
NF-κB: A Druggable Target in Acute Myeloid Leukemia

 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
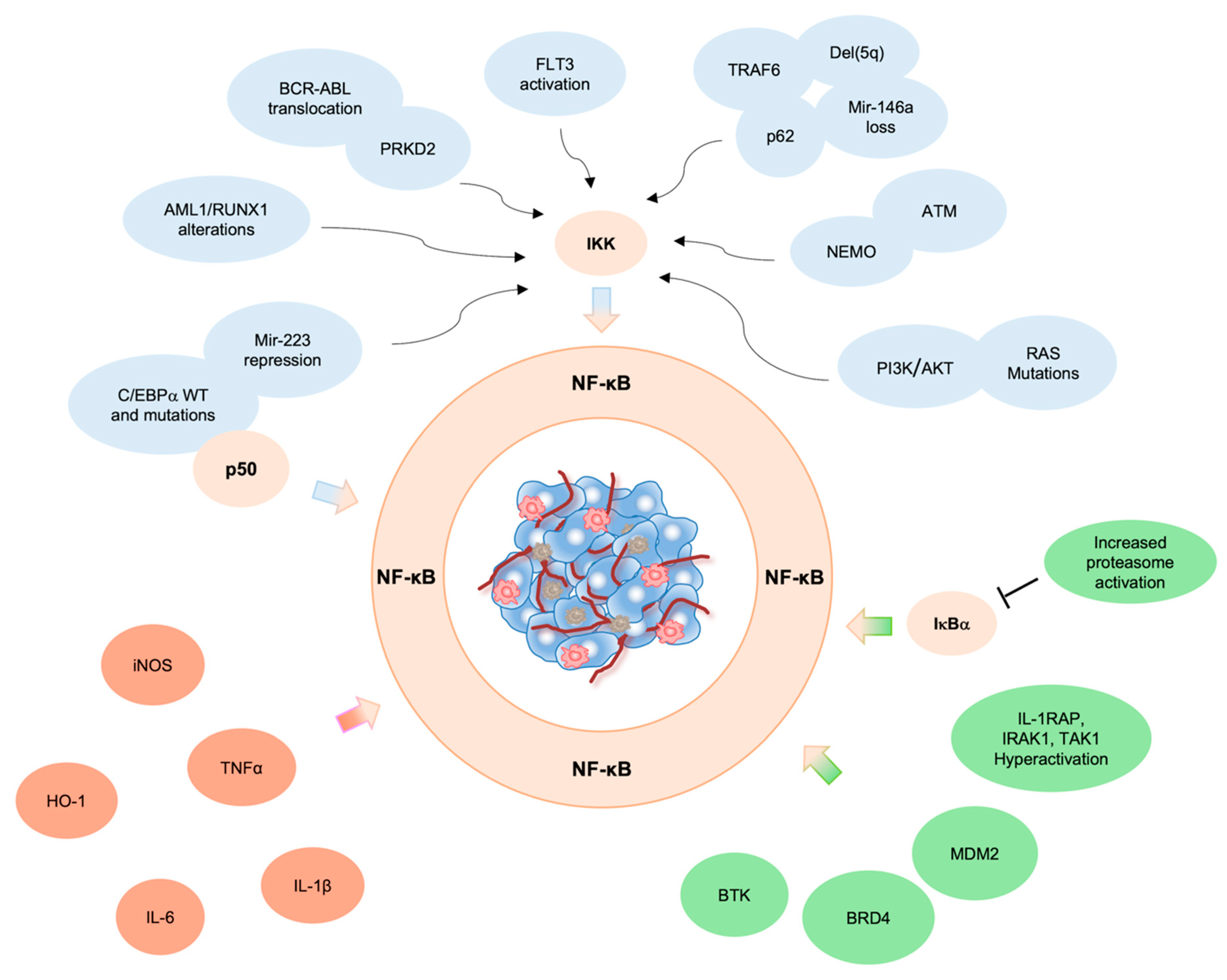
2. NF-κB Pathway in AML Pathogenesis
3. Genetic Alterations Drive NF-κB Constitutive Activation in AML
3.1. ATM
3.2. BCR::ABL
3.3. RUNX1
3.4. C/EBPα
3.5. q Deletion
3.6. RAS
4. Pro-Inflammatory Microenvironment and NF-κB Activation in Leukemic Cells
5. Aberrant NF-κB Activation by NF-κB Regulators/Interactors
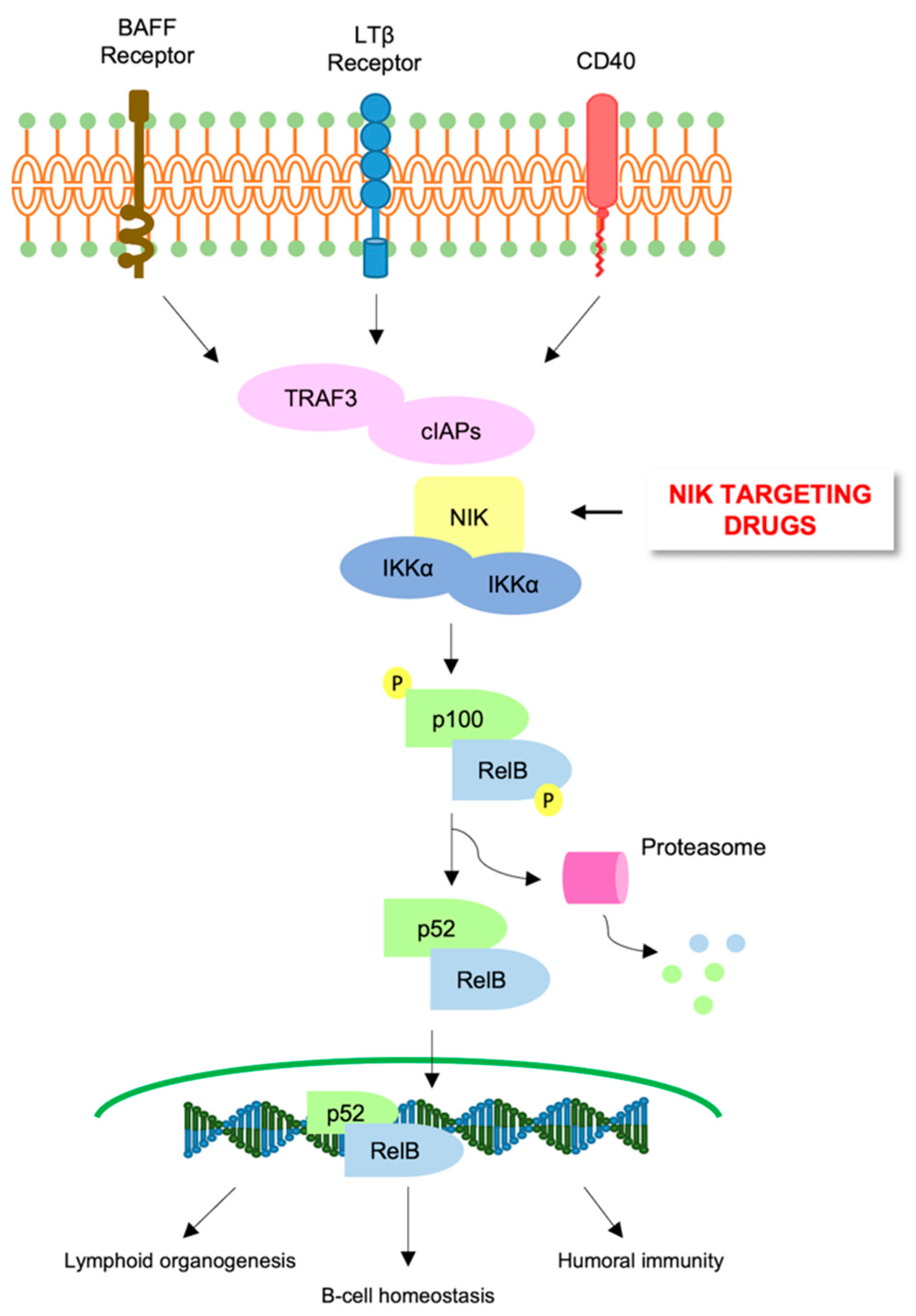
6. Alternative NF-κB Pathway in AML
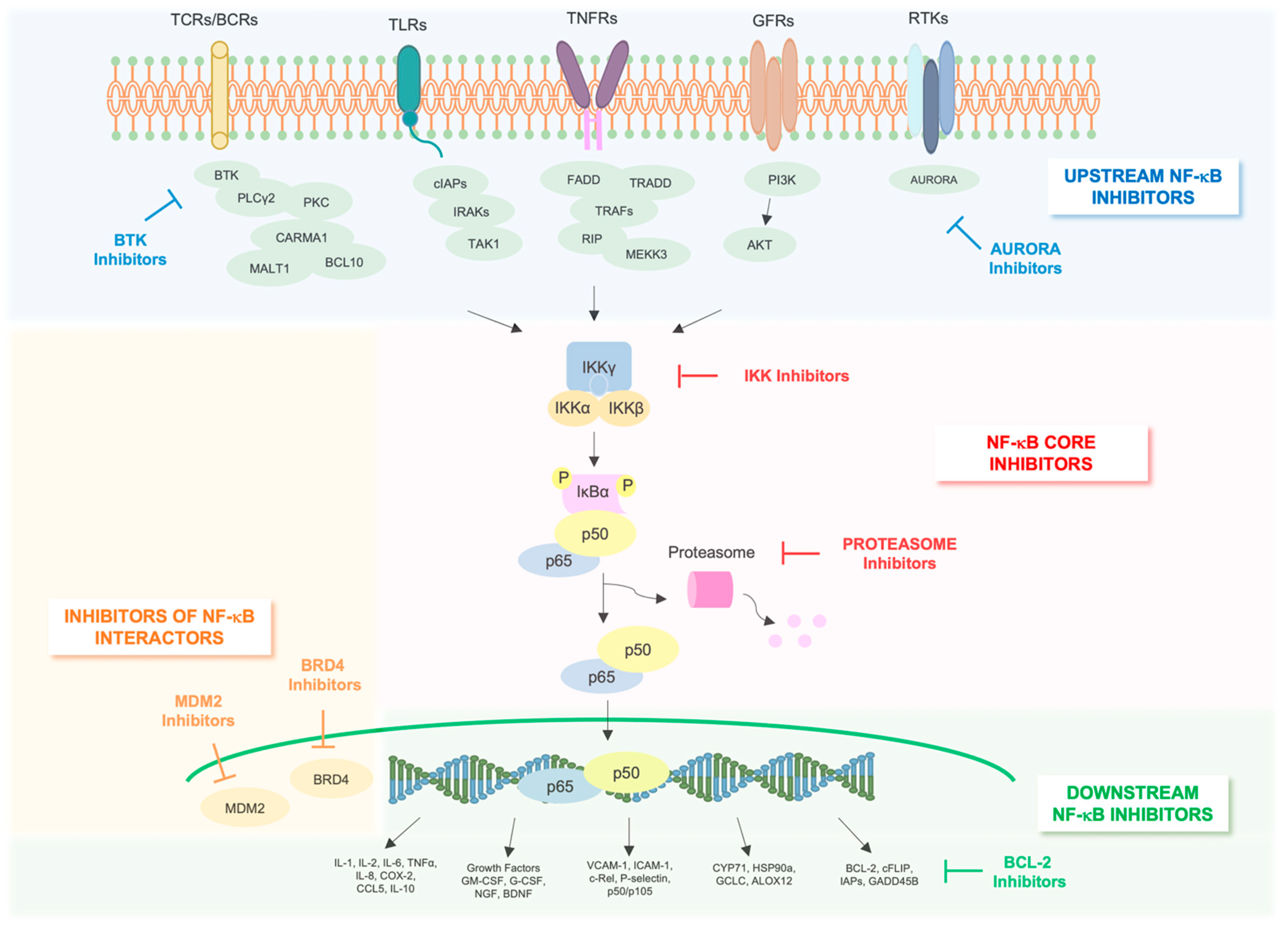
7. Targeting NF-κB Pathway in AML
8. NF-κB Core Pathway Inhibitors
8.1. Proteasome Inhibitors
8.2. IKK Inhibitors
9. Indirect NF-κB Inhibitors
10. Inhibitors of Upstream Regulators of NF-κB
10.1. Tyrosine Kinases Inhibitors (TKI)
10.2. Serine-Threonine Kinases Inhibitors
10.3. IL-1β Inhibitors
11. Inhibitors of NF-κB Interactome
11.1. BRD4 Inhibitors
11.2. MDM2 Inhibitors
12. Inhibitors of Downstream Effectors of NF-κB
BCL-2 Inhibitors
13. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cancer Facts & Figures 2022. Available online: https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2022.html (accessed on 1 July 2022).
- Löwenberg, B.; Downing, J.R.; Burnett, A. Acute Myeloid Leukemia. N. Engl. J. Med. 1999, 341, 1051–1062. [Google Scholar] [CrossRef]
- Kreso, A.; Dick, J.E. Evolution of the Cancer Stem Cell Model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef] [Green Version]
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [Green Version]
- Dombret, H.; Gardin, C. An Update of Current Treatments for Adult Acute Myeloid Leukemia. Blood 2016, 127, 53–61. [Google Scholar] [CrossRef]
- Short, N.J.; Rytting, M.E.; Cortes, J.E. Acute Myeloid Leukaemia. Lancet 2018, 392, 593–606. [Google Scholar] [CrossRef]
- Chen, K.T.J.; Gilabert-Oriol, R.; Bally, M.B.; Leung, A.W.Y. Recent Treatment Advances and the Role of Nanotechnology, Combination Products, and Immunotherapy in Changing the Therapeutic Landscape of Acute Myeloid Leukemia. Pharm. Res. 2019, 36, 125. [Google Scholar] [CrossRef] [Green Version]
- Begalli, F.; Bennett, J.; Capece, D.; Verzella, D.; D’Andrea, D.; Tornatore, L.; Franzoso, G. Unlocking the NF-ΚB Conundrum: Embracing Complexity to Achieve Specificity. Biomedicines 2017, 5, 50. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-ΚB Pathway for the Therapy of Diseases: Mechanism and Clinical Study. Signal Transduct. Target. Ther. 2020, 5, 209. [Google Scholar] [CrossRef] [PubMed]
- Bottero, V.; Withoff, S.; Verma, I.M. NF-KappaB and the Regulation of Hematopoiesis. Cell Death Differ. 2006, 13, 785–797. [Google Scholar] [CrossRef] [Green Version]
- Capece, D.; Verzella, D.; Di Francesco, B.; Alesse, E.; Franzoso, G.; Zazzeroni, F. NF-ΚB and Mitochondria Cross Paths in Cancer: Mitochondrial Metabolism and Beyond. Semin. Cell Dev. Biol. 2020, 98, 118–128. [Google Scholar] [CrossRef]
- Bennett, J.; Capece, D.; Begalli, F.; Verzella, D.; D’Andrea, D.; Tornatore, L.; Franzoso, G. NF-ΚB in the Crosshairs: Rethinking an Old Riddle. Int. J. Biochem. Cell Biol. 2018, 95, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Baud, V.; Karin, M. Is NF-KappaB a Good Target for Cancer Therapy? Hopes and Pitfalls. Nature reviews. Drug Discov. 2009, 8, 33–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oeckinghaus, A.; Ghosh, S. The NF-KappaB Family of Transcription Factors and Its Regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Capece, D.; D’Andrea, D.; Verzella, D.; Tornatore, L.; Begalli, F.; Bennett, J.; Zazzeroni, F.; Franzoso, G. Turning an Old GADDget into a Troublemaker. Cell Death Differ. 2018, 25, 642–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-ΚB Signaling in Inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labbozzetta, M.; Notarbartolo, M.; Poma, P. Can NF-ΚB Be Considered a Valid Drug Target in Neoplastic Diseases? Our Point of View. Int. J. Mol. Sci. 2020, 21, 3070. [Google Scholar] [CrossRef]
- Bosman, M.C.J.; Schuringa, J.J.; Vellenga, E. Constitutive NF-ΚB Activation in AML: Causes and Treatment Strategies. Crit. Rev. Oncol./Hematol. 2016, 98, 35–44. [Google Scholar] [CrossRef]
- Gasparini, C.; Celeghini, C.; Monasta, L.; Zauli, G. NF-ΚB Pathways in Hematological Malignancies. Cell. Mol. Life Sci. CMLS 2014, 71, 2083–2102. [Google Scholar] [CrossRef]
- Guzman, M.L.; Neering, S.J.; Upchurch, D.; Grimes, B.; Howard, D.S.; Rizzieri, D.A.; Luger, S.M.; Jordan, C.T. Nuclear Factor-KappaB Is Constitutively Activated in Primitive Human Acute Myelogenous Leukemia Cells. Blood 2001, 98, 2301–2307. [Google Scholar] [CrossRef]
- Baumgartner, B.; Weber, M.; Quirling, M.; Fischer, C.; Page, S.; Adam, M.; Von Schilling, C.; Waterhouse, C.; Schmid, C.; Neumeier, D.; et al. Increased IkappaB Kinase Activity Is Associated with Activated NF-KappaB in Acute Myeloid Blasts. Leukemia 2002, 16, 2062–2071. [Google Scholar] [CrossRef] [Green Version]
- Mehta, S.V.; Shukla, S.N.; Vora, H.H. Overexpression of Bcl2 Protein Predicts Chemoresistance in Acute Myeloid Leukemia: Its Correlation with FLT3. Neoplasma 2013, 60, 666–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, T.-Y.W.; Wu, P.-Y.; Wu, T.-J.; Hou, H.-A.; Chou, W.-C.; Teng, C.-L.J.; Lin, C.-R.; Chen, J.-M.M.; Lin, T.-Y.; Su, H.-C.; et al. Aurora a and NF-ΚB Survival Pathway Drive Chemoresistance in Acute Myeloid Leukemia via the TRAF-Interacting Protein TIFA. Cancer Res. 2017, 77, 494–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Notarbartolo, M.; Cervello, M.; Dusonchet, L.; Cusimano, A.; D’Alessandro, N. Resistance to Diverse Apoptotic Triggers in Multidrug Resistant HL60 Cells and Its Possible Relationship to the Expression of P-Glycoprotein, Fas and of the Novel Anti-Apoptosis Factors IAP (Inhibitory of Apoptosis Proteins). Cancer Lett. 2002, 180, 91–101. [Google Scholar] [CrossRef]
- Notarbartolo, M.; Cervello, M.; Giannitrapani, L.; Meli, M.; Poma, P.; Dusonchet, L.; Montalto, G.; D’Alessandro, N. Expression of IAPs and Alternative Splice Variants in Hepatocellular Carcinoma Tissues and Cells. Ann. N. Y. Acad. Sci. 2004, 1028, 289–293. [Google Scholar] [CrossRef]
- Tamm, I.; Kornblau, S.M.; Segall, H.; Krajewski, S.; Welsh, K.; Kitada, S.; Scudiero, D.A.; Tudor, G.; Qui, Y.H.; Monks, A.; et al. Expression and Prognostic Significance of IAP-Family Genes in Human Cancers and Myeloid Leukemias. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2000, 6, 1796–1803. [Google Scholar]
- Hrdinka, M.; Yabal, M. Inhibitor of Apoptosis Proteins in Human Health and Disease. Genes Immun. 2019, 20, 641–650. [Google Scholar] [CrossRef]
- Marchand, T.; Pinho, S. Leukemic Stem Cells: From Leukemic Niche Biology to Treatment Opportunities. Front. Immunol. 2021, 12, 775128. [Google Scholar] [CrossRef]
- Zhou, J.; Chooi, J.Y.; Ching, Y.Q.; Quah, J.Y.; Toh, S.H.M.; Ng, Y.; Tan, T.Z.; Chng, W.J. NF-ΚB Promotes the Stem-like Properties of Leukemia Cells by Activation of LIN28B. World J. Stem Cells 2018, 10, 34–42. [Google Scholar] [CrossRef]
- Grosjean-Raillard, J.; Tailler, M.; Adès, L.; Perfettini, J.-L.; Fabre, C.; Braun, T.; De Botton, S.; Fenaux, P.; Kroemer, G. ATM Mediates Constitutive NF-KappaB Activation in High-Risk Myelodysplastic Syndrome and Acute Myeloid Leukemia. Oncogene 2009, 28, 1099–1109. [Google Scholar] [CrossRef] [Green Version]
- Shiloh, Y.; Ziv, Y. The ATM Protein Kinase: Regulating the Cellular Response to Genotoxic Stress, and More. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef]
- Grosjean-Raillard, J.; Adès, L.; Boehrer, S.; Tailler, M.; Fabre, C.; Braun, T.; De Botton, S.; Israel, A.; Fenaux, P.; Kroemer, G. Flt3 Receptor Inhibition Reduces Constitutive NFkappaB Activation in High-Risk Myelodysplastic Syndrome and Acute Myeloid Leukemia. Apoptosis Int. J. Program. Cell Death 2008, 13, 1148–1161. [Google Scholar] [CrossRef] [PubMed]
- Neuendorff, N.R.; Burmeister, T.; Dörken, B.; Westermann, J. BCR-ABL-Positive Acute Myeloid Leukemia: A New Entity? Analysis of Clinical and Molecular Features. Ann. Hematol. 2016, 95, 1211–1221. [Google Scholar] [CrossRef] [PubMed]
- Neuendorff, N.R.; Hemmati, P.; Arnold, R.; Ihlow, J.; Dörken, B.; Müller-Tidow, C.; Westermann, J. BCR-ABL(+) Acute Myeloid Leukemia: Are We Always Dealing with a High-Risk Disease? Blood Adv. 2018, 2, 1409–1411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariotti, B.; Meconi, F.; Palmieri, R.; De Bellis, E.; Lavorgna, S.; Ottone, T.; Martini, V.; Lo-Coco, F.; Cicconi, L. Acute Myeloid Leukemia with Concomitant BCR-ABL and NPM1 Mutations. Case Rep. Hematol. 2019, 2019, 6707506. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.-Y.; Van Etten, R.A. IKK-Dependent Activation of NF-ΚB Contributes to Myeloid and Lymphoid Leukemogenesis by BCR-ABL1. Blood 2014, 123, 2401–2411. [Google Scholar] [CrossRef] [Green Version]
- Mihailovic, T.; Marx, M.; Auer, A.; Van Lint, J.; Schmid, M.; Weber, C.; Seufferlein, T. Protein Kinase D2 Mediates Activation of Nuclear Factor KappaB by Bcr-Abl in Bcr-Abl+ Human Myeloid Leukemia Cells. Cancer Res. 2004, 64, 8939–8944. [Google Scholar] [CrossRef] [Green Version]
- Schnittger, S.; Dicker, F.; Kern, W.; Wendland, N.; Sundermann, J.; Alpermann, T.; Haferlach, C.; Haferlach, T. RUNX1 Mutations Are Frequent in de Novo AML with Noncomplex Karyotype and Confer an Unfavorable Prognosis. Blood 2011, 117, 2348–2357. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, H.; Shimizu, K.; Kozu, T.; Maseki, N.; Kaneko, Y.; Ohki, M. T(8;21) Breakpoints on Chromosome 21 in Acute Myeloid Leukemia Are Clustered within a Limited Region of a Single Gene, AML1. Proc. Natl. Acad. Sci. USA 1991, 88, 10431–10434. [Google Scholar] [CrossRef] [Green Version]
- Marcucci, G.; Caligiuri, M.A.; Bloomfield, C.D. Molecular and Clinical Advances in Core Binding Factor Primary Acute Myeloid Leukemia: A Paradigm for Translational Research in Malignant Hematology. Cancer Investig. 2000, 18, 768–780. [Google Scholar] [CrossRef]
- Fazi, F.; Racanicchi, S.; Zardo, G.; Starnes, L.M.; Mancini, M.; Travaglini, L.; Diverio, D.; Ammatuna, E.; Cimino, G.; Lo-Coco, F.; et al. Epigenetic Silencing of the Myelopoiesis Regulator MicroRNA-223 by the AML1/ETO Oncoprotein. Cancer Cell 2007, 12, 457–466. [Google Scholar] [CrossRef]
- Fukao, T.; Fukuda, Y.; Kiga, K.; Sharif, J.; Hino, K.; Enomoto, Y.; Kawamura, A.; Nakamura, K.; Takeuchi, T.; Tanabe, M. An Evolutionarily Conserved Mechanism for MicroRNA-223 Expression Revealed by MicroRNA Gene Profiling. Cell 2007, 129, 617–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Morgan, M.J.; Choksi, S.; Zhang, Y.; Kim, Y.-S.; Liu, Z. MicroRNAs Modulate the Noncanonical Transcription Factor NF-KappaB Pathway by Regulating Expression of the Kinase IKKalpha during Macrophage Differentiation. Nat. Immunol. 2010, 11, 799–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, M.; Shimabe, M.; Watanabe-Okochi, N.; Arai, S.; Yoshimi, A.; Shinohara, A.; Nishimoto, N.; Kataoka, K.; Sato, T.; Kumano, K.; et al. AML1/RUNX1 Functions as a Cytoplasmic Attenuator of NF-ΚB Signaling in the Repression of Myeloid Tumors. Blood 2011, 118, 6626–6637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paz-Priel, I.; Friedman, A. C/EBPα Dysregulation in AML and ALL. Crit. Rev. Oncog. 2011, 16, 93–102. [Google Scholar] [CrossRef]
- Roe, J.-S.; Vakoc, C.R. C/EBPα: Critical at the Origin of Leukemic Transformation. J. Exp. Med. 2014, 211, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Grardel, N.; Roumier, C.; Soenen, V.; Lai, J.L.; Plantier, I.; Gheveart, C.; Cosson, A.; Fenaux, P.; Preudhomme, C. Acute Myeloblastic Leukemia (AML) with Inv (16)(P13;Q22) and the Rare I Type CBFbeta-MYH11 Transcript: Report of Two New Cases. Leukemia 2002, 16, 150–151. [Google Scholar] [CrossRef] [Green Version]
- Green, C.L.; Koo, K.K.; Hills, R.K.; Burnett, A.K.; Linch, D.C.; Gale, R.E. Prognostic Significance of CEBPA Mutations in a Large Cohort of Younger Adult Patients with Acute Myeloid Leukemia: Impact of Double CEBPA Mutations and the Interaction with FLT3 and NPM1 Mutations. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 2739–2747. [Google Scholar] [CrossRef]
- Paz-Priel, I.; Cai, D.H.; Wang, D.; Kowalski, J.; Blackford, A.; Liu, H.; Heckman, C.A.; Gombart, A.F.; Koeffler, H.P.; Boxer, L.M.; et al. CCAAT/Enhancer Binding Protein Alpha (C/EBPalpha) and C/EBPalpha Myeloid Oncoproteins Induce Bcl-2 via Interaction of Their Basic Regions with Nuclear Factor-KappaB P50. Mol. Cancer Res. MCR 2005, 3, 585–596. [Google Scholar] [CrossRef] [Green Version]
- Paz-Priel, I.; Ghosal, A.K.; Kowalski, J.; Friedman, A.D. C/EBPalpha or C/EBPalpha Oncoproteins Regulate the Intrinsic and Extrinsic Apoptotic Pathways by Direct Interaction with NF-KappaB P50 Bound to the Bcl-2 and FLIP Gene Promoters. Leukemia 2009, 23, 365–374. [Google Scholar] [CrossRef]
- Pulikkan, J.A.; Peramangalam, P.S.; Dengler, V.; Ho, P.A.; Preudhomme, C.; Meshinchi, S.; Christopeit, M.; Nibourel, O.; Müller-Tidow, C.; Bohlander, S.K.; et al. C/EBPα Regulated MicroRNA-34a Targets E2F3 during Granulopoiesis and Is down-Regulated in AML with CEBPA Mutations. Blood 2010, 116, 5638–5649. [Google Scholar] [CrossRef] [Green Version]
- Ebert, B.L. Deletion 5q in Myelodysplastic Syndrome: A Paradigm for the Study of Hemizygous Deletions in Cancer. Leukemia 2009, 23, 1252–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, J.; Barker, B.; Bolanos, L.; Liu, X.; Jerez, A.; Makishima, H.; Christie, S.; Chen, X.; Rao, D.S.; Grimes, H.L.; et al. Myeloid Malignancies with Chromosome 5q Deletions Acquire a Dependency on an Intrachromosomal NF-ΚB Gene Network. Cell Rep. 2014, 8, 1328–1338. [Google Scholar] [CrossRef] [Green Version]
- Taganov, K.D.; Boldin, M.P.; Chang, K.-J.; Baltimore, D. NF-KappaB-Dependent Induction of MicroRNA MiR-146, an Inhibitor Targeted to Signaling Proteins of Innate Immune Responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef] [Green Version]
- Boldin, M.P.; Taganov, K.D.; Rao, D.S.; Yang, L.; Zhao, J.L.; Kalwani, M.; Garcia-Flores, Y.; Luong, M.; Devrekanli, A.; Xu, J.; et al. MiR-146a Is a Significant Brake on Autoimmunity, Myeloproliferation, and Cancer in Mice. J. Exp. Med. 2011, 208, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.L.; Rao, D.S.; Boldin, M.P.; Taganov, K.D.; O’Connell, R.M.; Baltimore, D. NF-KappaB Dysregulation in MicroRNA-146a-Deficient Mice Drives the Development of Myeloid Malignancies. Proc. Natl. Acad. Sci. USA 2011, 108, 9184–9189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaupre, D.M.; Kurzrock, R. RAS and Leukemia: From Basic Mechanisms to Gene-Directed Therapy. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1999, 17, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Reuter, C.W.; Morgan, M.A.; Bergmann, L. Targeting the Ras Signaling Pathway: A Rational, Mechanism-Based Treatment for Hematologic Malignancies? Blood 2000, 96, 1655–1669. [Google Scholar] [CrossRef]
- Ward, A.F.; Braun, B.S.; Shannon, K.M. Targeting Oncogenic Ras Signaling in Hematologic Malignancies. Blood 2012, 120, 3397–3406. [Google Scholar] [CrossRef] [Green Version]
- Birkenkamp, K.U.; Geugien, M.; Schepers, H.; Westra, J.; Lemmink, H.H.; Vellenga, E. Constitutive NF-KappaB DNA-Binding Activity in AML Is Frequently Mediated by a Ras/PI3-K/PKB-Dependent Pathway. Leukemia 2004, 18, 103–112. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S.; Harigae, H.; Ishii, K.K.; Inomata, M.; Fujiwara, T.; Yokoyama, H.; Ishizawa, K.; Kameoka, J.; Licht, J.D.; Sasaki, T.; et al. Over-Expression of Flt3 Induces NF-KappaB Pathway and Increases the Expression of IL-6. Leuk. Res. 2005, 29, 893–899. [Google Scholar] [CrossRef]
- Shanmugam, R.; Gade, P.; Wilson-Weekes, A.; Sayar, H.; Suvannasankha, A.; Goswami, C.; Li, L.; Gupta, S.; Cardoso, A.A.; Baghdadi, T.A.; et al. A Noncanonical Flt3ITD/NF-ΚB Signaling Pathway Represses DAPK1 in Acute Myeloid Leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 360–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [Green Version]
- Terzić, J.; Grivennikov, S.; Karin, E.; Karin, M. Inflammation and Colon Cancer. Gastroenterology 2010, 138, 2101–2114.e5. [Google Scholar] [CrossRef]
- Rushworth, S.A.; MacEwan, D.J. HO-1 Underlies Resistance of AML Cells to TNF-Induced Apoptosis. Blood 2008, 111, 3793–3801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heasman, S.-A.; Zaitseva, L.; Bowles, K.M.; Rushworth, S.A.; Macewan, D.J. Protection of Acute Myeloid Leukaemia Cells from Apoptosis Induced by Front-Line Chemotherapeutics Is Mediated by Haem Oxygenase-1. Oncotarget 2011, 2, 658–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, S.Y.; Garbán, H.J. Regulation of Apoptosis-Related Genes by Nitric Oxide in Cancer. Nitric Oxide Biol. Chem. 2008, 19, 170–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandão, M.M.; Soares, E.; Salles, T.S.; Saad, S.T. Expression of Inducible Nitric Oxide Synthase Is Increased in Acute Myeloid Leukaemia. Acta Haematol. 2001, 106, 95–99. [Google Scholar] [CrossRef]
- Zhang, B.; Ho, Y.W.; Huang, Q.; Maeda, T.; Lin, A.; Lee, S.-U.; Hair, A.; Holyoake, T.L.; Huettner, C.; Bhatia, R. Altered Microenvironmental Regulation of Leukemic and Normal Stem Cells in Chronic Myelogenous Leukemia. Cancer Cell 2012, 21, 577–592. [Google Scholar] [CrossRef] [Green Version]
- Jacamo, R.; Chen, Y.; Wang, Z.; Ma, W.; Zhang, M.; Spaeth, E.L.; Wang, Y.; Battula, V.L.; Mak, P.Y.; Schallmoser, K.; et al. Reciprocal Leukemia-Stroma VCAM-1/VLA-4-Dependent Activation of NF-ΚB Mediates Chemoresistance. Blood 2014, 123, 2691–2702. [Google Scholar] [CrossRef]
- Park, M.H.; Hong, J.T. Roles of NF-ΚB in Cancer and Inflammatory Diseases and Their Therapeutic Approaches. Cells 2016, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Vlahopoulos, S.A. Aberrant Control of NF-ΚB in Cancer Permits Transcriptional and Phenotypic Plasticity, to Curtail Dependence on Host Tissue: Molecular Mode. Cancer Biol. Med. 2017, 14, 254–270. [Google Scholar] [CrossRef] [Green Version]
- Puissant, A.; Medyouf, H. Walking the Tightrope: Balancing Delicate Inflammation Response to Eradicate Acute Myeloid Leukemia. Cancer Discov. 2022, 12, 1617–1619. [Google Scholar] [CrossRef] [PubMed]
- Ellegast, J.M.; Alexe, G.; Hamze, A.; Lin, S.; Uckelmann, H.J.; Rauch, P.J.; Pimkin, M.; Ross, L.S.; Dharia, N.V.; Robichaud, A.L.; et al. Unleashing Cell-Intrinsic Inflammation as a Strategy to Kill AML Blasts. Cancer Discov. 2022, 12, 1760–1781. [Google Scholar] [CrossRef] [PubMed]
- Volk, A.; Li, J.; Xin, J.; You, D.; Zhang, J.; Liu, X.; Xiao, Y.; Breslin, P.; Li, Z.; Wei, W.; et al. Co-Inhibition of NF-ΚB and JNK Is Synergistic in TNF-Expressing Human AML. J. Exp. Med. 2014, 211, 1093–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csizmar, C.M.; Kim, D.-H.; Sachs, Z. The Role of the Proteasome in AML. Blood Cancer J. 2016, 6, e503. [Google Scholar] [CrossRef] [Green Version]
- Kagoya, Y.; Yoshimi, A.; Kataoka, K.; Nakagawa, M.; Kumano, K.; Arai, S.; Kobayashi, H.; Saito, T.; Iwakura, Y.; Kurokawa, M. Positive Feedback between NF-ΚB and TNF-α Promotes Leukemia-Initiating Cell Capacity. J. Clin. Investig. 2014, 124, 528–542. [Google Scholar] [CrossRef]
- Li, J.; Volk, A.; Zhang, J.; Cannova, J.; Dai, S.; Hao, C.; Hu, C.; Sun, J.; Xu, Y.; Wei, W.; et al. Sensitizing Leukemia Stem Cells to NF-ΚB Inhibitor Treatment in Vivo by Inactivation of Both TNF and IL-1 Signaling. Oncotarget 2016, 8, 8420–8435. [Google Scholar] [CrossRef] [Green Version]
- Grondona, P.; Bucher, P.; Schulze-Osthoff, K.; Hailfinger, S.; Schmitt, A. NF-ΚB Activation in Lymphoid Malignancies: Genetics, Signaling, and Targeted Therapy. Biomedicines 2018, 6, 38. [Google Scholar] [CrossRef] [Green Version]
- Xiao, G.; Fu, J. NF-ΚB and Cancer: A Paradigm of Yin-Yang. Am. J. Cancer Res. 2011, 1, 192–221. [Google Scholar]
- Fang, J.; Rhyasen, G.; Bolanos, L.; Rasch, C.; Varney, M.; Wunderlich, M.; Goyama, S.; Jansen, G.; Cloos, J.; Rigolino, C.; et al. Cytotoxic Effects of Bortezomib in Myelodysplastic Syndrome/Acute Myeloid Leukemia Depend on Autophagy-Mediated Lysosomal Degradation of TRAF6 and Repression of PSMA1. Blood 2012, 120, 858–867. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, M.M.; Kurtz, S.E.; Abdelhamed, S.; Mahmood, S.; Davare, M.A.; Kaempf, A.; Elferich, J.; McDermott, J.E.; Liu, T.; Payne, S.H.; et al. Inhibition of Interleukin-1 Receptor-Associated Kinase-1 Is a Therapeutic Strategy for Acute Myeloid Leukemia Subtypes. Leukemia 2018, 32, 2374–2387. [Google Scholar] [CrossRef] [PubMed]
- Bosman, M.C.J.; Schepers, H.; Jaques, J.; Brouwers-Vos, A.Z.; Quax, W.J.; Schuringa, J.J.; Vellenga, E. The TAK1-NF-ΚB Axis as Therapeutic Target for AML. Blood 2014, 124, 3130–3140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhyasen, G.W.; Bolanos, L.; Starczynowski, D.T. Differential IRAK Signaling in Hematologic Malignancies. Exp. Hematol. 2013, 41, 1005–1007. [Google Scholar] [CrossRef] [Green Version]
- Sawaguchi, Y.; Yamazaki, R.; Nishiyama, Y.; Mae, M.; Abe, A.; Nishiyama, H.; Nishisaka, F.; Ibuki, T.; Sasai, T.; Matsuzaki, T. Novel Pan-Pim Kinase Inhibitors with Imidazopyridazine and Thiazolidinedione Structure Exert Potent Antitumor Activities. Front. Pharmacol. 2021, 12, 672536. [Google Scholar] [CrossRef]
- Liu, Z.; Han, M.; Ding, K.; Fu, R. The Role of Pim Kinase in Immunomodulation. Am. J. Cancer Res. 2020, 10, 4085–4097. [Google Scholar]
- Wang, Y.; Xiu, J.; Ren, C.; Yu, Z. Protein Kinase PIM2: A Simple PIM Family Kinase with Complex Functions in Cancer Metabolism and Therapeutics. J. Cancer 2021, 12, 2570–2581. [Google Scholar] [CrossRef]
- Nihira, K.; Ando, Y.; Yamaguchi, T.; Kagami, Y.; Miki, Y.; Yoshida, K. Pim-1 Controls NF-B Signalling by Stabilizing RelA/P65. Cell Death Differ. 2010, 17, 689–698. [Google Scholar] [CrossRef] [Green Version]
- Zhu, N.; Ramirez, L.M.; Lee, R.L.; Magnuson, N.S.; Bishop, G.A.; Gold, M.R. CD40 Signaling in B Cells Regulates the Expression of the Pim-1 Kinase via the NF-ΚB Pathway. J. Immunol. 2002, 168, 744–754. [Google Scholar] [CrossRef] [Green Version]
- Filippakopoulos, P.; Knapp, S. Targeting Bromodomains: Epigenetic Readers of Lysine Acetylation. Nat. Rev. Drug Discov. 2014, 13, 337–356. [Google Scholar] [CrossRef]
- Hajmirza, A.; Emadali, A.; Gauthier, A.; Casasnovas, O.; Gressin, R.; Callanan, M.B. BET Family Protein BRD4: An Emerging Actor in NFκB Signaling in Inflammation and Cancer. Biomedicines 2018, 6, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, B.; Yang, X.-D.; Zhou, M.-M.; Ozato, K.; Chen, L.-F. Brd4 Coactivates Transcriptional Activation of NF-KappaB via Specific Binding to Acetylated RelA. Mol. Cell. Biol. 2009, 29, 1375–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Z.; Huang, B.; Wu, X.; Zhang, H.; Qi, J.; Bradner, J.; Nair, S.; Chen, L.-F. Brd4 Maintains Constitutively Active NF-ΚB in Cancer Cells by Binding to Acetylated RelA. Oncogene 2014, 33, 2395–2404. [Google Scholar] [CrossRef] [Green Version]
- Smale, S.T. Hierarchies of NF-ΚB Target-Gene Regulation. Nat. Immunol. 2011, 12, 689–694. [Google Scholar] [CrossRef] [Green Version]
- Zuber, J.; Shi, J.; Wang, E.; Rappaport, A.R.; Herrmann, H.; Sison, E.A.; Magoon, D.; Qi, J.; Blatt, K.; Wunderlich, M.; et al. RNAi Screen Identifies Brd4 as a Therapeutic Target in Acute Myeloid Leukaemia. Nature 2011, 478, 524–528. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.D.; Lin, C.Y.; Duan, Q.; Griffin, G.; Federation, A.; Paranal, R.M.; Bair, S.; Newton, G.; Lichtman, A.; Kung, A.; et al. NF-ΚB Directs Dynamic Super Enhancer Formation in Inflammation and Atherogenesis. Mol. Cell 2014, 56, 219–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.-W.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of Inflammation by a Synthetic Histone Mimic. Nature 2010, 468, 1119–1123. [Google Scholar] [CrossRef]
- Gu, L.; Findley, H.W.; Zhou, M. MDM2 Induces NF-KappaB/P65 Expression Transcriptionally through Sp1-Binding Sites: A Novel, P53-Independent Role of MDM2 in Doxorubicin Resistance in Acute Lymphoblastic Leukemia. Blood 2002, 99, 3367–3375. [Google Scholar] [CrossRef] [Green Version]
- Thomasova, D.; Mulay, S.R.; Bruns, H.; Anders, H.-J. P53-Independent Roles of MDM2 in NF-ΚB Signaling: Implications for Cancer Therapy, Wound Healing, and Autoimmune Diseases. Neoplasia 2012, 14, 1097–1101. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, Y.; Goyama, S.; Liu, X.; Tamura, M.; Asada, S.; Tanaka, Y.; Fukuyama, T.; Wunderlich, M.; O’Brien, E.; Mizukawa, B.; et al. Antitumor Immunity Augments the Therapeutic Effects of P53 Activation on Acute Myeloid Leukemia. Nat. Commun. 2019, 10, 4869. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, D.J.; Orlowski, R.Z. The Immunoproteasome as a Target in Hematologic Malignancies. Semin. Hematol. 2012, 49, 258–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, W.; Kantarjian, H.; Bekele, B.; Donahue, A.C.; Zhang, X.; Zhang, Z.J.; O’Brien, S.; Estey, E.; Estrov, Z.; Cortes, J.; et al. Proteasome Enzymatic Activities in Plasma as Risk Stratification of Patients with Acute Myeloid Leukemia and Advanced-Stage Myelodysplastic Syndrome. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 3820–3826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niewerth, D.; Dingjan, I.; Cloos, J.; Jansen, G.; Kaspers, G. Proteasome Inhibitors in Acute Leukemia. Expert Rev. Anticancer Ther. 2013, 13, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Bonardi, F.; Fusetti, F.; Deelen, P.; van Gosliga, D.; Vellenga, E.; Schuringa, J.J. A Proteomics and Transcriptomics Approach to Identify Leukemic Stem Cell (LSC) Markers. Mol. Cell. Proteom. MCP 2013, 12, 626–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiu, Y.; Dong, Q.; Li, Q.; Li, F.; Borcherding, N.; Zhang, W.; Boyce, B.; Xue, H.-H.; Zhao, C. Stabilization of NF-ΚB-Inducing Kinase Suppresses MLL-AF9-Induced Acute Myeloid Leukemia. Cell Rep. 2018, 22, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Xiu, Y.; Fu, L.; Dong, Q.; Borcherding, N.; Wang, Y.; Li, Q.; De Silva, N.S.; Klein, U.; Boyce, B.F.; et al. TIFAB Accelerates MLL-AF9−Induced Acute Myeloid Leukemia through Upregulation of HOXA9. iScience 2021, 24, 103425. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Signaling to NF-KappaB. Genes Dev. 2004, 18, 2195–2224. [Google Scholar] [CrossRef] [Green Version]
- Ramadass, V.; Vaiyapuri, T.; Tergaonkar, V. Small Molecule NF-ΚB Pathway Inhibitors in Clinic. Int. J. Mol. Sci. 2020, 21, 5164. [Google Scholar] [CrossRef]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome Inhibitors in Cancer Therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef]
- Dai, Y.; Chen, S.; Wang, L.; Pei, X.-Y.; Kramer, L.B.; Dent, P.; Grant, S. Bortezomib Interacts Synergistically with Belinostat in Human Acute Myeloid Leukaemia and Acute Lymphoblastic Leukaemia Cells in Association with Perturbations in NF-ΚB and Bim. Br. J. Haematol. 2011, 153, 222–235. [Google Scholar] [CrossRef] [Green Version]
- Tomlinson, B.K.; Tuscano, J.M.; Abedi, M.; Welborn, J.; Arora, M.; O’Donnell, R.T.; Wun, T.; Jonas, B.A. A Phase II Study of Bortezomib in Combination with Pegylated Liposomal Doxorubicin for Acute Myeloid Leukemia. Am. J. Hematol. 2019, 94, E291–E294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roboz, G.J.; Mandrekar, S.J.; Desai, P.; Laumann, K.; Walker, A.R.; Wang, E.S.; Kolitz, J.E.; Powell, B.L.; Attar, E.C.; Stock, W.; et al. Randomized Trial of 10 Days of Decitabine 6 Bortezomib in Untreated Older Patients with AML: CALGB 11002 (Alliance). Blood Adv. 2018, 2, 3608–3617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muus, P.; Langemeijer, S.; van Bijnen, S.; Blijlevens, N.; de Witte, T. A Phase I Clinical Trial to Study the Safety of Treatment with Tipifarnib Combined with Bortezomib in Patients with Advanced Stages of Myelodysplastic Syndrome and Oligoblastic Acute Myeloid Leukemia. Leuk. Res. 2021, 105, 106573. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, U.; Ganesan, S.; Alex, A.A.; Palani, H.; David, S.; Balasundaram, N.; Venkatraman, A.; Thenmozhi, M.; Jeyaseelan, L.; Korula, A.; et al. A Phase II Study Evaluating the Role of Bortezomib in the Management of Relapsed Acute Promyelocytic Leukemia Treated Upfront with Arsenic Trioxide. Cancer Med. 2020, 9, 2603–2610. [Google Scholar] [CrossRef]
- Aplenc, R.; Meshinchi, S.; Sung, L.; Alonzo, T.; Choi, J.; Fisher, B.; Gerbing, R.; Hirsch, B.; Horton, T.; Kahwash, S.; et al. Bortezomib with Standard Chemotherapy for Children with Acute Myeloid Leukemia Does Not Improve Treatment Outcomes: A Report from the Children’s Oncology Group. Haematologica 2020, 105, 1879–1886. [Google Scholar] [CrossRef]
- Liu, S.; Wu, L.-C.; Pang, J.; Santhanam, R.; Schwind, S.; Wu, Y.-Z.; Hickey, C.J.; Yu, J.; Becker, H.; Maharry, K.; et al. Sp1/NFkappaB/HDAC/MiR-29b Regulatory Network in KIT-Driven Myeloid Leukemia. Cancer Cell 2010, 17, 333–347. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Liu, Z.; Xie, Z.; Pang, J.; Yu, J.; Lehmann, E.; Huynh, L.; Vukosavljevic, T.; Takeki, M.; Klisovic, R.B.; et al. Bortezomib Induces DNA Hypomethylation and Silenced Gene Transcription by Interfering with Sp1/NF-KappaB-Dependent DNA Methyltransferase Activity in Acute Myeloid Leukemia. Blood 2008, 111, 2364–2373. [Google Scholar] [CrossRef]
- Bosman, M.C.J.; Schuringa, J.J.; Quax, W.J.; Vellenga, E. Bortezomib Sensitivity of Acute Myeloid Leukemia CD34(+) Cells Can Be Enhanced by Targeting the Persisting Activity of NF-ΚB and the Accumulation of MCL-1. Exp. Hematol. 2013, 41, 530–538.e1. [Google Scholar] [CrossRef]
- Horton, T.M.; Pati, D.; Plon, S.E.; Thompson, P.A.; Bomgaars, L.R.; Adamson, P.C.; Ingle, A.M.; Wright, J.; Brockman, A.H.; Paton, M.; et al. A Phase 1 Study of the Proteasome Inhibitor Bortezomib in Pediatric Patients with Refractory Leukemia: A Children’s Oncology Group Study. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007, 13, 1516–1522. [Google Scholar] [CrossRef] [Green Version]
- Guzman, M.L.; Swiderski, C.F.; Howard, D.S.; Grimes, B.A.; Rossi, R.M.; Szilvassy, S.J.; Jordan, C.T. Preferential Induction of Apoptosis for Primary Human Leukemic Stem Cells. Proc. Natl. Acad. Sci. USA 2002, 99, 16220–16225. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Qin, Y.; Zhou, J.; Ruan, J.; Xiong, F.; Dong, J.; Huang, X.; Yu, Z.; Gao, S. Bortezomib Suppresses Self-Renewal and Leukemogenesis of Leukemia Stem Cell by NF-ĸB-Dependent Inhibition of CDK6 in MLL-Rearranged Myeloid Leukemia. J. Cell. Mol. Med. 2021, 25, 3124–3135. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, A.A.; Zuvanich, E.G.; Khan, D.A.; Mushtaq, S.; Silswal, N.; Qureshi, N. Proteasome Inhibitors Modulate Anticancer and Anti-Proliferative Properties via NF-KB Signaling, and Ubiquitin-Proteasome Pathways in Cancer Cell Lines of Different Organs. Lipids Health Dis. 2018, 17, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayanan, S.; Cai, C.-Y.; Assaraf, Y.G.; Guo, H.-Q.; Cui, Q.; Wei, L.; Huang, J.-J.; Ashby, C.R.J.; Chen, Z.-S. Targeting the Ubiquitin-Proteasome Pathway to Overcome Anti-Cancer Drug Resistance. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer. Chemother. 2020, 48, 100663. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.; Edwards, J.; Pepper, C.; Mackay, S. Inhibitory-ΚB Kinase (IKK) α and Nuclear Factor-ΚB (NFκB)-Inducing Kinase (NIK) as Anti-Cancer Drug Targets. Cells 2018, 7, 176. [Google Scholar] [CrossRef] [Green Version]
- Frelin, C.; Imbert, V.; Griessinger, E.; Peyron, A.-C.; Rochet, N.; Philip, P.; Dageville, C.; Sirvent, A.; Hummelsberger, M.; Bérard, E.; et al. Targeting NF-KappaB Activation via Pharmacologic Inhibition of IKK2-Induced Apoptosis of Human Acute Myeloid Leukemia Cells. Blood 2005, 105, 804–811. [Google Scholar] [CrossRef]
- Jordheim, L.P.; Plesa, A.; Dreano, M.; Cros-Perrial, E.; Keime, C.; Herveau, S.; Demangel, D.; Vendrell, J.A.; Dumontet, C. Sensitivity and Gene Expression Profile of Fresh Human Acute Myeloid Leukemia Cells Exposed Ex Vivo to AS602868. Cancer Chemother. Pharmacol. 2011, 68, 97–105. [Google Scholar] [CrossRef] [Green Version]
- Burke, J.R.; Pattoli, M.A.; Gregor, K.R.; Brassil, P.J.; MacMaster, J.F.; McIntyre, K.W.; Yang, X.; Iotzova, V.S.; Clarke, W.; Strnad, J.; et al. BMS-345541 Is a Highly Selective Inhibitor of I Kappa B Kinase That Binds at an Allosteric Site of the Enzyme and Blocks NF-Kappa B-Dependent Transcription in Mice. J. Biol. Chem. 2003, 278, 1450–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battula, V.L.; Nguyen, K.; Sun, J.; Pitner, M.K.; Yuan, B.; Bartholomeusz, C.; Hail, N.; Andreeff, M. IKK Inhibition by BMS-345541 Suppresses Breast Tumorigenesis and Metastases by Targeting GD2+ Cancer Stem Cells. Oncotarget 2017, 8, 36936–36949. [Google Scholar] [CrossRef] [Green Version]
- Reikvam, H. Inhibition of NF-ΚB Signaling Alters Acute Myelogenous Leukemia Cell Transcriptomics. Cells 2020, 9, 1677. [Google Scholar] [CrossRef]
- Harikumar, K.B.; Kunnumakkara, A.B.; Ahn, K.S.; Anand, P.; Krishnan, S.; Guha, S.; Aggarwal, B.B. Modification of the Cysteine Residues in IkappaBalpha Kinase and NF-KappaB (P65) by Xanthohumol Leads to Suppression of NF-KappaB-Regulated Gene Products and Potentiation of Apoptosis in Leukemia Cells. Blood 2009, 113, 2003–2013. [Google Scholar] [CrossRef] [Green Version]
- Guzman, M.L.; Rossi, R.M.; Karnischky, L.; Li, X.; Peterson, D.R.; Howard, D.S.; Jordan, C.T. The Sesquiterpene Lactone Parthenolide Induces Apoptosis of Human Acute Myelogenous Leukemia Stem and Progenitor Cells. Blood 2005, 105, 4163–4169. [Google Scholar] [CrossRef] [PubMed]
- Ghantous, A.; Sinjab, A.; Herceg, Z.; Darwiche, N. Parthenolide: From Plant Shoots to Cancer Roots. Drug Discov. Today 2013, 18, 894–905. [Google Scholar] [CrossRef] [PubMed]
- Guzman, M.L.; Rossi, R.M.; Neelakantan, S.; Li, X.; Corbett, C.A.; Hassane, D.C.; Becker, M.W.; Bennett, J.M.; Sullivan, E.; Lachowicz, J.L.; et al. An Orally Bioavailable Parthenolide Analog Selectively Eradicates Acute Myelogenous Leukemia Stem and Progenitor Cells. Blood 2007, 110, 4427–4435. [Google Scholar] [CrossRef] [PubMed]
- Darwish, N.H.E.; Sudha, T.; Godugu, K.; Bharali, D.J.; Elbaz, O.; El-Ghaffar, H.A.A.; Azmy, E.; Anber, N.; Mousa, S.A. Novel Targeted Nano-Parthenolide Molecule against NF-KB in Acute Myeloid Leukemia. Molecules 2019, 24, 2103. [Google Scholar] [CrossRef] [Green Version]
- Sudha, T.; Bharali, D.J.; Yalcin, M.; Darwish, N.H.; Coskun, M.D.; Keating, K.A.; Lin, H.-Y.; Davis, P.J.; Mousa, S.A. Targeted Delivery of Cisplatin to Tumor Xenografts via the Nanoparticle Component of Nano-Diamino-Tetrac. Nanomedicine 2017, 12, 195–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janganati, V.; Ponder, J.; Thakkar, S.; Jordan, C.T.; Crooks, P.A. Succinamide Derivatives of Melampomagnolide B and Their Anti-Cancer Activities. Bioorg. Med. Chem. 2017, 25, 3694–3705. [Google Scholar] [CrossRef]
- Hehner, S.P.; Heinrich, M.; Bork, P.M.; Vogt, M.; Ratter, F.; Lehmann, V.; Schulze-Osthoff, K.; Dröge, W.; Schmitz, M.L. Sesquiterpene Lactones Specifically Inhibit Activation of NF-Kappa B by Preventing the Degradation of I Kappa B-Alpha and I Kappa B-Beta. J. Biol. Chem. 1998, 273, 1288–1297. [Google Scholar] [CrossRef] [Green Version]
- Pei, S.; Minhajuddin, M.; Callahan, K.P.; Balys, M.; Ashton, J.M.; Neering, S.J.; Lagadinou, E.D.; Corbett, C.; Ye, H.; Liesveld, J.L.; et al. Targeting Aberrant Glutathione Metabolism to Eradicate Human Acute Myelogenous Leukemia Cells. J. Biol. Chem. 2013, 288, 33542–33558. [Google Scholar] [CrossRef] [Green Version]
- Janganati, V.; Ponder, J.; Balasubramaniam, M.; Bhat-Nakshatri, P.; Bar, E.E.; Nakshatri, H.; Jordan, C.T.; Crooks, P.A. MMB Triazole Analogs Are Potent NF-ΚB Inhibitors and Anti-Cancer Agents against Both Hematological and Solid Tumor Cells. Eur. J. Med. Chem. 2018, 157, 562–581. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-H.; Koo, T.H.; Yoon, H.; Jung, H.S.; Jin, H.Z.; Lee, K.; Hong, Y.-S.; Lee, J.J. Inhibition of NF-Kappa B Activation through Targeting I Kappa B Kinase by Celastrol, a Quinone Methide Triterpenoid. Biochem. Pharmacol. 2006, 72, 1311–1321. [Google Scholar] [CrossRef]
- Pazhang, Y.; Jaliani, H.Z.; Imani, M.; Dariushnejad, H. Synergism between NF-Kappa B Inhibitor, Celastrol, and XIAP Inhibitor, Embelin, in an Acute Myeloid Leukemia Cell Line, HL-60. J. Cancer Res. Ther. 2016, 12, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Rhee, M.H.; Kim, E.; Cho, J.Y. BAY 11-7082 Is a Broad-Spectrum Inhibitor with Anti-Inflammatory Activity against Multiple Targets. Mediat. Inflamm. 2012, 2012, 416036. [Google Scholar] [CrossRef] [PubMed]
- Strickson, S.; Campbell, D.G.; Emmerich, C.H.; Knebel, A.; Plater, L.; Ritorto, M.S.; Shpiro, N.; Cohen, P. The Anti-Inflammatory Drug BAY 11-7082 Suppresses the MyD88-Dependent Signalling Network by Targeting the Ubiquitin System. Biochem. J. 2013, 451, 427–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omsland, M.; Bruserud, Ø.; Gjertsen, B.T.; Andresen, V. Tunneling Nanotube (TNT) Formation Is Downregulated by Cytarabine and NF-ΚB Inhibition in Acute Myeloid Leukemia (AML). Oncotarget 2017, 8, 7946–7963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hewlings, S.J.; Kalman, D.S. Curcumin: A Review of Its Effects on Human Health. Foods 2017, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.-H.; Dai, H.-P.; Shen, Q.; Ji, O.; Zhang, Q.; Zhai, Y.-L. Curcumin Induces Apoptosis and Suppresses Invasion through MAPK and MMP Signaling in Human Monocytic Leukemia SHI-1 Cells. Pharm. Biol. 2016, 54, 1303–1311. [Google Scholar] [CrossRef]
- Siegelin, M.D. Inhibition of the Mitochondrial Hsp90 Chaperone Network: A Novel, Efficient Treatment Strategy for Cancer? Cancer Lett. 2013, 333, 133–146. [Google Scholar] [CrossRef]
- Walsby, E.J.; Lazenby, M.; Pepper, C.J.; Knapper, S.; Burnett, A.K. The HSP90 Inhibitor NVP-AUY922-AG Inhibits the PI3K and IKK Signalling Pathways and Synergizes with Cytarabine in Acute Myeloid Leukaemia Cells. Br. J. Haematol. 2013, 161, 57–67. [Google Scholar] [CrossRef]
- Lancet, J.E.; Gojo, I.; Burton, M.; Quinn, M.; Tighe, S.M.; Kersey, K.; Zhong, Z.; Albitar, M.X.; Bhalla, K.; Hannah, A.L.; et al. Phase I Study of the Heat Shock Protein 90 Inhibitor Alvespimycin (KOS-1022, 17-DMAG) Administered Intravenously Twice Weekly to Patients with Acute Myeloid Leukemia. Leukemia 2010, 24, 699–705. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.-H.; Wang, K.; Fu, X.-L.; Zhou, P.-J.; Liu, Z.; Xu, D.-D.; Wang, Y.-F.; Yang, D.-P.; Xie, Q.-L.; Liu, Q.-Y. Hsp90 Inhibitor Induces KG-1a Cell Differentiation and Apoptosis via Akt/NF-ΚB Signaling. Oncol. Rep. 2017, 38, 1517–1524. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Li, C.; Zhu, X. FLT3 Inhibitors in Acute Myeloid Leukemia. J. Hematol. Oncol. 2018, 11, 133. [Google Scholar] [CrossRef] [PubMed]
- Wander, S.A.; Levis, M.J.; Fathi, A.T. The Evolving Role of FLT3 Inhibitors in Acute Myeloid Leukemia: Quizartinib and beyond. Ther. Adv. Hematol. 2014, 5, 65–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antar, A.I.; Otrock, Z.K.; Jabbour, E.; Mohty, M.; Bazarbachi, A. FLT3 Inhibitors in Acute Myeloid Leukemia: Ten Frequently Asked Questions. Leukemia 2020, 34, 682–696. [Google Scholar] [CrossRef] [PubMed]
- Griessinger, E.; Imbert, V.; Lagadec, P.; Gonthier, N.; Dubreuil, P.; Romanelli, A.; Dreano, M.; Peyron, J.-F. AS602868, a Dual Inhibitor of IKK2 and FLT3 to Target AML Cells. Leukemia 2007, 21, 877–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Lu, J.; Wang, Y.; Bai, S.; Wang, Y.; Wang, L.; Sheng, G. Combined Effects of FLT3 and NF-ΚB Selective Inhibitors on Acute Myeloid Leukemia in Vivo. J. Biochem. Mol. Toxicol. 2012, 26, 35–43. [Google Scholar] [CrossRef]
- Gallogly, M.M.; Lazarus, H.M.; Cooper, B.W. Midostaurin: A Novel Therapeutic Agent for Patients with FLT3-Mutated Acute Myeloid Leukemia and Systemic Mastocytosis. Ther. Adv. Hematol. 2017, 8, 245–261. [Google Scholar] [CrossRef]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Döhner, K.; Marcucci, G.; et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef]
- Levis, M. Midostaurin Approved for FLT3-Mutated AML. Blood 2017, 129, 3403–3406. [Google Scholar] [CrossRef]
- Edwards, D.K.; Javidi-Sharifi, N.; Rofelty, A.; Rosenfeld, C.; Roth-Carter, R.; Tardi, P.; Mayer, L.; Tyner, J.W. Effective Combination of CPX-351 with FLT3 Inhibitors in AML Blasts Harboring the FLT3-ITD Mutation. Blood 2016, 128, 5124. [Google Scholar] [CrossRef]
- Renneville, A.; Roumier, C.; Biggio, V.; Nibourel, O.; Boissel, N.; Fenaux, P.; Preudhomme, C. Cooperating Gene Mutations in Acute Myeloid Leukemia: A Review of the Literature. Leukemia 2008, 22, 915–931. [Google Scholar] [CrossRef] [Green Version]
- Tomasson, M.H.; Xiang, Z.; Walgren, R.; Zhao, Y.; Kasai, Y.; Miner, T.; Ries, R.E.; Lubman, O.; Fremont, D.H.; McLellan, M.D.; et al. Somatic Mutations and Germline Sequence Variants in the Expressed Tyrosine Kinase Genes of Patients with de Novo Acute Myeloid Leukemia. Blood 2008, 111, 4797–4808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rushworth, S.A.; Murray, M.Y.; Zaitseva, L.; Bowles, K.M.; MacEwan, D.J. Identification of Bruton’s Tyrosine Kinase as a Therapeutic Target in Acute Myeloid Leukemia. Blood 2014, 123, 1229–1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singer, J.W.; Fleischman, A.; Al-Fayoumi, S.; Mascarenhas, J.O.; Yu, Q.; Agarwal, A. Inhibition of Interleukin-1 Receptor-Associated Kinase 1 (IRAK1) as a Therapeutic Strategy. Oncotarget 2018, 9, 33416–33439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhyasen, G.W.; Starczynowski, D.T. IRAK Signalling in Cancer. Br. J. Cancer 2015, 112, 232–237. [Google Scholar] [CrossRef] [Green Version]
- Goldenson, B.; Crispino, J.D. The Aurora Kinases in Cell Cycle and Leukemia. Oncogene 2015, 34, 537–545. [Google Scholar] [CrossRef] [Green Version]
- Carmena, M.; Earnshaw, W.C. The Cellular Geography of Aurora Kinases. Nat. Rev. Mol. Cell Biol. 2003, 4, 842–854. [Google Scholar] [CrossRef]
- Ye, D.; Garcia-Manero, G.; Kantarjian, H.M.; Xiao, L.; Vadhan-Raj, S.; Fernandez, M.H.; Nguyen, M.H.; Medeiros, L.J.; Bueso-Ramos, C.E. Analysis of Aurora Kinase A Expression in CD34(+) Blast Cells Isolated from Patients with Myelodysplastic Syndromes and Acute Myeloid Leukemia. J. Hematop. 2009, 2, 2–8. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Ikezoe, T.; Nishioka, C.; Nobumoto, A.; Udaka, K.; Yokoyama, A. CD34+/CD38− Acute Myelogenous Leukemia Cells Aberrantly Express Aurora Kinase A. Int. J. Cancer 2013, 133, 2706–2719. [Google Scholar] [CrossRef]
- Briassouli, P.; Chan, F.; Savage, K.; Reis-Filho, J.S.; Linardopoulos, S. Aurora-A Regulation of Nuclear Factor-KappaB Signaling by Phosphorylation of IkappaBalpha. Cancer Res. 2007, 67, 1689–1695. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.-F.; Luo, S.-K.; Xu, J.; Li, J.; Xu, D.-R.; Wang, L.-H.; Yan, M.; Wang, X.-R.; Wan, X.-B.; Zheng, F.-M.; et al. Aurora Kinase Inhibitory VX-680 Increases Bax/Bcl-2 Ratio and Induces Apoptosis in Aurora-A-High Acute Myeloid Leukemia. Blood 2008, 111, 2854–2865. [Google Scholar] [CrossRef] [Green Version]
- Kantarjian, H.; Thomas, D.; Jorgensen, J.; Kebriaei, P.; Jabbour, E.; Rytting, M.; York, S.; Ravandi, F.; Garris, R.; Kwari, M.; et al. Results of Inotuzumab Ozogamicin, a CD22 Monoclonal Antibody, in Refractory and Relapsed Acute Lymphocytic Leukemia. Cancer 2013, 119, 2728–2736. [Google Scholar] [CrossRef]
- Moore, A.S.; Faisal, A.; de Castro, D.G.; Bavetsias, V.; Sun, C.; Atrash, B.; Valenti, M.; de Haven Brandon, A.; Avery, S.; Mair, D.; et al. Selective FLT3 Inhibition of FLT3-ITD+ Acute Myeloid Leukaemia Resulting in Secondary D835Y Mutation: A Model for Emerging Clinical Resistance Patterns. Leukemia 2012, 26, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.S.; Faisal, A.; Mak, G.W.Y.; Miraki-Moud, F.; Bavetsias, V.; Valenti, M.; Box, G.; Hallsworth, A.; de Haven Brandon, A.; Xavier, C.P.R.; et al. Quizartinib-Resistant FLT3-ITD Acute Myeloid Leukemia Cells Are Sensitive to the FLT3-Aurora Kinase Inhibitor CCT241736. Blood Adv. 2020, 4, 1478–1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjørnstad, R.; Aesoy, R.; Bruserud, Y.; Brenner, A.K.; Giraud, F.; Dowling, T.H.; Gausdal, G.; Moreau, P.; Døskeland, S.O.; Anizon, F.; et al. A Kinase Inhibitor with Anti-PIM Kinase Activity Is a Potent and Selective Cytotoxic Agent toward Acute Myeloid Leukemia. Mol. Cancer Ther. 2019, 18, 567–578. [Google Scholar] [CrossRef] [Green Version]
- Keeton, E.K.; McEachern, K.; Dillman, K.S.; Palakurthi, S.; Cao, Y.; Grondine, M.R.; Kaur, S.; Wang, S.; Chen, Y.; Wu, A.; et al. AZD1208, a Potent and Selective Pan-Pim Kinase Inhibitor, Demonstrates Efficacy in Preclinical Models of Acute Myeloid Leukemia. Blood 2014, 123, 905–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinelli, G.; Santoro, A.; Gambacorti-passerini, C.; Polo, S.V.; Scott, R.; Mukherjee, S.; Levy, M.Y.; Wierzbowska, A.; Calbacho, M. Poster Session Phase 1/2 Study of SEL24/MEN1703, a First-in-Class Dual PIM/FLT3 Kinase Inhibitor, in Patients with IDH1/2 -Mutated Acute Myeloid Leukemia: The DIAMOND-01 Trial. EHA 2022, 357384, P520. [Google Scholar]
- Czardybon, W.; Windak, R.; Gołas, A.; Gałezowski, M.; Sabiniarz, A.; Dolata, I.; Salwińska, M.; Guzik, P.; Zawadzka, M.; Gabor-Worwa, E.; et al. A Novel, Dual Pan-PIM/FLT3 Inhibitor SEL24 Exhibits Broad Therapeutic Potential in Acute Myeloid Leukemia. Oncotarget 2018, 9, 16917–16931. [Google Scholar] [CrossRef] [Green Version]
- Estrov, Z.; Manna, S.K.; Harris, D.; Van, Q.; Estey, E.H.; Kantarjian, H.M.; Talpaz, M.; Aggarwal, B.B. Phenylarsine Oxide Blocks Interleukin-1beta-Induced Activation of the Nuclear Transcription Factor NF-KappaB, Inhibits Proliferation, and Induces Apoptosis of Acute Myelogenous Leukemia Cells. Blood 1999, 94, 2844–2853. [Google Scholar] [CrossRef]
- Estrov, Z.; Shishodia, S.; Faderl, S.; Harris, D.; Van, Q.; Kantarjian, H.M.; Talpaz, M.; Aggarwal, B.B. Resveratrol Blocks Interleukin-1beta-Induced Activation of the Nuclear Transcription Factor NF-KappaB, Inhibits Proliferation, Causes S-Phase Arrest, and Induces Apoptosis of Acute Myeloid Leukemia Cells. Blood 2003, 102, 987–995. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, H.; Cerny-Reiterer, S.; Gleixner, K.V.; Blatt, K.; Herndlhofer, S.; Rabitsch, W.; Jäger, E.; Mitterbauer-Hohendanner, G.; Streubel, B.; Selzer, E.; et al. CD34(+)/CD38(−) Stem Cells in Chronic Myeloid Leukemia Express Siglec-3 (CD33) and Are Responsive to the CD33-Targeting Drug Gemtuzumab/Ozogamicin. Haematologica 2012, 97, 219–226. [Google Scholar] [CrossRef]
- Alqahtani, A.; Choucair, K.; Ashraf, M.; Hammouda, D.M.; Alloghbi, A.; Khan, T.; Senzer, N.; Nemunaitis, J. Bromodomain and Extra-Terminal Motif Inhibitors: A Review of Preclinical and Clinical Advances in Cancer Therapy. Future Sci. OA 2019, 5, FSO372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albrecht, B.K.; Gehling, V.S.; Hewitt, M.C.; Vaswani, R.G.; Côté, A.; Leblanc, Y.; Nasveschuk, C.G.; Bellon, S.; Bergeron, L.; Campbell, R.; et al. Identification of a Benzoisoxazoloazepine Inhibitor (CPI-0610) of the Bromodomain and Extra-Terminal (BET) Family as a Candidate for Human Clinical Trials. J. Med. Chem. 2016, 59, 1330–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes-Garau, D.; Ribeiro, M.L.; Roué, G. Pharmacological Targeting of BET Bromodomain Proteins in Acute Myeloid Leukemia and Malignant Lymphomas: From Molecular Characterization to Clinical Applications. Cancers 2019, 11, 1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pericole, F.V.; Lazarini, M.; de Paiva, L.B.; Duarte, A.d.S.S.; Vieira Ferro, K.P.; Niemann, F.S.; Roversi, F.M.; Olalla Saad, S.T. BRD4 Inhibition Enhances Azacitidine Efficacy in Acute Myeloid Leukemia and Myelodysplastic Syndromes. Front. Oncol. 2019, 9, 16. [Google Scholar] [CrossRef] [Green Version]
- Kojima, K.; Konopleva, M.; Samudio, I.J.; Shikami, M.; Cabreira-Hansen, M.; McQueen, T.; Ruvolo, V.; Tsao, T.; Zeng, Z.; Vassilev, L.T.; et al. MDM2 Antagonists Induce P53-Dependent Apoptosis in AML: Implications for Leukemia Therapy. Blood 2005, 106, 3150–3159. [Google Scholar] [CrossRef] [Green Version]
- Latif, A.-L.; Newcombe, A.; Li, S.; Gilroy, K.; Robertson, N.A.; Lei, X.; Stewart, H.J.S.; Cole, J.; Terradas, M.T.; Rishi, L.; et al. BRD4-Mediated Repression of P53 Is a Target for Combination Therapy in AML. Nat. Commun. 2021, 12, 241. [Google Scholar] [CrossRef]
- Godwin, P.; Baird, A.M.; Heavey, S.; Barr, M.P.; O’Byrne, K.J.; Gately, K. Targeting Nuclear Factor-Kappa B to Overcome Resistance to Chemotherapy. Front. Oncol. 2013, 3, 120. [Google Scholar] [CrossRef] [Green Version]
- Pahl, H.L. Activators and Target Genes of Rel/NF-KappaB Transcription Factors. Oncogene 1999, 18, 6853–6866. [Google Scholar] [CrossRef] [Green Version]
- Warren, C.F.A.; Wong-Brown, M.W.; Bowden, N.A. BCL-2 Family Isoforms in Apoptosis and Cancer. Cell Death Dis. 2019, 10, 177. [Google Scholar] [CrossRef] [Green Version]
- Pan, R.; Hogdal, L.J.; Benito, J.M.; Bucci, D.; Han, L.; Borthakur, G.; Cortes, J.; DeAngelo, D.J.; Debose, L.; Mu, H.; et al. Selective BCL-2 Inhibition by ABT-199 Causes on-Target Cell Death in Acute Myeloid Leukemia. Cancer Discov. 2014, 4, 362–375. [Google Scholar] [CrossRef] [Green Version]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a Potent and Selective BCL-2 Inhibitor, Achieves Antitumor Activity While Sparing Platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Vogler, M.; Dinsdale, D.; Dyer, M.J.S.; Cohen, G.M. ABT-199 Selectively Inhibits BCL2 but Not BCL2L1 and Efficiently Induces Apoptosis of Chronic Lymphocytic Leukaemic Cells but Not Platelets. Br. J. Haematol. 2013, 163, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Leverson, J.D.; Phillips, D.C.; Mitten, M.J.; Boghaert, E.R.; Diaz, D.; Tahir, S.K.; Belmont, L.D.; Nimmer, P.; Xiao, Y.; Ma, X.M.; et al. Exploiting Selective BCL-2 Family Inhibitors to Dissect Cell Survival Dependencies and Define Improved Strategies for Cancer Therapy. Sci. Transl. Med. 2015, 7, 279ra40. [Google Scholar] [CrossRef] [PubMed]
- Pollyea, D.A.; Stevens, B.M.; Jones, C.L.; Winters, A.; Pei, S.; Minhajuddin, M.; D’Alessandro, A.; Culp-Hill, R.; Riemondy, K.A.; Gillen, A.E.; et al. Venetoclax with Azacitidine Disrupts Energy Metabolism and Targets Leukemia Stem Cells in Patients with Acute Myeloid Leukemia. Nat. Med. 2018, 24, 1859–1866. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Venetoclax Combined with Decitabine or Azacitidine in Treatment-Naive, Elderly Patients with Acute Myeloid Leukemia. Blood 2019, 133, 7–17. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Francesco, B.; Verzella, D.; Capece, D.; Vecchiotti, D.; Di Vito Nolfi, M.; Flati, I.; Cornice, J.; Di Padova, M.; Angelucci, A.; Alesse, E.; et al. NF-κB: A Druggable Target in Acute Myeloid Leukemia. Cancers 2022, 14, 3557. https://doi.org/10.3390/cancers14143557
Di Francesco B, Verzella D, Capece D, Vecchiotti D, Di Vito Nolfi M, Flati I, Cornice J, Di Padova M, Angelucci A, Alesse E, et al. NF-κB: A Druggable Target in Acute Myeloid Leukemia. Cancers. 2022; 14(14):3557. https://doi.org/10.3390/cancers14143557
Chicago/Turabian StyleDi Francesco, Barbara, Daniela Verzella, Daria Capece, Davide Vecchiotti, Mauro Di Vito Nolfi, Irene Flati, Jessica Cornice, Monica Di Padova, Adriano Angelucci, Edoardo Alesse, and et al. 2022. "NF-κB: A Druggable Target in Acute Myeloid Leukemia" Cancers 14, no. 14: 3557. https://doi.org/10.3390/cancers14143557