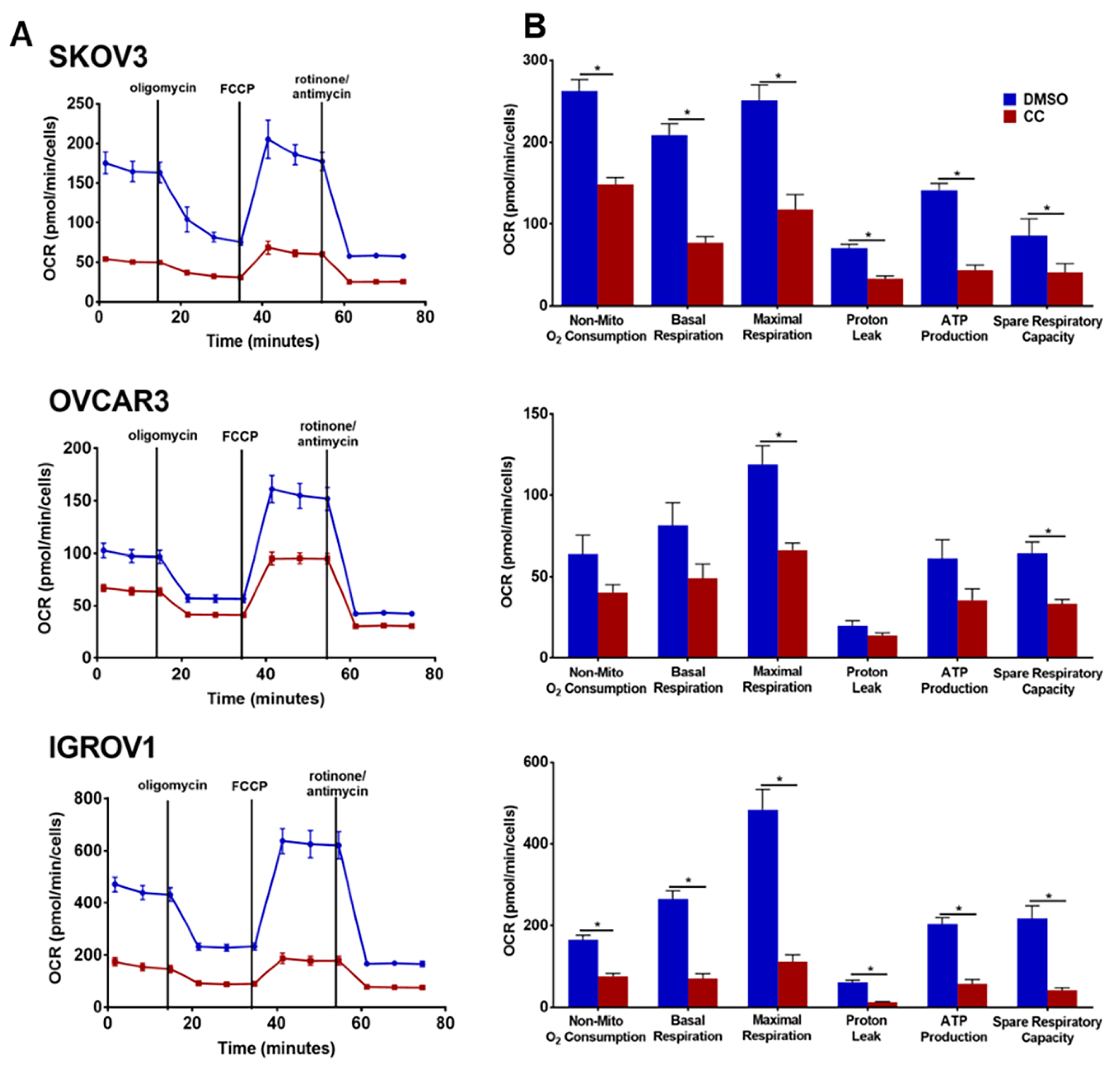
3.6. Compound C Suppresses Cellular Bioenergetics
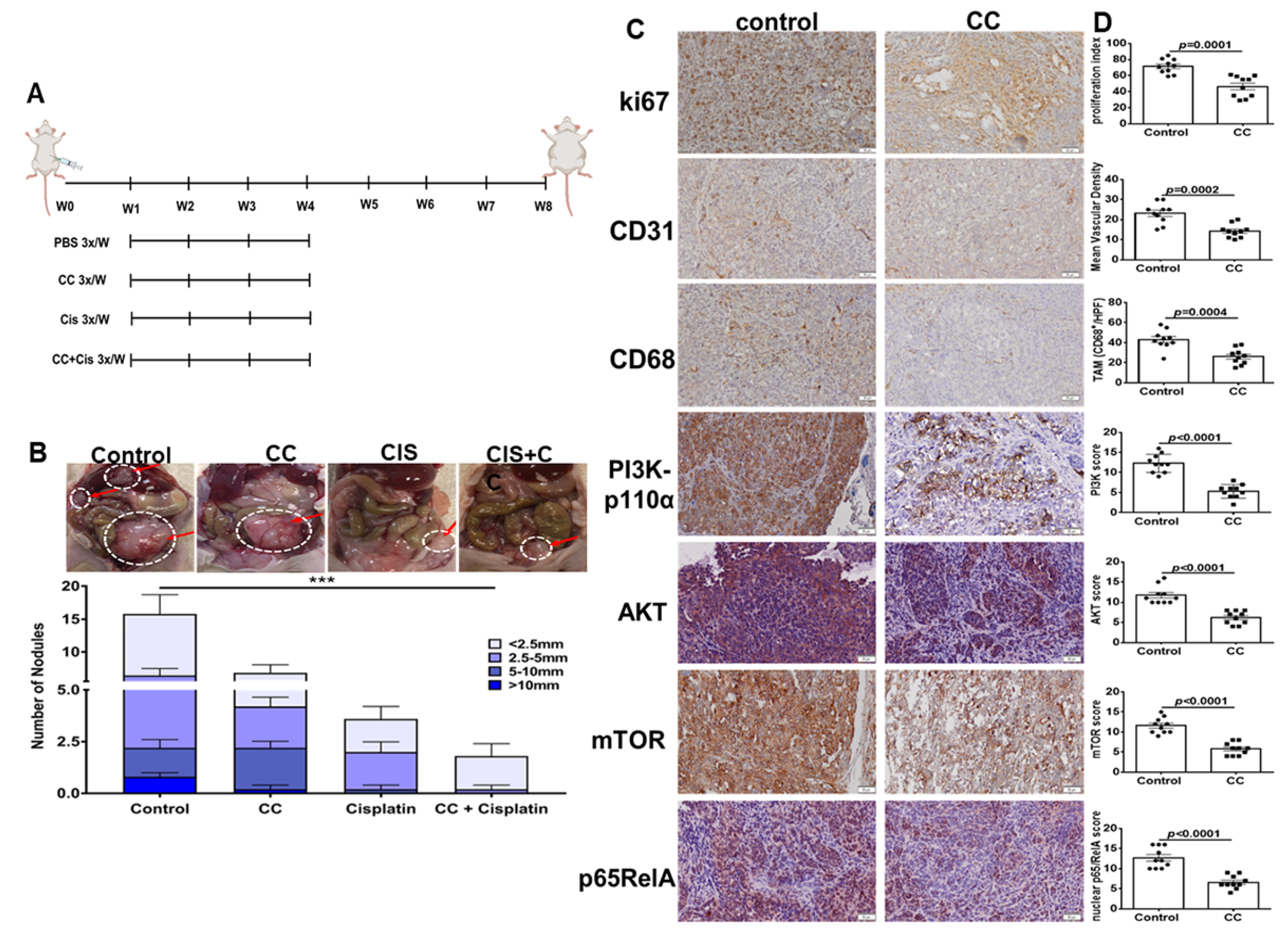
3.7. Combination Therapy of Compound C and Cisplatin, Reduced Tumor Burden in SKOV3 Xenografts in Athymic Nude Mice
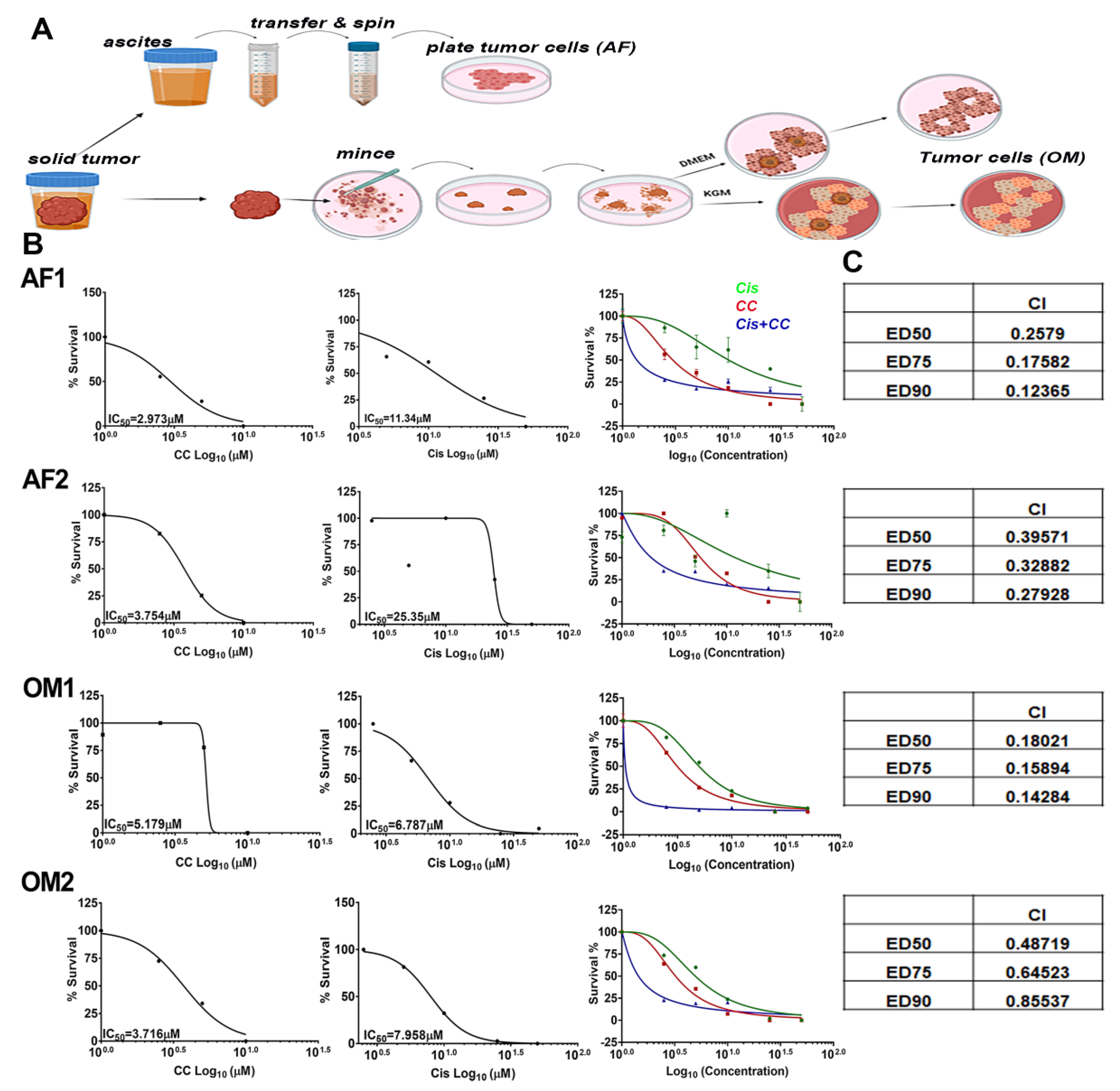
3.8. Compound C Synergized with Cisplatin in Platinum-Resistant Patient Derived OvCa Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Rojas, V.; Hirshfield, K.M.; Ganesan, S.; Rodriguez-Rodriguez, L. Molecular Characterization of Epithelial Ovarian Cancer: Implications for Diagnosis and Treatment. Int. J. Mol. Sci. 2016, 17, 2113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitajima, S.; Lee, K.L.; Hikasa, H.; Sun, W.; Huang, R.Y.; Yang, H.; Matsunaga, S.; Yamaguchi, T.; Araki, M.; Kato, H.; et al. Hypoxia-inducible factor-1alpha promotes cell survival during ammonia stress response in ovarian cancer stem-like cells. Oncotarget 2017, 8, 114481–114494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, D.; Berchuck, A.; Birrer, M.; Chien, J.; Cramer, D.W.; Dao, F.; Dhir, R.; DiSaia, P.; Gabra, H.; Glenn, P.; et al. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.Y.C.; Lheureux, S.; Karakasis, K.; Burnier, J.V.; Bruce, J.P.; Clouthier, D.L.; Danesh, A.; Quevedo, R.; Dowar, M.; Hanna, Y.; et al. Landscape of genomic alterations in high-grade serous ovarian cancer from exceptional long- and short-term survivors. Genome Med. 2018, 10, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheaib, B.; Auguste, A.; Leary, A. The PI3K/Akt/mTOR pathway in ovarian cancer: Therapeutic opportunities and challenges. Chin. J. Cancer 2015, 34, 4–16. [Google Scholar] [CrossRef] [Green Version]
- Gasparri, M.L.; Bardhi, E.; Ruscito, I.; Papadia, A.; Farooqi, A.A.; Marchetti, C.; Bogani, G.; Ceccacci, I.; Mueller, M.D.; Benedetti Panici, P. PI3K/AKT/mTOR Pathway in Ovarian Cancer Treatment: Are We on the Right Track? Geburtshilfe Frauenheilkd 2017, 77, 1095–1103. [Google Scholar] [CrossRef] [Green Version]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [Green Version]
- Al-Hassan, N.N.; Behzadian, A.; Caldwell, R.; Ivanova, V.S.; Syed, V.; Motamed, K.; Said, N.A. Differential roles of uPAR in peritoneal ovarian carcinomatosis. Neoplasia 2012, 14, 259–270. [Google Scholar] [CrossRef]
- John, B.; Naczki, C.; Patel, C.; Ghoneum, A.; Qasem, S.; Salih, Z.; Said, N. Regulation of the bi-directional cross-talk between ovarian cancer cells and adipocytes by SPARC. Oncogene 2019, 38, 4366–4383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Said, N.; Socha, M.J.; Olearczyk, J.J.; Elmarakby, A.A.; Imig, J.D.; Motamed, K. Normalization of the ovarian cancer microenvironment by SPARC. Mol. Cancer Res. 2007, 5, 1015–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Said, N.A.; Elmarakby, A.A.; Imig, J.D.; Fulton, D.J.; Motamed, K. SPARC ameliorates ovarian cancer-associated inflammation. Neoplasia 2008, 10, 1092–1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Said, N.A.; Najwer, I.; Socha, M.J.; Fulton, D.J.; Mok, S.C.; Motamed, K. SPARC inhibits LPA-mediated mesothelial-ovarian cancer cell crosstalk. Neoplasia 2007, 9, 23–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savant, S.S.; Sriramkumar, S.; O’Hagan, H.M. The Role of Inflammation and Inflammatory Mediators in the Development, Progression, Metastasis, and Chemoresistance of Epithelial Ovarian Cancer. Cancers 2018, 10, 251. [Google Scholar] [CrossRef] [Green Version]
- Hagemann, T.; Robinson, S.C.; Thompson, R.G.; Charles, K.; Kulbe, H.; Balkwill, F.R. Ovarian cancer cell-derived migration inhibitory factor enhances tumor growth, progression, and angiogenesis. Mol. Cancer Ther. 2007, 6, 1993–2002. [Google Scholar] [CrossRef] [Green Version]
- Oh, K.; Moon, H.G.; Lee, D.S.; Yoo, Y.B. Tissue transglutaminase-interleukin-6 axis facilitates peritoneal tumor spreading and metastasis of human ovarian cancer cells. Lab. Anim. Res. 2015, 31, 188–197. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Castranova, V.; Shi, X. New insights into the role of nuclear factor-kappaB in cell growth regulation. Am. J. Pathol. 2001, 159, 387–397. [Google Scholar] [CrossRef]
- Dan, H.C.; Cooper, M.J.; Cogswell, P.C.; Duncan, J.A.; Ting, J.P.; Baldwin, A.S. Akt-dependent regulation of NF-{kappa}B is controlled by mTOR and Raptor in association with IKK. Genes Dev. 2008, 22, 1490–1500. [Google Scholar] [CrossRef] [Green Version]
- Covarrubias, A.J.; Aksoylar, H.I.; Horng, T. Control of macrophage metabolism and activation by mTOR and Akt signaling. Semin. Immunol. 2015, 27, 286–296. [Google Scholar] [CrossRef]
- Troutman, T.D.; Bazan, J.F.; Pasare, C. Toll-like receptors, signaling adapters and regulation of the pro-inflammatory response by PI3K. Cell Cycle 2012, 11, 3559–3567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, L.; Tan, S.; Zhou, Y.; Lin, J.; Wang, H.; Oyang, L.; Tian, Y.; Liu, L.; Su, M.; Wang, H.; et al. Role of the NFkappaB-signaling pathway in cancer. Onco. Targets Ther. 2018, 11, 2063–2073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieman, K.M.; Romero, I.L.; van Houten, B.; Lengyel, E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim. Biophys. Acta 2013, 1831, 1533–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naczki, C.; John, B.; Patel, C.; Lafferty, A.; Ghoneum, A.; Afify, H.; White, M.; Davis, A.; Jin, G.; Kridel, S.; et al. SPARC Inhibits Metabolic Plasticity in Ovarian Cancer. Cancers 2018, 10, 385. [Google Scholar] [CrossRef] [Green Version]
- Isakovic, A.; Harhaji, L.; Stevanovic, D.; Markovic, Z.; Sumarac-Dumanovic, M.; Starcevic, V.; Micic, D.; Trajkovic, V. Dual antiglioma action of metformin: Cell cycle arrest and mitochondria-dependent apoptosis. Cell Mol. Life Sci. 2007, 64, 1290–1302. [Google Scholar] [CrossRef]
- Garulli, C.; Kalogris, C.; Pietrella, L.; Bartolacci, C.; Andreani, C.; Falconi, M.; Marchini, C.; Amici, A. Dorsomorphin reverses the mesenchymal phenotype of breast cancer initiating cells by inhibition of bone morphogenetic protein signaling. Cell Signal 2014, 26, 352–362. [Google Scholar] [CrossRef]
- Bain, J.; Plater, L.; Elliott, M.; Shpiro, N.; Hastie, C.J.; McLauchlan, H.; Klevernic, I.; Arthur, J.S.; Alessi, D.R.; Cohen, P. The selectivity of protein kinase inhibitors: A further update. Biochem. J. 2007, 408, 297–315. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, Y.; Hsiao, E.C.; Sami, S.; Lancero, M.; Schlieve, C.R.; Nguyen, T.; Yano, K.; Nagahashi, A.; Ikeya, M.; Matsumoto, Y.; et al. BMP-SMAD-ID promotes reprogramming to pluripotency by inhibiting p16/INK4A-dependent senescence. Proc. Natl. Acad. Sci. USA 2016, 113, 13057–13062. [Google Scholar] [CrossRef] [Green Version]
- Bai, L.; Chang, H.M.; Cheng, J.C.; Chu, G.; Leung, P.C.K.; Yang, G. ALK2/ALK3-BMPR2/ACVR2A Mediate BMP2-Induced Downregulation of Pentraxin 3 Expression in Human Granulosa-Lutein Cells. Endocrinology 2017, 158, 3501–3511. [Google Scholar] [CrossRef]
- Pedersen, P.L. Tumor mitochondria and the bioenergetics of cancer cells. Prog. Exp. Tumor Res. 1978, 22, 190–274. [Google Scholar] [CrossRef] [PubMed]
- Ali, J.L.; Lagasse, B.J.; Minuk, A.J.; Love, A.J.; Moraya, A.I.; Lam, L.; Arthur, G.; Gibson, S.B.; Morrison, L.C.; Werbowetski-Ogilvie, T.E.; et al. Differential cellular responses induced by dorsomorphin and LDN-193189 in chemotherapy-sensitive and chemotherapy-resistant human epithelial ovarian cancer cells. Int. J. Cancer 2015, 136, E455–E469. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.T.; Lim, S.H.; Lee, B.; Kang, I.; Yeo, E.J. Compound C Inhibits B16-F1 Tumor Growth in a Syngeneic Mouse Model Via the Blockage of Cell Cycle Progression and Angiogenesis. Cancers 2019, 11, 823. [Google Scholar] [CrossRef] [Green Version]
- Gündüz, D.; Klewer, M.; Bauer, P.; Tanislav, C.; Sedding, D.; Rohrbach, S.; Schulz, R.; Aslam, M. Compound C inhibits in vitro angiogenesis and ameliorates thrombin-induced endothelial barrier failure. Eur. J. Pharmacol. 2015, 768, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Said, N.; Motamed, K. Absence of host-secreted protein acidic and rich in cysteine (SPARC) augments peritoneal ovarian carcinomatosis. Am. J. Pathol. 2005, 167, 1739–1752. [Google Scholar] [CrossRef] [Green Version]
- Said, N.; Najwer, I.; Motamed, K. Secreted protein acidic and rich in cysteine (SPARC) inhibits integrin-mediated adhesion and growth factor-dependent survival signaling in ovarian cancer. Am. J. Pathol. 2007, 170, 1054–1063. [Google Scholar] [CrossRef] [Green Version]
- Hirst, J.; Pathak, H.B.; Hyter, S.; Pessetto, Z.Y.; Ly, T.; Graw, S.; Koestler, D.C.; Krieg, A.J.; Roby, K.F.; Godwin, A.K. Licofelone Enhances the Efficacy of Paclitaxel in Ovarian Cancer by Reversing Drug Resistance and Tumor Stem-like Properties. Cancer Res. 2018, 78, 4370–4385. [Google Scholar] [CrossRef] [Green Version]
- Said, N.; Frierson, H.F.; Sanchez-Carbayo, M.; Brekken, R.A.; Theodorescu, D. Loss of SPARC in bladder cancer enhances carcinogenesis and progression. J. Clin. Investig. 2013, 123, 751–766. [Google Scholar] [CrossRef] [Green Version]
- Rafehi, H.; Orlowski, C.; Georgiadis, G.T.; Ververis, K.; El-Osta, A.; Karagiannis, T.C. Clonogenic assay: Adherent cells. J. Vis. Exp. 2011, 49, e2573. [Google Scholar] [CrossRef] [Green Version]
- Peng, J.; Yoshioka, Y.; Mandai, M.; Matsumura, N.; Baba, T.; Yamaguchi, K.; Hamanishi, J.; Kharma, B.; Murakami, R.; Abiko, K.; et al. The BMP signaling pathway leads to enhanced proliferation in serous ovarian cancer—A potential therapeutic target. Mol. Carcinog. 2016, 55, 335–345. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.J.; Liu, Z.; Wang, L.; Shin, E.; Loda, M.F.; Roberts, T.M. The oncogenic properties of mutant p110alpha and p110beta phosphatidylinositol 3-kinases in human mammary epithelial cells. Proc. Natl. Acad. Sci. USA 2005, 102, 18443–18448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boehm, J.S.; Zhao, J.J.; Yao, J.; Kim, S.Y.; Firestein, R.; Dunn, I.F.; Sjostrom, S.K.; Garraway, L.A.; Weremowicz, S.; Richardson, A.L.; et al. Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell 2007, 129, 1065–1079. [Google Scholar] [CrossRef] [Green Version]
- Morgenstern, J.P.; Land, H. Advanced mammalian gene transfer: High titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 1990, 18, 3587–3596. [Google Scholar] [CrossRef] [Green Version]
- Naviaux, R.K.; Costanzi, E.; Haas, M.; Verma, I.M. The pCL vector system: Rapid production of helper-free, high-titer, recombinant retroviruses. J. Virol. 1996, 70, 5701–5705. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Walker, E.H.; Pacold, M.E.; Perisic, O.; Stephens, L.; Hawkins, P.T.; Wymann, M.P.; Williams, R.L. Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol. Cell 2000, 6, 909–919. [Google Scholar] [CrossRef]
- Yang, H.; Rudge, D.G.; Koos, J.D.; Vaidialingam, B.; Yang, H.J.; Pavletich, N.P. mTOR kinase structure, mechanism and regulation. Nature 2013, 497, 217–223. [Google Scholar] [CrossRef]
- Stroud, J.C.; Oltman, A.; Han, A.; Bates, D.L.; Chen, L. Structural basis of HIV-1 activation by NF-kappaB--a higher-order complex of p50:RelA bound to the HIV-1 LTR. J. Mol. Biol. 2009, 393, 98–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.J.; Barham, W.; Saskowski, J.; Tikhomirov, O.; Chen, L.; Lee, H.J.; Yull, F.; Khabele, D. Tracking NF-kappaB activity in tumor cells during ovarian cancer progression in a syngeneic mouse model. J. Ovarian Res. 2013, 6, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrington, B.S.; Annunziata, C.M. NF-kappaB Signaling in Ovarian Cancer. Cancers 2019, 11, 1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghoneum, A.; Abdulfattah, A.Y.; Said, N. Targeting the PI3K/AKT/mTOR/NFkappaB Axis in Ovarian Cancer. J. Cell Immunol. 2020, 2, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Ghoneum, A.; Said, N. PI3K-AKT-mTOR and NFkappaB Pathways in Ovarian Cancer: Implications for Targeted Therapeutics. Cancers 2019, 11, 949. [Google Scholar] [CrossRef] [Green Version]
- Davidowitz, R.A.; Iwanicki, M.P.; Brugge, J.S. In vitro mesothelial clearance assay that models the early steps of ovarian cancer metastasis. J. Vis. Exp. 2012, 60, e3888. [Google Scholar] [CrossRef] [Green Version]
- Davidowitz, R.A.; Selfors, L.M.; Iwanicki, M.P.; Elias, K.M.; Karst, A.; Piao, H.; Ince, T.A.; Drage, M.G.; Dering, J.; Konecny, G.E.; et al. Mesenchymal gene program-expressing ovarian cancer spheroids exhibit enhanced mesothelial clearance. J. Clin. Investig. 2014, 124, 2611–2625. [Google Scholar] [CrossRef] [Green Version]
- Nowak, M.; Klink, M. The Role of Tumor-Associated Macrophages in the Progression and Chemoresistance of Ovarian Cancer. Cells 2020, 9, 1299. [Google Scholar] [CrossRef]
- Ghoneum, A.; Almousa, S.; Warren, B.; Abdulfattah, A.Y.; Shu, J.; Abouelfadl, H.; Gonzalez, D.; Livingston, C.; Said, N. Exploring the clinical value of tumor microenvironment in platinum-resistant ovarian cancer. Semin. Cancer Biol. 2021, 77, 83–98. [Google Scholar] [CrossRef]
- Accordi, B.; Galla, L.; Milani, G.; Curtarello, M.; Serafin, V.; Lissandron, V.; Viola, G.; te Kronnie, G.; de Maria, R.; Petricoin, E.F., 3rd; et al. AMPK inhibition enhances apoptosis in MLL-rearranged pediatric B-acute lymphoblastic leukemia cells. Leukemia 2013, 27, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Ghanaatgar-Kasbi, S.; Amerizadeh, F.; Rahmani, F.; Hassanian, S.M.; Khazaei, M.; Ferns, G.A.; Avan, A. AMP-kinase inhibitor dorsomorphin reduces the proliferation and migration behavior of colorectal cancer cells by targeting the AKT/mTOR pathway. IUBMB Life 2019, 71, 1929–1936. [Google Scholar] [CrossRef] [PubMed]
- Jaschke, N.; Kleymann, A.; Hofbauer, L.C.; Gobel, A.; Rachner, T.D. Dorsomorphin: A novel inhibitor of Dickkopf-1 in breast cancer. Biochem. Biophys. Res. Commun. 2020, 524, 360–365. [Google Scholar] [CrossRef]
- Liu, X.; Chhipa, R.R.; Nakano, I.; Dasgupta, B. The AMPK inhibitor compound C is a potent AMPK-independent antiglioma agent. Mol. Cancer Ther. 2014, 13, 596–605. [Google Scholar] [CrossRef] [Green Version]
- Majd, S.; Koblar, S.; Power, J. Compound C enhances tau phosphorylation at Serine396 via PI3K activation in an AMPK and rapamycin independent way in differentiated SH-SY5Y cells. Neurosci. Lett. 2018, 670, 53–61. [Google Scholar] [CrossRef]
- Patil, K.; Joseph, S.; Shah, J.; Mukherjee, S. An integrated in silico analysis highlighted angiogenesis regulating miRNA-mRNA network in PCOS pathophysiology. J. Assist. Reprod. Genet. 2022, 39, 427–440. [Google Scholar] [CrossRef]
- Wang, J.; Yang, L.; Liang, F.; Chen, Y.; Yang, G. Integrin alpha x stimulates cancer angiogenesis through PI3K/Akt signaling-mediated VEGFR2/VEGF-A overexpression in blood vessel endothelial cells. J. Cell Biochem. 2019, 120, 1807–1818. [Google Scholar] [CrossRef]
- Shariati, M.; Meric-Bernstam, F. Targeting AKT for cancer therapy. Expert Opin. Investig. Drugs 2019, 28, 977–988. [Google Scholar] [CrossRef]
- Karar, J.; Maity, A. PI3K/AKT/mTOR Pathway in Angiogenesis. Front. Mol. Neurosci. 2011, 4, 51. [Google Scholar] [CrossRef] [Green Version]
- Tabruyn, S.P.; Griffioen, A.W. NF-κB: A new player in angiostatic therapy. Angiogenesis 2008, 11, 101–106. [Google Scholar] [CrossRef]
- Schmidt, D.; Textor, B.; Pein, O.T.; Licht, A.H.; Andrecht, S.; Sator-Schmitt, M.; Fusenig, N.E.; Angel, P.; Schorpp-Kistner, M. Critical role for NF-κB-induced JunB in VEGF regulation and tumor angiogenesis. EMBO J. 2007, 26, 710–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esmat, A.; Alzahrani, A.M.; Alharthy, B.T.; Ramadan, W.S.; Sattar Ahmad, M.A.A. Potential Nephroprotective Effect of Dorsomorphin Homolog 1 (DMH1) in a rat model of diabetic nephropathy. Eur. Rev. Med. Pharmacol. Sci. 2022, 26, 2489–2500. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Zhang, Y.; Chen, J.; Wu, P.; Dong, Y.; Wang, Y. Neuroprotective effects of liraglutide against inflammation through the AMPK/NF-κB pathway in a mouse model of Parkinson’s disease. Metab. Brain Dis. 2022, 37, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Hao, J.; Wu, Y.; Liu, C.; Shimizu, K.; Li, R.; Zhang, C. Protective effects of heterophyllin B against bleomycin-induced pulmonary fibrosis in mice via AMPK activation. Eur. J. Pharmacol. 2022, 921, 174825. [Google Scholar] [CrossRef]
- Lin, T.; Wang, X.L.; Zettervall, S.L.; Cai, Y.; Guzman, R.J. Dorsomorphin homologue 1, a highly selective small-molecule bone morphogenetic protein inhibitor, suppresses medial artery calcification. J. Vasc. Surg. 2017, 66, 586–593. [Google Scholar] [CrossRef]
- Hao, J.; Ho, J.N.; Lewis, J.A.; Karim, K.A.; Daniels, R.N.; Gentry, P.R.; Hopkins, C.R.; Lindsley, C.W.; Hong, C.C. In Vivo Structure−Activity Relationship Study of Dorsomorphin Analogues Identifies Selective VEGF and BMP Inhibitors. ACS Chem. Biol. 2010, 5, 245–253. [Google Scholar] [CrossRef] [Green Version]
- Lindholm, H.T.; Parmar, N.; Drurey, C.; Campillo Poveda, M.; Vornewald, P.M.; Ostrop, J.; Díez-Sanchez, A.; Maizels, R.M.; Oudhoff, M.J. BMP signaling in the intestinal epithelium drives a critical feedback loop to restrain IL-13-driven tuft cell hyperplasia. Sci. Immunol. 2022, 7, eabl6543. [Google Scholar] [CrossRef]
- Eder, A.M.; Sasagawa, T.; Mao, M.; Aoki, J.; Mills, G.B. Constitutive and Lysophosphatidic Acid (LPA)-induced LPA Production: Role of Phospholipase D and Phospholipase A2. Clin. Cancer Res. 2000, 6, 2482–2491. [Google Scholar]
- Mills, G.B.; Moolenaar, W.H. The emerging role of lysophosphatidic acid in cancer. Nat. Rev. Cancer 2003, 3, 582–591. [Google Scholar] [CrossRef]
- Ghoneum, A.; Gonzalez, D.; Abdulfattah, A.Y.; Said, N. Metabolic Plasticity in Ovarian Cancer Stem Cells. Cancers 2020, 12, 1267. [Google Scholar] [CrossRef]
- Rinne, N.; Christie, E.L.; Ardasheva, A.; Kwok, C.H.; Demchenko, N.; Low, C.; Tralau-Stewart, C.; Fotopoulou, C.; Cunnea, P. Targeting the PI3K/AKT/mTOR pathway in epithelial ovarian cancer, therapeutic treatment options for platinum-resistant ovarian cancer. Cancer Drug Resist. 2021, 4, 573–595. [Google Scholar] [CrossRef] [PubMed]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghoneum, A.; Gonzalez, D.; Afify, H.; Shu, J.; Hegarty, A.; Adisa, J.; Kelly, M.; Lentz, S.; Salsbury, F.; Said, N. Compound C Inhibits Ovarian Cancer Progression via PI3K-AKT-mTOR-NFκB Pathway. Cancers 2022, 14, 5099. https://doi.org/10.3390/cancers14205099
Ghoneum A, Gonzalez D, Afify H, Shu J, Hegarty A, Adisa J, Kelly M, Lentz S, Salsbury F, Said N. Compound C Inhibits Ovarian Cancer Progression via PI3K-AKT-mTOR-NFκB Pathway. Cancers. 2022; 14(20):5099. https://doi.org/10.3390/cancers14205099
Chicago/Turabian StyleGhoneum, Alia, Daniela Gonzalez, Hesham Afify, Junjun Shu, Abigail Hegarty, Jemima Adisa, Michael Kelly, Samuel Lentz, Freddie Salsbury, and Neveen Said. 2022. "Compound C Inhibits Ovarian Cancer Progression via PI3K-AKT-mTOR-NFκB Pathway" Cancers 14, no. 20: 5099. https://doi.org/10.3390/cancers14205099