
The Differential Metabolic Signature of Breast Cancer Cellular Response to Olaparib Treatment

 , ,
, ,  and
and
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials
Cell Lines and Chemicals
3. Methods
3.1. Cell Viability Assays
3.2. Immunostaining for γH2AX and p53BP1
3.3. Sample Preparation and Metabolite Extraction
3.4. Liquid Chromatography Tandem Mass Spectrometry (LC-MS/MS)
3.5. Mass Spectrometry Data Processing
3.6. Pathway Analysis with MetaboAnalyst
3.7. Statistical Analysis
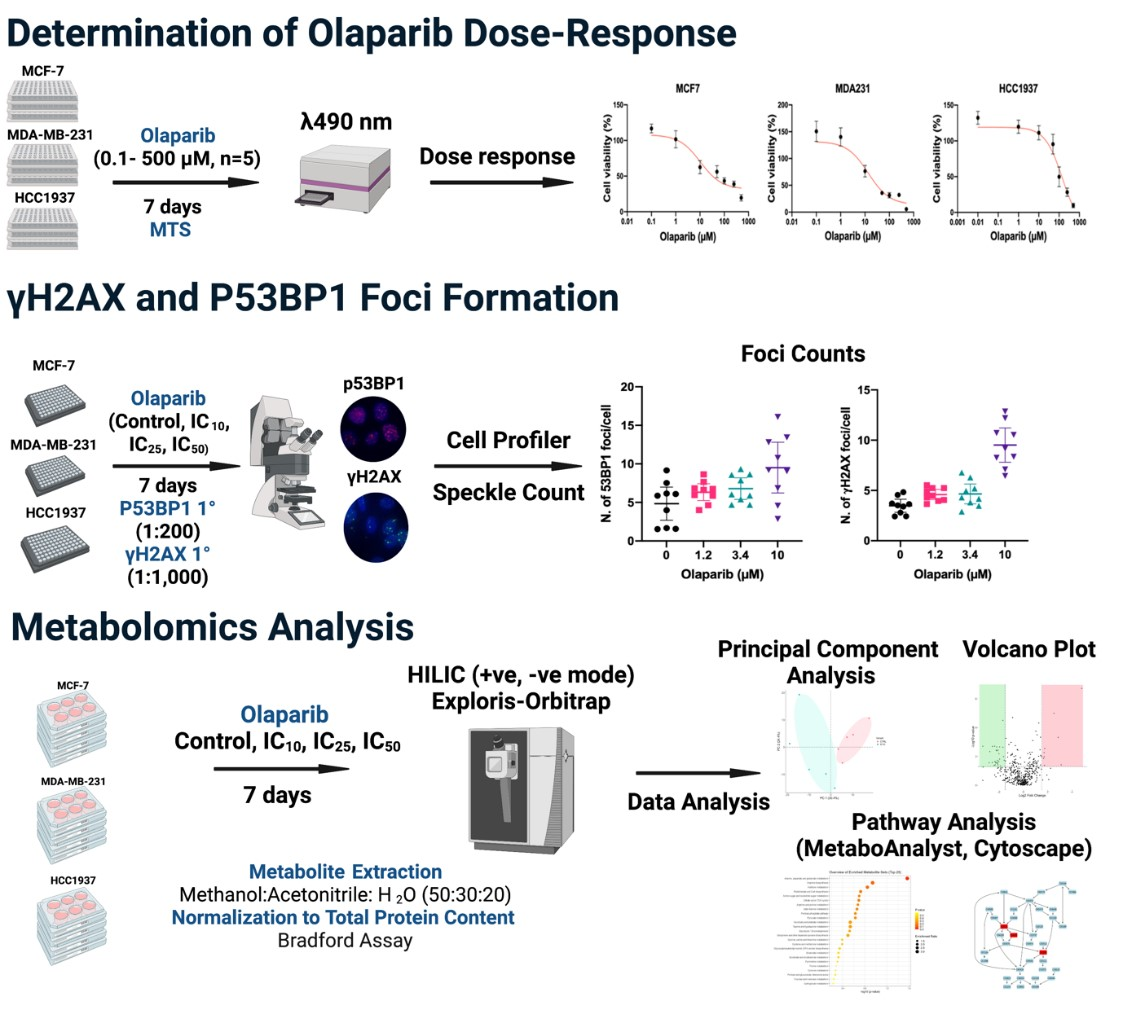
4. Results
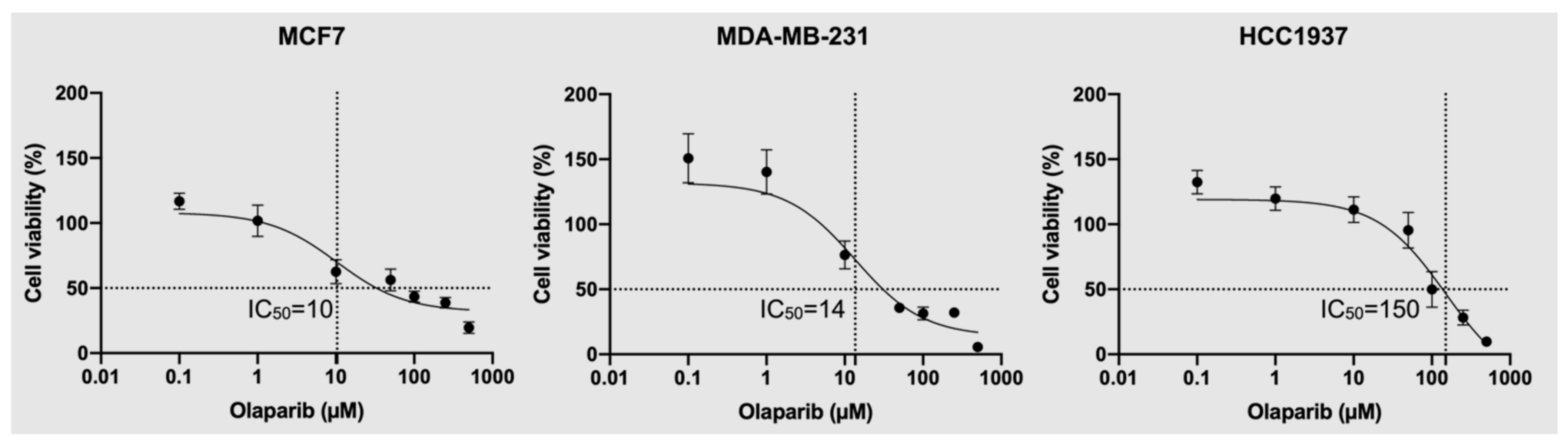
4.1. Olaparib Sensitivity Analysis
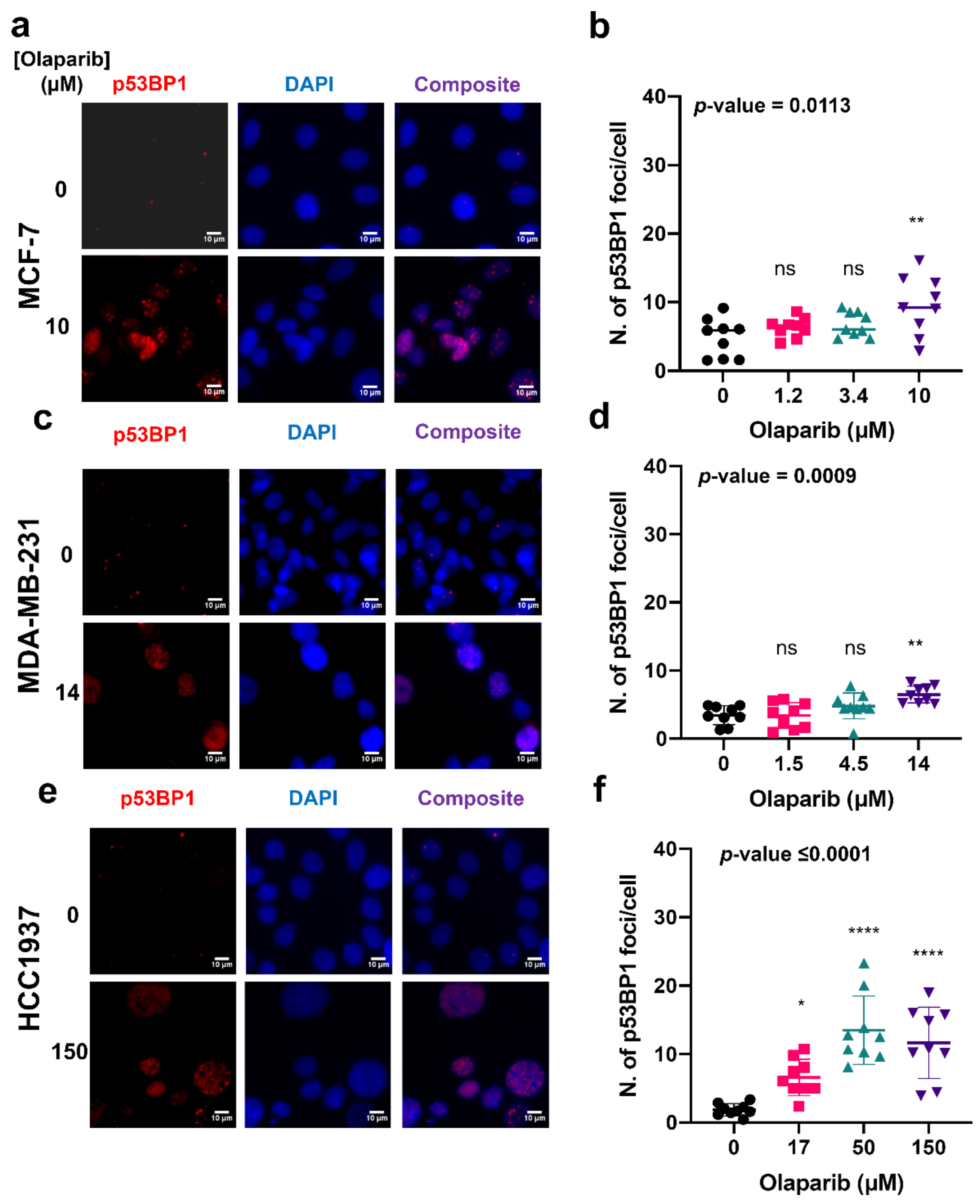
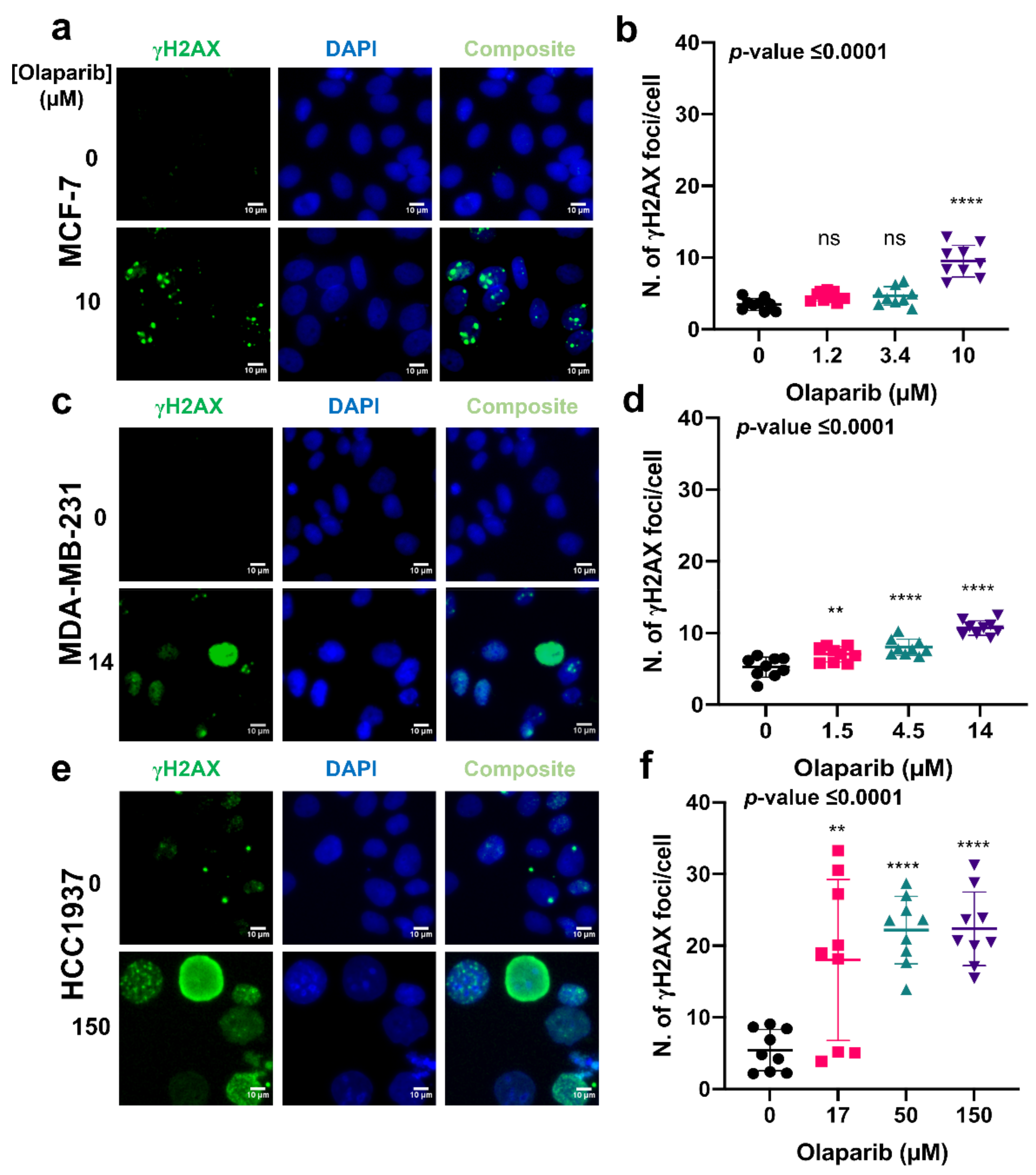
4.2. Exposure to Olaparib Induces Dose-Dependent Formation of γH2AX and 53BP1 Foci in Breast Cancer Cells
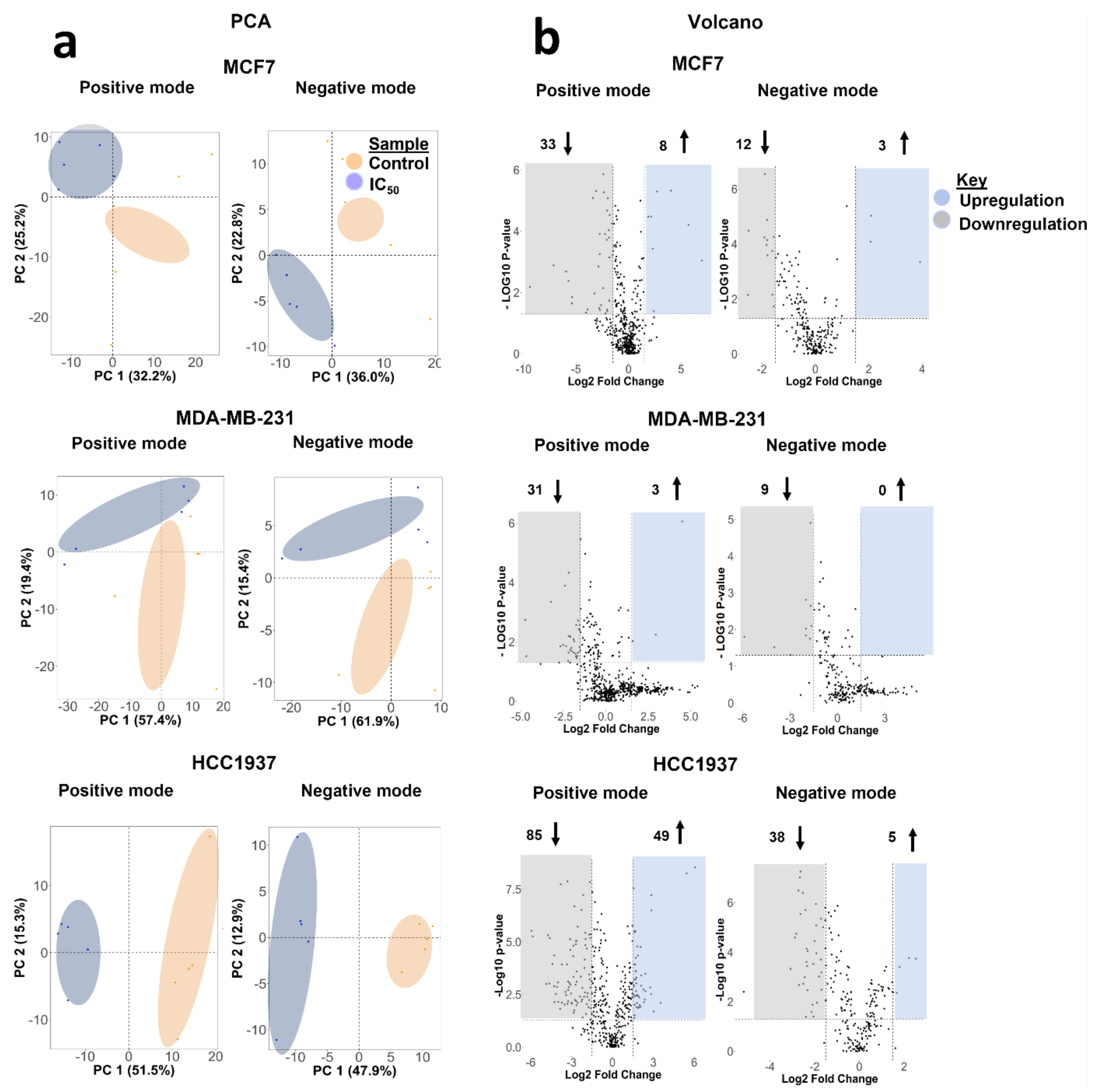
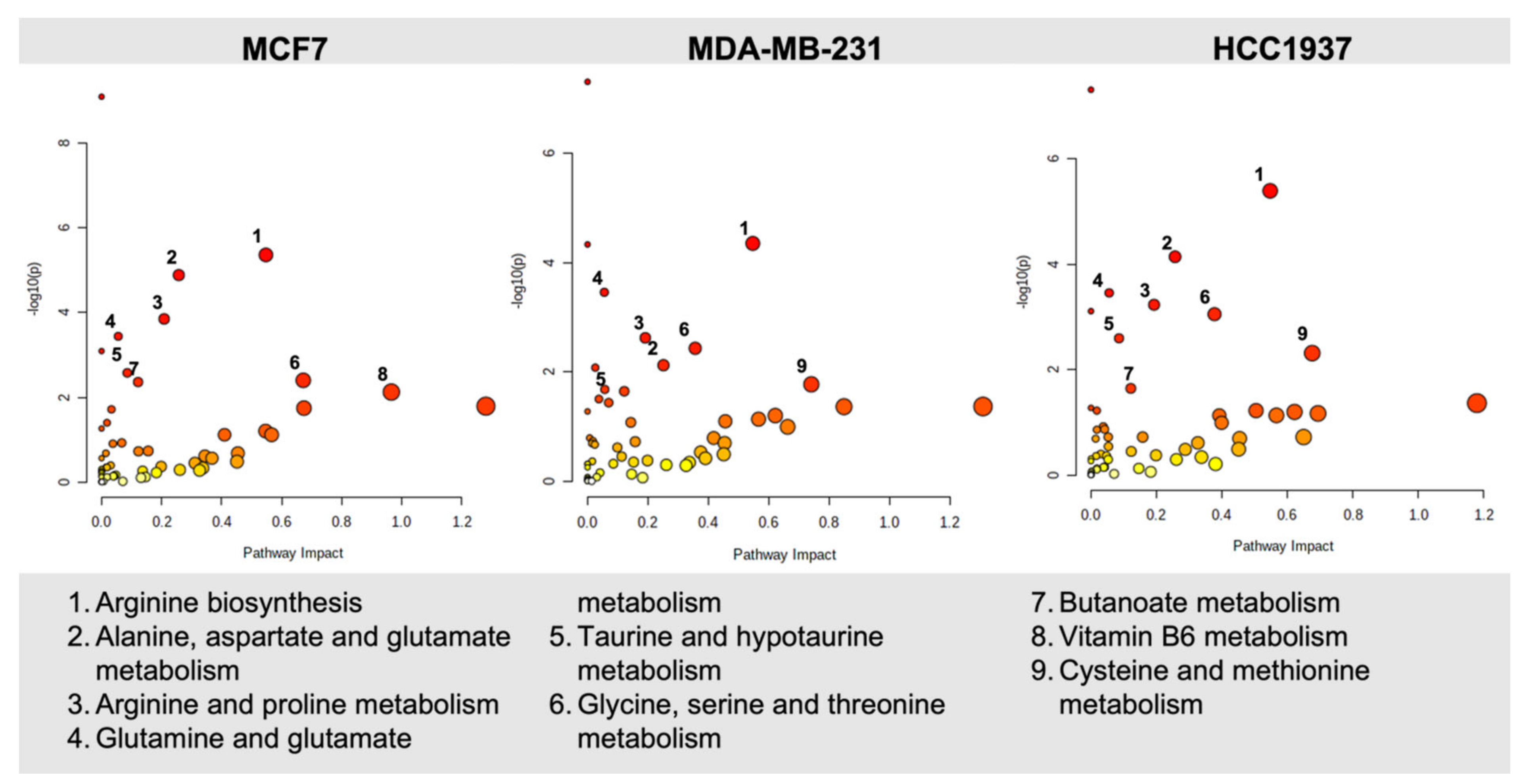
4.3. Biomolecular Pathways Altered in Response to Olaparib Exposure Vary across Different Cell Lines
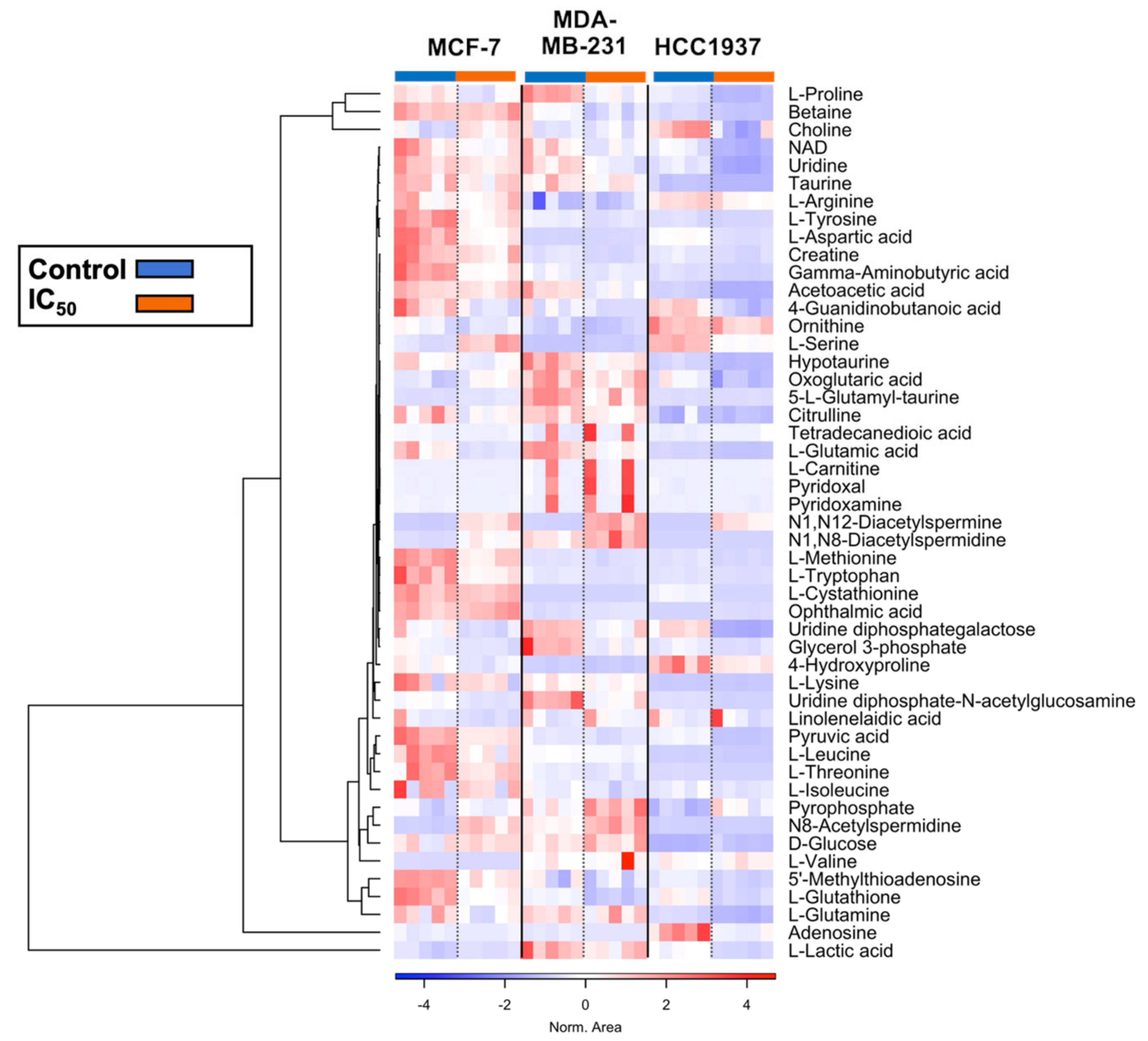
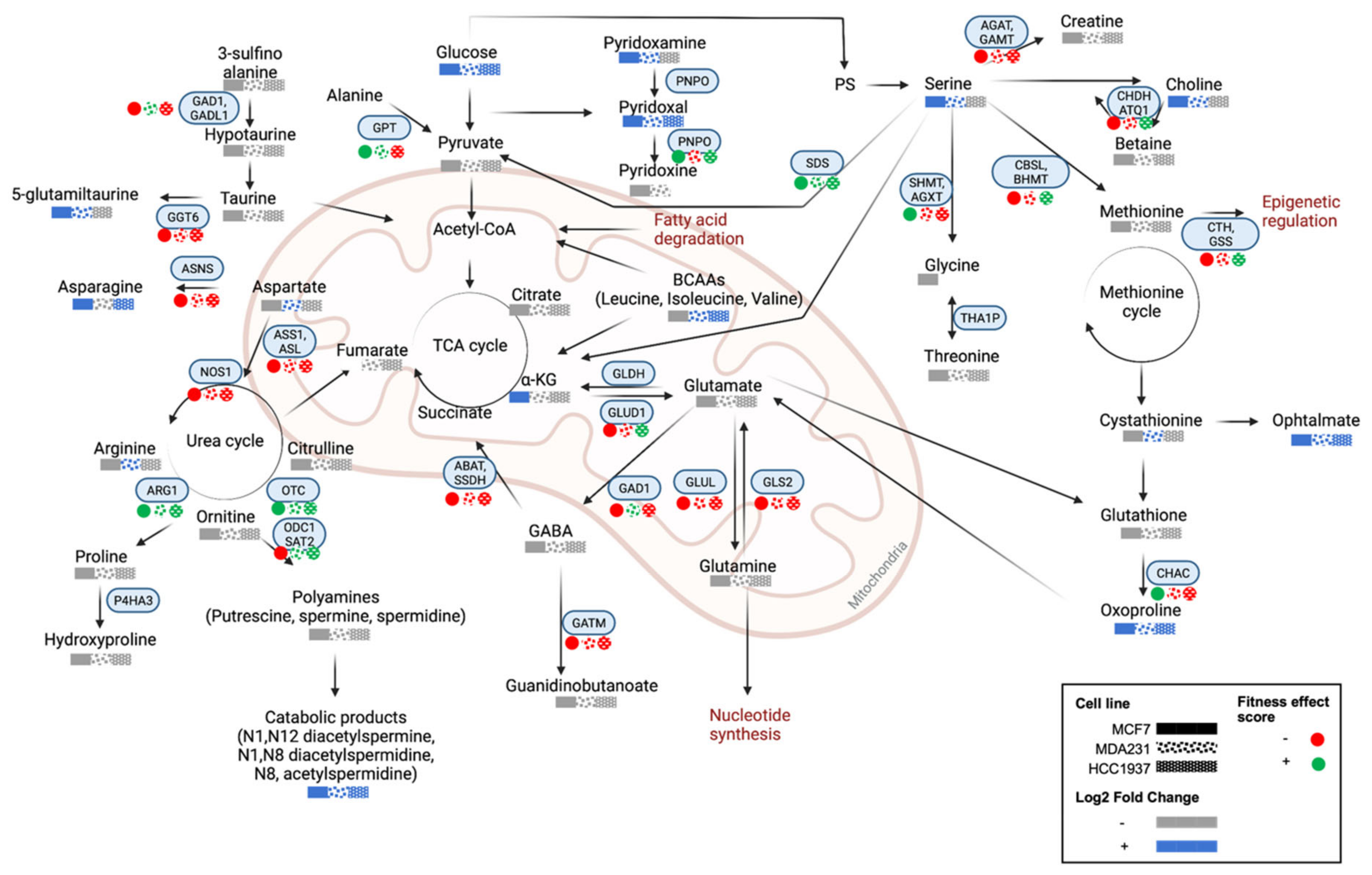
4.4. Amino Acid and Lipid Metabolism Are Significantly Altered in Response to Olaparib Exposure
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gueble, S.E.; Bindra, R.S. Oncometabolites as Regulators of DNA Damage Response and Repair. Semin. Radiat. Oncol. 2021, 32, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Zhou, P.-K. DNA damage repair: Historical perspectives, mechanistic pathways and clinical translation for targeted cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 254. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Kawaguchi, K.; Toi, M. DNA damage repair functions and targeted treatment in breast cancer. Breast Cancer 2020, 27, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Ottini, L.; Rizzolo, P.; Silvestri, V.; Falchetti, M. Inherited and acquired alterations in development of breast cancer. Appl. Clin. Genet. 2011, 4, 145–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godet, I.; Gilkes, D.M. BRCA1 and BRCA2 mutations and treatment strategies for breast cancer. Integr. Cancer Sci. Ther. 2017, 4. [Google Scholar] [CrossRef] [Green Version]
- O’Neil, N.; Bailey, M.L.; Hieter, P. Synthetic lethality and cancer. Nat. Rev. Genet. 2017, 18, 613–623. [Google Scholar] [CrossRef]
- Li, S.; Topatana, W.; Juengpanich, S.; Cao, J.; Hu, J.; Zhang, B.; Ma, D.; Cai, X.; Chen, M. Development of synthetic lethality in cancer: Molecular and cellular classification. Signal Transduct. Target. Ther. 2020, 5, 241. [Google Scholar] [CrossRef]
- Lord, C.J.; Tutt, A.N.; Ashworth, A. Synthetic Lethality and Cancer Therapy: Lessons Learned from the Development of PARP Inhibitors. Annu. Rev. Med. 2015, 66, 455–470. [Google Scholar] [CrossRef]
- Cortesi, L.; Rugo, H.S.; Jackisch, C. An Overview of PARP Inhibitors for the Treatment of Breast Cancer. Target. Oncol. 2021, 16, 255–282. [Google Scholar] [CrossRef]
- FDA. FDA Approves Olaparib for Adjuvant Treatment of High-Risk Early Breast Cancer 2022. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-olaparib-adjuvant-treatment-high-risk-early-breast-cancer (accessed on 20 May 2022).
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Maria, R.M.; Altei, W.F.; Selistre-De-Araujo, H.S.; Colnago, L.A. Impact of chemotherapy on metabolic reprogramming: Characterization of the metabolic profile of breast cancer MDA-MB-231 cells using 1 H HR-MAS NMR spectroscopy. J. Pharm. Biomed. Anal. 2017, 146, 324–328. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, N.; Das, G. Metabolic Reprogramming in Breast Cancer and Its Therapeutic Implications. Cells 2019, 8, 89. [Google Scholar] [CrossRef] [Green Version]
- Palaskas, N.; Larson, S.M.; Schultz, N.; Komisopoulou, E.; Wong, J.; Rohle, D.; Campos, C.; Yannuzzi, N.; Osborne, J.R.; Linkov, I.; et al. 18F-fluorodeoxy-glucose positron emission tomography marks MYC-overexpressing human basal-like breast cancers. Cancer Res. 2011, 71, 5164–5174. [Google Scholar] [CrossRef] [Green Version]
- Turgeon, M.; Perry, N.J.S.; Poulogiannis, G. DNA Damage, Repair, and Cancer Metabolism. Front. Oncol. 2018, 8, 15. [Google Scholar] [CrossRef] [Green Version]
- Bhute, V.J.; Ma, Y.; Bao, X.; Palecek, S.P. The Poly (ADP-Ribose) Polymerase Inhibitor Veliparib and Radiation Cause Significant Cell Line Dependent Metabolic Changes in Breast Cancer Cells. Sci. Rep. 2016, 6, 36061. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Lin, C.-C.; Spasojevic, I.; Iversen, E.S.; Chi, J.-T.; Marks, J.R. A joint analysis of metabolomics and genetics of breast cancer. Breast Cancer Res. 2014, 16, 415. [Google Scholar] [CrossRef] [Green Version]
- Huang, A.; Garraway, L.A.; Ashworth, A.; Weber, B. Synthetic lethality as an engine for cancer drug target discovery. Nat. Rev. Drug Discov. 2019, 19, 23–38. [Google Scholar] [CrossRef]
- Mah, L.; Karagiannis, T.C. Gammah2ax: A sensitive molecular marker of DNA damage and repair. Leukemia 2010, 24, 679–686. [Google Scholar] [CrossRef] [Green Version]
- Schultz, L.B.; Chehab, N.H.; Malikzay, A.; Halazonetis, T.D. P53 Binding Protein 1 (53bp1) Is an Early Participant in the Cellular Response to DNA Double-Strand Breaks. J. Cell Biol. 2000, 151, 1381–1390. [Google Scholar] [CrossRef] [Green Version]
- Dempster, J.M.; Boyle, I.; Vazquez, F.; Root, D.E.; Boehm, J.S.; Hahn, W.C.; Tsherniak, A.; McFarland, J.M. Chronos: A cell population dynamics model of CRISPR experiments that improves inference of gene fitness effects. Genome Biol. 2021, 22, 343. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- AstraZeneca. Lynparza Approved in the US as Adjuvant Treatment for Patients with Germline BRCA-Mutated HER2-Negative High-Risk Early Breast Cancer 2022. Available online: https://www.astrazeneca.com/media-centre/press-releases/2022/lynparza-approved-in-the-us-as-adjuvant-treatment-for-patients-with-germline-brca-mutated-her2-negative-high-risk-early-breast-cancer.html (accessed on 25 May 2022).
- Keung, M.Y.; Wu, Y.; Badar, F.; Vadgama, J.V. Response of Breast Cancer Cells to PARP Inhibitors Is Independent of BRCA Status. J. Clin. Med. 2020, 9, 940. [Google Scholar] [CrossRef] [Green Version]
- Davies, H.; Glodzik, D.; Morganella, S.; Yates, L.R.; Staaf, J.; Zou, X.; Ramakrishna, M.; Martin, S.; Boyault, S.; Sieuwerts, A.M.; et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 2017, 23, 517–525. [Google Scholar] [CrossRef]
- McGrail, D.J.; Lin, C.C.-J.; Garnett, J.; Liu, Q.; Mo, W.; Dai, H.; Lu, Y.; Yu, Q.; Ju, Z.; Yin, J.; et al. Improved prediction of PARP inhibitor response and identification of synergizing agents through use of a novel gene expression signature generation algorithm. NPJ Syst. Biol. Appl. 2017, 3, 8. [Google Scholar] [CrossRef] [Green Version]
- Harbeck, N.; Penault-Lorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nature 2019, 5, 6. [Google Scholar]
- Hart, C.D.; Tenori, L.; Luchinat, C.; Di Leo, A. Metabolomics in Breast Cancer: Current Status and Perspectives. Nov. Biomark. Contin. Breast Cancer 2016, 882, 217–234. [Google Scholar] [CrossRef]
- Bhute, V.J.; Palecek, S.P. Metabolic responses induced by DNA damage and poly (ADP-ribose) polymerase (PARP) inhibition in MCF-7 cells. Metabolomics 2015, 11, 1779–1791. [Google Scholar] [CrossRef] [Green Version]
- Gajan, A.; Sarma, A.; Kim, S.; Gurdziel, K.; Wu, G.S.; Shekhar, M.P. Analysis of Adaptive Olaparib Resistance Effects on Cisplatin Sensitivity in Triple Negative Breast Cancer Cells. Front. Oncol. 2021, 11, 2857. [Google Scholar] [CrossRef]
- Najumudeen, A.K.; CRUK Rosetta Grand Challenge Consortium; Ceteci, F.; Fey, S.K.; Hamm, G.; Steven, R.T.; Hall, H.; Nikula, C.J.; Dexter, A.; Murta, T.; et al. The amino acid transporter SLC7A5 is required for efficient growth of KRAS-mutant colorectal cancer. Nat. Genet. 2021, 53, 16–26. [Google Scholar] [CrossRef]
- Tennant, D.A.; Frezza, C.; MacKenzie, E.D.; Nguyen, Q.D.; Zheng, L.; Selak, M.A.; Roberts, D.L.; Dive, C.; Watson, D.G.; Aboagye, E.O.; et al. Reactivating HIF prolyl hydroxylases under hypoxia results in metabolic catastrophe and cell death. Oncogene 2009, 28, 4009–4021. [Google Scholar] [CrossRef] [Green Version]
- Scalia, M.; Satriano, C.; Greca, R.; Stella, A.M.G.; Rizzarelli, E.; Spina-Purrello, V. PARP-1 Inhibitors DPQ and PJ-34 Negatively Modulate Proinflammatory Commitment of Human Glioblastoma Cells. Neurochem. Res. 2012, 38, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Sulkowski, P.L.; Corso, C.D.; Robinson, N.D.; Scanlon, S.E.; Purshouse, K.R.; Bai, H.; Liu, Y.; Sundaram, R.K.; Hegan, D.C.; Fons, N.R.; et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci. Transl. Med. 2017, 9, eaal2463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, S.; Li, W.Y.; Tseng, A.; Beerman, I.; Elia, A.J.; Bendall, S.C.; Lemonnier, F.; Kron, K.J.; Cescon, D.W.; Hao, Z.; et al. Mutant IDH1 Downregulates ATM and Alters DNA Repair and Sensitivity to DNA Damage Independent of TET2. Cancer Cell 2016, 30, 337–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulkowski, P.L.; Oeck, S.; Dow, J.; Economos, N.G.; Mirfakhraie, L.; Liu, Y.; Noronha, K.; Bao, X.; Li, J.; Shuch, B.M.; et al. Oncometabolites suppress DNA repair by disrupting local chromatin signalling. Nature 2020, 582, 586–591. [Google Scholar] [CrossRef] [PubMed]
- Possemato, R.; Marks, K.M.; Shaul, Y.D.; Pacold, M.E.; Kim, D.; Birsoy, K.; Sethumadhavan, S.; Woo, H.-K.; Jang, H.G.; Jha, A.K.; et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 2011, 476, 346–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBerardinis, R.; Cheng, T. Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010, 29, 313–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okazaki, A.; Gameiro, P.A.; Christodoulou, D.; Laviollette, L.; Schneider, M.; Chaves, F.; Stemmer-Rachamimov, A.; Yazinski, S.A.; Lee, R.; Stephanopoulos, G.; et al. Glutaminase and poly(ADP-ribose) polymerase inhibitors suppress pyrimidine synthesis and VHL-deficient renal cancers. J. Clin. Investig. 2017, 127, 1631–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef]
- Høyer-Hansen, M.; Jäättelä, M. AMP-Activated Protein Kinase: A Universal Regulator of Autophagy? Autophagy 2007, 3, 381–383. [Google Scholar] [CrossRef] [Green Version]
- Cao, W.; Li, J.; Hao, Q.; Vadgama, J.V.; Wu, Y. AMP-activated protein kinase: A potential therapeutic target for triple-negative breast cancer. Breast Cancer Res. 2019, 21, 29. [Google Scholar] [CrossRef] [Green Version]
- Xie, G.; Zhou, B.; Zhao, A.; Qiu, Y.; Zhao, X.; Garmire, L.; Shvetsov, Y.B.; Yu, H.; Yen, Y.; Jia, W. Lowered circulating aspartate is a metabolic feature of human breast cancer. Oncotarget 2015, 6, 33369–33381. [Google Scholar] [CrossRef] [Green Version]
- Stegink, L.; Filer, L.J., Jr.; Brummel, M.C.; Baker, G.L.; Krause, W.L.; Bell, E.F.; Ziegler, E.E. Plasma amino acid concentrations and amino acid ratios in normal adults and adults heterozygous for phenylketonuria ingesting a hamburger and milk shake meal. Am. J. Clin. Nutr. 1991, 53, 670–675. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Lee, A.G.; Briones-Martin-Del-Campo, M.; Conn, C.S.; Simpson, D.R.; Scott, A.I.; Le, A.; Cowan, T.M.; Ruggero, D.; Sweet-Cordero, E.A. Oncogenic KRAS Regulates Amino Acid Homeostasis and Asparagine Biosynthesis via ATF4 and Alters Sensitivity to L-Asparaginase. Cancer Cell 2018, 33, 91–107.e6. [Google Scholar] [CrossRef] [Green Version]
- Knott, S.R.V.; Wagenblast, E.; Khan, S.; Kim, S.Y.; Soto, M.; Wagner, M.; Turgeon, M.-O.; Fish, L.; Erard, N.; Gable, A.L.; et al. Asparagine bioavailability governs metastasis in a model of breast cancer. Nature 2018, 554, 378–381. [Google Scholar] [CrossRef]
- Krall, A.S.; Xu, S.; Graeber, T.G.; Braas, D.; Christofk, H.R. Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nat. Commun. 2016, 7, 11457. [Google Scholar] [CrossRef] [Green Version]
- Kim, I.; Manni, A.; Lynch, J.; Demers, L. Polyamine involvement in the secretion and action of TGF-α in hormone sensitive human breast cancer cells in culture. Breast Cancer Res. Treat. 1991, 18, 83–91. [Google Scholar] [CrossRef]
- Lee, C.-Y.; Su, G.-C.; Huang, W.-C.; Ko, M.-Y.; Yeh, H.-Y.; Chang, G.-D.; Lin, S.-J.; Chi, P. Promotion of homology-directed DNA repair by polyamines. Nat. Commun. 2019, 10, 65. [Google Scholar] [CrossRef]
- Zhang, X.; Lin, Y.; Gillies, R.J. Tumor pH and Its Measurement. J. Nucl. Med. 2010, 51, 1167–1170. [Google Scholar] [CrossRef] [Green Version]
- Balgi, A.D.; Diering, G.H.; Donohue, E.; Lam, K.; Fonseca, B.D.; Zimmerman, C.; Numata, M.; Roberge, M. Regulation of mTORC1 Signaling by pH. PLoS ONE 2011, 6, e21549. [Google Scholar] [CrossRef]
- Lien, E.C.; Lyssiotis, C.A.; Juvekar, A.; Hu, H.; Asara, J.M.; Cantley, L.C.; Toker, A. Glutathione biosynthesis is a metabolic vulnerability in PI(3)K/Akt-driven breast cancer. Nat. Cell Biol. 2016, 18, 572–578. [Google Scholar] [CrossRef] [Green Version]
- Beckonert, O.; Bonk, U.; Leibfritz, D. Visualizing metabolic changes in breast-cancer tissue using1H-NMR spectroscopy and self-organizing maps. NMR Biomed. 2003, 16, 1–11. [Google Scholar] [CrossRef]
- Cao, M.D.; Döpkens, M.; Krishnamachary, B.; Vesuna, F.; Gadiya, M.M.; Loenning, P.E.; Bhujwalla, Z.M.; Gribbestad, I.S.; Glunde, K. Glycerophosphodiester phosphodiesterase domain containing 5 (GDPD5) expression correlates with malignant choline phospholipid metabolite profiles in human breast cancer. NMR Biomed. 2012, 25, 1033–1042. [Google Scholar] [CrossRef] [Green Version]
- Deshpande, R.; Mansara, P.; Suryavanshi, S.; Kaul-Ghanekar, R. Alpha-linolenic acid regulates the growth of breast and cervical cancercell lines through regulation of NO release and induction of lipidperoxidation. J. Mol. Biochem. 2013, 2, 6–17. [Google Scholar]
- Kim, J.-Y.; Park, H.D.; Park, E.; Chon, J.-W.; Park, Y.K. Growth-Inhibitory and Proapoptotic Effects of Alpha-Linolenic Acid on Estrogen-Positive Breast Cancer Cells. Ann. N. Y. Acad. Sci. 2009, 1171, 190–195. [Google Scholar] [CrossRef]






 ), MDA-MB-231 (
), MDA-MB-231 (  ), and HCC1937 (
), and HCC1937 (  ). Fitness effect score is based on the Chronos algorithm.
), MDA-MB-231 ( ), and HCC1937 ( ). Fitness effect score is based on the Chronos algorithm.
). Fitness effect score is based on the Chronos algorithm.
), MDA-MB-231 ( ), and HCC1937 ( ). Fitness effect score is based on the Chronos algorithm.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berardi, D.; Hunter, Y.; van den Driest, L.; Farrell, G.; Rattray, N.J.W.; Rattray, Z. The Differential Metabolic Signature of Breast Cancer Cellular Response to Olaparib Treatment. Cancers 2022, 14, 3661. https://doi.org/10.3390/cancers14153661
Berardi D, Hunter Y, van den Driest L, Farrell G, Rattray NJW, Rattray Z. The Differential Metabolic Signature of Breast Cancer Cellular Response to Olaparib Treatment. Cancers. 2022; 14(15):3661. https://doi.org/10.3390/cancers14153661
Chicago/Turabian StyleBerardi, Domenica, Yasmin Hunter, Lisa van den Driest, Gillian Farrell, Nicholas J. W. Rattray, and Zahra Rattray. 2022. "The Differential Metabolic Signature of Breast Cancer Cellular Response to Olaparib Treatment" Cancers 14, no. 15: 3661. https://doi.org/10.3390/cancers14153661