
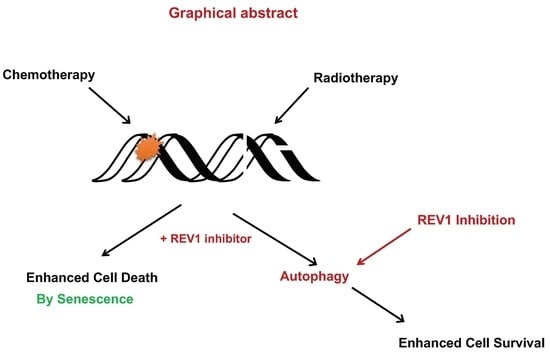
REV1 Inhibition Enhances Radioresistance and Autophagy

 , , ,
, , ,  , ,
, ,  , , and
, , and
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
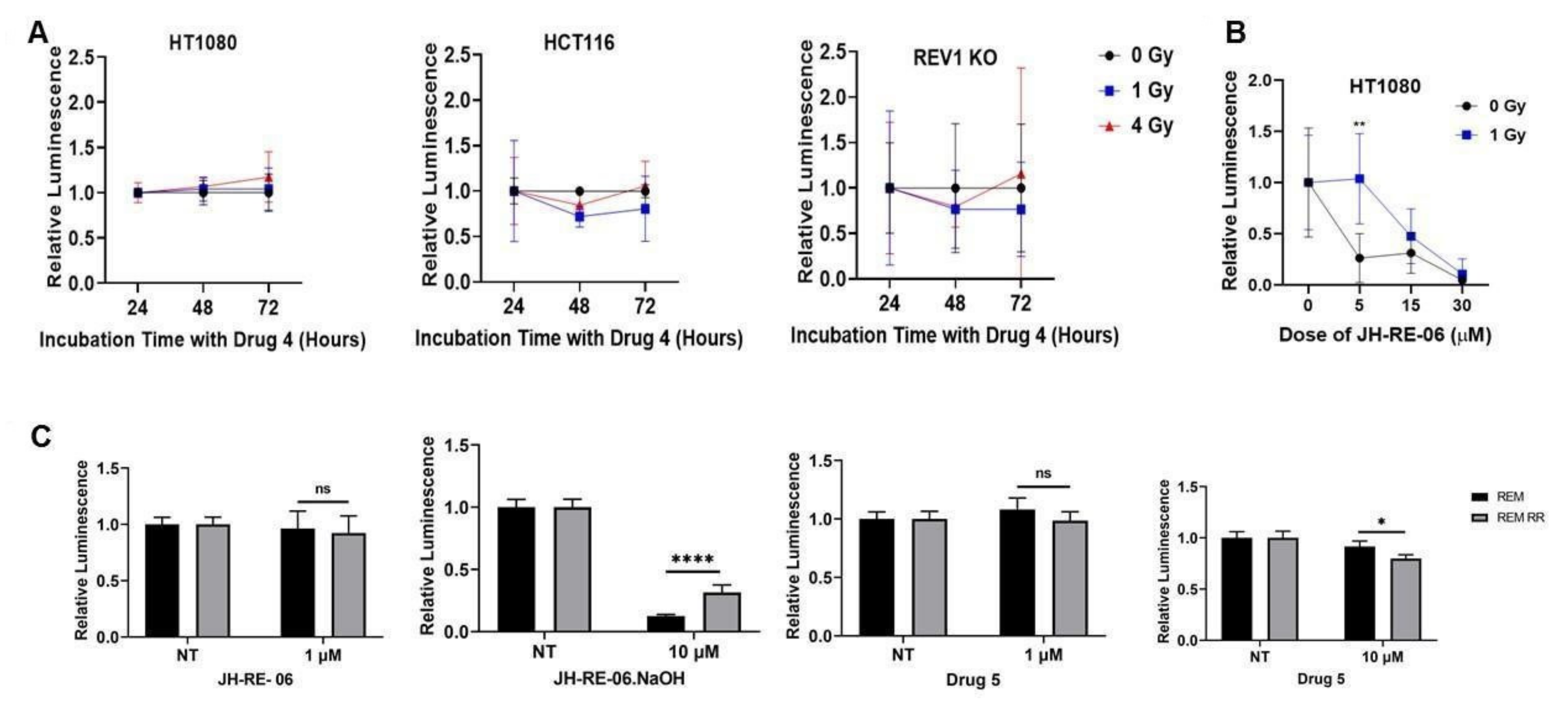
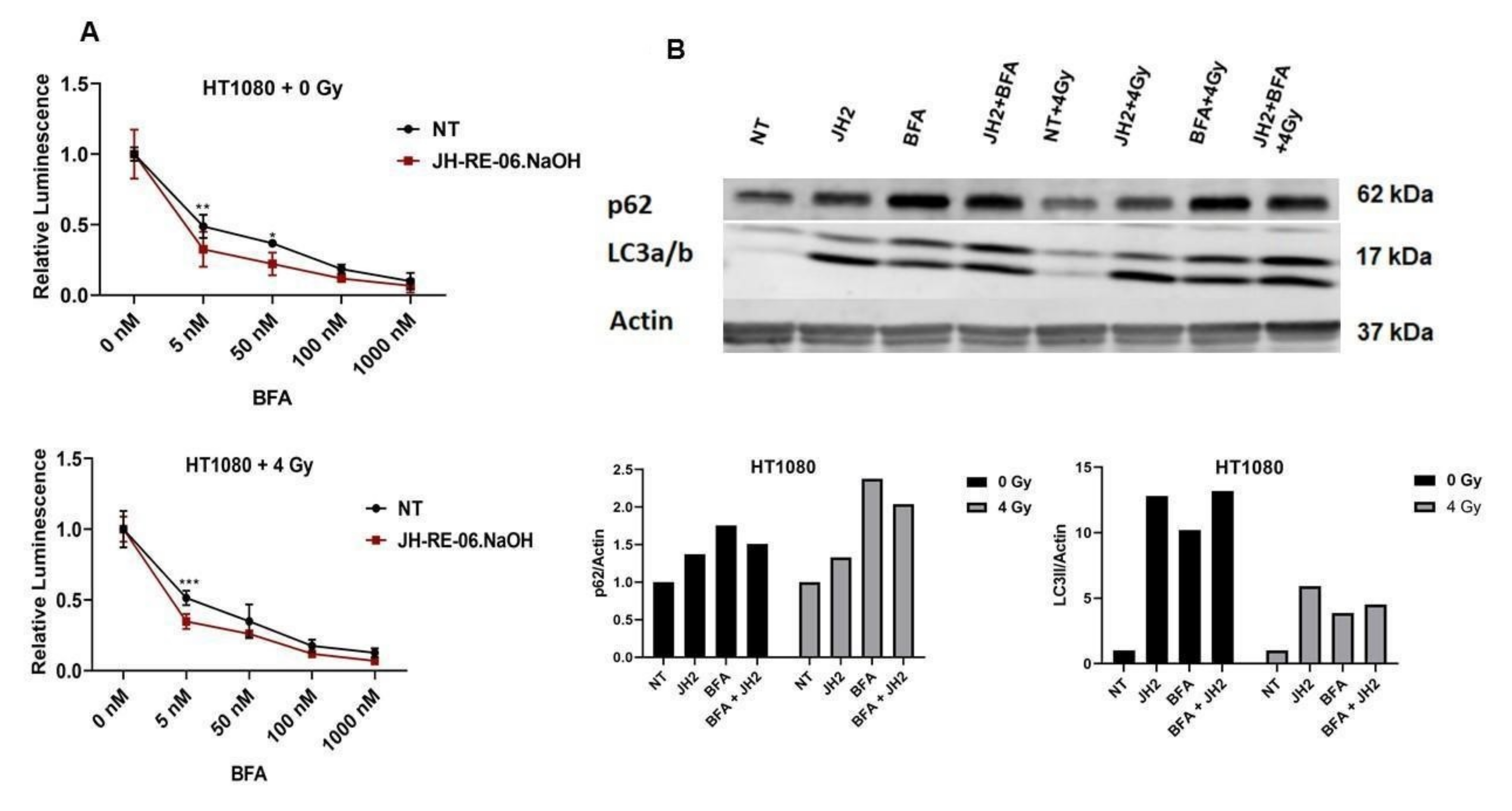
2.1. REV1 Inhibition Does Not Sensitize Cancer Cells to IR
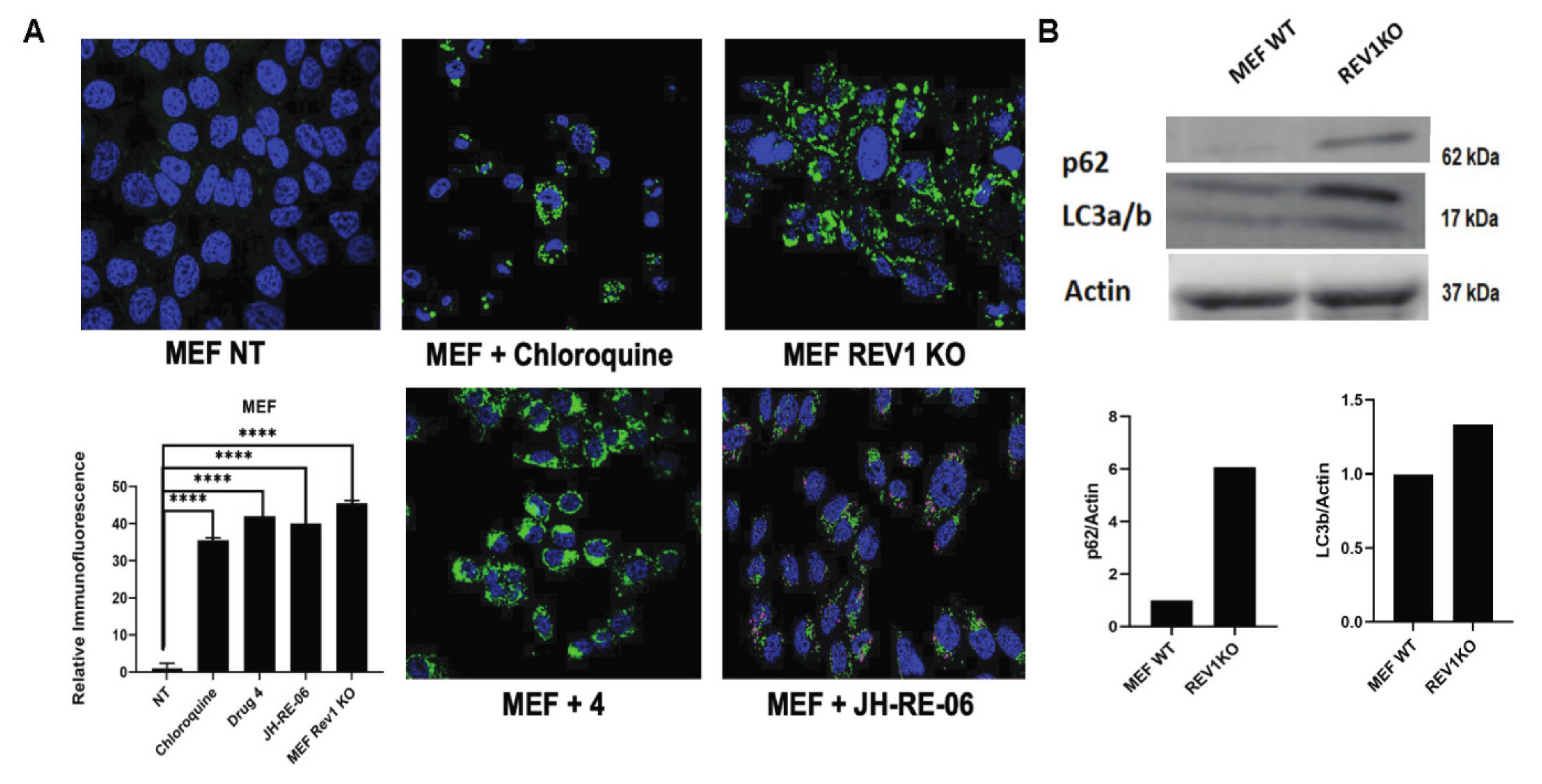
2.2. REV1 Inhibition Triggers Autophagy, a Radioresistance Biomarker
3. Discussion
4. Methods
4.1. Mammalian Cell Culturing
4.2. Drug Inhibitors
4.3. Cytotoxicity Assay
4.4. Colony Survival Assay
4.5. Immunofluorescence Detection of Autophagy
4.6. Western Blot Analysis
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yamanaka, K.; Chatterjee, N.; Hemann, M.T.; Walker, G.C. Inhibition of mutagenic translesion synthesis: A possible strategy for improving chemotherapy? PLoS Genet. 2017, 13, e1006842. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, N.; D’Souza, S.; Shabab, M.; Harris, C.A.; Hilinski, G.J.; Verdine, G.L.; Walker, G.C. A stapled POL kappa peptide targets REV1 to inhibit mutagenic translesion synthesis. Environ. Mol. Mutagen. 2020, 61, 830–836. [Google Scholar] [CrossRef]
- Wojtaszek, J.L.; Chatterjee, N.; Najeeb, J.; Ramos, A.; Lee, M.; Bian, K.; Xue, J.Y.; Fenton, B.A.; Park, H.; Li, D.; et al. A Small Molecule Targeting Mutagenic Translesion Synthesis Improves Chemotherapy. Cell 2019, 178, 152–159.e11. [Google Scholar] [CrossRef]
- Sail, V.; Rizzo, A.A.; Chatterjee, N.; Dash, R.C.; Ozen, Z.; Walker, G.C.; Korzhnev, D.M.; Hadden, M.K. Identification of Small Molecule Translesion Synthesis Inhibitors That Target the Rev1-CT/RIR Protein-Protein Interaction. ACS Chem. Biol. 2017, 12, 1903–1912. [Google Scholar] [CrossRef] [PubMed]
- Dash, R.C.; Ozen, Z.; McCarthy, K.R.; Chatterjee, N.; Harris, C.A.; Rizzo, A.A.; Walker, G.C.; Korzhnev, D.M.; Hadden, M.K. Virtual Pharmacophore Screening Identifies Small-Molecule Inhibitors of the Rev1-CT/RIR Protein-Protein Interaction. ChemMedChem 2019, 14, 1610–1617. [Google Scholar] [CrossRef]
- Wojtaszek, J.; Liu, J.; D’Souza, S.; Wang, S.; Xue, Y.; Walker, G.C.; Zhou, P. Multifaceted recognition of vertebrate Rev1 by translesion polymerases zeta and kappa. J. Biol. Chem. 2012, 287, 26400–26408. [Google Scholar] [CrossRef] [Green Version]
- Pozhidaeva, A.; Pustovalova, Y.; D’Souza, S.; Bezsonova, I.; Walker, G.C.; Korzhnev, D.M. NMR structure and dynamics of the C-terminal domain from human Rev1 and its complex with Rev1 interacting region of DNA polymerase eta. Biochemistry 2012, 51, 5506–5520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, N.W.M.; Harris, C.A.; Lee, M.; Jonas, J.; Lien, E.C.; Heiden, M.G.V.; Hong, J.; Zhou, P.; Hemann, M.T.; Walker, G.C. REV1 inhibitor JH-RE-06 enhances tumor cell response to chemotherapy by triggering senescence. Proc. Natl. Acad. Sci. USA 2020, 117, 28918–28921. [Google Scholar] [CrossRef] [PubMed]
- Atwell, D.; Elks, J.; Cahill, K.; Hearn, N.; Vignarajah, D.; Lagopoulos, J.; Min, M. A Review of Modern Radiation Therapy Dose Escalation in Locally Advanced Head and Neck Cancer. Clin. Oncol. (R. Coll. Radiol.) 2020, 32, 330–341. [Google Scholar] [CrossRef]
- Willers, H.; Azzoli, C.G.; Santivasi, W.L.; Xia, F. Basic mechanisms of therapeutic resistance to radiation and chemotherapy in lung cancer. Cancer J. 2013, 19, 200–207. [Google Scholar] [CrossRef] [Green Version]
- Rattray, A.J.; Shafer, B.K.; McGill, C.B.; Strathern, J.N. The roles of REV3 and RAD57 in double-strand-break-repair-induced mutagenesis of Saccharomyces cerevisiae. Genetics 2002, 162, 1063–1077. [Google Scholar] [CrossRef] [PubMed]
- Kolas, N.K.; Durocher, D. DNA repair: DNA polymerase zeta and Rev1 break in. Curr. Biol. 2006, 16, R296–R299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaachouay, H.; Ohneseit, P.; Toulany, M.; Kehlbach, R.; Multhoff, G.; Rodemann, H.P. Autophagy contributes to resistance of tumor cells to ionizing radiation. Radiother. Oncol. 2011, 99, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-G.; Bae, J.-H.; Kim, J.-A.; Heo, K.; Yang, K.; Yi, J.M. Combination Effect of Epigenetic Regulation and Ionizing Radiation in Colorectal Cancer Cells. PLoS ONE 2014, 9, e105405. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Moretti, L.; Mitchell, L.; Jung, D.K.; Lu, B. Endoplasmic reticulum stress mediates radiation-induced autophagy by perk-eIF2α in caspase-3/7-deficient cells. Oncogene 2010, 29, 3241–3251. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Wang, X.; Hu, S.; Ying, X.; Wang, P.; Zhang, X.; Wang, J.; Wang, H.; Wang, Y. miR-21-mediated Radioresistance Occurs via Promoting Repair of DNA Double Strand Breaks. J. Biol. Chem. 2017, 292, 3531–3540, retraction in J. Biol. Chem. 2020, 295, 6250. [Google Scholar] [CrossRef] [Green Version]
- Murphy, J.D.; Lucas, D.R.; Somnay, Y.R.; Hamstra, D.A.; Ray, M.E. Gemcitabine-mediated radiosensitization of human soft tissue sarcoma. Transl. Oncol. 2008, 1, 50–56. [Google Scholar] [CrossRef] [Green Version]
- Gray, M.; Turnbull, A.K.; Ward, C.; Meehan, J.; Martinez-Perez, C.; Bonello, M.; Pang, L.Y.; Langdon, S.P.; Kunkler, I.H.; Murray, A.; et al. Development and characterisation of acquired radioresistant breast cancer cell lines. Radiat. Oncol. 2019, 14, 64. [Google Scholar] [CrossRef] [Green Version]
- Gray, M.; Turnbull, A.K.; Meehan, J.; Martinez-Perez, C.; Kay, C.; Pang, L.Y.; Argyle, D.J. Comparative Analysis of the Development of Acquired Radioresistance in Canine and Human Mammary Cancer Cell Lines. Front. Vet. Sci. 2020, 7, 439. [Google Scholar] [CrossRef] [PubMed]
- Bouhamdani, N.; Comeau, D.; Cormier, K.; Turcotte, S. STF-62247 accumulates in lysosomes and blocks late stages of autophagy to selectively target von Hippel-Lindau-inactivated cells. Am. J. Physiol. Cell Physiol. 2019, 316, C605–C620. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Wu, J.; Dang, Y.; Su, W.; Liu, C.; Ma, H.; Shan, Y.; Pei, Y.; Wan, B.; Guo, J.; Yu, L. Molecular cloning and characterization of rat LC3A and LC3B--two novel markers of autophagosome. Biochem. Biophys. Res. Commun. 2006, 339, 437–442. [Google Scholar] [CrossRef]
- Mauvezin, C.; Neufeld, T.P. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy 2015, 11, 1437–1438. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Hicks, J.K.; Chute, C.L.; Brennan, J.R.; Ahn, J.Y.; Glover, T.W.; Canman, C.E. REV1 and polymerase ζ facilitate homologous recombination repair. Nucleic Acids Res. 2012, 40, 682–691. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Mi, Y.; Mueller, T.; Kreibich, S.; Williams, E.G.; Van Drogen, A.; Borel, C.; Frank, M.; Germain, P.L.; Bludau, I.; et al. Multi-omic measurements of heterogeneity in HeLa cells across laboratories. Nat. Biotechnol. 2019, 37, 314–322. [Google Scholar] [CrossRef]
- Adey, A.; Burton, J.N.; Kitzman, J.O.; Hiatt, J.B.; Lewis, A.P.; Martin, B.K.; Qiu, R.; Lee, C.; Shendure, J. The haplotype-resolved genome and epigenome of the aneuploid HeLa cancer cell line. Nature 2013, 500, 207–211. [Google Scholar] [CrossRef]
- Nam, H.Y.; Han, M.W.; Chang, H.W.; Kim, S.Y.; Kim, S.W. Prolonged autophagy by MTOR inhibitor leads radioresistant cancer cells into senescence. Autophagy 2013, 9, 1631–1632. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ikeh, K.E.; Lamkin, E.N.; Crompton, A.; Deutsch, J.; Fisher, K.J.; Gray, M.; Argyle, D.J.; Lim, W.Y.; Korzhnev, D.M.; Hadden, M.K.; et al. REV1 Inhibition Enhances Radioresistance and Autophagy. Cancers 2021, 13, 5290. https://doi.org/10.3390/cancers13215290
Ikeh KE, Lamkin EN, Crompton A, Deutsch J, Fisher KJ, Gray M, Argyle DJ, Lim WY, Korzhnev DM, Hadden MK, et al. REV1 Inhibition Enhances Radioresistance and Autophagy. Cancers. 2021; 13(21):5290. https://doi.org/10.3390/cancers13215290
Chicago/Turabian StyleIkeh, Kanayo E., Erica N. Lamkin, Andrew Crompton, Jamie Deutsch, Kira J. Fisher, Mark Gray, David J. Argyle, Won Y. Lim, Dmitry M. Korzhnev, M. Kyle Hadden, and et al. 2021. "REV1 Inhibition Enhances Radioresistance and Autophagy" Cancers 13, no. 21: 5290. https://doi.org/10.3390/cancers13215290