
Dynamic CD8+ T Cell Cooperation with Macrophages and Monocytes for Successful Cancer Immunotherapy


{kind=link}
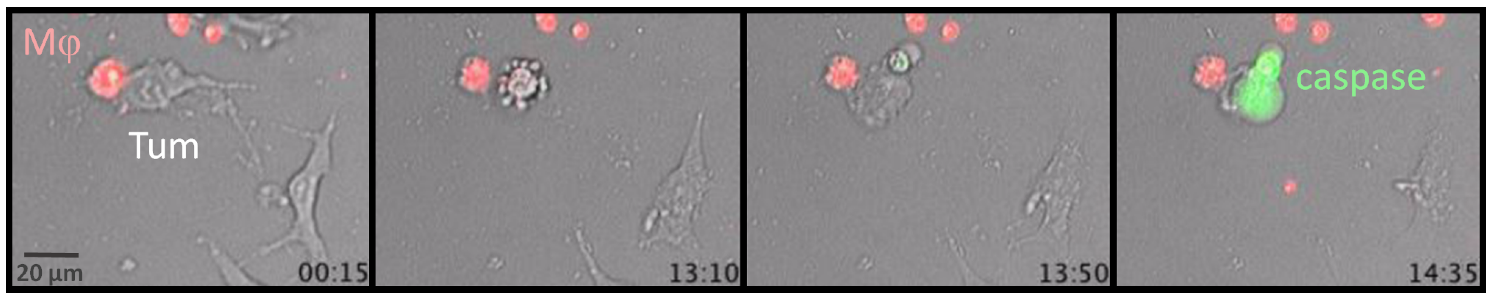{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Contribution of Activated Macrophages and Monocytes to T-Cell Infiltration
2.1. Cytokine Burst and Inflammasome-Induced Pyroptosis of Macrophages Stimulate T-Cell Recruitment
2.2. Additional Elements Regulating T-Cell Infiltration by Activated Macrophages and Monocytes
3. Regulation of Macrophages and Monocytes Killing Activities by CD8+ T Cells
3.1. Direct Killing of Tumor Cells by Macrophages and Monocytes through NO, ROS and TNFa
3.2. Tumoricidal TAMs Receive Activating Signals from TILs
4. Local Reactivation of T Cells by Macrophage Antigen Presentation
4.1. Cross-Presentation by Macrophages in Lymphoid Organs Stimulates a First Wave of Effector CD8+ T Cells
4.2. TAMs are Abundant and Efficient Antigen-Ingesting Cells that Interact with T Cells at the Tumor Site
5. TIL and TAM Cooperation in the Frame of Human Cancer Immunotherapy
6. Concluding Remarks and Future Directions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- HoptionCann, S.A.; van Netten, J.P.; van Netten, C. Dr William Coley and Tumour Regression: A Place in History or in the Future. Postgrad. Med. J. 2003, 79, 672–680. [Google Scholar]
- Lum, H.D.; Buhtoiarov, I.N.; Schmidt, B.E.; Berke, G.; Paulnock, D.M.; Sondel, P.M.; Rakhmilevich, A.L. Tumoristatic Effects of Anti-CD40 MAb-Activated Macrophages Involve Nitric Oxide and Tumour Necrosis Factor-Alpha. Immunology 2006, 118, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Ohkuri, T.; Kosaka, A.; Ishibashi, K.; Kumai, T.; Hirata, Y.; Ohara, K.; Nagato, T.; Oikawa, K.; Aoki, N.; Harabuchi, Y.; et al. Intratumoral Administration of CGAMP Transiently Accumulates Potent Macrophages for Anti-Tumor Immunity at a Mouse Tumor Site. Cancer Immunol. Immunother. 2017, 66, 705–716. [Google Scholar] [CrossRef]
- Thoreau, M.; Penny, H.L.; Tan, K.; Regnier, F.; Weiss, J.M.; Lee, B.; Johannes, L.; Dransart, E.; Le Bon, A.; Abastado, J.-P.; et al. Vaccine-Induced Tumor Regression Requires a Dynamic Cooperation between T Cells and Myeloid Cells at the Tumor Site. Oncotarget 2015, 6, 27832–27846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, J.M.; Guérin, M.V.; Regnier, F.; Renault, G.; Galy-Fauroux, I.; Vimeux, L.; Feuillet, V.; Peranzoni, E.; Thoreau, M.; Trautmann, A.; et al. The STING Agonist DMXAA Triggers a Cooperation between T Lymphocytes and Myeloid Cells That Leads to Tumor Regression. Oncoimmunology 2017, 6, e1346765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trautmann, L.; Janbazian, L.; Chomont, N.; Said, E.A.; Gimmig, S.; Bessette, B.; Boulassel, M.-R.; Delwart, E.; Sepulveda, H.; Balderas, R.S.; et al. Upregulation of PD-1 Expression on HIV-Specific CD8+ T Cells Leads to Reversible Immune Dysfunction. Nat. Med. 2006, 12, 1198–1202. [Google Scholar] [CrossRef]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring Function in Exhausted CD8 T Cells during Chronic Viral Infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef]
- Sagoo, P.; Garcia, Z.; Breart, B.; Lemaître, F.; Michonneau, D.; Albert, M.L.; Levy, Y.; Bousso, P. In Vivo Imaging of Inflammasome Activation Reveals a Subcapsular Macrophage Burst Response That Mobilizes Innate and Adaptive Immunity. Nat. Med. 2016, 22, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Brown, A.S.; Yang, C.; Fung, K.Y.; Bachem, A.; Bourges, D.; Bedoui, S.; Hartland, E.L.; van Driel, I.R. Cooperation between Monocyte-Derived Cells and Lymphoid Cells in the Acute Response to a Bacterial Lung Pathogen. PLoS Pathog. 2016, 12, e1005691. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 Inflammasome in Dendritic Cells Induces IL-1β–Dependent Adaptive Immunity against Tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef]
- Wang, W.; Hu, D.; Wu, C.; Feng, Y.; Li, A.; Liu, W.; Wang, Y.; Chen, K.; Tian, M.; Xiao, F.; et al. STING Promotes NLRP3 Localization in ER and Facilitates NLRP3 Deubiquitination to Activate the Inflammasome upon HSV-1 Infection. PLoSPathog. 2020, 16, e1008335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loyher, P.-L.; Hamon, P.; Laviron, M.; Meghraoui-Kheddar, A.; Goncalves, E.; Deng, Z.; Torstensson, S.; Bercovici, N.; Baudesson de Chanville, C.; Combadière, B.; et al. Macrophages of Distinct Origins Contribute to Tumor Development in the Lung. J. Exp. Med. 2018, 215, 2536–2553. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; Scott, C.L. Does Niche Competition Determine the Origin of Tissue-Resident Macrophages? Nat. Rev. Immunol. 2017, 17, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Guerin, M.V.; Regnier, F.; Feuillet, V.; Vimeux, L.; Weiss, J.M.; Bismuth, G.; Altan-Bonnet, G.; Guilbert, T.; Thoreau, M.; Finisguerra, V.; et al. TGFβ Blocks IFNα/β Release and Tumor Rejection in Spontaneous Mammary Tumors. Nat. Commun. 2019, 10, 4131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulder, K.; Patel, A.A.; Kong, W.T.; Piot, C.; Halitzki, E.; Dunsmore, G.; Khalilnezhad, S.; Irac, S.E.; Dubuisson, A.; Chevrier, M.; et al. Cross-Tissue Single-Cell Landscape of Human Monocytes and Macrophages in Health and Disease. Immunity 2021, 54, 1883–1900.e5. [Google Scholar] [CrossRef]
- Laviron, M.; Petit, M.; Weber-Delacroix, E.; Combes, A.J.; Arkal, A.R.; Barthélémy, S.; Courau, T.; Hume, D.A.; Combadière, C.; Krummel, M.F.; et al. Tumor-Associated Macrophage Heterogeneity Is Driven by Tissue Territories in Breast Cancer. Cell Rep. 2022, 39, 110865. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic Cell Death in Cancer and Infectious Disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Olekhnovitch, R.; Bousso, P. Induction, Propagation, and Activity of Host Nitric Oxide: Lessons from Leishmania Infection. Trends Parasitol. 2015, 31, 653–664. [Google Scholar] [CrossRef]
- Postat, J.; Olekhnovitch, R.; Lemaître, F.; Bousso, P. A Metabolism-Based Quorum Sensing Mechanism Contributes to Termination of Inflammatory Responses. Immunity 2018, 49, 654–665.e5. [Google Scholar] [CrossRef] [Green Version]
- Klug, F.; Prakash, H.; Huber, P.E.; Seibel, T.; Bender, N.; Halama, N.; Pfirschke, C.; Voss, R.H.; Timke, C.; Umansky, L.; et al. Low-Dose Irradiation Programs Macrophage Differentiation to an INOS+/M1 Phenotype That Orchestrates Effective T Cell Immunotherapy. Cancer Cell 2013, 24, 589–602. [Google Scholar] [CrossRef] [Green Version]
- Sektioglu, I.M.; Carretero, R.; Bender, N.; Bogdan, C.; Garbi, N.; Umansky, V.; Umansky, L.; Urban, K.; von Knebel-Döberitz, M.; Somasundaram, V.; et al. Macrophage-Derived Nitric Oxide Initiates T-Cell Diapedesis and Tumor Rejection. Oncoimmunology 2016, 5, e1204506. [Google Scholar] [CrossRef] [PubMed]
- Dong Kim, K.; Zhao, J.; Auh, S.; Yang, X.; Du, P.; Tang, H.; Fu, Y.-X. Adaptive Immune CellsTemper Initial InnateResponses. Nat. Med. 2007, 13, 1248–1252. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Madan, R.; Karp, C.L.; Braciale, T.J. Effector T Cells Control Lung Inflammation during Acute Influenza Virus Infection by Producing IL-10. Nat. Med. 2009, 15, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Palmer, E.M.; Holbrook, B.C.; Arimilli, S.; Parks, G.D.; Alexander-Miller, M.A. IFNγ-Producing, Virus-Specific CD8+ Effector Cells Acquire the Ability to Produce IL-10 as a Result of Entry into the Infected Lung Environment. Virology 2010, 404, 225–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Zhao, J.; Mangalam, A.K.; Channappanavar, R.; Fett, C.; Meyerholz, D.K.; Agnihothram, S.; Baric, R.S.; David, C.S.; Perlman, S. Airway Memory CD4+ T Cells Mediate Protective Immunity against Emerging Respiratory Coronaviruses. Immunity 2016, 44, 1379–1391. [Google Scholar] [CrossRef] [Green Version]
- Giavridis, T.; van der Stegen, S.J.C.; Eyquem, J.; Hamieh, M.; Piersigilli, A.; Sadelain, M. CAR T Cell-Induced Cytokine Release Syndrome Is Mediated by Macrophages and Abated by IL-1 Blockade. Nat. Med. 2018, 24, 731–738. [Google Scholar] [CrossRef]
- Weiss, G.; Schaible, U.E. Macrophage Defense Mechanisms against Intracellular Bacteria. Immunol. Rev. 2015, 264, 182–203. [Google Scholar] [CrossRef] [Green Version]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 Agonists Alter Tumor Stroma and Show Efficacy Against Pancreatic Carcinoma in Mice and Humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef] [Green Version]
- Bonnotte, B.; Larmonier, N.; Favre, N.; Fromentin, A.; Moutet, M.; Martin, M.; Gurbuxani, S.; Solary, E.; Chauffert, B.; Martin, F. Identification of Tumor-Infiltrating Macrophages as the Killers of Tumor Cells After Immunization in a Rat Model System. J. Immunol. 2001, 167, 5077–5083. [Google Scholar] [CrossRef] [Green Version]
- Bercovici, N.; Guérin, M.V.; Trautmann, A.; Donnadieu, E. The Remarkable Plasticity of Macrophages: A Chance to Fight Cancer. Front. Immunol. 2019, 10, 1563. [Google Scholar] [CrossRef] [Green Version]
- Imaizumi, K.; Kawabe, T.; Ichiyama, S.; Kikutani, H.; Yagita, H.; Shimokata, K.; Hasegawa, Y. Enhancement of Tumoricidal Activity of Alveolar Macrophages via CD40-CD40 Ligand Interaction. Am. J. Physiol.-Lung Cell. Mol. Physiol. 1999, 277, L49–L57. [Google Scholar] [CrossRef] [PubMed]
- Serbina, N.V.; Salazar-Mather, T.P.; Biron, C.A.; Kuziel, W.A.; Pamer, E.G. TNF/INOS-Producing Dendritic Cells Mediate Innate Immune Defense against Bacterial Infection. Immunity 2003, 19, 59–70. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Kees, T.; Almeida, A.S.; Liu, B.; He, X.-Y.; Ng, D.; Han, X.; Spector, D.L.; McNeish, I.A.; Gimotty, P.; et al. Activating a Collaborative Innate-Adaptive Immune Response to Control Metastasis. Cancer Cell 2021, 39, 1361–1374.e9. [Google Scholar] [CrossRef] [PubMed]
- Grandjean, C.L.; Garcia, Z.; Lemaître, F.; Bréart, B.; Bousso, P. Imaging the Mechanisms of Anti-CD20 Therapy in Vivo Uncovers Spatiotemporal Bottlenecks in Antibody-Dependent Phagocytosis. Sci. Adv. 2021, 7, eabd6167. [Google Scholar] [CrossRef]
- Qu, T.; Li, B.; Wang, Y. Targeting CD47/SIRPα as a Therapeutic Strategy, Where We Are and Where We Are Headed. Biomark. Res. 2022, 10, 20. [Google Scholar] [CrossRef]
- Chen, S.-H.; Dominik, P.K.; Stanfield, J.; Ding, S.; Yang, W.; Kurd, N.; Llewellyn, R.; Heyen, J.; Wang, C.; Melton, Z.; et al. Dual Checkpoint Blockade of CD47 and PD-L1 Using an Affinity-Tuned Bispecific Antibody Maximizes Antitumor Immunity. J. Immunother. Cancer 2021, 9, e003464. [Google Scholar] [CrossRef]
- Zhang, W.; Huang, Q.; Xiao, W.; Zhao, Y.; Pi, J.; Xu, H.; Zhao, H.; Xu, J.; Evans, C.E.; Jin, H. Advances in Anti-Tumor Treatments Targeting the CD47/SIRPα Axis. Front. Immunol. 2020, 11, 18. [Google Scholar] [CrossRef] [Green Version]
- Tveita, A.A.; Schjesvold, F.H.; Sundnes, O.; Haabeth, O.A.W.; Haraldsen, G.; Bogen, B. Indirect CD4+ T-Cell-Mediated Elimination of MHC II(NEG) Tumor Cells Is Spatially Restricted and Fails to Prevent Escape of Antigen-Negative Cells. Eur. J. Immunol. 2014, 44, 2625–2637. [Google Scholar] [CrossRef]
- Müller, E.; Speth, M.; Christopoulos, P.F.; Lunde, A.; Avdagic, A.; Øynebråten, I.; Corthay, A. Both Type I and Type II Interferons Can Activate Antitumor M1 Macrophages When Combined with TLR Stimulation. Front. Immunol. 2018, 9, 2520. [Google Scholar] [CrossRef]
- Müller, E.; Christopoulos, P.F.; Halder, S.; Lunde, A.; Beraki, K.; Speth, M.; Øynebråten, I.; Corthay, A. Toll-Like Receptor Ligands and Interferon-γ Synergize for Induction of Antitumor M1 Macrophages. Front. Immunol. 2017, 8, 1383. [Google Scholar] [CrossRef]
- Soudja, S.M.; Chandrabos, C.; Yakob, E.; Veenstra, M.; Palliser, D.; Lauvau, G. Memory-T-Cell-Derived Interferon-γ Instructs Potent Innate Cell Activation for Protective Immunity. Immunity 2014, 40, 974–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, R.; Alexander, P. Cooperation of Immune Lymphoid Cells with Macrophages in Tumour Immunity. Nature 1970, 228, 620–622. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, M.E.; Bornes, L.; Dijkgraaf, F.E.; Philips, D.; Pardieck, I.N.; Toebes, M.; Thommen, D.S.; van Rheenen, J.; Schumacher, T.N.M. Long-Distance Modulation of Bystander Tumor Cells by CD8 + T-Cell-Secreted IFN-γ. Nat. Cancer 2020, 1, 291–301. [Google Scholar] [CrossRef] [Green Version]
- Thibaut, R.; Bost, P.; Milo, I.; Cazaux, M.; Lemaître, F.; Garcia, Z.; Amit, I.; Breart, B.; Cornuot, C.; Schwikowski, B.; et al. Bystander IFN-γ Activity Promotes Widespread and Sustained Cytokine Signaling Altering the Tumor Microenvironment. Nat. Cancer 2020, 1, 302–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollenbaugh, J.A.; Reome, J.; Dobrzanski, M.; Dutton, R.W. The Rate of the CD8-Dependent Initial Reduction in Tumor Volume Is Not Limited by Contact-Dependent Perforin, Fas Ligand, or TNF-Mediated Cytolysis. J. Immunol. 2004, 173, 1738–1743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Sluis, T.C.; Sluijter, M.; van Duikeren, S.; West, B.L.; Melief, C.J.M.; Arens, R.; van der Burg, S.H.; van Hall, T. Therapeutic Peptide Vaccine-Induced CD8 T Cells Strongly Modulate Intratumoral Macrophages Required for Tumor Regression. Cancer Immunol. Res. 2015, 3, 1042–1051. [Google Scholar] [CrossRef] [Green Version]
- Bogen, B.; Fauskanger, M.; Haabeth, O.A.; Tveita, A. CD4+ T Cells Indirectly Kill Tumor Cells via Induction of Cytotoxic Macrophages in Mouse Models. Cancer Immunol. Immunother. 2019, 68, 1865–1873. [Google Scholar] [CrossRef]
- Chmielewski, M.; Kopecky, C.; Hombach, A.A.; Abken, H. IL-12 Release by Engineered T Cells Expressing Chimeric Antigen Receptors Can Effectively Muster an Antigen-Independent Macrophage Response on Tumor Cells That Have Shut Down Tumor Antigen Expression. Cancer Res. 2011, 71, 5697–5706. [Google Scholar] [CrossRef] [Green Version]
- Enders, M.; Franken, L.; Philipp, M.-S.; Kessler, N.; Baumgart, A.-K.; Eichler, M.; Wiertz, E.J.H.; Garbi, N.; Kurts, C. Splenic Red Pulp Macrophages Cross-Prime Early Effector CTL That Provide Rapid Defense against Viral Infections. J. Immunol. 2020, 204, 87–100. [Google Scholar] [CrossRef]
- Asano, K.; Nabeyama, A.; Miyake, Y.; Qiu, C.-H.; Kurita, A.; Tomura, M.; Kanagawa, O.; Fujii, S.; Tanaka, M. CD169-Positive Macrophages Dominate Antitumor Immunity by Crosspresenting Dead Cell-Associated Antigens. Immunity 2011, 34, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Shiota, T.; Miyasato, Y.; Ohnishi, K.; Yamamoto-Ibusuki, M.; Yamamoto, Y.; Iwase, H.; Takeya, M.; Komohara, Y. The Clinical Significance of CD169-Positive Lymph Node Macrophage in Patients with Breast Cancer. PLoS ONE 2016, 11, e0166680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broz, M.L.; Binnewies, M.; Boldajipour, B.; Nelson, A.E.; Pollack, J.L.; Erle, D.J.; Barczak, A.; Rosenblum, M.D.; Daud, A.; Barber, D.L.; et al. Dissecting the Tumor Myeloid Compartment Reveals Rare Activating Antigen-Presenting Cells Critical for T Cell Immunity. Cancer Cell 2014, 26, 638–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelhardt, J.J.; Boldajipour, B.; Beemiller, P.; Pandurangi, P.; Sorensen, C.; Werb, Z.; Egeblad, M.; Krummel, M.F. Marginating Dendritic Cells of the Tumor Microenvironment Cross-Present Tumor Antigens and Stably Engage Tumor-Specific T Cells. Cancer Cell 2012, 21, 402–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmon, H.; Idoyaga, J.; Rahman, A.; Leboeuf, M.; Remark, R.; Jordan, S.; Casanova-Acebes, M.; Khudoynazarova, M.; Agudo, J.; Tung, N.; et al. Expansion and Activation of CD103+ Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 2016, 44, 924–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kersten, K.; Hu, K.H.; Combes, A.J.; Samad, B.; Harwin, T.; Ray, A.; Rao, A.A.; Cai, E.; Marchuk, K.; Artichoker, J.; et al. Spatiotemporal Co-Dependency between Macrophages and Exhausted CD8+ T Cells in Cancer. Cancer Cell 2022, 40, 624–638.e9. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-F.; Fouquet, S.; Chapon, M.; Salmon, H.; Regnier, F.; Labroquère, K.; Badoual, C.; Damotte, D.; Validire, P.; Maubec, E.; et al. Early T Cell Signalling Is Reversibly Altered in PD-1+ T Lymphocytes Infiltrating Human Tumors. PLoS ONE 2011, 6, e17621. [Google Scholar] [CrossRef] [PubMed]
- Chu, K.-L.; Batista, N.V.; Girard, M.; Watts, T.H. Monocyte-Derived Cells in Tissue-Resident Memory T Cell Formation. J. Immunol. 2020, 204, 477–485. [Google Scholar] [CrossRef]
- Dieu-Nosjean, M.-C.; Goc, J.; Giraldo, N.A.; Sautès-Fridman, C.; Fridman, W.H. Tertiary Lymphoid Structures in Cancer and Beyond. Trends Immunol. 2014, 35, 571–580. [Google Scholar] [CrossRef]
- Corthay, A.; Skovseth, D.K.; Lundin, K.U.; Røsjø, E.; Omholt, H.; Hofgaard, P.O.; Haraldsen, G.; Bogen, B. Primary Antitumor Immune Response Mediated by CD4+ T Cells. Immunity 2005, 22, 371–383. [Google Scholar] [CrossRef] [Green Version]
- Alspach, E.; Lussier, D.M.; Miceli, A.P.; Kizhvatov, I.; DuPage, M.; Luoma, A.M.; Meng, W.; Lichti, C.F.; Esaulova, E.; Vomund, A.N.; et al. MHC-II Neoantigens Shape Tumour Immunity and Response to Immunotherapy. Nature 2019, 574, 696–701. [Google Scholar] [CrossRef]
- Fridman, W.H.; Zitvogel, L.; Sautès-Fridman, C.; Kroemer, G. The Immune Contexture in Cancer Prognosis and Treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef] [PubMed]
- Peranzoni, E.; Ingangi, V.; Masetto, E.; Pinton, L.; Marigo, I. Myeloid Cells as Clinical Biomarkers for Immune Checkpoint Blockade. Front. Immunol. 2020, 11, 1590. [Google Scholar] [CrossRef]
- Hammerl, D.; Martens, J.W.M.; Timmermans, M.; Smid, M.; Trapman-Jansen, A.M.; Foekens, R.; Isaeva, O.I.; Voorwerk, L.; Balcioglu, H.E.; Wijers, R.; et al. Spatial Immunophenotypes Predict Response to Anti-PD1 Treatment and Capture Distinct Paths of T Cell Evasion in Triple Negative Breast Cancer. Nat. Commun. 2021, 12, 5668. [Google Scholar] [CrossRef]
- Hwang, S.; Kwon, A.-Y.; Jeong, J.-Y.; Kim, S.; Kang, H.; Park, J.; Kim, J.-H.; Han, O.J.; Lim, S.M.; An, H.J. Immune Gene Signatures for Predicting Durable Clinical Benefit of Anti-PD-1 Immunotherapy in Patients with Non-Small Cell Lung Cancer. Sci. Rep. 2020, 10, 643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- House, I.G.; Savas, P.; Lai, J.; Chen, A.X.Y.; Oliver, A.J.; Teo, Z.L.; Todd, K.L.; Henderson, M.A.; Giuffrida, L.; Petley, E.V.; et al. Macrophage-Derived CXCL9 and CXCL10 Are Required for Antitumor Immune Responses Following Immune Checkpoint Blockade. Clin. Cancer Res. 2020, 26, 487–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Chen, H.; Mo, H.; Hu, X.; Gao, R.; Zhao, Y.; Liu, B.; Niu, L.; Sun, X.; Yu, X.; et al. Single-Cell Analyses Reveal Key Immune Cell Subsets Associated with Response to PD-L1 Blockade in Triple-Negative Breast Cancer. Cancer Cell 2021, 39, 1578–1593.e8. [Google Scholar] [CrossRef]
- Bischoff, P.; Trinks, A.; Obermayer, B.; Pett, J.P.; Wiederspahn, J.; Uhlitz, F.; Liang, X.; Lehmann, A.; Jurmeister, P.; Elsner, A.; et al. Single-Cell RNA Sequencing Reveals Distinct Tumor Microenvironmental Patterns in Lung Adenocarcinoma. Oncogene 2021, 40, 6748–6758. [Google Scholar] [CrossRef]
- Ardighieri, L.; Missale, F.; Bugatti, M.; Gatta, L.B.; Pezzali, I.; Monti, M.; Gottardi, S.; Zanotti, L.; Bignotti, E.; Ravaggi, A.; et al. Infiltration by CXCL10 Secreting Macrophages Is Associated with Antitumor Immunity and Response to Therapy in Ovarian Cancer Subtypes. Front. Immunol. 2021, 12, 690201. [Google Scholar] [CrossRef]
- Bassez, A.; Vos, H.; Van Dyck, L.; Floris, G.; Arijs, I.; Desmedt, C.; Boeckx, B.; VandenBempt, M.; Nevelsteen, I.; Lambein, K.; et al. A Single-Cell Map of Intratumoral Changes during Anti-PD1 Treatment of Patients with Breast Cancer. Nat. Med. 2021, 27, 820–832. [Google Scholar] [CrossRef]
- Marcovecchio, P.M.; Thomas, G.; Salek-Ardakani, S. CXCL9-Expressing Tumor-Associated Macrophages: New Players in the Fight against Cancer. J. Immunother. Cancer 2021, 9, e002045. [Google Scholar] [CrossRef]
- Romano, E.; Kusio-Kobialka, M.; Foukas, P.G.; Baumgaertner, P.; Meyer, C.; Ballabeni, P.; Michielin, O.; Weide, B.; Romero, P.; Speiser, D.E. Ipilimumab-Dependent Cell-Mediated Cytotoxicity of Regulatory T Cells Ex Vivo by Nonclassical Monocytes in Melanoma Patients. Proc. Natl. Acad. Sci. USA 2015, 112, 6140–6145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krieg, C.; Nowicka, M.; Guglietta, S.; Schindler, S.; Hartmann, F.J.; Weber, L.M.; Dummer, R.; Robinson, M.D.; Levesque, M.P.; Becher, B. High-Dimensional Single-Cell Analysis Predicts Response to Anti-PD-1 Immunotherapy. Nat. Med. 2018, 24, 144–153. [Google Scholar] [CrossRef]
- Bocanegra, A.; Fernandez-Hinojal, G.; Zuazo-Ibarra, M.; Arasanz, H.; Garcia-Granda, M.; Hernandez, C.; Ibañez, M.; Hernandez-Marin, B.; Martinez-Aguillo, M.; Lecumberri, M.; et al. PD-L1 Expression in Systemic Immune Cell Populations as a Potential Predictive Biomarker of Responses to PD-L1/PD-1 Blockade Therapy in Lung Cancer. Int. J. Mol. Sci. 2019, 20, 1631. [Google Scholar] [CrossRef] [Green Version]
- Vilain, R.E.; Menzies, A.M.; Wilmott, J.S.; Kakavand, H.; Madore, J.; Guminski, A.; Liniker, E.; Kong, B.Y.; Cooper, A.J.; Howle, J.R.; et al. Dynamic Changes in PD-L1 Expression and Immune Infiltrates Early During Treatment Predict Response to PD-1 Blockade in Melanoma. Clin. Cancer Res. 2017, 23, 5024–5033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.H.; Sun, L.; Mochizuki, A.Y.; Reynoso, J.G.; Orpilla, J.; Chow, F.; Kienzler, J.C.; Everson, R.G.; Nathanson, D.A.; Bensinger, S.J.; et al. Neoadjuvant PD-1 Blockade Induces T Cell and CDC1 Activation but Fails to Overcome the Immunosuppressive Tumor Associated Macrophages in Recurrent Glioblastoma. Nat. Commun. 2021, 12, 6938. [Google Scholar] [CrossRef] [PubMed]
- Bi, K.; He, M.X.; Bakouny, Z.; Kanodia, A.; Napolitano, S.; Wu, J.; Grimaldi, G.; Braun, D.A.; Cuoco, M.S.; Mayorga, A.; et al. Tumor and Immune Reprogramming during Immunotherapy in Advanced Renal Cell Carcinoma. Cancer Cell 2021, 39, 649–661.e5. [Google Scholar] [CrossRef]
- Cindy Yang, S.Y.; Lien, S.C.; Wang, B.X.; Clouthier, D.L.; Hanna, Y.; Cirlan, I.; Zhu, K.; Bruce, J.P.; El Ghamrasni, S.; Iafolla, M.A.J.; et al. Pan-Cancer Analysis of Longitudinal Metastatic Tumors Reveals Genomic Alterations and Immune Landscape Dynamics Associated with Pembrolizumab Sensitivity. Nat. Commun. 2021, 12, 5137. [Google Scholar] [CrossRef]
- Cottrell, T.R.; Thompson, E.D.; Forde, P.M.; Stein, J.E.; Duffield, A.S.; Anagnostou, V.; Rekhtman, N.; Anders, R.A.; Cuda, J.D.; Illei, P.B.; et al. Pathologic Features of Response to Neoadjuvant Anti-PD-1 in Resected Non-Small-Cell Lung Carcinoma: A Proposal for Quantitative Immune-Related Pathologic Response Criteria (IrPRC). Ann. Oncol. 2018, 29, 1853–1860. [Google Scholar] [CrossRef]
- Krishna, C.; DiNatale, R.G.; Kuo, F.; Srivastava, R.M.; Vuong, L.; Chowell, D.; Gupta, S.; Vanderbilt, C.; Purohit, T.A.; Liu, M.; et al. Single-Cell Sequencing Links Multiregional Immune Landscapes and Tissue-Resident T Cells in CcRCC to Tumor Topology and Therapy Efficacy. Cancer Cell 2021, 39, 662–677.e6. [Google Scholar] [CrossRef]
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guérin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages Impede CD8 T Cells from Reaching Tumor Cells and Limit the Efficacy of Anti-PD-1 Treatment. Proc. Natl. Acad. Sci. USA 2018, 115, E4041–E4050. [Google Scholar] [CrossRef] [Green Version]
- Gruosso, T.; Gigoux, M.; Manem, V.S.K.; Bertos, N.; Zuo, D.; Perlitch, I.; Saleh, S.M.I.; Zhao, H.; Souleimanova, M.; Johnson, R.M.; et al. Spatially Distinct Tumor Immune Microenvironments Stratify Triple-Negative Breast Cancers. J. Clin. Invest. 2019, 129, 1785–1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duraiswamy, J.; Turrini, R.; Minasyan, A.; Barras, D.; Crespo, I.; Grimm, A.J.; Casado, J.; Genolet, R.; Benedetti, F.; Wicky, A.; et al. Myeloid Antigen-Presenting Cell Niches Sustain Antitumor T Cells and License PD-1 Blockade via CD28 Costimulation. Cancer Cell 2021, 39, 1623–1642.e20. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vermare, A.; Guérin, M.V.; Peranzoni, E.; Bercovici, N. Dynamic CD8+ T Cell Cooperation with Macrophages and Monocytes for Successful Cancer Immunotherapy. Cancers 2022, 14, 3546. https://doi.org/10.3390/cancers14143546
Vermare A, Guérin MV, Peranzoni E, Bercovici N. Dynamic CD8+ T Cell Cooperation with Macrophages and Monocytes for Successful Cancer Immunotherapy. Cancers. 2022; 14(14):3546. https://doi.org/10.3390/cancers14143546
Chicago/Turabian StyleVermare, Anaïs, Marion V. Guérin, Elisa Peranzoni, and Nadège Bercovici. 2022. "Dynamic CD8+ T Cell Cooperation with Macrophages and Monocytes for Successful Cancer Immunotherapy" Cancers 14, no. 14: 3546. https://doi.org/10.3390/cancers14143546