
Immunotherapy as a Turning Point in the Treatment of Acute Myeloid Leukemia

 , , , ,
, , , ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Antibody-Based Therapy
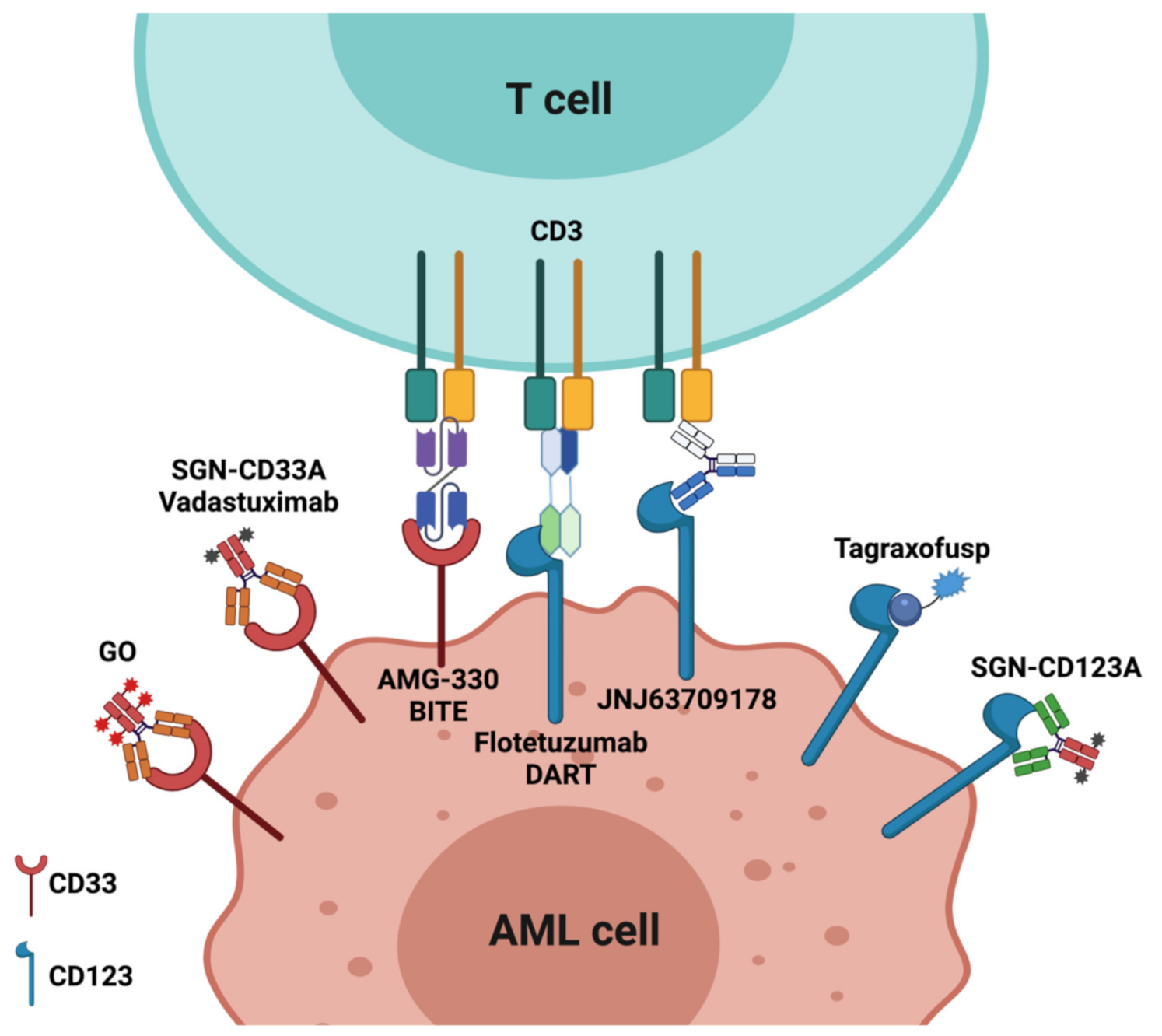
2.1. Antibody–Drug Conjugates (ADCs)
2.1.1. CD33
2.1.2. CD123
2.2. Bispecific Antibodies
2.3. Monoclonal Antibodies Directed against Human Leukemia Stem Cells (LSCs)
2.4. Fc-Engineered Antibodies
3. Immune Checkpoint Inhibitors
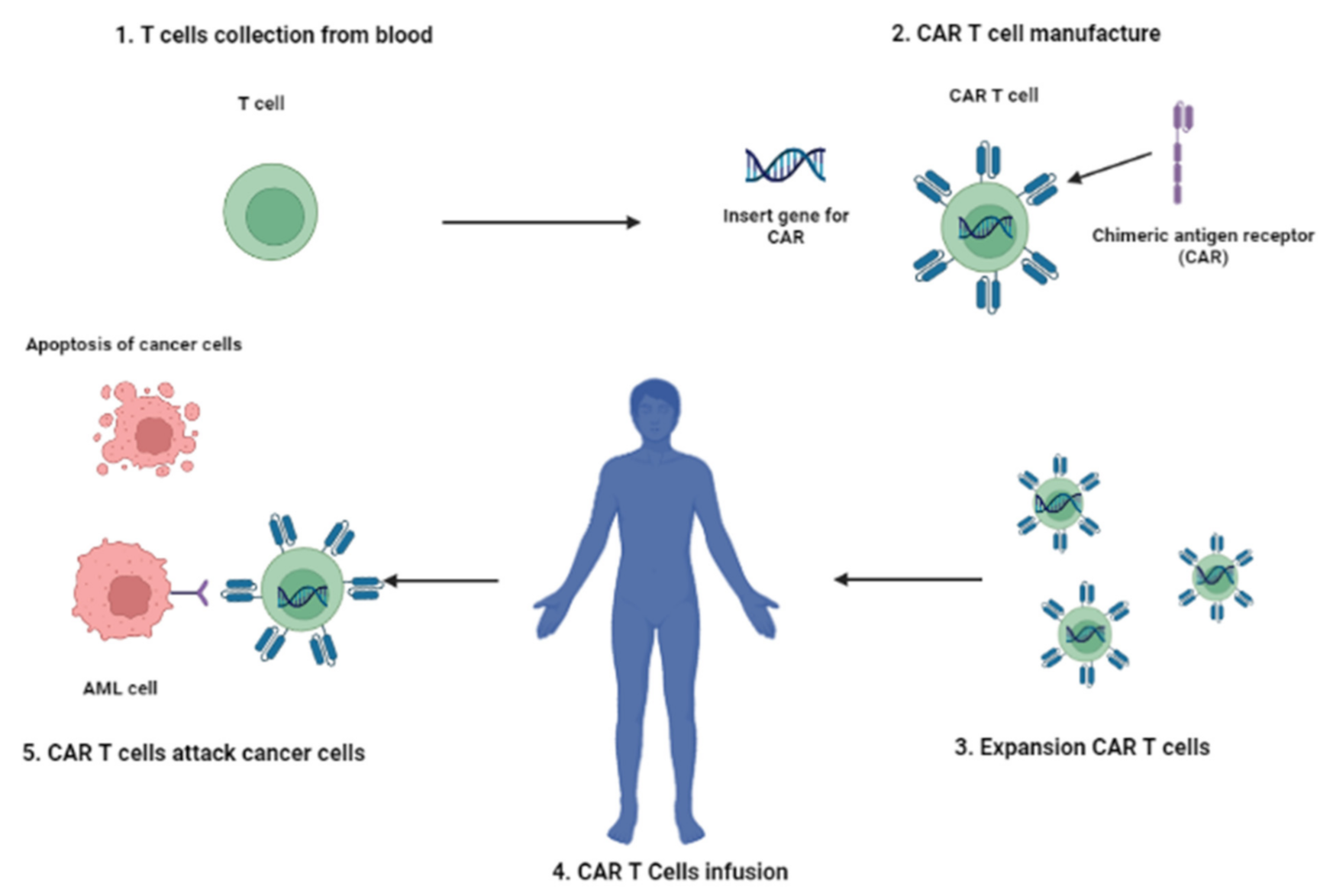
4. CAR-T Cell Therapy
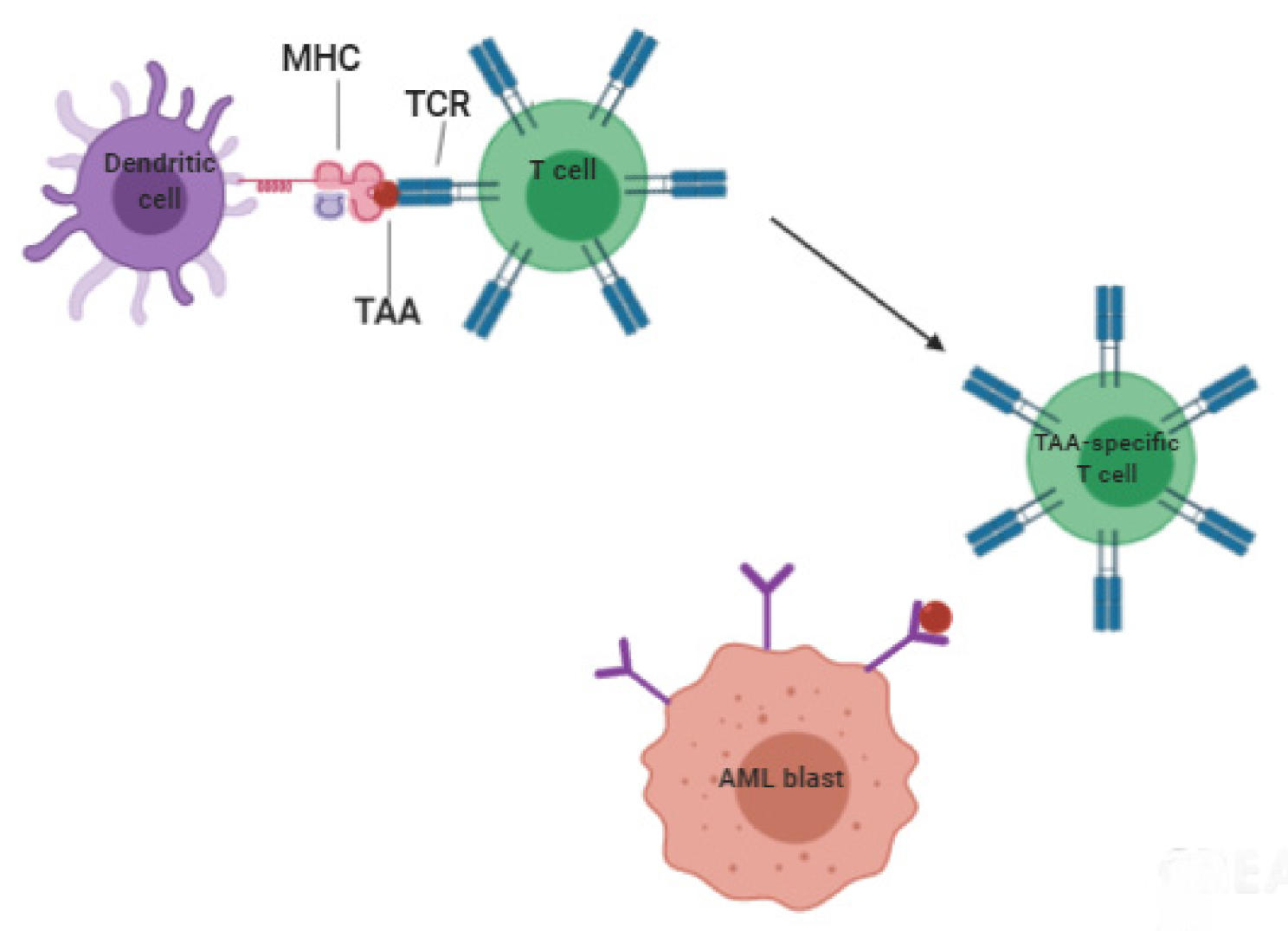
5. Vaccine-Based Therapies
6. Future Perspectives
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nardi, V.; Hasserjian, R.P. Genetic Testing in Acute Myeloid Leukemia and Myelodysplastic Syndromes. Surg. Pathol. Clin. 2016, 9, 143–163. [Google Scholar] [CrossRef] [PubMed]
- Del Principe, M.I.; Buccisano, F.; Soddu, S.; Maurillo, L.; Cefalo, M.; Piciocchi, A.; Consalvo, M.I.; Paterno, G.; Sarlo, C.; De Bellis, E.; et al. Involvement of central nervous system in adult patients with acute myeloid leukemia: Incidence and impact on outcome. Semin. Hematol. 2018, 55, 209–214. [Google Scholar] [CrossRef]
- Del Principe, M.I.; Maurillo, L.; Buccisano, F.; Sconocchia, G.; Cefalo, M.; De Santis, G.; Di Veroli, A.; Ditto, C.; Nasso, D.; Postorino, M.; et al. Central nervous system involvement in adult acute lymphoblastic leukemia: Diagnostic tools, prophylaxis and therapy. Mediterr. J. Hematol. Infect. Dis. 2014, 6, e2014075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Estey, E.H.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Burnett, A.K.; Dombret, H.; Fenaux, P.; Grimwade, D.; Larson, R.A.; et al. Diagnosis and management of acute myeloid leukemia in adults: Recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 2010, 115, 453–474. [Google Scholar] [CrossRef]
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [Green Version]
- Van Der Bruggen, P.; Traversari, C.; Chomez, P.; Lurquin, C.; De Plaen, E.; Van Den Eynde, B.; Knuth, A.; Boon, T. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 1991, 254, 1643–1647. [Google Scholar] [CrossRef]
- Newsome, B.W.; Ernstoff, M.S. The clinical pharmacology of therapeutic monoclonal antibodies in the treatment of malignancy; have the magic bullets arrived? Br. J. Clin. Pharmacol. 2008, 66, 6–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Barsan, V.; Ramakrishna, S.; Davis, K.L. Immunotherapy for the Treatment of Acute Lymphoblastic Leukemia. Curr. Oncol. Rep. 2020, 22, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Lichtenegger, F.S.; Schnorfeil, F.M.; Hiddemann, W.; Subklewe, M. Current strategies in immunotherapy for acute myeloid leukemia. Immunotherapy 2013, 5, 63–78. [Google Scholar] [CrossRef] [PubMed]
- Lichtenegger, F.S.; Krupka, C.; Köhnke, T.; Subklewe, M. Immunotherapy for Acute Myeloid Leukemia. Semin. Hematol. 2015, 52, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Buccisano, F.; Maurillo, L.; Piciocchi, A.; Del Principe, M.I.; Picardi, A.; Cerretti, R.; Cudillo, L.; De Angelis, G.; Sarlo, C.; Cefalo, M.; et al. Pre-transplant persistence of minimal residual disease does not contraindicate allogeneic stem cell transplantation for adult patients with acute myeloid leukemia. Bone Marrow Transplant. 2016, 52, 473–475. [Google Scholar] [CrossRef]
- Van Rhenen, A.; Feller, N.; Kelder, A.; Westra, A.H.; Rombouts, E.; Zweegman, S.; Van Der Pol, M.A.; Waisfisz, Q.; Ossenkoppele, G.J.; Schuurhuis, G.J. High Stem Cell Frequency in Acute Myeloid Leukemia at Diagnosis Predicts High Minimal Residual Disease and Poor Survival. Clin. Cancer Res. 2005, 11, 6520–6527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos, N.R.; Mo, C.C.; Karp, J.E.; Hourigan, C.S. Current Approaches in the Treatment of Relapsed and Refractory Acute Myeloid Leukemia. J. Clin. Med. 2015, 4, 665–695. [Google Scholar] [CrossRef] [Green Version]
- Porter, R.R. Separation and Isolation of Fractions of Rabbit Gamma-Globulin containing the Antibody and Antigenic Combining Sites. Nat. Cell Biol. 1958, 182, 670–671. [Google Scholar] [CrossRef]
- Houshmand, P.; Zlotnik, A. Targeting tumor cells. Curr. Opin. Cell Biol. 2003, 15, 640–644. [Google Scholar] [CrossRef]
- Segal, D.M.; Sconocchia, G.; Titus, J.A.; Jost, C.R.; Kurucz, I. Alternative Triggering Molecules and Single Chain Bispecific Antibodies. J. Hematotherapy 1995, 4, 377–382. [Google Scholar] [CrossRef]
- Redman, J.; Hill, E.; AlDeghaither, D.; Weiner, L. Mechanisms of action of therapeutic antibodies for cancer. Mol. Immunol. 2015, 67, 28–45. [Google Scholar] [CrossRef] [Green Version]
- Laszlo, G.S.; Estey, E.H.; Walter, R.B. The past and future of CD33 as therapeutic target in acute myeloid leukemia. Blood Rev. 2014, 28, 143–153. [Google Scholar] [CrossRef]
- Hinman, L.M.; Hamann, P.R.; Wallace, R.; Menendez, A.T.; Durr, F.E.; Upeslacis, J. Preparation and characterization of mon-oclonal antibody conjugates of the calicheamicins: A novel and potent family of antitumor antibiotics. Cancer Res. 1993, 53, 3336–3342. [Google Scholar]
- Bross, P.F.; Beitz, J.; Chen, G.; Chen, X.H.; Duffy, E.; Kieffer, L.; Roy, S.; Sridhara, R.; Rahman, A.; Williams, G.; et al. Ap-proval summary: Gemtuzumabozogamicin in relapsed acute myeloid leukemia. Clin. Cancer Res. 2001, 7, 1490–1496. [Google Scholar] [PubMed]
- Godwin, C.D.; Gale, R.P.; Walter, R.B. Gemtuzumabozogamicin in acute myeloid leukemia. Leukemia 2017, 31, 1855–1868. [Google Scholar] [CrossRef] [PubMed]
- Gbolahan, O.B.; Zeidan, A.M.; Stahl, M.; Abu Zaid, M.; Farag, S.; Paczesny, S.; Konig, H. Immunotherapeutic Concepts to Target Acute Myeloid Leukemia: Focusing on the Role of Monoclonal Antibodies, Hypomethylating Agents and the Leukemic Microenvironment. Int. J. Mol. Sci. 2017, 18, 1660. [Google Scholar] [CrossRef] [Green Version]
- Masarova, L.; Kantarjian, H.; Garcia-Mannero, G.; Ravandi, F.; Sharma, P.; Daver, N. Harnessing the Immune System Against Leukemia: Monoclonal Antibodies and Checkpoint Strategies for AML. Adv. Exp. Med. Biol. 2017, 995, 73–95. [Google Scholar] [CrossRef]
- Petersdorf, S.H.; Kopecky, K.J.; Slovak, M.; Willman, C.; Nevill, T.; Brandwein, J.; Larson, R.A.; Erba, H.P.; Stiff, P.J.; Stuart, R.K.; et al. A phase 3 study of gemtuzumabozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 2013, 121, 4854–4860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hourigan, C.S.; Karp, J.E. Minimal residual disease in acute myeloid leukaemia. Nat. Rev. Clin. Oncol. 2013, 10, 460–471. [Google Scholar] [CrossRef]
- Kell, W.J. A feasibility study of simultaneous administration of gemtuzumabozogamicin with intensive chemotherapy in induction and consolidation in younger patients with acute myeloid leukemia. Blood 2003, 102, 4277–4283. [Google Scholar] [CrossRef]
- Burnett, A.K.; Hills, R.K.; Milligan, D.; Kjeldsen, L.; Kell, J.; Russell, N.H.; Yin, J.A.; Hunter, A.; Goldstone, A.H.; Wheatley, K. Identification of Patients With Acute Myeloblastic Leukemia Who Benefit From the Addition of GemtuzumabOzogamicin: Results of the MRC AML15 Trial. J. Clin. Oncol. 2011, 29, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Burnett, A.K.; Russell, N.H.; Hills, R.; Kell, J.; Freeman, S.; Kjeldsen, L.; Hunter, A.E.; Yin, J.; Craddock, C.; Dufva, I.H.; et al. Addition of GemtuzumabOzogamicin to Induction Chemotherapy Improves Survival in Older Patients With Acute Myeloid Leukemia. J. Clin. Oncol. 2012, 30, 3924–3931. [Google Scholar] [CrossRef] [PubMed]
- Castaigne, S.; Pautas, C.; Terré, C.; Raffoux, E.; Bordessoule, D.; Bastie, J.-N.; Legrand, O.; Thomas, X.; Turlure, P.; Reman, O.; et al. Effect of gemtuzumabozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): A randomised, open-label, phase 3 study. Lancet 2012, 379, 1508–1516. [Google Scholar] [CrossRef]
- Hills, R.K.; Castaigne, S.; Appelbaum, F.R.; Delaunay, J.; Petersdorf, S.; Othus, M.; Estey, E.H.; Dombret, H.; Chevret, S.; Ifrah, N.; et al. Addition of gemtuzumabozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: A meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014, 15, 986–996. [Google Scholar] [CrossRef] [Green Version]
- Baron, J.; Wang, E.S. Gemtuzumabozogamicin for the treatment of acute myeloid leukemia. Expert Rev. Clin. Pharmacol. 2018, 11, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, B.C.; Tanaka, T.N.; Balaian, L.; Bashey, A.; Guzdar, A.; Li, H.; Messer, K.; Ball, E.D. A Phase I/II Trial of the Combination of Azacitidine and GemtuzumabOzogamicin for Treatment of Relapsed Acute Myeloid Leukemia. Clin. LymphomaMyeloma Leuk. 2018, 18, 346–352.e5. [Google Scholar] [CrossRef] [PubMed]
- Nand, S.; Othus, M.; Godwin, J.; Willman, C.L.; Norwood, T.H.; Howard, D.S.; Coutre, S.E.; Erba, H.P.; Appelbaum, F.R. A phase 2 trial of azacitidine and gemtuzumabozogamicin therapy in older patients with acute myeloid leukemia. Blood 2013, 122, 3432–3439. [Google Scholar] [CrossRef] [Green Version]
- Fathi, A.T.; Erba, H.P.; Lancet, J.E.; Stein, E.M.; Ravandi, F.; Faderl, S.; Walter, R.B.; Advani, A.S.; DeAngelo, D.; Kovacsovics, T.J.; et al. A phase 1 trial of vadastuximabtalirine combined with hypomethylating agents in patients with CD33-positive AML. Blood 2018, 132, 1125–1133. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, L.; Nomdedeu, J.F.; López, O.; Carnicer, M.J.; Bellido, M.; Aventín, A.; Brunet, S.; Sierra, J. Interleukin-3 receptor alpha chain (CD123) is widely expressed in hematologic malignancies. Haematologica 2001, 86, 1261–1269. [Google Scholar]
- Sconocchia, G.; Keyvanfar, K.; El Ouriaghli, F.; Grube, M.; Rezvani, K.; Fujiwara, H.; McCoy, J.P.; Hensel, N.; Barrett, A.J.; McCoy, J.P., Jr. Phenotype and function of a CD56+ peripheral blood monocyte. Leukemia 2004, 19, 69–76. [Google Scholar] [CrossRef]
- Li, F.; Sutherland, M.K.; Yu, C.; Walter, R.B.; Westendorf, L.; Valliere-Douglass, J.; Pan, L.; Cronkite, A.; Sussman, D.; Klussman, K.; et al. Characterization of SGN-CD123A, A Potent CD123-Directed Antibody–Drug Conjugate for Acute Myeloid Leukemia. Mol. Cancer Ther. 2018, 17, 554–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mani, R.; Goswami, S.; Gopalakrishnan, B.; Ramaswamy, R.; Wasmuth, R.; Tranh, M.; Mo, X.; Gordon, A.; Bucci, D.; Lucas, D.M.; et al. The interleukin-3 receptor CD123 targeted SL-401 mediates potent cytotoxic activity against CD34+CD123+ cells from acute myeloid leukemia/myelodysplastic syndrome patients and healthy donors. Haematologica 2018, 103, 1288–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pemmaraju, N.; Lane, A.A.; Sweet, K.L.; Stein, A.S.; Vasu, S.; Blum, W.; Rizzieri, D.A.; Wang, E.S.; Duvic, M.; Sloan, J.M.; et al. Tagraxofusp in BlasticPlasmacytoid Dendritic-Cell Neoplasm. N. Engl. J. Med. 2019, 380, 1628–1637. [Google Scholar] [CrossRef] [PubMed]
- Nisonoff, A.; Rivers, M. Recombination of a mixture of univalent antibody fragments of different specificity. Arch. Biochem. Biophys. 1961, 93, 460–462. [Google Scholar] [CrossRef]
- Staerz, U.D.; Kanagawa, O.; Bevan, M.J. Hybrid antibodies can target sites for attack by T cells. Nat. Cell Biol. 1985, 314, 628–631. [Google Scholar] [CrossRef]
- Perez, P.; Hoffman, R.W.; Shaw, S.; Bluestone, J.A.; Segal, D.M. Specific targeting of cytotoxic T cells by anti-T3 linked to anti-target cell antibody. Nat. Cell Biol. 1985, 316, 354–356. [Google Scholar] [CrossRef] [PubMed]
- de Gast, G.C.; Haagen, I.-A.; van Houten, A.A.; Klein, S.C.; Duits, A.J.; de Weger, R.A.; Vroom, T.M.; Clark, M.; Phillips, J.; van Dijk, A.J.G.; et al. CD8 T cell activation after intravenous administration of CD3×CD19 bispecific antibody in patients with non-Hodgkin lymphoma. Cancer Immunol. Immunother. 1995, 40, 390–396. [Google Scholar] [CrossRef]
- Hartmann, F.; Renner, C.; Jung, W.; Deisting, C.; Juwana, M.; Eichentopf, B.; Kloft, M.; Pfreundschuh, M. Treatment of re-fractory Hodgkin’s disease with an anti-CD16/CD30 bispecific antibody. Blood 1997, 89, 2042–2047. [Google Scholar] [CrossRef]
- Walter, R.B. Biting back: BiTE antibodies as a promising therapy for acute myeloid leukemia. Expert Rev. Hematol. 2014, 7, 317–319. [Google Scholar] [CrossRef]
- Laszlo, G.S.; Gudgeon, C.J.; Harrington, K.H.; Dell’Aringa, J.; Newhall, K.J.; Means, G.D.; Sinclair, A.M.; Kischel, R.; Frankel, S.R.; Walter, R.B. Cellular determinants for preclinical activity of a novel CD33/CD3 bispecific T-cell engager (BiTE) antibody, AMG 330, against human AML. Blood 2014, 123, 554–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, L.H.; Lum, L.G. Treatment of hematological malignancies with T cell redirected bispecific antibodies: Current status and future needs. Expert Opin. Biol. Ther. 2019, 19, 707–720. [Google Scholar] [CrossRef]
- Walter, R.B.; Laszlo, G.S.; Lionberger, J.M.; Pollard, J.A.; Harrington, K.H.; Gudgeon, C.J.; Othus, M.; Rafii, S.; Meshinchi, S.; Appelbaum, F.R.; et al. Heterogeneity of Clonal Expansion and Maturation-Linked Mutation Acquisition in Hematopoietic Progenitors in Human Acute Myeloid Leukemia. Leukemia 2014, 28, 1969–1977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assi, R.; Kantarjian, H.; Ravandi, F.; Daver, N. Immune therapies in acute myeloid leukemia. Curr. Opin. Hematol. 2018, 25, 136–145. [Google Scholar] [CrossRef]
- Daver, N.; Alotaibi, A.S.; Bücklein, V.; Subklewe, M. T-cell-based immunotherapy of acute myeloid leukemia: Current concepts and future developments. Leukemia 2021, 35, 1843–1863. [Google Scholar] [CrossRef] [PubMed]
- Rader, C. DARTs take aim at BiTEs. Blood 2011, 117, 4403–4404. [Google Scholar] [CrossRef] [PubMed]
- Al-Hussaini, M.; Rettig, M.P.; Ritchey, J.K.; Karpova, D.; Uy, G.L.; Eissenberg, L.G.; Gao, F.; Eades, W.C.; Bonvini, E.; Chichili, G.R.; et al. Targeting CD123 in acute myeloid leukemia using a T-cell–directed dual-affinity retargeting platform. Blood 2016, 127, 122–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uy, G.L.; Aldoss, I.; Foster, M.C.; Sayre, P.H.; Wieduwilt, M.J.; Advani, A.S.; Godwin, J.E.; Arellano, M.L.; Sweet, K.L.; Emadi, A.; et al. Flotetuzumab as salvage immunotherapy for refractory acute myeloid leukemia. Blood 2021, 137, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Guy, D.G.; Uy, G.L. Bispecific Antibodies for the Treatment of Acute Myeloid Leukemia. Curr. Hematol. Malig. Rep. 2018, 13, 417–425. [Google Scholar] [CrossRef]
- Bakker, A.B.H.; Oudenrijn, S.V.D.; Bakker, A.Q.; Feller, N.; Van Meijer, M.; Bia, J.A.; Jongeneelen, M.A.C.; Visser, T.J.; Bijl, N.; Geuijen, C.A.W.; et al. C-Type Lectin-Like Molecule-1. Cancer Res. 2004, 64, 8443–8450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Loo, P.F.; Hangalapura, B.N.; Thordardottir, S.; Gibbins, J.; Veninga, H.; Hendriks, L.J.A.; Kramer, A.; Roovers, R.C.; Leenders, M.; De Kruif, J.; et al. MCLA-117, a CLEC12AxCD3 bispecific antibody targeting a leukaemic stem cell antigen, induces T cell-mediated AML blast lysis. Expert Opin. Biol. Ther. 2019, 19, 721–733. [Google Scholar] [CrossRef]
- Jin, L.; Lee, E.M.; Ramshaw, H.; Busfield, S.J.; Peoppl, A.G.; Wilkinson, L.; Guthridge, M.A.; Thomas, D.; Barry, E.F.; Boyd, A.; et al. Monoclonal Antibody-Mediated Targeting of CD123, IL-3 Receptor α Chain, Eliminates Human Acute Myeloid Leukemic Stem Cells. Cell Stem Cell 2009, 5, 31–42. [Google Scholar] [CrossRef]
- Majeti, R. Monoclonal antibody therapy directed against human acute myeloid leukemia stem cells. Oncogene 2010, 30, 1009–1019. [Google Scholar] [CrossRef] [Green Version]
- Van Rhenen, A.; van Dongen, G.A.M.S.; Kelder, A.; Rombouts, E.J.; Feller, N.; Moshaver, B.; Walsum, M.S.-V.; Zweegman, S.; Ossenkoppele, G.J.; Schuurhuis, G.J. The novel AML stem cell–associated antigen CLL-1 aids in discrimination between normal and leukemic stem cells. Blood 2007, 110, 2659–2666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, H.; Padmanabhan, I.S.; Parmar, S.; Gong, Y. Targeting CLL-1 for acute myeloid leukemia therapy. J. Hematol. Oncol. 2019, 12, 1–11. [Google Scholar] [CrossRef]
- Zhao, X.; Singh, S.; Pardoux, C.; Zhao, J.; Hsi, E.D.; Abo, A.; Korver, W. Targeting C-type lectin-like molecule-1 for antibody-mediated immunotherapy in acute myeloid leukemia. Haematologica 2009, 95, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Cate, B.; De Bruyn, M.; Wei, Y.; Bremer, E.; Helfrich, W.; Mourabet, M.; El-Hachem, S.; Harrison, J.; Binion, D. Targeted Elimination of Leukemia Stem Cells; a New Therapeutic Approach in Hemato-Oncology. Curr. Drug Targets 2010, 11, 95–110. [Google Scholar] [CrossRef]
- Sy, M.-S. Mechanisms regulating the binding activity of CD44 to hyaluronic acid. Front. Biosci. 1998, 3, d631–d636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Q.; Yang, C.; Gao, F. The state of CD44 activation in cancer progression and therapeutic targeting. FEBS J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Sconocchia, G.; Titus, J.A.; Segal, D.M. CD44 is a cytotoxic triggering molecule in human peripheral blood NK cells. J. Immunol. 1994, 153, 5473–5481. [Google Scholar] [PubMed]
- Pericle, F.; Sconocchia, G.; Titus, J.A.; Segal, D.M. CD44 is a cytotoxic triggering molecule on human polymorphonuclear cells. J. Immunol. 1996, 157, 4657–4663. [Google Scholar] [PubMed]
- Sconocchia, G.; Campagnano, L.; Adorno, D.; Iacona, A.; Cococcetta, N.Y.; Boffo, V.; Amadori, S.; Casciani, C.U. CD44 liga-tion on peripheralbloodpolymorphonuclearcellsinduces interleukin-6 production. Blood 2001, 97, 3621–3627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, L.; Hope, K.J.; Zhai, Q.; Smadja-Joffe, F.; Dick, J. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat. Med. 2006, 12, 1167–1174. [Google Scholar] [CrossRef]
- Legras, S.; Gunthert, U.; Stauder, R.; Curt, F.; Oliferenko, S.; Kluin-Nelemans, H.C.; Marie, J.P.; Proctor, S.; Jasmin, C.; Smad-ja-Joffe, F. A strong expression of CD44-6v correlates with shorter survival of patients with acute myeloid leukemia. Blood 1998, 91, 3401–3413. [Google Scholar] [CrossRef] [PubMed]
- Erb, U.; Megaptche, A.P.; Gu, X.; Büchler, M.W.; Zöller, M. CD44 standard and CD44v10 isoform expression on leukemia cells distinctly influences niche embedding of hematopoietic stem cells. J. Hematol. Oncol. 2014, 7, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, E.J. Integrin-associated protein (CD47) and its ligands. Trends Cell Biol. 2001, 11, 130–135. [Google Scholar] [CrossRef]
- Seiffert, M.; Cant, C.; Chen, Z.; Rappold, I.; Brugger, W.; Kanz, L.; Brown, E.J.; Ullrich, A.; Buhring, H.J. Human sig-nal-regulatory protein is expressed on normal, but not on subsets of leukemic myeloid cells and mediates cellular adhesion involving its counterreceptor CD47. Blood 1999, 94, 3633–3643. [Google Scholar] [CrossRef]
- Seiffert, M.; Brossart, P.; Cant, C.; Cella, M.; Colonna, M.; Brugger, W.; Kanz, L.; Ullrich, A.; Buhring, H.J. Signal-regulatory protein α (SIRPα) but not SIRPβ is involved in T-cell activation, binds to CD47 with high affinity, and is expressed on immature CD34+CD38−hematopoietic cells. Blood 2001, 97, 2741–2749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deuse, T.; Hu, X.; Agbor-Enoh, S.; Jang, M.K.; Alawi, M.; Saygi, C.; Gravina, A.; Tediashvili, G.; Nguyen, V.Q.; Liu, Y.; et al. The SIRPα–CD47 immune checkpoint in NK cells. J. Exp. Med. 2021, 218, 218. [Google Scholar] [CrossRef]
- Pietsch, E.C.; Dong, J.; Cardoso, R.; Zhang, X.; Chin, D.; Hawkins, R.; Dinh, T.; Zhou, M.; Strake, B.; Feng, P.-H.; et al. Anti-leukemic activity and tolerability of anti-human CD47 monoclonal antibodies. Blood Cancer J. 2017, 7, e536. [Google Scholar] [CrossRef] [PubMed]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D.; Van Rooijen, N.; Weissman, I.L. CD47 Is an Adverse Prognostic Factor and Therapeutic Antibody Target on Human Acute Myeloid Leukemia Stem Cells. Cell 2009, 138, 286–299. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, S.; Jamieson, C.H.; Pang, W.W.; Park, C.Y.; Chao, M.P.; Majeti, R.; Traver, D.; Van Rooijen, N.; Weissman, I.L. CD47 Is Upregulated on Circulating Hematopoietic Stem Cells and Leukemia Cells to Avoid Phagocytosis. Cell 2009, 138, 271–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sikic, B.I.; Lakhani, N.; Patnaik, A.; Shah, S.A.; Chandana, S.R.; Rasco, D.; Colevas, A.D.; O’Rourke, T.; Narayanan, S.; Papadopoulos, K.; et al. First-in-Human, First-in-Class Phase I Trial of the Anti-CD47 Antibody Hu5F9-G4 in Patients With Advanced Cancers. J. Clin. Oncol. 2019, 37, 946–953. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M. Checkpoint Blockade in Lymphoma. J. Clin. Oncol. 2021, 39, 525–533. [Google Scholar] [CrossRef] [PubMed]
- He, S.Z.; Busfield, S.; Ritchie, D.S.; Hertzberg, M.S.; Durrant, S.; Lewis, I.D.; Marlton, P.; McLachlan, A.J.; Kerridge, I.; Bradstock, K.F.; et al. A Phase 1 study of the safety, pharmacokinetics and anti-leukemic activity of the anti-CD123 monoclonal antibody CSL360 in relapsed, refractory or high-risk acute myeloid leukemia. Leuk Lymphoma 2015, 56, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Jalil, A.R.; Andrechak, J.C.; Discher, D.E. Macrophage checkpoint blockade: Results from initial clinical trials, binding analyses, and CD47-SIRPα structure–function. Antib. Ther. 2020, 3, 80–94. [Google Scholar] [CrossRef] [Green Version]
- Zeidan, M.A.M.; DeAngelo, D.J.; Palmer, J.M.; Seet, C.S.; Tallman, M.S.; Wei, X.; Li, Y.F.; Hock, R.N.; Burgess, M.R.; Hege, K.; et al. A Phase I Study of CC-90002, a Monoclonal Antibody Targeting CD47, in Patients with Relapsed and/or Refractory (R/R) Acute Myeloid Leukemia (AML) and High-Risk Myelodysplastic Syndromes (MDS): Final Results. Blood 2019, 134, 1320. [Google Scholar] [CrossRef]
- Kubasch, A.S.; Schulze, F.; Giagounidis, A.; Götze, K.S.; Krönke, J.; Sockel, K.; Middeke, J.M.; Chermat, F.; Gloaguen, S.; Puttrich, M.; et al. Single agent talacotuzumab demonstrates limited efficacy but considerable toxicity in elderly high-risk MDS or AML patients failing hypomethylating agents. Leukemia 2020, 34, 1182–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montesinos, P.; Roboz, G.J.; Bulabois, C.-E.; Subklewe, M.; Platzbecker, U.; Ofran, Y.; Papayannidis, C.; Wierzbowska, A.; Shin, H.J.; Doronin, V.; et al. Safety and efficacy of talacotuzumab plus decitabine or decitabine alone in patients with acute myeloid leukemia not eligible for chemotherapy: Results from a multicenter, randomized, phase 2/3 study. Leukemia 2021, 35, 62–74. [Google Scholar] [CrossRef]
- Hutmacher, C.; Volta, L.; Rinaldi, F.; Murer, P.; Myburgh, R.; Manz, M.; Neri, D. Development of a novel fully-human anti-CD123 antibody to target acute myeloid leukemia. Leuk. Res. 2019, 84, 106178. [Google Scholar] [CrossRef] [PubMed]
- Vasu, S.; Karl-Heinz, H.; Cheney, C.; Gopalakrishnan, B.; Mani, R.; Lozanski, G.; Mo, X.; Groh, V.; Whitman, S.P.; Konopitzky, R.; et al. Decitabine enhances anti-CD33 monoclonal antibody BI 836858–mediated natural killer ADCC against AML blasts. Blood 2016, 127, 2879–2889. [Google Scholar] [CrossRef]
- Krupka, C.; Lichtenegger, F.S.; Köhnke, T.; Bögeholz, J.; Bücklein, V.; Roiss, M.; Altmann, T.; Do, T.U.; Dusek, R.; Wilson, K.; et al. Targeting CD157 in AML using a novel, Fc-engineered antibody construct. Oncotarget 2017, 8, 35707–35717. [Google Scholar] [CrossRef] [Green Version]
- Weixler, B.; Cremonesi, E.; Sorge, R.; Muraro, M.G.; Delko, T.; Nebiker, C.; Däster, S.; Governa, V.; Amicarella, F.; Soysal, S.; et al. OX40 expression enhances the prognostic significance of CD8 positive lymphocyte infiltration in colorectal cancer. Oncotarget 2015, 6, 37588–37599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, Density, and Location of Immune Cells Within Human Colorectal Tumors Predict Clinical Outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allavena, P.; Sica, A.; Solinas, G.; Porta, C.; Mantovani, A. The inflammatory micro-environment in tumor progression: The role of tumor-associated macrophages. Crit. Rev. Oncol. Hematol. 2008, 66, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sconocchia, G.; Eppenberger, S.; Spagnoli, G.C.; Tornillo, L.; Droeser, R.; Caratelli, S.; Ferrelli, F.; Coppola, A.; Arriga, R.; Lauro, D.; et al. NK cells and T cells cooperate during the clinical course of colorectal cancer. OncoImmunology 2014, 3, e952197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortez, M.A.; Masrorpour, F.; Ivan, C.; Zhang, J.; Younes, A.I.; Lu, Y.; Estecio, M.R.; Barsoumian, H.B.; Menon, H.; Caetano, M.D.S.; et al. Bone morphogenetic protein 7 promotes resistance to immunotherapy. Nat. Commun. 2020, 11, 4840. [Google Scholar] [CrossRef] [PubMed]
- Sconocchia, T.; Hochgerner, M.; Schwarzenberger, E.; Tam-Amersdorfer, C.; Borek, I.; Benezeder, T.; Bauer, T.; Zyulina, V.; Painsi, C.; Passegger, C.; et al. Bone morphogenetic protein signaling regulates skin inflammation via modulating dendritic cell function. J. Allergy Clin. Immunol. 2021, 147, 1810–1822. [Google Scholar] [CrossRef]
- Sconocchia, T.; Sconocchia, G. Regulation of the immune system in health and disease by members of the bone morphogenetic protein family. Front. Immunol. 2021, 12, 802346. [Google Scholar] [CrossRef]
- Antin, J.H. Graft-versus-leukemia: No longer an epiphenomenon. Blood 1993, 82, 2273–2277. [Google Scholar] [CrossRef] [Green Version]
- Davids, M.S.; Kim, H.T.; Bachireddy, P.; Costello, C.; Liguori, R.; Savell, A.; Lukez, A.P.; Avigan, D.; Chen, Y.-B.; McSweeney, P.; et al. Ipilimumab for Patients with Relapse after Allogeneic Transplantation. N. Engl. J. Med. 2016, 375, 143–153. [Google Scholar] [CrossRef]
- Lamble, A.J.; Lind, E.F. Targeting the Immune Microenvironment in Acute Myeloid Leukemia: A Focus on T Cell Immunity. Front. Oncol. 2018, 8, 213. [Google Scholar] [CrossRef]
- Ismail, M.M.; Abdulateef, N.A.B. Bone marrow T-cell percentage: A novel prognostic indicator in acute myeloid leukemia. Int. J. Hematol. 2016, 105, 453–464. [Google Scholar] [CrossRef]
- Lipson, E.J.; Drake, C.G. Ipilimumab: An Anti-CTLA-4 Antibody for Metastatic Melanoma. Clin. Cancer Res. 2011, 17, 6958–6962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.-J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef] [PubMed]
- Ryu, R.; Ward, K.E. Atezolizumab for the First-Line Treatment of Non-small Cell Lung Cancer (NSCLC): Current Status and Future Prospects. Front. Oncol. 2018, 8, 277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 Blockade with Nivolumab in Relapsed or Refractory Hodgkin’s Lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armand, P.; Shipp, M.A.; Ribrag, V.; Michot, J.-M.; Zinzani, P.L.; Kuruvilla, J.; Snyder, E.S.; Ricart, A.D.; Balakumaran, A.; Rose, S.; et al. Programmed Death-1 Blockade WithPembrolizumab in Patients With Classical Hodgkin Lymphoma After BrentuximabVedotin Failure. J. Clin. Oncol. 2016, 34, 3733–3739. [Google Scholar] [CrossRef]
- Galanina, N.; Kline, J.; Bishop, M.R. Emerging role of checkpoint blockade therapy in lymphoma. Ther. Adv. Hematol. 2017, 8, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Fevery, S.; Billiau, A.D.; Sprangers, B.; Rutgeerts, O.; Lenaerts, C.; Goebels, J.; Landuyt, W.; Kasran, A.; Boon, L.; Sagaert, X.; et al. CTLA-4 blockade in murine bone marrow chimeras induces a host-derived antileukemic effect without graft-versus-host disease. Leukemia 2007, 21, 1451–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koestner, W.; Hapke, M.; Herbst, J.; Klein, C.; Welte, K.; Fruehauf, J.; Flatley, A.; Vignali, D.A.; Hardtke-Wolenski, M.; Jaeckel, E.; et al. PD-L1 blockade effectively restores strong graft-versus-leukemia effects without graft-versus-host disease after delayed adoptive transfer of T-cell receptor gene-engineered allogeneic CD8+ T cells. Blood 2011, 117, 1030–1041. [Google Scholar] [CrossRef]
- Liu, Y.; Bewersdorf, J.P.; Stahl, M.; Zeidan, A.M. Immunotherapy in acute myeloid leukemia and myelodysplastic syndromes: The dawn of a new era? Blood Rev. 2019, 34, 67–83. [Google Scholar] [CrossRef]
- Daver, N.; Garcia-Manero, G.; Basu, S.; Boddu, P.C.; Alfayez, M.; Cortes, J.E.; Konopleva, M.; Ravandi-Kashani, F.; Jabbour, E.; Kadia, T.M.; et al. Efficacy, Safety, and Biomarkers of Response to Azacitidine and Nivolumab in Relapsed/Refractory Acute Myeloid Leukemia: A Nonrandomized, Open-Label, Phase II Study. Cancer Discov. 2019, 9, 370–383. [Google Scholar] [CrossRef] [Green Version]
- Das, M.; Zhu, C.; Kuchroo, V.K. Tim-3 and its role in regulating anti-tumor immunity. Immunol. Rev. 2017, 276, 97–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, I.G.; Yasinska, I.M.; Sakhnevych, S.S.; Fiedler, W.; Wellbrock, J.; Bardelli, M.; Varani, L.; Hussain, R.; Siligardi, G.; Ceccone, G.; et al. The Tim-3-galectin-9 Secretory Pathway is Involved in the Immune Escape of Human Acute Myeloid Leukemia Cells. EBioMedicine 2017, 22, 44–57. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Munger, M.; Veenstra, R.G.; Weigel, B.J.; Hirashima, M.; Munn, D.; Murphy, W.J.; Azuma, M.; Anderson, A.C.; Kuchroo, V.K.; et al. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood 2011, 117, 4501–4510. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Sun, L.; Li, C.-F.; Wang, Y.-H.; Yao, J.; Li, H.; Yan, M.; Chang, W.-C.; Hsu, J.-M.; Cha, J.-H.; et al. Galectin-9 interacts with PD-1 and TIM-3 to regulate T cell death and is a target for cancer immunotherapy. Nat. Commun. 2021, 12, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric Antigen Receptor–Modified T Cells for Acute Lymphoid Leukemia. N. Engl. J. Med. 2013, 18, 1509–1518. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.-S.; Wang, Y.; Lv, H.-Y.; Han, Q.-W.; Fan, H.; Guo, B.; Wang, L.-L.; Han, W.-D. Treatment of CD33-directed Chimeric Antigen Receptor-modified T Cells in One Patient with Relapsed and Refractory Acute Myeloid Leukemia. Mol. Ther. 2015, 23, 184–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Ranganathan, R.; Jiang, S.; Fang, C.; Sun, J.; Kim, S.; Newick, K.; Lo, A.; June, C.H.; Zhao, Y.; et al. A Chimeric Switch-Receptor Targeting PD1 Augments the Efficacy of Second-Generation CAR T Cells in Advanced Solid Tumors. Cancer Res. 2016, 76, 1578–1590. [Google Scholar] [CrossRef] [Green Version]
- Bonini, C.; Brenner, M.K.; Heslop, H.E.; Morgan, R.A. Genetic Modification of T Cells. Biol. Blood Marrow Transplant. 2011, 17, S15–S20. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.A.; Ashraf, M.U.; Aman, A.K.; Bae, Y.S. Advances in personalized therapy: Co-targeting intracellular immune checkpoints in controlling acute myelòoid leukemia. In Recent Developments in Medicine and Medical Research; Karaman, R., Ed.; BP International: Jerusalem, Israel, 2021; Volume 14, pp. 108–151. [Google Scholar]
- Negrin, R.S. Graft-versus-host disease versus graft-versus-leukemia. Hematology 2015, 2015, 225–230. [Google Scholar] [CrossRef] [Green Version]
- Goswami, M. Novel Antigen Targets for Immunotherapy of Acute Myeloid Leukemia. Curr. Drug Targets 2017, 18, 296–303. [Google Scholar] [CrossRef]
- Fujiwara, H.; Melenhorst, J.J.; El Ouriaghli, F.; Kajigaya, S.; Grube, M.; Sconocchia, G.; Rezvani, K.; Price, D.; Hensel, N.F.; Douek, D.C.; et al. In vitro Induction of Myeloid Leukemia–Specific CD4 and CD8 T Cells by CD40 Ligand—Activated B Cells Gene Modified to Express Primary Granule Proteins. Clin. Cancer Res. 2005, 11, 4495–4503. [Google Scholar] [CrossRef] [Green Version]
- Zuhrie, S.R.; Harris, R.; Freeman, C.B.; Maciver, J.E.; Geary, C.G.; Delamore, I.W.; Tooth, J.A. Immunotherapy alone vs no maintenance treatment in acute myelogenous leukaemia. Br. J. Cancer 1980, 41, 372–377. [Google Scholar] [CrossRef] [Green Version]
- Omura, G.A.; Vogler, W.R.; Lefante, J.; Silberman, H.; Knospe, W.; Gordon, D.; Jarrell, R. Treatment of acute myelogenous leukemia: Influence of three induction regimens and maintenance with chemotherapy or BCG immunotherapy. Cancer 1982, 49, 1530–1536. [Google Scholar] [CrossRef] [Green Version]
- Summerfield, G.P.; Gibbs, T.J.; Bellingham, A.J. Immunotherapy using BCG during remission induction and as the sole form of maintenance in acute myeloid leukaemia. Br. J. Cancer 1979, 40, 736–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, R.; Zuhrie, S.R.; Freeman, C.B.; Taylor, G.M.; Maciver, J.E.; Geary, C.G.; Delamore, I.W.; Hull, P.J.; Tooth, J.A. Active immunotherapy in acute myelogenous leukaemia and the induction of second and subsequent remissions. Br. J. Cancer 1978, 37, 282–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.; Li, Y. The role of peptide and DNA vaccines in myeloid leukemia immunotherapy. Cancer Cell Int. 2013, 13, 13. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, M.; Casalegno-Garduño, R.; Xu, X.; Schmitt, A. Peptide vaccines for patients with acute myeloid leukemia. Expert Rev. Vaccines 2009, 8, 1415–1425. [Google Scholar] [CrossRef]
- Goldman, J.M.; Druker, B.J. Chronic myeloid leukemia: Current treatment options. Blood 2001, 98, 2039–2042. [Google Scholar] [CrossRef] [Green Version]
- Rezvani, K.; Grube, M.; Brenchley, J.M.; Sconocchia, G.; Fujiwara, H.; Price, D.A.; Gostick, E.; Yamada, K.; Melenhorst, J.; Childs, R.; et al. Functional leukemia-associated antigen-specific memory CD8+ T cells exist in healthy individuals and in patients with chronic myelogenous leukemia before and after stem cell transplantation. Blood 2003, 102, 2892–2900. [Google Scholar] [CrossRef] [PubMed]
- Sergeeva, A.; Alatrash, G.; He, H.; Ruisaard, K.; Lu, S.; Wygant, J.; McIntyre, B.W.; Ma, Q.; Li, D.; John, L.S.; et al. An anti–PR1/HLA-A2 T-cell receptor–like antibody mediates complement-dependent cytotoxicity against acute myeloid leukemia progenitor cells. Blood 2011, 117, 4262–4272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stasi, A.E.; Jimenez, A.M.; Eminagawa, K.; Eal-Obaidi, M.; Erezvani, K. Review of the Results of WT1 Peptide Vaccination Strategies for Myelodysplastic Syndromes and Acute Myeloid Leukemia from Nine Different Studies. Front. Immunol. 2015, 6, 36. [Google Scholar] [CrossRef] [Green Version]
- Brayer, J.; Lancet, J.E.; Powers, J.; List, A.; Balducci, L.; Komrokji, R.; Pinilla-Ibarz, J. WT1 vaccination in AML and MDS: A pilot trial with synthetic analog peptides. Am. J. Hematol. 2015, 90, 602–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maslak, P.G.; Dao, T.; Bernal, Y.; Chanel, S.M.; Zhang, R.; Frattini, M.; Rosenblat, T.; Jurcic, J.G.; Brentjens, R.J.; Arcila, M.E.; et al. Phase 2 trial of a multivalent WT1 peptide vaccine (galinpepimut-S) in acute myeloid leukemia. Blood Adv. 2018, 2, 224–234. [Google Scholar] [CrossRef]
- Bonaccorsi, I.; Pezzino, G.; Morandi, B.; Ferlazzo, G. Novel perspectives on dendritic cell-based immunotherapy of cancer. Immunol. Lett. 2013, 155, 6–10. [Google Scholar] [CrossRef]
- Sabado, R.L.; Balan, S.; Bhardwaj, N. Dendritic cell-based immunotherapy. Cell Res. 2017, 27, 74–95. [Google Scholar] [CrossRef] [Green Version]
- Schürch, C.; Riether, C.; Ochsenbein, A.F. Dendritic Cell-Based Immunotherapy for Myeloid Leukemias. Front. Immunol. 2013, 4, 496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Acker, H.H.; Versteven, M.; Lichtenegger, F.S.; Roex, G.; Campillo-Davo, D.; Lion, E.; Subklewe, M.; Van Tendeloo, V.F.; Berneman, Z.N.; Anguille, S. Dendritic Cell-Based Immunotherapy of Acute Myeloid Leukemia. J. Clin. Med. 2019, 8, 579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anguille, S.; Willemen, Y.; Lion, E.; Smits, E.; Berneman, Z. Dendritic cell vaccination in acute myeloid leukemia. Cytotherapy 2012, 14, 647–656. [Google Scholar] [CrossRef]
- Houtenbos, I.; Westers, T.M.; Dijkhuis, A.; De Gruijl, T.D.; Ossenkoppele, G.J.; Van De Loosdrecht, A.A. Leukemia-Specific T-Cell Reactivity Induced by Leukemic Dendritic Cells Is Augmented by 4-1BB Targeting. Clin. Cancer Res. 2007, 13, 307–315. [Google Scholar] [CrossRef] [Green Version]
- Curti, A.; Pandolfi, S.; Valzasina, B.; Aluigi, M.; Isidori, A.; Ferri, E.; Salvestrini, V.; Bonanno, G.; Rutella, S.; Durelli, I.; et al. Modulation of tryptophan catabolism by human leukemic cells results in the conversion of CD25− into CD25+ T regulatory cells. Blood 2006, 109, 2871–2877. [Google Scholar] [CrossRef] [PubMed]
- Berneman, Z.N.; Van De Velde, A.; Anguille, S.; Willemen, Y.; Schroyens, W.A.; Gadisseur, A.P.; Vrelust, I.; Nijs, G.; Stein, B.; Cools, N.; et al. Prevention Of Relapse In Acute Myeloid Leukemia By Dendritic Cell Vaccination: Report on a Phase II Study With 29 Patients. Blood 2013, 122, 236. [Google Scholar] [CrossRef]
- Kumar, S.; Nagpal, R.; Kumar, A.; Ashraf, M.; Bae, Y.-S. Immunotherapeutic Potential of m6A-Modifiers and MicroRNAs in Controlling Acute Myeloid Leukaemia. Biomedicines 2021, 9, 690. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Drug | Target | Phase | ClinicalTrials.gov Identifier | Estimated Enrollment Number | Disease Conditions | Status |
|---|---|---|---|---|---|---|
| AMG 330 | CD33/CD3 | 1 | NCT02520427 | 256 | R/R AML/MRD Positive AML/MDS | Active/Recruiting |
| MGD006 | CD123/CD3 | 1/2 | NCT02152956 | 330 | Primary Induction Failure (PIF) or Early-Relapse (ER) AML | Active/Recruiting |
| JNJ-63709178 | CD123/CD3 | 1 | NCT02715011 | 62 | R/R AML | Recruitment Completed |
| MCLA 117 | CLL1/CD3 | 1 | NCT03038230 | 62 | R/R AML | Active/NotRecruiting |
| Target | Phase | ClinicalTrials.gov Identifier | Estimated Enrollment Number | Status | Disease Conditions | Intervention/Treatment |
|---|---|---|---|---|---|---|
| CD33, CD38, CD123, CD56, MucI, CLL1 | 1/2 | NCT03222674 | 10 | Unknown | R/R AML | Muc1/CLL1/CD33/CD38/CD56/CD123-specific gene-engineered T cells |
| CD33, CD38, CD56, CD117, CD123, CD34, Muc1 | 1 | NCT03291444 | 30 | Active/Recruiting | ALL/R/R AML/MDS | CAR-T cells/Eps8or WT1 peptide-specific dendritic cells |
| CD123 | 1 | NCT02159495 | 42 | Active/Recruiting | R/R AML or BPDCN | Cyclophosphamide/autologous or allogenic CD123CAR-CD28-CD3zeta-EGFRt-expressing T lymphocytes/fludarabine phosphate |
| CD123 | 1 | NCT03114670 | 20 | Unknown | Adult relapsed AML following allogeneic HSCT | CD123CAR-41BB-CD3zeta-EGFRt-expressing T cells |
| CD123 | 1 | NCT03190278 | 65 | Active/Recruiting | R/R AML | UCART123v1.2(allogeneic engineered Tcells expressing anti-CD123 chimeric antigen receptor) |
| CD123 | 1/2 | NCT03556982 | 10 | Unknown | R/R AML | Fludarabine-cyclophosphamide chemotherapy followed by infusion of allogeneic or autologous CD123-targeted CAR-T cells |
| CD123 | 1 | NCT03766126 | 12 | Active/Not recruiting | Adult R/R AML | Fludarabine-cyclophosphamide chemotherapy followed by infusion of anti-CD123 CAR-T (autologous lentivirally transduced) (CD123CAR-41BB-CD3) |
| CD123,CLL1 | 2/3 | NCT03631576 | 20 | Active/Recruiting | R/R AML | CD123/CLL1 CAR-T cell therapy |
| CD123 | 1 | NCT03796390 | 15 | Unknown | R/R AML | Chemotherapy/CD123 CAR-T cells (autologous lentivirally transduced) |
| CD44 | 1/2 | NCT04097301 | 58 | Active/Recruiting | R/R AML, MM | CD44v6 CAR-Tcells (MLM-CAR44.1 Tcells), cyclophosphamide, and fludarabine |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aureli, A.; Marziani, B.; Sconocchia, T.; Del Principe, M.I.; Buzzatti, E.; Pasqualone, G.; Venditti, A.; Sconocchia, G. Immunotherapy as a Turning Point in the Treatment of Acute Myeloid Leukemia. Cancers 2021, 13, 6246. https://doi.org/10.3390/cancers13246246
Aureli A, Marziani B, Sconocchia T, Del Principe MI, Buzzatti E, Pasqualone G, Venditti A, Sconocchia G. Immunotherapy as a Turning Point in the Treatment of Acute Myeloid Leukemia. Cancers. 2021; 13(24):6246. https://doi.org/10.3390/cancers13246246
Chicago/Turabian StyleAureli, Anna, Beatrice Marziani, Tommaso Sconocchia, Maria Ilaria Del Principe, Elisa Buzzatti, Gianmario Pasqualone, Adriano Venditti, and Giuseppe Sconocchia. 2021. "Immunotherapy as a Turning Point in the Treatment of Acute Myeloid Leukemia" Cancers 13, no. 24: 6246. https://doi.org/10.3390/cancers13246246