Acetyl-CoA Synthetase 2 as a Therapeutic Target in Tumor Metabolism

Abstract
:Simple Summary
Abstract
1. Introduction
2. ACSS2
3. ACSS2 and Tumor Prognosis
3.1. Breast Cancer
3.2. Glioblastoma (GBM)
3.3. Hepatocellular Carcinoma (HCC)
3.4. Myeloma
3.5. Prostate Cancer and Bladder Cancer
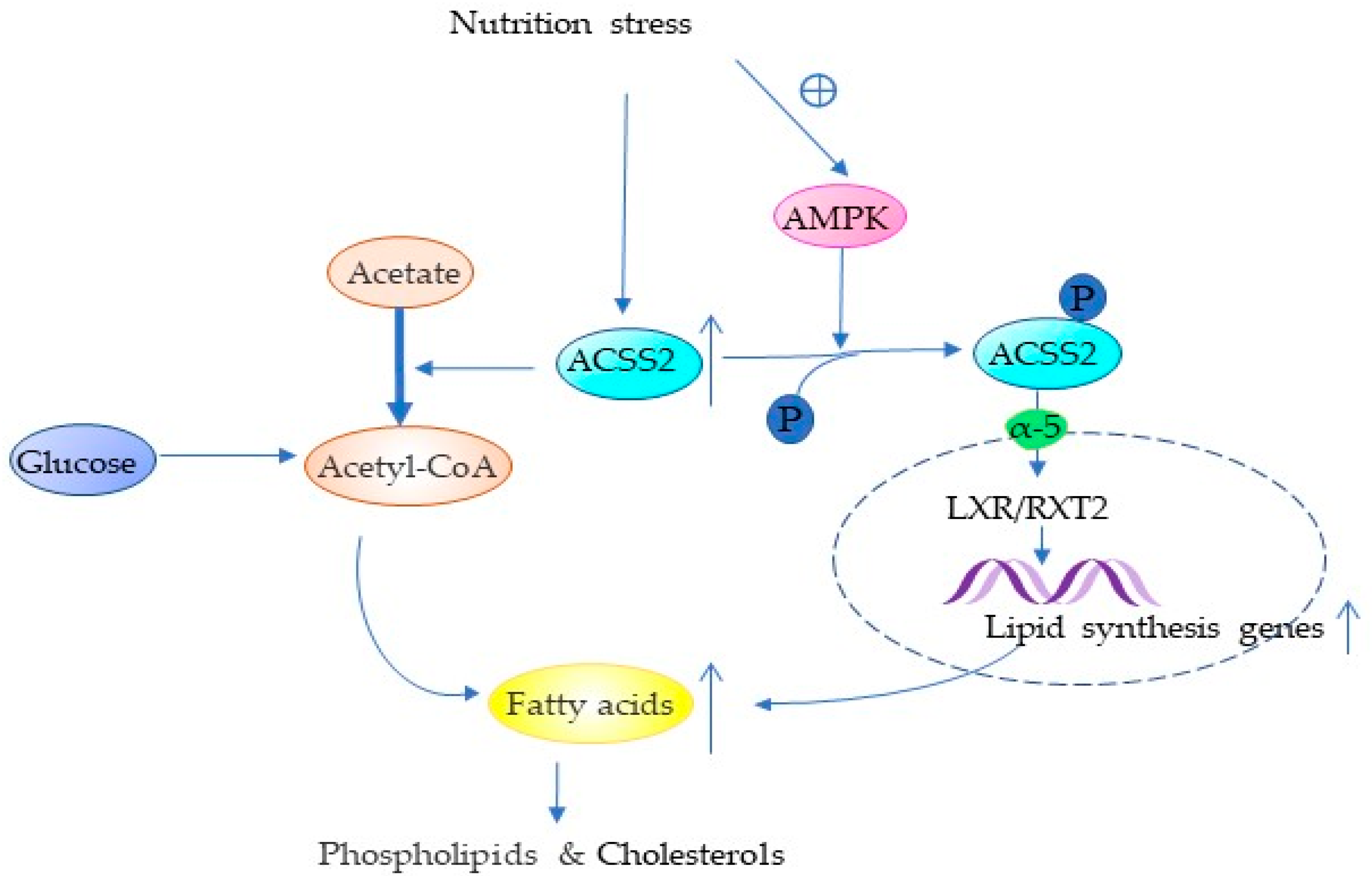
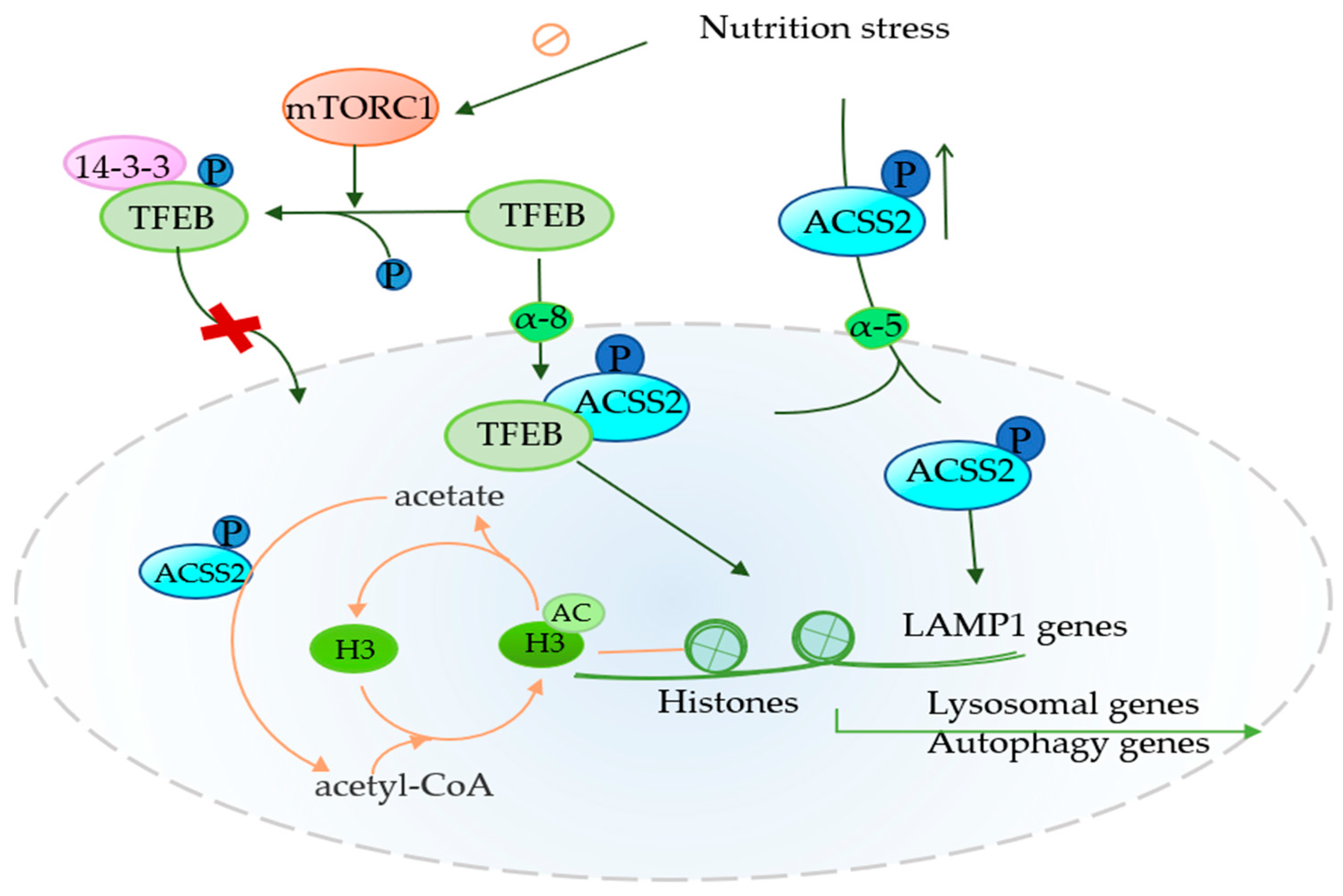
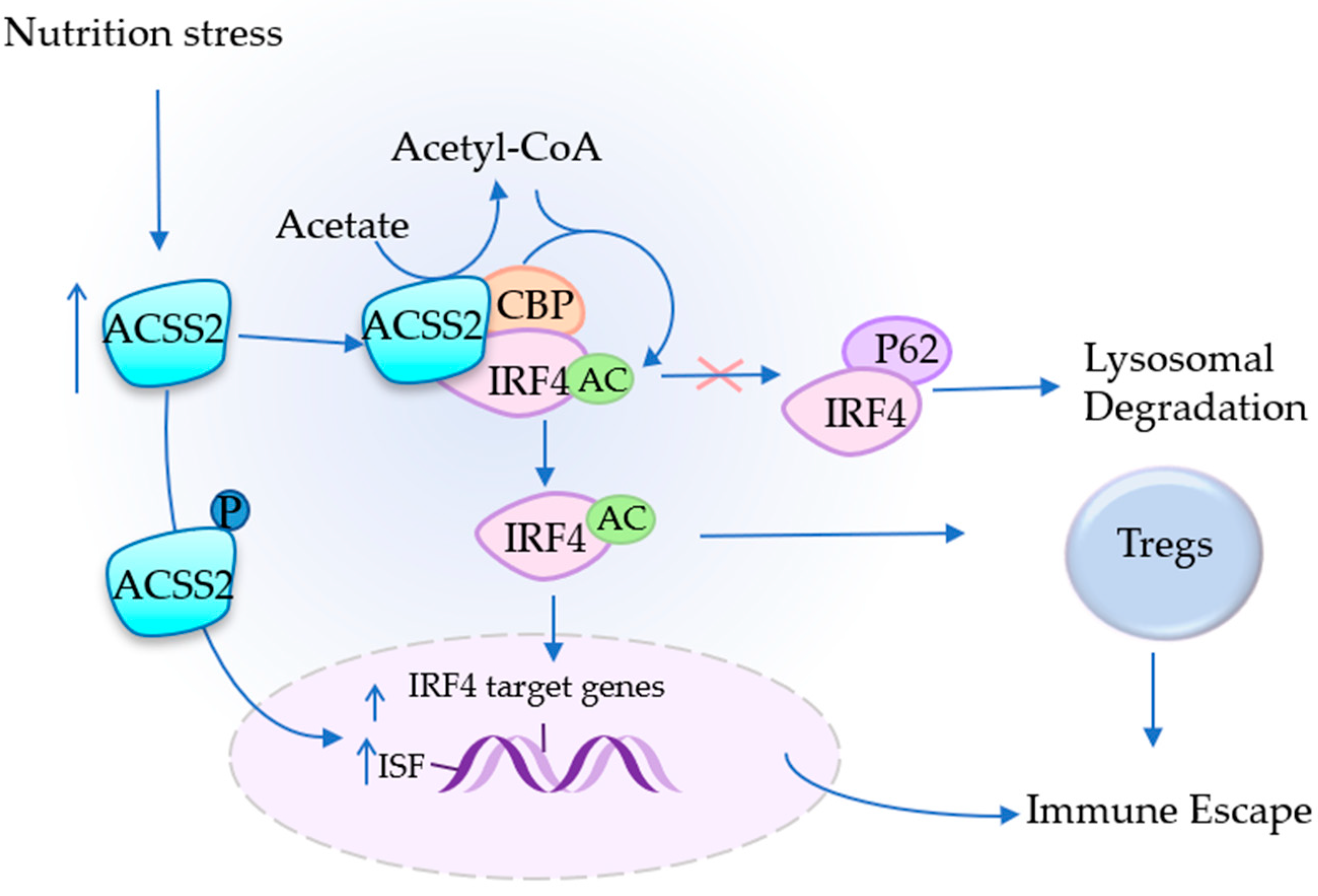
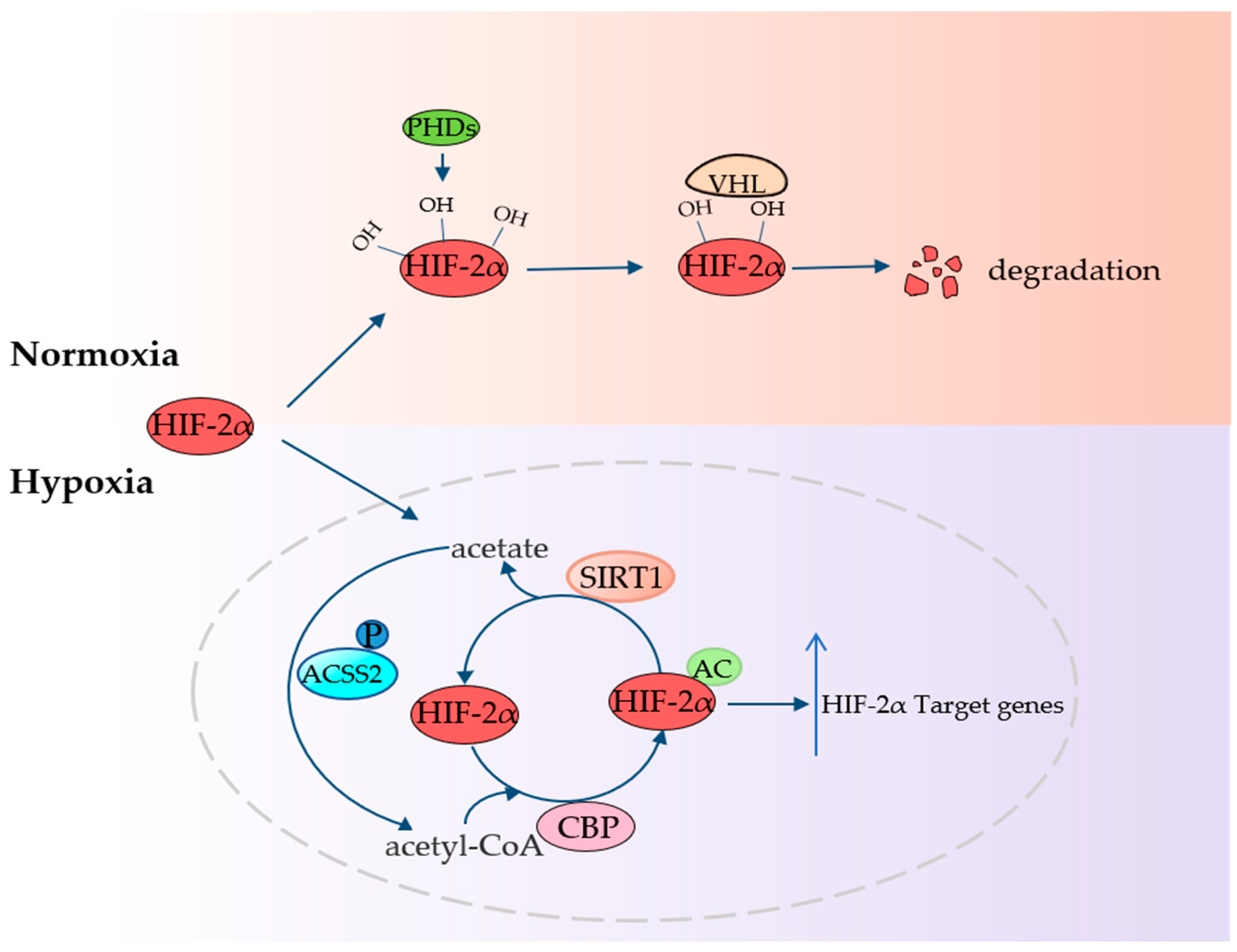
4. ACSS2 Plays an Important Role in Tumor Promotion
4.1. ACSS2 Promotes Lipid Synthesis
4.2. Role of ACSS2 in Autophagy
4.3. ACSS2 Assists in Immune Escape
4.4. ACSS2 Fights Hypoxia
5. Prospect of Targeted Inhibition of ACSS2
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Jing, X.; Yang, F.; Shao, C.; Wei, K.; Xie, M.; Shen, H.; Shu, Y. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol. Cancer 2019, 18, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Tameemi, W.; Dale, T.P.; Al-Jumaily, R.M.K.; Forsyth, N.R. Hypoxia-Modified Cancer Cell Metabolism. Front. Cell Dev. Biol. 2019, 7, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.C.; Qian, Y.; Yu, J. Interplay between epigenetics and metabolism in oncogenesis: Mechanisms and therapeutic approaches. Oncogene 2017, 36, 3359–3374. [Google Scholar] [CrossRef] [PubMed]
- Kamphorst, J.J.; Chung, M.K.; Fan, J.; Rabinowitz, J.D. Quantitative analysis of acetyl-CoA production in hypoxic cancer cells reveals substantial contribution from acetate. Cancer Metab. 2014, 2, 23. [Google Scholar] [CrossRef] [PubMed]
- Schug, Z.T.; Peck, B.; Jones, D.T.; Zhang, Q.; Grosskurth, S.; Alam, I.S.; Goodwin, L.M.; Smethurst, E.; Mason, S.; Blyth, K.; et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell 2015, 27, 57–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schug, Z.T.; Vande Voorde, J.; Gottlieb, E. The metabolic fate of acetate in cancer. Nat. Rev. Cancer 2016, 16, 708–717. [Google Scholar] [CrossRef]
- Comerford, S.A.; Huang, Z.; Du, X.; Wang, Y.; Cai, L.; Witkiewicz, A.K.; Walters, H.; Tantawy, M.N.; Fu, A.; Manning, H.C.; et al. Acetate dependence of tumors. Cell 2014, 159, 1591–1602. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Shao, F.; Shi, S.; Feng, X.; Wang, W.; Wang, Y.; Guo, W.; Wang, J.; Gao, S.; Gao, Y.; et al. Prognostic Impact of Metabolism Reprogramming Markers Acetyl-CoA Synthetase 2 Phosphorylation and Ketohexokinase-A Expression in Non-Small-Cell Lung Carcinoma. Front. Oncol. 2019, 9, 1123. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; He, J.; Jia, Z.; Yan, Z.; Yang, J. Acetyl-CoA synthetase 2 enhances tumorigenesis and is indicative of a poor prognosis for patients with renal cell carcinoma. Urol. Oncol. Semin. Orig. Investig. 2018, 36, 243.e9–243.e20. [Google Scholar] [CrossRef]
- Huang, Z.; Zhang, M.; Plec, A.A.; Estill, S.J.; Cai, L.; Repa, J.J.; McKnight, S.L.; Tu, B.P. ACSS2 promotes systemic fat storage and utilization through selective regulation of genes involved in lipid metabolism. Proc. Natl. Acad. Sci. USA 2018, 115, E9499–E9506. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Lin, S.H.; Ren, F.; Li, J.T.; Chen, J.J.; Yao, C.B.; Yang, H.B.; Jiang, S.X.; Yan, G.Q.; Wang, D.; et al. Acetate functions as an epigenetic metabolite to promote lipid synthesis under hypoxia. Nat. Commun. 2016, 7, 11960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Yu, W.; Qian, X.; Xia, Y.; Zheng, Y.; Lee, J.-H.; Li, W.; Lyu, J.; Rao, G.; Zhang, X.; et al. Nucleus-Translocated ACSS2 Promotes Gene Transcription for Lysosomal Biogenesis and Autophagy. Mol. Cell 2017, 66, 684–697.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bidkhori, G.; Benfeitas, R.; Klevstig, M.; Zhang, C.; Nielsen, J.; Uhlen, M.; Boren, J.; Mardinoglu, A. Metabolic network-based stratification of hepatocellular carcinoma reveals three distinct tumor subtypes. Proc. Natl. Acad. Sci. USA 2018, 115, E11874–E11883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Nie, Z.W.; Zhou, D.J.; Cui, X.S. Acetyl-CoA synthases are essential for maintaining histone acetylation under metabolic stress during zygotic genome activation in pigs. J. Cell. Physiol. 2021, 236, 6948–6962. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Zhang, J.; Nie, Q. Inferring latent temporal progression and regulatory networks from cross-sectional transcriptomic data of cancer samples. PLoS Comput. Biol. 2021, 17, e1008379. [Google Scholar] [CrossRef]
- Mashimo, T.; Pichumani, K.; Vemireddy, V.; Hatanpaa, K.J.; Singh, D.K.; Sirasanagandla, S.; Nannepaga, S.; Piccirillo, S.G.; Kovacs, Z.; Foong, C.; et al. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell 2014, 159, 1603–1614. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, Y.; Araki, A.; Maruta, H.; Takahashi, Y.; Yamashita, H. Molecular cloning of rat acss3 and characterization of mammalian propionyl-CoA synthetase in the liver mitochondrial matrix. J. Biochem. 2017, 161, 279–289. [Google Scholar]
- Zhang, J.; Duan, H.; Feng, Z.; Han, X.; Gu, C. Acetyl-CoA synthetase 3 promotes bladder cancer cell growth under metabolic stress. Oncogenesis 2020, 9, 46. [Google Scholar] [CrossRef]
- Zhou, L.; Song, Z.; Hu, J.; Liu, L.; Hou, Y.; Zhang, X.; Yang, X.; Chen, K. ACSS3 represses prostate cancer progression through downregulating lipid droplet-associated protein PLIN3. Theranostics 2021, 11, 841–860. [Google Scholar] [CrossRef]
- Xu, H.; Luo, J.; Ma, G.; Zhang, X.; Yao, D.; Li, M.; Loor, J.J. Acyl-CoA synthetase short-chain family member 2 (ACSS2) is regulated by SREBP-1 and plays a role in fatty acid synthesis in caprine mammary epithelial cells. J. Cell. Physiol. 2018, 233, 1005–1016. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. The SREBP pathway: Regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 1997, 89, 331–340. [Google Scholar] [CrossRef] [Green Version]
- Luong, A.; Hannah, V.C.; Brown, M.S.; Goldstein, J.L. Molecular characterization of human acetyl-CoA synthetase, an enzyme regulated by sterol regulatory element-binding proteins. J. Biol. Chem. 2000, 275, 26458–26466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallows, W.C.; Lee, S.; Denu, J.M. Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc. Natl. Acad. Sci. USA 2006, 103, 10230–10235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moffett, J.R.; Puthillathu, N.; Vengilote, R.; Jaworski, D.M.; Namboodiri, A.M. Acetate Revisited: A Key Biomolecule at the Nexus of Metabolism, Epigenetics, and Oncogenesis-Part 2: Acetate and ACSS2 in Health and Disease. Front. Physiol. 2020, 11, 580171. [Google Scholar] [CrossRef] [PubMed]
- Bulusu, V.; Tumanov, S.; Michalopoulou, E.; van den Broek, N.J.; MacKay, G.; Nixon, C.; Dhayade, S.; Schug, Z.T.; Vande Voorde, J.; Blyth, K.; et al. Acetate Recapturing by Nuclear Acetyl-CoA Synthetase 2 Prevents Loss of Histone Acetylation during Oxygen and Serum Limitation. Cell Rep. 2017, 18, 647–658. [Google Scholar] [CrossRef] [Green Version]
- Nagati, J.S.; Xu, M.; Garcia, T.; Comerford, S.A.; Hammer, R.E.; Garcia, J.A. A substitution mutation in a conserved domain of mammalian acetate-dependent acetyl CoA synthetase 2 results in destabilized protein and impaired HIF-2 signaling. PLoS ONE 2019, 14, e0225105. [Google Scholar] [CrossRef]
- Li, Z.; Liu, H.; He, J.; Wang, Z.; Yin, Z.; You, G.; Wang, Z.; Davis, R.E.; Lin, P.; Bergsagel, P.L.; et al. Acetyl-CoA Synthetase 2: A Critical Linkage in Obesity-Induced Tumorigenesis in Myeloma. Cell Metab. 2021, 33, 78–93.e7. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- DeSantis, C.E.; Ma, J.; Gaudet, M.M.; Newman, L.A.; Miller, K.D.; Goding Sauer, A.; Jemal, A.; Siegel, R.L. Breast cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 438–451. [Google Scholar] [CrossRef]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13 Pt 1, 4429–4434. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Pi, H.; Liao, L.; Tan, M.; Deng, P.; Yue, Y.; Xi, Y.; Tian, L.; Xie, J.; Chen, M.; et al. Cadmium promotes breast cancer cell proliferation, migration and invasion by inhibiting ACSS2/ATG5-mediated autophagy. Environ. Pollut. 2021, 273, 116504. [Google Scholar] [CrossRef] [PubMed]
- Mi, L.; Zhou, Y.; Wu, D.; Tao, Q.; Wang, X.; Zhu, H.; Gao, X.; Wang, J.; Ling, R.; Deng, J.; et al. ACSS2/AMPK/PCNA pathway-driven proliferation and chemoresistance of esophageal squamous carcinoma cells under nutrient stress. Mol. Med. Rep. 2019, 20, 5286–5296. [Google Scholar] [CrossRef] [PubMed]
- Yun, M.; Bang, S.H.; Kim, J.W.; Park, J.Y.; Kim, K.S.; Lee, J.D. The importance of acetyl coenzyme A synthetase for 11C-acetate uptake and cell survival in hepatocellular carcinoma. J. Nucl. Med. 2009, 50, 1222–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.-H.; Huang, S.; Zhu, L.; Yang, Q.; Yang, X.-M.; Gu, J.-R.; Zhang, Z.-G.; Nie, H.-Z.; Li, J. Alternative transcription start site selection in ACSS2 controls its nuclear localization and promotes ribosome biosynthesis in hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2019, 514, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Kong, Y.; Cao, M.; Zhou, H.; Li, H.; Cui, Y.; Fang, F.; Zhang, W.; Li, J.; Zhu, X.; et al. Decreased expression of acetyl-CoA synthase 2 promotes metastasis and predicts poor prognosis in hepatocellular carcinoma. Cancer Sci. 2017, 108, 1338–1346. [Google Scholar] [CrossRef]
- Yao, L.; Guo, X.; Gui, Y. Acetyl-CoA Synthetase 2 Promotes Cell Migration and Invasion of Renal Cell Carcinoma by Upregulating Lysosomal-Associated Membrane Protein 1 Expression. Cell. Physiol. Biochem. 2018, 45, 984–992. [Google Scholar] [CrossRef]
- Lee, M.Y.; Yeon, A.; Shahid, M.; Cho, E.; Sairam, V.; Figlin, R.; Kim, K.-H.; Kim, J. Reprogrammed lipid metabolism in bladder cancer with cisplatin resistance. Oncotarget 2018, 9, 13231–13243. [Google Scholar] [CrossRef] [Green Version]
- Wen, H.; Lee, S.; Zhu, W.-G.; Lee, O.-J.; Yun, S.J.; Kim, J.; Park, S. Glucose-derived acetate and ACSS2 as key players in cisplatin resistance in bladder cancer. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2019, 1864, 413–421. [Google Scholar] [CrossRef]
- Li, C.-J.; Chiu, Y.-H.; Chang, C.; Chang, Y.-C.I.; Sheu, J.J.-C.; Chiang, A.-J. Acetyl Coenzyme A Synthase 2 Acts as a Prognostic Biomarker Associated with Immune Infiltration in Cervical Squamous Cell Carcinoma. Cancers 2021, 13, 3125. [Google Scholar] [CrossRef]
- Wen, P.Y.; Weller, M.; Lee, E.Q.; Alexander, B.M.; Barnholtz-Sloan, J.S.; Barthel, F.P.; Batchelor, T.T.; Bindra, R.S.; Chang, S.M.; Chiocca, E.A.; et al. Glioblastoma in adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro-Oncology 2020, 22, 1073–1113. [Google Scholar] [CrossRef]
- Fine, H.A. Bevacizumab in glioblastoma-still much to learn. N. Engl. J. Med. 2014, 370, 764–765. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Craig, A.J.; von Felden, J.; Garcia-Lezana, T.; Sarcognato, S.; Villanueva, A. Tumour evolution in hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Hoshida, Y. Molecular heterogeneity in hepatocellular carcinoma. Hepat. Oncol. 2018, 5, HEP10. [Google Scholar] [CrossRef] [Green Version]
- Hideshima, T.; Mitsiades, C.; Tonon, G.; Richardson, P.G.; Anderson, K.C. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat. Rev. Cancer 2007, 7, 585–598. [Google Scholar] [CrossRef]
- Silberstein, J.; Tuchman, S.; Grant, S.J. What Is Multiple Myeloma? JAMA 2022, 327, 497. [Google Scholar] [CrossRef]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [Green Version]
- Teo, M.Y.; Rathkopf, D.E.; Kantoff, P. Treatment of Advanced Prostate Cancer. Annu. Rev. Med. 2019, 70, 479–499. [Google Scholar] [CrossRef]
- Oyama, N.; Akino, H.; Kanamaru, H.; Suzuki, Y.; Muramoto, S.; Yonekura, Y.; Sadato, N.; Yamamoto, K.; Okada, K. 11C-Acetate PET Imaging of Prostate Cancer. J. Nucl. Med. 2002, 43, 181. [Google Scholar]
- Spick, C.; Herrmann, K.; Czernin, J. Evaluation of Prostate Cancer with 11C-Acetate PET/CT. J. Nucl. Med. 2016, 57 (Suppl. 3), 30S–37S. [Google Scholar] [CrossRef] [Green Version]
- Folger, O.; Jerby, L.; Frezza, C.; Gottlieb, E.; Ruppin, E.; Shlomi, T. Predicting selective drug targets in cancer through metabolic networks. Mol. Syst. Biol. 2011, 7, 501. [Google Scholar] [CrossRef] [PubMed]
- Bian, X.; Liu, R.; Meng, Y.; Xing, D.; Xu, D.; Lu, Z. Lipid metabolism and cancer. J. Exp. Med. 2020, 218, e20201606. [Google Scholar] [CrossRef] [PubMed]
- Röhrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Maxfield, F.R. MEMBRANE DOMAINS. Annu. Rev. Cell Dev. Biol. 2004, 20, 839–866. [Google Scholar] [CrossRef] [PubMed]
- Pomorski, T.; Hrafnsdóttir, S.; Devaux, P.F.; Meer, G.V. Lipid distribution and transport across cellular membranes. Semin. Cell Dev. Biol. 2001, 12, 139–148. [Google Scholar] [CrossRef] [PubMed]
- van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef]
- Maxfield, F.R. Plasma membrane microdomains. Curr. Opin. Cell Biol. 2002, 14, 483–487. [Google Scholar] [CrossRef]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [Green Version]
- Guo, D.; Bell, E.H.; Chakravarti, A. Lipid metabolism emerges as a promising target for malignant glioma therapy. CNS Oncol. 2013, 2, 289–299. [Google Scholar] [CrossRef]
- Li, H.; Feng, Z.; He, M.-L. Lipid metabolism alteration contributes to and maintains the properties of cancer stem cells. Theranostics 2020, 10, 7053–7069. [Google Scholar] [CrossRef]
- Efeyan, A.; Comb, W.C.; Sabatini, D.M. Nutrient-sensing mechanisms and pathways. Nature 2015, 517, 302–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.A.; Wei, J.; Nguyen, T.-L.M.; Shi, H.; Su, W.; Palacios, G.; Dhungana, Y.; Chapman, N.M.; Long, L.; Saravia, J.; et al. Lipid signalling enforces functional specialization of Treg cells in tumours. Nature 2021, 591, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, Y.; Furukawa, T.; Yoshii, H.; Mori, T.; Kiyono, Y.; Waki, A.; Kobayashi, M.; Tsujikawa, T.; Kudo, T.; Okazawa, H.; et al. Cytosolic acetyl-CoA synthetase affected tumor cell survival under hypoxia: The possible function in tumor acetyl-CoA/acetate metabolism. Cancer Sci. 2009, 100, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, D.; Simon, M.C. Hypoxia, lipids, and cancer: Surviving the harsh tumor microenvironment. Trends Cell Biol. 2014, 24, 472–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Zhang, J.; Sampieri, K.; Clohessy, J.G.; Mendez, L.; Gonzalez-Billalabeitia, E.; Liu, X.-S.; Lee, Y.-R.; Fung, J.; Katon, J.M.; et al. An aberrant SREBP-dependent lipogenic program promotes metastatic prostate cancer. Nat. Genet. 2018, 50, 206–218. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Qian, X.; Lu, Z. Local histone acetylation by ACSS2 promotes gene transcription for lysosomal biogenesis and autophagy. Autophagy 2017, 13, 1790–1791. [Google Scholar] [CrossRef] [Green Version]
- Sakakibara, I.; Fujino, T.; Ishii, M.; Tanaka, T.; Shimosawa, T.; Miura, S.; Zhang, W.; Tokutake, Y.; Yamamoto, J.; Awano, M.; et al. Fasting-induced hypothermia and reduced energy production in mice lacking acetyl-CoA synthetase 2. Cell Metab. 2009, 9, 191–202. [Google Scholar] [CrossRef] [Green Version]
- Mews, P.; Donahue, G.; Drake, A.M.; Luczak, V.; Abel, T.; Berger, S.L. Acetyl-CoA synthetase regulates histone acetylation and hippocampal memory. Nature 2017, 546, 381–386. [Google Scholar] [CrossRef] [Green Version]
- Yoshii, Y.; Furukawa, T.; Saga, T.; Fujibayashi, Y. Acetate/acetyl-CoA metabolism associated with cancer fatty acid synthesis: Overview and application. Cancer Lett. 2015, 356 Pt A, 211–216. [Google Scholar] [CrossRef]
- Yoshii, Y.; Waki, A.; Furukawa, T.; Kiyono, Y.; Mori, T.; Yoshii, H.; Kudo, T.; Okazawa, H.; Welch, M.J.; Fujibayashi, Y. Tumor uptake of radiolabeled acetate reflects the expression of cytosolic acetyl-CoA synthetase: Implications for the mechanism of acetate PET. Nucl. Med. Biol. 2009, 36, 771–777. [Google Scholar] [CrossRef] [Green Version]
- Son, Y.L.; Lee, Y.C. Molecular determinants of the interactions between SRC-1 and LXR/RXR heterodimers. FEBS Lett. 2010, 584, 3862–3866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, L.; Ye, F.; Gui, C.; Luo, H.; Cai, J.; Shen, J.; Chen, K.; Shen, X.; Jiang, H. Ligand-binding regulation of LXR/RXR and LXR/PPAR heterodimerizations: SPR technology-based kinetic analysis correlated with molecular dynamics simulation. Protein Sci. A Publ. Protein Soc. 2005, 14, 812–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaravadi, R.K.; Kimmelman, A.C.; Debnath, J. Targeting Autophagy in Cancer: Recent Advances and Future Directions. Cancer Discov. 2019, 9, 1167–1181. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 12. [Google Scholar] [CrossRef]
- Alessandrini, F.; Pezze, L.; Ciribilli, Y. LAMPs: Shedding light on cancer biology. Semin. Oncol. 2017, 44, 239–253. [Google Scholar] [CrossRef]
- Eskelinen, E.-L. Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Mol. Asp. Med. 2006, 27, 495–502. [Google Scholar] [CrossRef]
- Fehrenbacher, N.; Bastholm, L.; Kirkegaard-Sørensen, T.; Rafn, B.; Bøttzauw, T.; Nielsen, C.; Weber, E.; Shirasawa, S.; Kallunki, T.; Jäättelä, M. Sensitization to the Lysosomal Cell Death Pathway by Oncogene-Induced Down-regulation of Lysosome-Associated Membrane Proteins 1 and 2. Cancer Res. 2008, 68, 6623–6633. [Google Scholar] [CrossRef] [Green Version]
- Fu, H.; Subramanian, R.R.; Masters, S.C. 14-3-3 proteins: Structure, function, and regulation. Annu. Rev. Pharm. Toxicol. 2000, 40, 617–647. [Google Scholar] [CrossRef]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.; Lee, J.-S.; Inn, K.-S.; Gack, M.U.; Li, Q.; Roberts, E.A.; Vergne, I.; Deretic, V.; Feng, P.; Akazawa, C.; et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat. Cell Biol. 2008, 10, 776–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, Y.; Klucken, J.; Patterson, C.; Hyman, B.T.; McLean, P.J. The co-chaperone carboxyl terminus of Hsp70-interacting protein (CHIP) mediates alpha-synuclein degradation decisions between proteasomal and lysosomal pathways. J. Biol. Chem. 2005, 280, 23727–23734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Settembre, C.; De Cegli, R.; Mansueto, G.; Saha, P.K.; Vetrini, F.; Visvikis, O.; Huynh, T.; Carissimo, A.; Palmer, D.; Klisch, T.J.; et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol. 2013, 15, 647–658. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Zhou, X.; Wu, Q.; Zhou, L.; Zhang, Z.; Zhang, R.; Deng, C.; Zhang, X. Acetyl CoA synthase 2 potentiates ATG5-induced autophagy against neuronal apoptosis after subarachnoid hemorrhage. J. Mol. Histol. 2022, 53, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Yi, L.; Deng, P.; Wang, L.; Yue, Y.; Wang, H.; Tian, L.; Xie, J.; Chen, M.; Luo, Y.; et al. Rapamycin antagonizes cadmium-induced breast cancer cell proliferation and metastasis through directly modulating ACSS2. Ecotoxicol. Environ. Saf. 2021, 224, 112626. [Google Scholar] [CrossRef]
- Emens, L.A.; Silverstein, S.C.; Khleif, S.; Marincola, F.M.; Galon, J. Toward integrative cancer immunotherapy: Targeting the tumor microenvironment. J. Transl. Med. 2012, 10, 70. [Google Scholar] [CrossRef]
- Savage, P.A.; Klawon, D.E.J.; Miller, C.H. Regulatory T Cell Development. Annu. Rev. Immunol. 2020, 38, 421–453. [Google Scholar] [CrossRef] [Green Version]
- Alvisi, G.; Brummelman, J.; Puccio, S.; Mazza, E.M.; Tomada, E.P.; Losurdo, A.; Zanon, V.; Peano, C.; Colombo, F.S.; Scarpa, A.; et al. IRF4 instructs effector Treg differentiation and immune suppression in human cancer. J. Clin. Investig. 2020, 130, 3137–3150. [Google Scholar] [CrossRef] [Green Version]
- Agnarelli, A.; Chevassut, T.; Mancini, E.J. IRF4 in multiple myeloma—Biology, disease and therapeutic target. Leuk. Res. 2018, 72, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, A.L.; Emre, N.C.T.; Lamy, L.; Ngo, V.N.; Wright, G.; Xiao, W.; Powell, J.; Dave, S.; Yu, X.; Zhao, H.; et al. IRF4 addiction in multiple myeloma. Nature 2008, 454, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Ma, D.; Cheng, B.; Fang, Q.; Kuang, X.; Yu, K.; Wang, W.; Hu, B.; Wang, J. Crucial role of HO-1/IRF4-dependent apoptosis induced by panobinostat and lenalidomide in multiple myeloma. Exp. Cell Res. 2018, 363, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.; Lim, J.S. Essential role of interferon regulatory factor 4 (IRF4) in immune cell development. Arch. Pharm. Res. 2016, 39, 1548–1555. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Ling, R.; Zhou, Y.; Chen, D.J. Knockdown of ACSS2 inhibits invasion and migration of cervical cancer cells induced by nutrient stress and its mechanism. Chin. J. Cell. Mol. Immunol. 2019, 35, 926–931. [Google Scholar]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 583084. [Google Scholar] [CrossRef] [PubMed]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef] [PubMed]
- Rankin, E.B.; Giaccia, A.J. Hypoxic control of metastasis. Science 2016, 352, 175–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaakkola, P.; Mole David, R.; Tian, Y.-M.; Wilson Michael, I.; Gielbert, J.; Gaskell Simon, J.; Kriegsheim Alexander, V.; Hebestreit Holger, F.; Mukherji, M.; Schofield Christopher, J.; et al. Targeting of HIF-α to the von Hippel-Lindau Ubiquitylation Complex by O2-Regulated Prolyl Hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef]
- Loboda, A.; Jozkowicz, A.; Dulak, J. HIF-1 and HIF-2 transcription factors-similar but not identical. Mol. Cells 2010, 29, 435–442. [Google Scholar] [CrossRef]
- Chen, R.; Xu, M.; Nagati, J.; Garcia, J.A. Coordinate regulation of stress signaling and epigenetic events by Acss2 and HIF-2 in cancer cells. PLoS ONE 2017, 12, e0190241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; Xu, M.; Nagati, J.S.; Hogg, R.T.; Das, A.; Gerard, R.D.; Garcia, J.A. The acetate/ACSS2 switch regulates HIF-2 stress signaling in the tumor cell microenvironment. PLoS ONE 2015, 10, e0116515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Hill, H.; Christie, A.; Kim, M.S.; Holloman, E.; Pavia-Jimenez, A.; Homayoun, F.; Ma, Y.; Patel, N.; Yell, P.; et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 2016, 539, 112–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, K.D.; Pniewski, K.; Perry, C.E.; Papp, S.B.; Shaffer, J.D.; Velasco-Silva, J.N.; Casciano, J.C.; Aramburu, T.M.; Srikanth, Y.V.V.; Cassel, J.; et al. Targeting ACSS2 with a Transition-State Mimetic Inhibits Triple-Negative Breast Cancer Growth. Cancer Res. 2021, 81, 1252–1264. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Mohseni, R.; Lucas, K.J.; Gutierrez, J.A.; Perry, R.G.; Trotter, J.F.; Rahimi, R.S.; Harrison, S.A.; Ajmera, V.; Wayne, J.D.; et al. TVB-2640 (FASN Inhibitor) for the Treatment of Nonalcoholic Steatohepatitis: FASCINATE-1, a Randomized, Placebo-Controlled Phase 2a Trial. Gastroenterology 2021, 161, 1475–1486. [Google Scholar] [CrossRef] [PubMed]
- Sabnis, R.W. Novel Substituted Tetrazoles as ACSS2 Inhibitors for Treating Cancer. ACS Med. Chem. Lett. 2021, 12, 1894–1895. [Google Scholar] [CrossRef] [PubMed]
- Sabnis, R.W. Amide-Substituted Condensed Pyridine Derivatives as ACSS2 Inhibitors for Treating Cancer. ACS Med. Chem. Lett. 2021, 12, 1870–1871. [Google Scholar] [CrossRef] [PubMed]
- Fontana, F.; Ge, X.; Su, X.; Hathi, D.; Xiang, J.; Cenci, S.; Civitelli, R.; Shoghi, K.I.; Akers, W.J.; D’Avignon, A.; et al. Evaluating Acetate Metabolism for Imaging and Targeting in Multiple Myeloma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 416–429. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Kim, D.; Kim, S.H.; Park, M.-a.; Chang, J.H.; Yun, M. The roles of 11C-acetate PET/CT in predicting tumor differentiation and survival in patients with cerebral glioma. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 1012. [Google Scholar] [CrossRef]
- Pantel, A.R.; Ackerman, D.; Lee, S.-C.; Mankoff, D.A.; Gade, T.P. Imaging Cancer Metabolism: Underlying Biology and Emerging Strategies. J. Nucl. Med. 2018, 59, 1340–1349. [Google Scholar] [CrossRef]
- Hur, H.; Kim, Y.B.; Ham, I.H.; Lee, D. Loss of ACSS2 expression predicts poor prognosis in patients with gastric cancer. J. Surg. Oncol. 2015, 112, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.M.; Kim, J.H.; Oh, H.J.; Park, H.E.; Lee, T.H.; Cho, N.Y.; Kang, G.H. Downregulation of acetyl-CoA synthetase 2 is a metabolic hallmark of tumor progression and aggressiveness in colorectal carcinoma. Mod. Pathol. 2017, 30, 267–277. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Systems | Tumors | ACSS2 Expression | Invasiveness | Survival | References |
|---|---|---|---|---|---|
| Nervous System | GBM | High | Up | Negative | [12,16] |
| Respiratory System | NSCLC | High | Up | Negative | [8] |
| Digestive System | EC | High | Up | Negative | [32] |
| HCC | High | Up | Negative | [33,34] | |
| High | Down | Positive | [35] | ||
| Urinary System | RCC | High | Up | Negative | [36] |
| Bladder Cancer | High | Up | Negative | [37,38] | |
| Reproductive System | Breast Cancer | High | Up | Negative | [7] |
| High | Down | Positive | [31] | ||
| Prostate Cancer | High | Up | Negative | [5] | |
| Cervical Cancer | High | Up | Negative | [39] | |
| Blood System | Myeloma | High | Up | Negative | [27] |
| Mechanisms and Metabolisms | Related Molecules and Cells | Effects | Cancers | References |
|---|---|---|---|---|
| Lipid metabolism | Acetate, FASN, LXR/RXR ways | Promotes lipid synthesis | NSCLC, BC, Melanoma | [5,7,25] |
| Autophagy | TFEB, Acetate, H3, LAMP1, TFEB | Promote autophagy | BC, GBM, RCC | [12,36] |
| Immunity | Acetate, IRF4, Tregs, TAMs, PD-L1 | Assists in immune escape | MM, CESC | [27,39] |
| Hypoxia | Acetate, HIF-2α | Fight hypoxia | Fibrosarcoma | [26] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, M.; Liu, N.; Wang, J.; Fu, S.; Wang, X.; Chen, D. Acetyl-CoA Synthetase 2 as a Therapeutic Target in Tumor Metabolism. Cancers 2022, 14, 2896. https://doi.org/10.3390/cancers14122896
Liu M, Liu N, Wang J, Fu S, Wang X, Chen D. Acetyl-CoA Synthetase 2 as a Therapeutic Target in Tumor Metabolism. Cancers. 2022; 14(12):2896. https://doi.org/10.3390/cancers14122896
Chicago/Turabian StyleLiu, Mengfang, Na Liu, Jinlei Wang, Shengqiao Fu, Xu Wang, and Deyu Chen. 2022. "Acetyl-CoA Synthetase 2 as a Therapeutic Target in Tumor Metabolism" Cancers 14, no. 12: 2896. https://doi.org/10.3390/cancers14122896