
Facilitating Drug Discovery in Breast Cancer by Virtually Screening Patients Using In Vitro Drug Response Modeling

 , and
, and
Abstract
:Simple Summary
Abstract
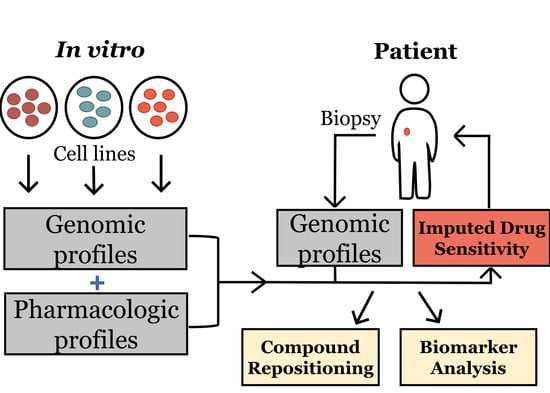
1. Introduction
2. Materials and Methods
2.1. Data Acquisition and Code Availability
2.2. Generating Models for Imputing Drug Response and Statistical Analysis
2.3. Criteria for Lead Compound Identifcation and Statistical Analysis
2.4. Gene-Set Enrichment Analysis
2.5. Obtaining Biomarker Associations between Imputed Drug Response and Nonsynonymous Somatic Mutations and GDSC ANOVA Biomarker Associations
2.6. In Vitro Cell Line Experiments
2.7. Xenograft Experiments
3. Results
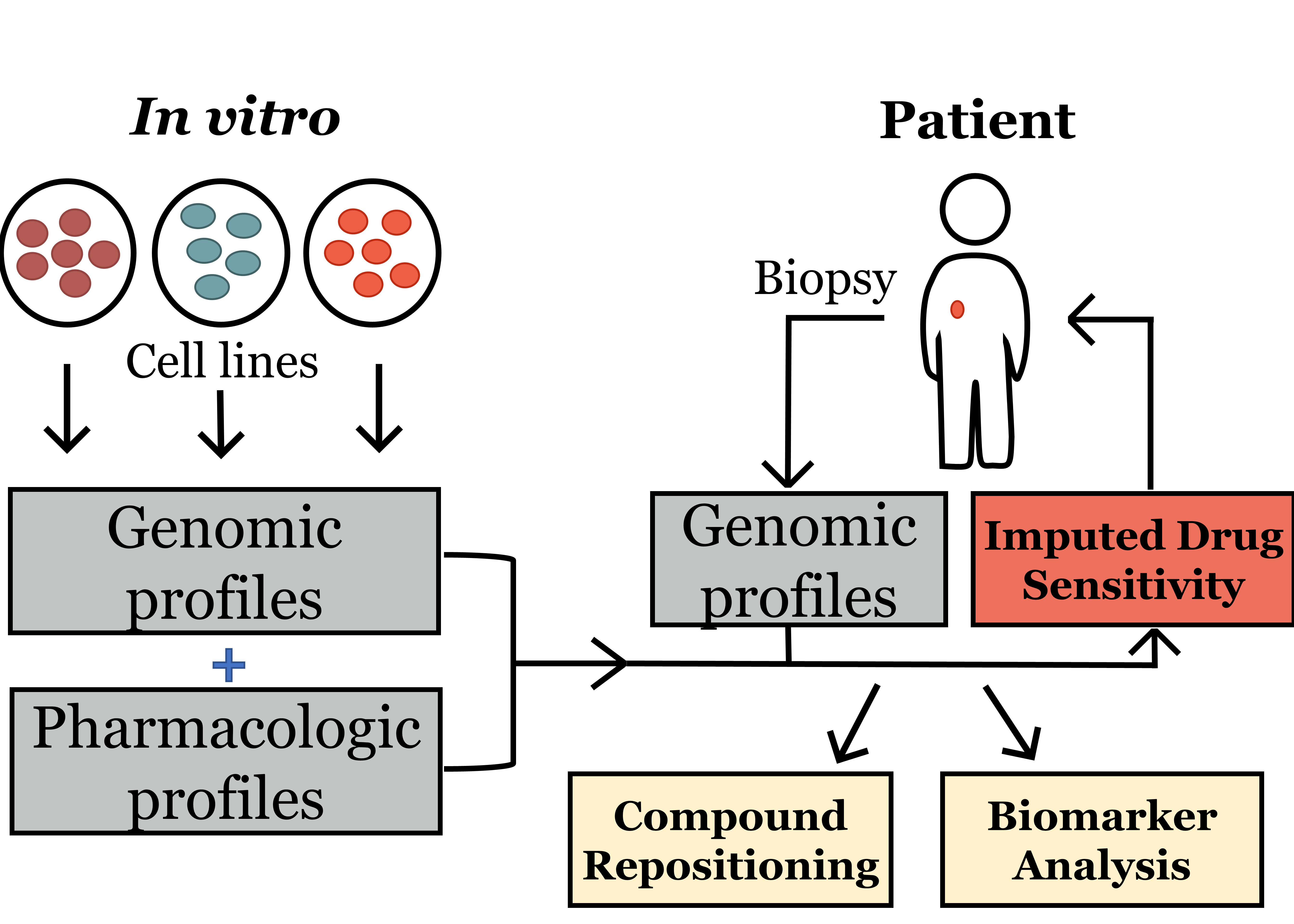
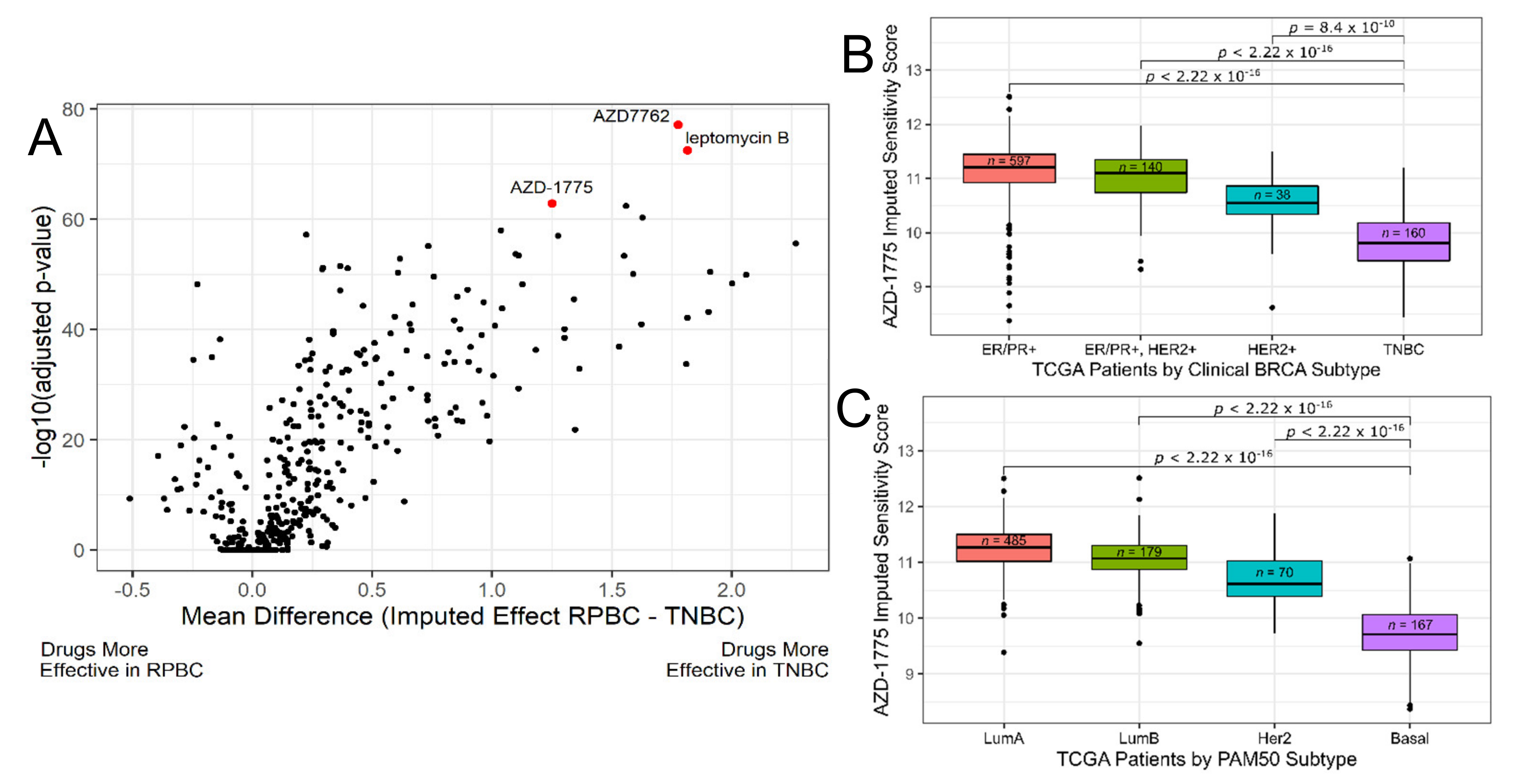
3.1. Discovery Phase: Imputing Patient Response to Medications Enables the Discovery of Candidate Drugs for TNBC
3.2. Discovery Phase: Identify Biomarkers for AZD-1775
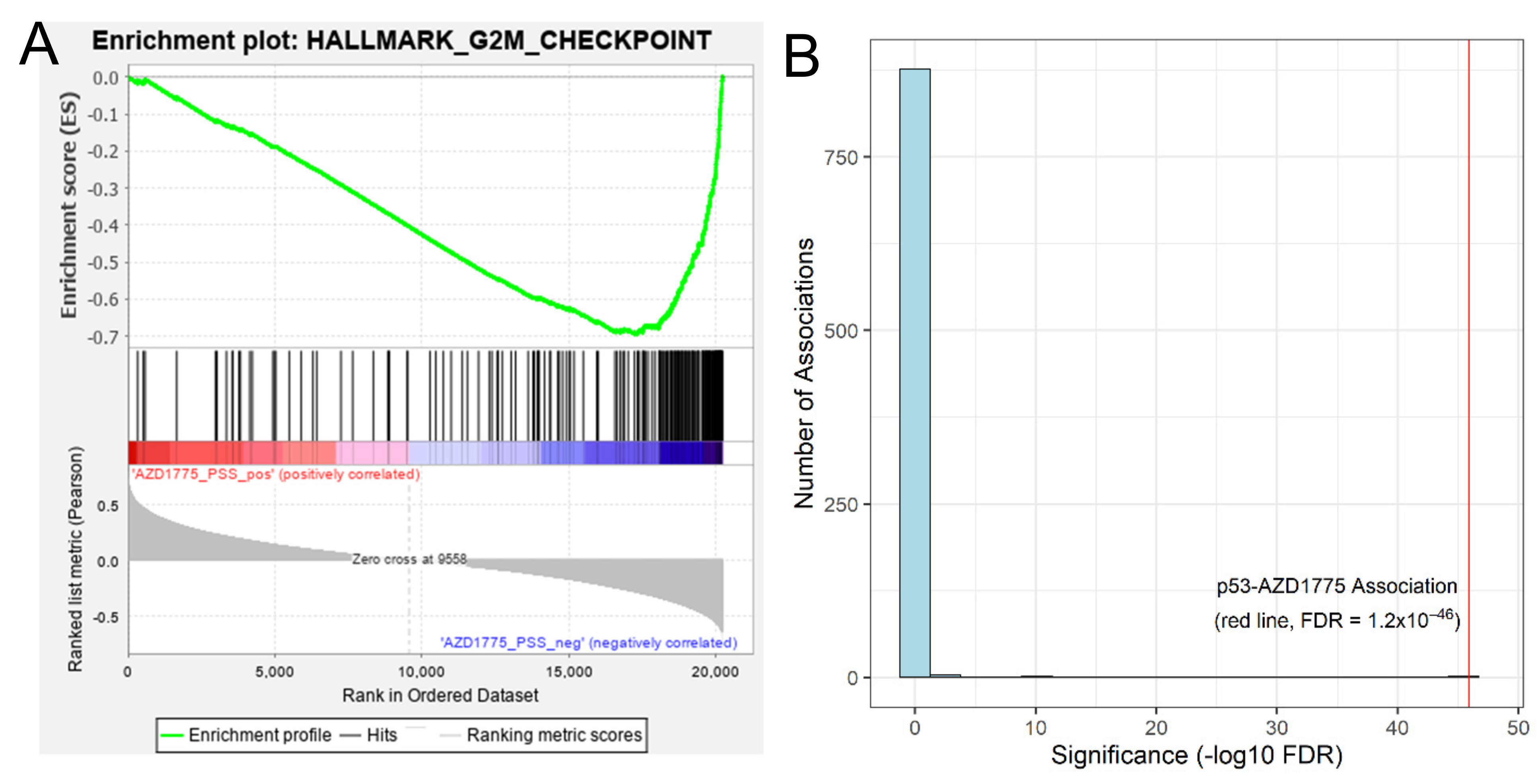
3.2.1. Proof-of-Concept: Tumors Predicted to Be Sensitive to AZD1775 Are Enriched with Cell Cycle Gene Sets
3.2.2. Imputation-Based Drug-Wide Association Analysis Reveals Potential Biomarkers for AZD-1775
3.3. Validation Phase: Measured Cell Line Response to AZD-1775 in an Independent In Vitro Dataset Validate Our Predictions
3.4. Validation Phase: In Vitro and In Vivo Assessment of Cellular Sensitivity to AZD-1775 in Combination with Standard-of-Care Paclitaxel
3.4.1. Single Agent use of AZD-1775 Is Able to Inhibit Growth of TNBC Cell Lines
3.4.2. AZD-1775 Alone and in Combination with Paclitaxel Inhibits MDA-MB-231 Xenograft Growth
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, X.; Yang, J.; Peng, L.; Sahin, A.A.; Huo, L.; Ward, K.C.; O’Regan, R.; Torres, M.A.; Meisel, J.L. Triple-negative breast cancer has worse overall survival and cause-specific survival than non-triple-negative breast cancer. Breast Cancer Res. Treat. 2017, 161, 279–287. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Wong, C.H.; Siah, K.W.; Lo, A.W. Estimation of clinical trial success rates and related parameters. Biostatistics 2019, 20, 273–286. [Google Scholar] [CrossRef]
- Hurle, M.R.; Yang, L.; Xie, Q.; Rajpal, D.K.; Sanseau, P.; Agarwal, P. Computational Drug Repositioning: From Data to Therapeutics. Clin. Pharmacol. Ther. 2013, 93, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Corsello, S.M.; Nagari, R.T.; Spangler, R.D.; Rossen, J.; Kocak, M.; Bryan, J.G.; Humeidi, R.; Peck, D.; Wu, X.; Tang, A.A.; et al. Discovering the anticancer potential of non-oncology drugs by systematic viability profiling. Nat. Rev. Cancer 2020, 1, 235–248. [Google Scholar] [CrossRef] [Green Version]
- Azuaje, F. Computational models for predicting drug responses in cancer research. Briefings Bioinform. 2016, 18, 820–829. [Google Scholar] [CrossRef] [PubMed]
- Geeleher, P.; Cox, N.J.; Huang, R.S. Clinical drug response can be predicted using baseline gene expression levels and in vitro drug sensitivity in cell lines. Genome Biol. 2014, 15, R47. [Google Scholar] [CrossRef] [Green Version]
- Geeleher, P.; Loboda, A.; Lenkala, D.; Wang, F.; Lacroix, B.; Karovic, S.; Wang, J.; Nebozhyn, M.; Chisamore, M.; Hardwick, J.; et al. Predicting Response to Histone Deacetylase Inhibitors Using High-Throughput Genomics. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Zhu, Y.; Tang, X.; Li, J.; Li, Y.; Zhong, Z.; Ding, G.; Li, Y. Integrated Analysis of Transcriptome in Cancer Patient-Derived Xenografts. PLoS ONE 2015, 10, e0124780. [Google Scholar] [CrossRef] [PubMed]
- Geeleher, P.; Zhang, Z.; Wang, F.; Gruener, R.F.; Nath, A.; Morrison, G.; Bhutra, S.; Grossman, R.L.; Huang, R.S. Discovering novel pharmacogenomic biomarkers by imputing drug response in cancer patients from large genomics studies. Genome Res. 2017, 27, 1743–1751. [Google Scholar] [CrossRef] [Green Version]
- Seashore-Ludlow, B.; Rees, M.G.; Cheah, J.H.; Cokol, M.; Price, E.V.; Coletti, M.E.; Jones, V.; Bodycombe, N.E.; Soule, C.K.; Gould, J.; et al. Harnessing Connectivity in a Large-Scale Small-Molecule Sensitivity Dataset. Cancer Discov. 2015, 5, 1210–1223. [Google Scholar] [CrossRef] [Green Version]
- (National Institute of Health) CTD2 Data Portal. Available online: https://ocg.cancer.gov/programs/ctd2/data-portal (accessed on 4 January 2019).
- (Broad Institute) Browse CCLE Data. Available online: https://portals.broadinstitute.org/ccle/data (accessed on 4 January 2019).
- Weinstein, J.N.; The Cancer Genome Atlas Research Network; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2012, 41, D955–D961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- (Wellcome Sanger Institute) Genomics of Drug Sensitivity in Cancer: Resources Download. Available online: https://www.cancerrxgene.org/downloads/bulk_download (accessed on 10 June 2020).
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostat. 2006, 8, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Wickham H ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4.
- Ling, A.; Gruener, R.F.; Fessler, J.; Huang, R.S. More than fishing for a cure: The promises and pitfalls of high throughput cancer cell line screens. Pharmacol. Ther. 2018, 191, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- (Wellcome Sanger Institute) Genomics of Drug Sensitivity in Cancer: Cancer Feature: TP53_mut. Available online: https://www.cancerrxgene.org/feature/TP53_mut/289/volcano (accessed on 5 February 2021).
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Ding, L.; Mardis, E.R.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Lok, S.W.; Whittle, J.R.; Vaillant, F.; Teh, C.E.; Lo, L.L.; Policheni, A.N.; Bergin, A.R.T.; Desai, J.; Ftouni, S.; Gandolfo, L.C.; et al. A Phase Ib Dose-Escalation and Expansion Study of the BCL2 Inhibitor Venetoclax Combined with Tamoxifen in ER and BCL2–Positive Metastatic Breast Cancer. Cancer Discov. 2018, 9, 354–369. [Google Scholar] [CrossRef]
- Hwang, S.-Y.; Park, S.; Kwon, Y. Recent therapeutic trends and promising targets in triple negative breast cancer. Pharmacol. Ther. 2019, 199, 30–57. [Google Scholar] [CrossRef] [PubMed]
- Leijen, S.; Van Geel, R.M.; Pavlick, A.C.; Tibes, R.; Rosen, L.; Razak, A.R.A.; Lam, R.; Demuth, T.; Rose, S.; Lee, M.A.; et al. Phase I Study Evaluating WEE1 Inhibitor AZD1775 As Monotherapy and in Combination With Gemcitabine, Cisplatin, or Carboplatin in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2016, 34, 4371–4380. [Google Scholar] [CrossRef] [PubMed]
- Hirai, H.; Iwasawa, Y.; Okada, M.; Arai, T.; Nishibata, T.; Kobayashi, M.; Kimura, T.; Kaneko, N.; Ohtani, J.; Yamanaka, K.; et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol. Cancer Ther. 2009, 8, 2992–3000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balafoutas, D.; Hausen, A.Z.; Mayer, S.; Hirschfeld, M.; Jaeger, M.; Denschlag, D.; Gitsch, G.; Jungbluth, A.; Stickeler, E. Cancer testis antigens and NY-BR-1 expression in primary breast cancer: Prognostic and therapeutic implications. BMC Cancer 2013, 13, 271. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Li, J.; Zhou, P.; Zhi, X. SPTBN1 and cancer, which links? J. Cell. Physiol. 2020, 235, 17–25. [Google Scholar] [CrossRef]
- Hussain, M.R.M.; Hoessli, D.C.; Fang, M. N-acetylgalactosaminyltransferases in cancer. Oncotarget 2016, 7, 54067–54081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do, K.; Wilsker, D.; Ji, J.; Zlott, J.; Freshwater, T.; Kinders, R.J.; Collins, J.; Chen, A.P.; Doroshow, J.H.; Kummar, S. Phase I Study of Single-Agent AZD1775 (MK-1775), a Wee1 Kinase Inhibitor, in Patients With Refractory Solid Tumors. J. Clin. Oncol. 2015, 33, 3409–3415. [Google Scholar] [CrossRef] [Green Version]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-Negative Breast Cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prat, A.; Adamo, B.; Cheang, M.C.; Anders, C.K.; Carey, L.A.; Perou, C.M. Molecular Characterization of Basal-Like and Non-Basal-Like Triple-Negative Breast Cancer. Oncologist 2013, 18, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Hoadley, K.A.; Yau, C.; Wolf, D.M.; Cherniack, A.D.; Tamborero, D.; Ng, S.; Leiserson, M.D.; Niu, B.; McLellan, M.D.; Uzunangelov, V.; et al. Multiplatform Analysis of 12 Cancer Types Reveals Molecular Classification within and across Tissues of Origin. Cell 2014, 158, 929–944. [Google Scholar] [CrossRef] [Green Version]
- Schätzle, L.-K.; Esfahani, A.H.; Schuppert, A. Methodological challenges in translational drug response modeling in cancer: A systematic analysis with FORESEE. PLoS Comput. Biol. 2014, 16, e1007803. [Google Scholar] [CrossRef] [Green Version]
- Albiges, L.; Goubar, A.; Scott, V.; Vicier, C.; Lefèbvre, C.; Alsafadi, S.; Commo, F.; Saghatchian, M.; Lazar, V.; Dessen, P.; et al. Chk1 as a new therapeutic target in triple-negative breast cancer. Breast 2014, 23, 250–258. [Google Scholar] [CrossRef]
- Bryant, C.; Rawlinson, R.; Massey, A.J. Chk1 Inhibition as a novel therapeutic strategy for treating triple-negative breast and ovarian cancers. BMC Cancer 2014, 14, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Witkiewicz, A.K.; Chung, S.; Brough, R.; Vail, P.; Franco, J.; Lord, C.J.; Knudsen, E.S. Targeting the Vulnerability of RB Tumor Suppressor Loss in Triple-Negative Breast Cancer. Cell Rep. 2018, 22, 1185–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, C.X.; Cai, S.; Li, S.; Ryan, C.E.; Guo, Z.; Schaiff, W.T.; Lin, L.; Hoog, J.; Goiffon, R.J.; Prat, A.; et al. Targeting Chk1 in p53-deficient triple-negative breast cancer is therapeutically beneficial in human-in-mouse tumor models. J. Clin. Investig. 2012, 122, 1541–1552. [Google Scholar] [CrossRef] [Green Version]
- Thompson, R.; Eastman, A. The cancer therapeutic potential of Chk1 inhibitors: How mechanistic studies impact on clinical trial design. Br. J. Clin. Pharmacol. 2013, 76, 358–369. [Google Scholar] [CrossRef] [Green Version]
- Ueda, A.; Oikawa, K.; Fujita, K.; Ishikawa, A.; Sato, E.; Ishikawa, T.; Kuroda, M.; Kanekura, K. Therapeutic potential of PLK1 inhibition in triple-negative breast cancer. Lab. Investig. 2019, 99, 1275–1286. [Google Scholar] [CrossRef] [PubMed]
- Cole, K.A.; Pal, S.; Kudgus, R.A.; Ijaz, H.; Liu, X.; Minard, C.G.; Pawel, B.R.; Maris, J.M.; Haas-Kogan, D.A.; Voss, S.D.; et al. Phase I Clinical Trial of the Wee1 Inhibitor Adavosertib (AZD1775) with Irinotecan in Children with Relapsed Solid Tumors: A COG Phase I Consortium Report (ADVL1312). Clin. Cancer Res. 2020, 26, 1213–1219. [Google Scholar] [CrossRef]
- Sanai, N.; Li, J.; Boerner, J.; Stark, K.; Wu, J.; Kim, S.; Derogatis, A.; Mehta, S.; Dhruv, H.D.; Heilbrun, L.K.; et al. Phase 0 Trial of AZD1775 in First-Recurrence Glioblastoma Patients. Clin. Cancer Res. 2018, 24, 3820–3828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Méndez, E.; Rodriguez, C.P.; Kao, M.C.; Raju, S.C.; Diab, A.; Harbison, R.A.; Konnick, E.Q.; Mugundu, G.M.; Santana-Davila, R.; Martins, R.; et al. A Phase I Clinical Trial of AZD1775 in Combination with Neoadjuvant Weekly Docetaxel and Cisplatin before Definitive Therapy in Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2018, 24, 2740–2748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leijen, S.; Van Geel, R.M.J.M.; Sonke, G.S.; De Jong, D.; Rosenberg, E.H.; Marchetti, S.; Pluim, D.; Van Werkhoven, E.; Rose, S.; Lee, M.A.; et al. Phase II Study of WEE1 Inhibitor AZD1775 Plus Carboplatin in Patients With TP53-Mutated Ovarian Cancer Refractory or Resistant to First-Line Therapy Within 3 Months. J. Clin. Oncol. 2016, 34, 4354–4361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, L.L.; Piwnica-Worms, H. Inactivation of the p34cdc2-cyclin B complex by the human WEE1 tyrosine kinase. Science 1992, 257, 1955–1957. [Google Scholar] [CrossRef] [PubMed]
- Geenen, J.J.; Schellens, J.H. Molecular Pathways: Targeting the Protein Kinase Wee1 in Cancer. Clin. Cancer Res. 2017, 23, 4540–4544. [Google Scholar] [CrossRef] [Green Version]
- Clausse, V.; Goloudina, A.R.; Uyanik, B.; Kochetkova, E.Y.; Richaud, S.; Fedorova, O.A.; Hammann, A.; Bardou, M.; Barlev, N.A.; Garrido, C.; et al. Wee1 inhibition potentiates Wip1-dependent p53-negative tumor cell death during chemotherapy. Cell Death Dis. 2016, 7, e2195. [Google Scholar] [CrossRef] [Green Version]
- Cuneo, K.C.; Morgan, M.A.; Davis, M.A.; Parcels, L.A.; Parcels, J.; Karnak, D.; Ryan, C.; Liu, N.; Maybaum, J.; Lawrence, T.S. Wee1 Kinase Inhibitor AZD1775 Radiosensitizes Hepatocellular Carcinoma Regardless of TP53 Mutational Status Through Induction of Replication Stress. Int. J. Radiat. Oncol. 2016, 95, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Diab, A.; Kao, M.; Kehrli, K.; Kim, H.Y.; Sidorova, J.; Mendez, E. Multiple Defects Sensitize p53-Deficient Head and Neck Cancer Cells to the WEE1 Kinase Inhibition. Mol. Cancer Res. 2019, 17, 1115–1128. [Google Scholar] [CrossRef] [Green Version]
- Hirai, H.; Arai, T.; Okada, M.; Nishibata, T.; Kobayashi, M.; Sakai, N.; Imagaki, K.; Ohtani, J.; Sakai, T.; Yoshizumi, T.; et al. MK-1775, a small molecule Wee1 inhibitor, enhances anti-tumor efficacy of various DNA-damaging agents, including 5-fluorouracil. Cancer Biol. Ther. 2010, 9, 514–522. [Google Scholar] [CrossRef] [Green Version]
- Mizuarai, S.; Yamanaka, K.; Itadani, H.; Arai, T.; Nishibata, T.; Hirai, H.; Kotani, H. Discovery of gene expression-based pharmacodynamic biomarker for a p53 context-specific anti-tumor drug Wee1 inhibitor. Mol. Cancer 2009, 8, 34. [Google Scholar] [CrossRef] [Green Version]
- Pappano, W.N.; Zhang, Q.; Tucker, L.A.; Tse, C.; Wang, J. Genetic inhibition of the atypical kinase Wee1 selectively drives apoptosis of p53 inactive tumor cells. BMC Cancer 2014, 14, 430. [Google Scholar] [CrossRef] [Green Version]
- Webster, P.J.; Littlejohns, A.T.; Gaunt, H.J.; Prasad, K.R.; Beech, D.J.; Burke, D.A. AZD1775 induces toxicity through double-stranded DNA breaks independently of chemotherapeutic agents in p53-mutated colorectal cancer cells. Cell Cycle 2017, 16, 2176–2182. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Shen, Q.; Tao, R.; Chang, W.; Li, R.; Xie, G.; Liu, W.; Zhang, P.; Tao, K. Wee1 inhibition can suppress tumor proliferation and sensitize p53 mutant colonic cancer cells to the anticancer effect of irinotecan. Mol. Med. Rep. 2017, 17, 3344–3349. [Google Scholar] [CrossRef]
- Lewis, C.W.; Jin, Z.; Macdonald, D.; Wei, W.; Qian, X.J.; Choi, W.S.; He, R.; Sun, X.; Chan, G. Prolonged mitotic arrest induced by Wee1 inhibition sensitizes breast cancer cells to paclitaxel. Oncotarget 2017, 8, 73705–73722. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Low, K.-H.; Alexander, A.; Jiang, Y.; Karakas, C.; Hess, K.R.; Carey, J.P.; Bui, T.N.; Vijayaraghavan, S.; Evans, K.W.; et al. Cyclin E Overexpression Sensitizes Triple-Negative Breast Cancer to Wee1 Kinase Inhibition. Clin. Cancer Res. 2018, 24, 6594–6610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, J.; Fang, H.; Yang, F.; Ji, W.; Guan, N.; Sun, Z.; Shi, Y.; Zhou, G.; Guan, X. Combined Inhibition of ATR and WEE1 as a Novel Therapeutic Strategy in Triple-Negative Breast Cancer. Neoplasia 2018, 20, 478–488. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mechanism of Action | # of Drugs in Top 10% | Total # of Drugs in Database | Drug(s) in Top 10% |
|---|---|---|---|
| CHK inhibitor * | 1 | 1 | AZD7762 |
| exportin antagonist | 1 | 1 | leptomycin B |
| WEE1 kinase inhibitor * | 1 | 1 | AZD-1775 |
| CDK inhibitor * | 5 | 6 | dinaciclib, alvocidib, SNS-032, PHA-793887, BRD-K30748066 |
| translation (eIF4F complex) inhibitor | 2 | 2 | CR-1-31B, SR-II-138A |
| PLK inhibitor * | 3 | 4 | GSK461364, BI-2536, rigosertib |
| proteasome inhibitor | 1 | 2 | MLN2238 |
| tubulin polymerization inhibitor * | 1 | 4 | docetaxel |
| phosphodiesterase inhibitor | 1 | 2 | ML030 |
| kinesin-like spindle protein inhibitor * | 1 | 1 | SB-743921 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gruener, R.F.; Ling, A.; Chang, Y.-F.; Morrison, G.; Geeleher, P.; Greene, G.L.; Huang, R.S. Facilitating Drug Discovery in Breast Cancer by Virtually Screening Patients Using In Vitro Drug Response Modeling. Cancers 2021, 13, 885. https://doi.org/10.3390/cancers13040885
Gruener RF, Ling A, Chang Y-F, Morrison G, Geeleher P, Greene GL, Huang RS. Facilitating Drug Discovery in Breast Cancer by Virtually Screening Patients Using In Vitro Drug Response Modeling. Cancers. 2021; 13(4):885. https://doi.org/10.3390/cancers13040885
Chicago/Turabian StyleGruener, Robert F., Alexander Ling, Ya-Fang Chang, Gladys Morrison, Paul Geeleher, Geoffrey L. Greene, and R. Stephanie Huang. 2021. "Facilitating Drug Discovery in Breast Cancer by Virtually Screening Patients Using In Vitro Drug Response Modeling" Cancers 13, no. 4: 885. https://doi.org/10.3390/cancers13040885