
Pharmacological Inhibition of PP2A Overcomes Nab-Paclitaxel Resistance by Downregulating MCL1 in Esophageal Squamous Cell Carcinoma (ESCC)

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
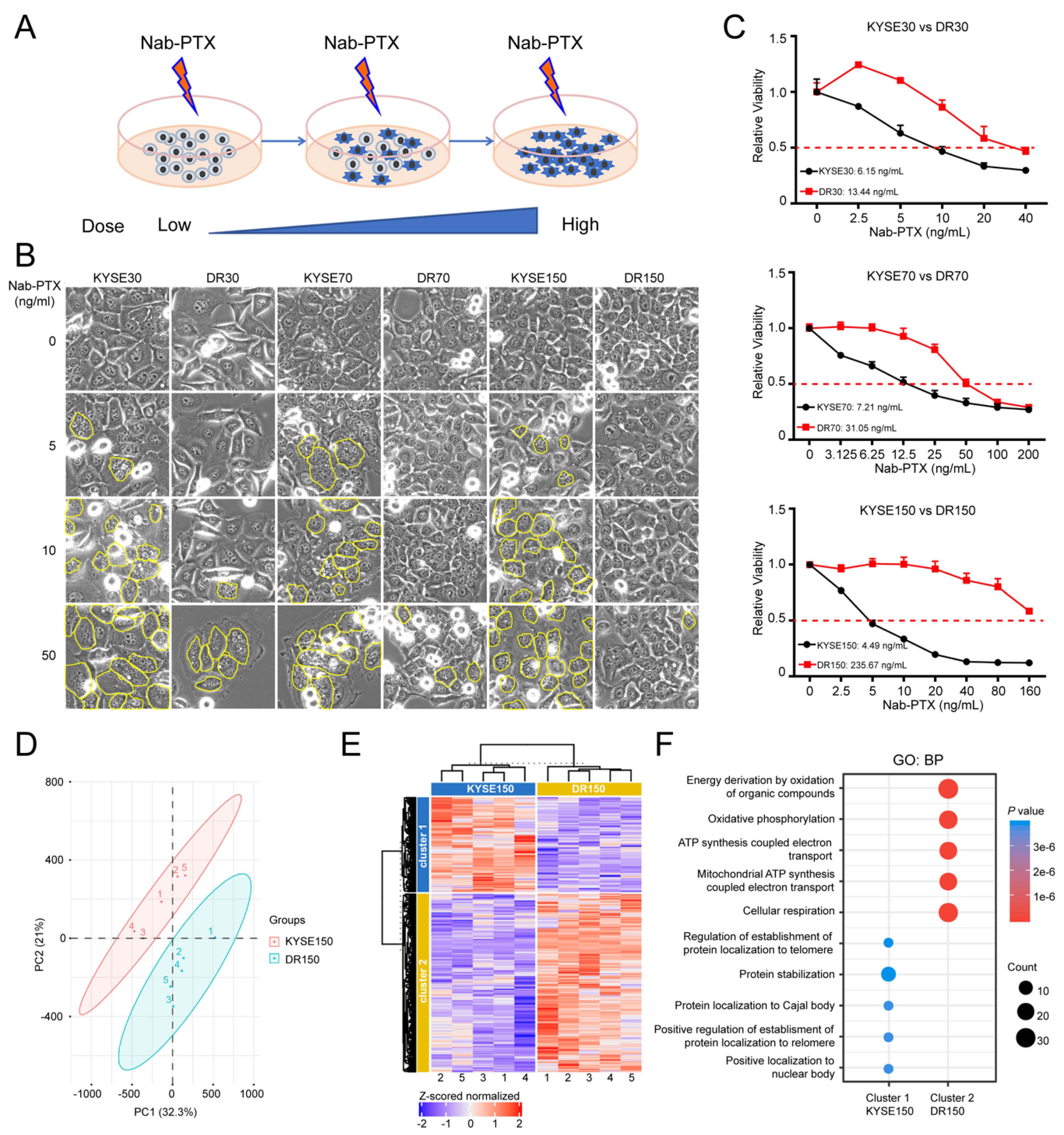
2.1. OXPHOS Is Elevated in Nab-PTX Resistant ESCC Cells
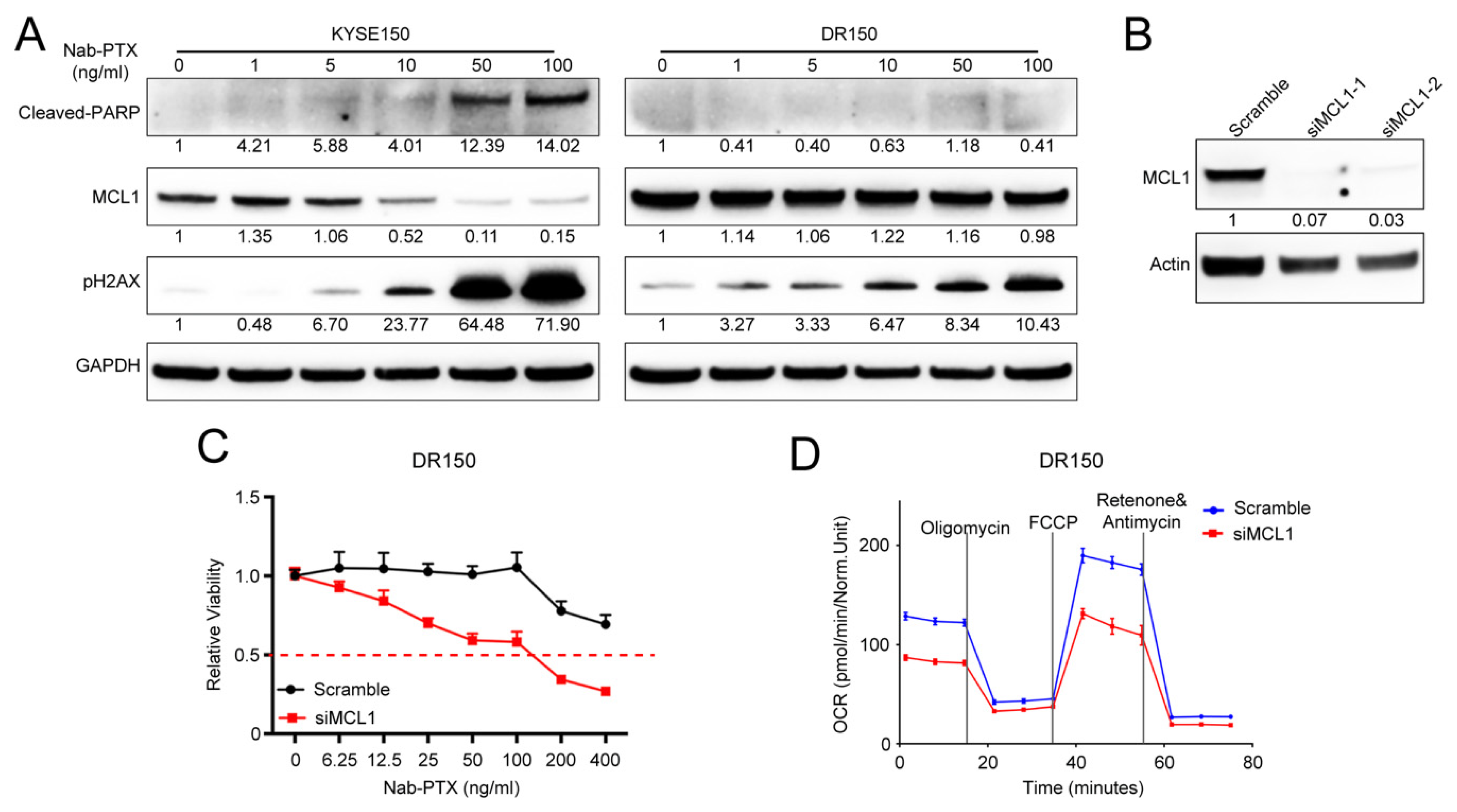
2.2. MCL1 Contributes to Nab-PTX Resistance in ESCC Cells
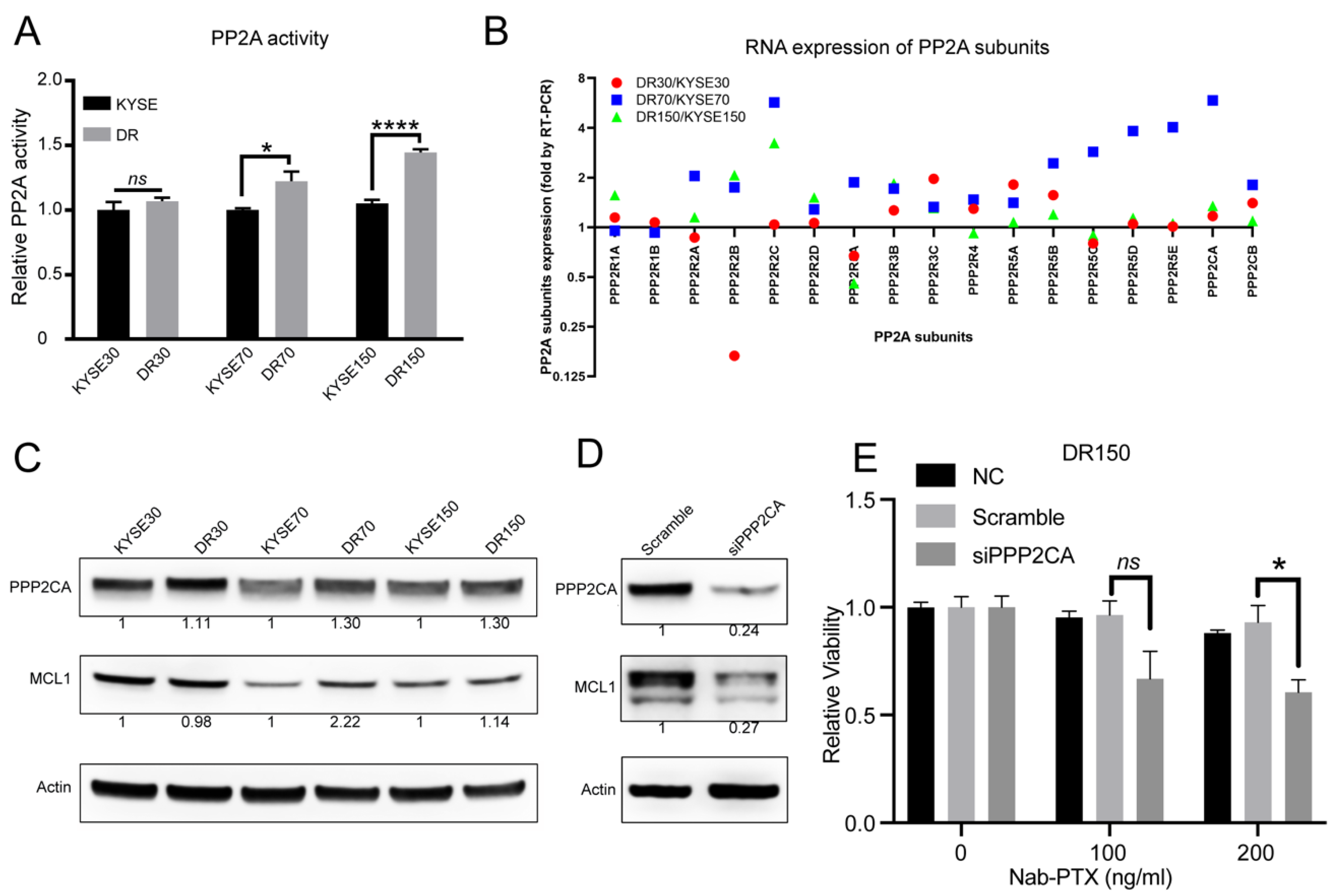
2.3. PP2A Activity Is Elevated in Nab-PTX Resistant ESCC Cells
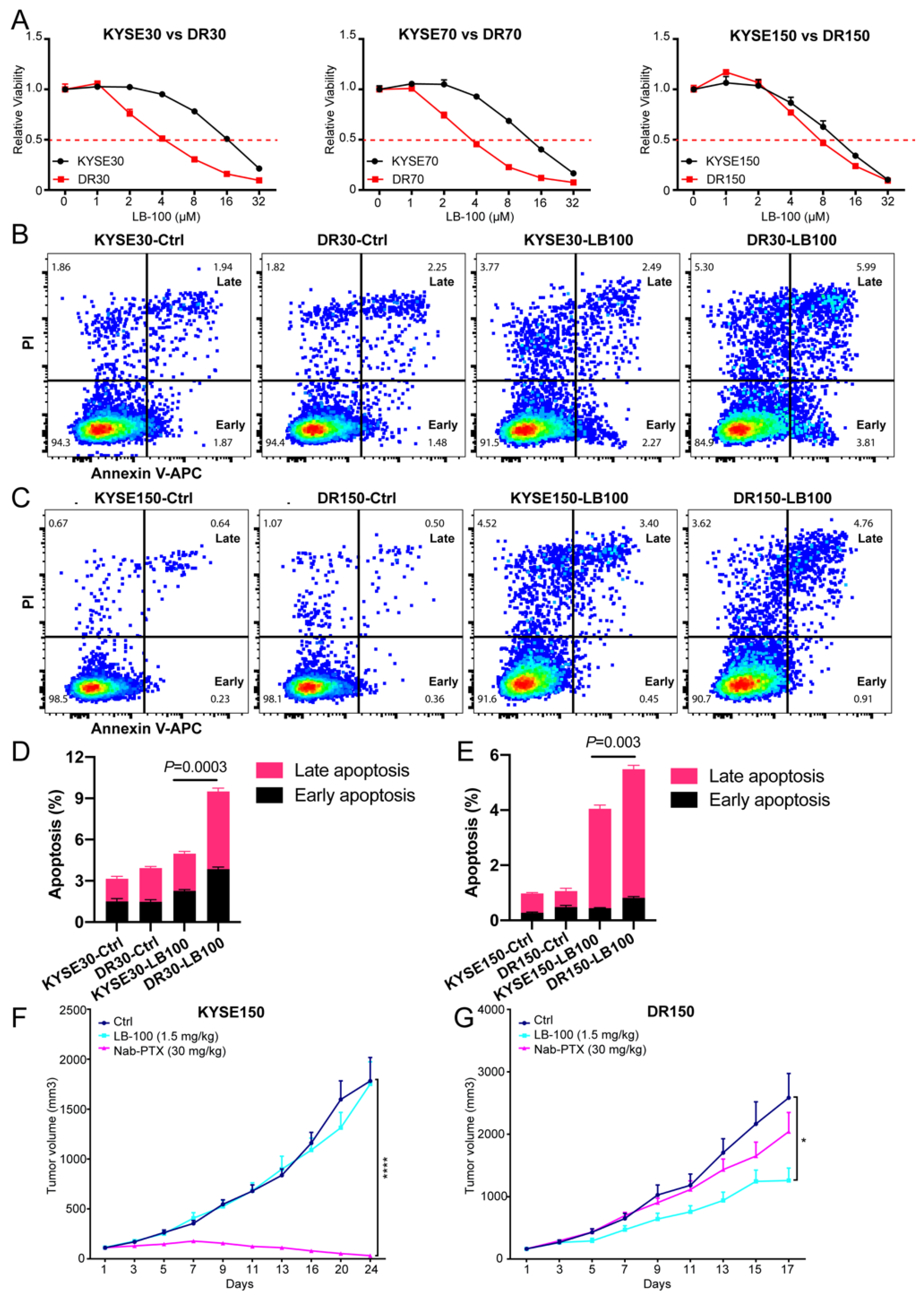
2.4. Pharmacological Inhibition of PP2A Inhibits the Growth of Nab-PTX Resistant ESCC Xenografts
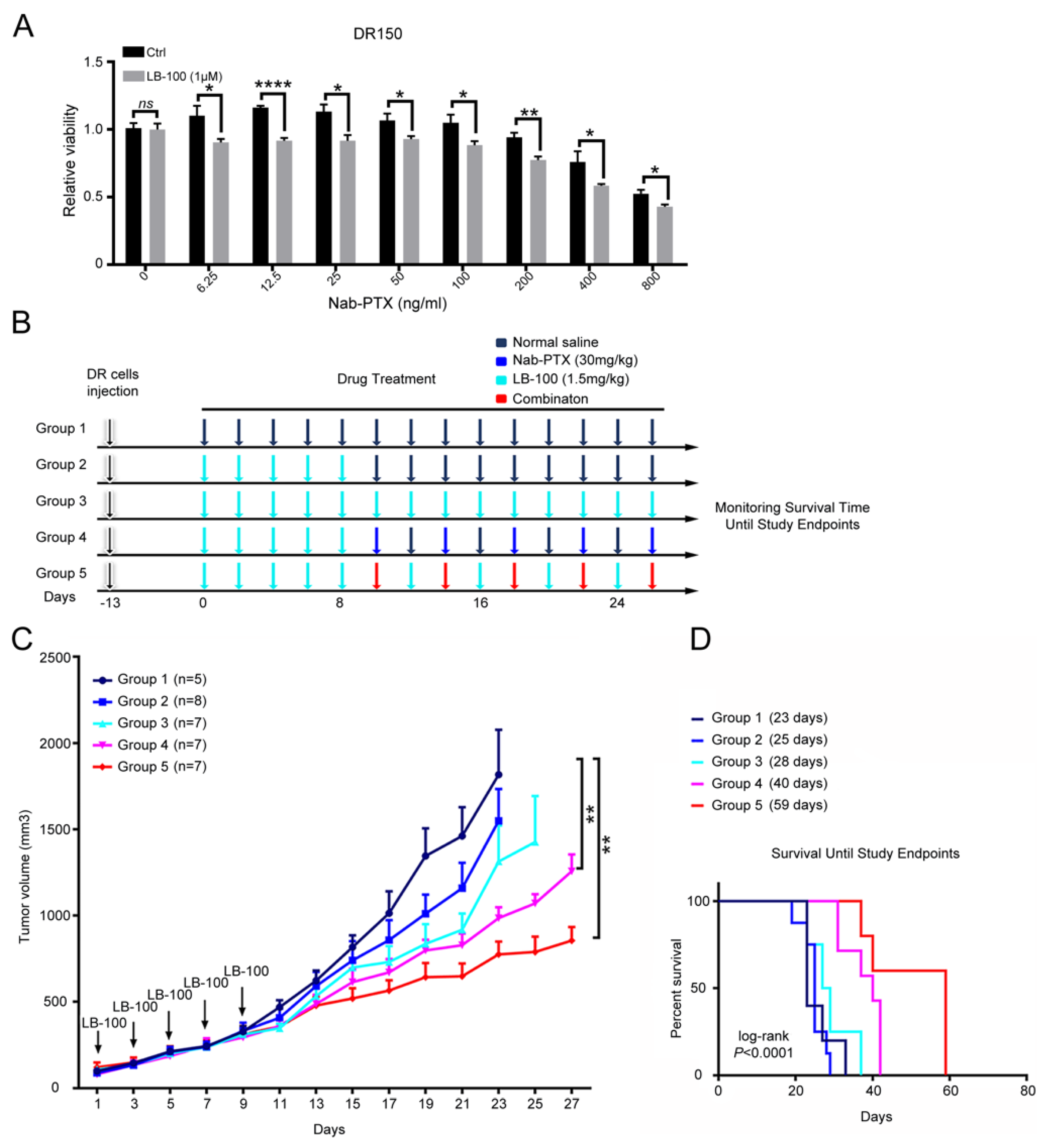
2.5. Pharmacological Inhibition of PP2A Resensitizes Nab-PTX Resistant ESCC to Nab-PTX
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Reagents
4.3. Development of nab-PTX Resistant Cell Lines and Cell Viability Assay
4.4. Proteomics Sample Preparation and LC-MS-Based Proteomics
4.5. Western Blot Analysis
4.6. PPP2CA, and MCL1 Knockdown with siRNA
4.7. Seahorse Analysis (XF Cell Mito Stress Test)
4.8. PP2A Activity Assay
4.9. Cell Apoptosis Assay
4.10. Animal Studies
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [Green Version]
- Arnold, M.; Soerjomataram, I.; Ferlay, J.; Forman, D. Global incidence of oesophageal cancer by histological subtype in 2012. Gut 2015, 64, 381–387. [Google Scholar] [CrossRef] [Green Version]
- Iizuka, T.; Kakegawa, T.; Ide, H.; Ando, N.; Watanabe, H.; Tanaka, O.; Takagi, I.; Isono, K.; Ishida, K.; Arimori, M.; et al. Phase II evaluation of cisplatin and 5-fluorouracil in advanced squamous cell carcinoma of the esophagus: A Japanese Esophageal Oncology Group Trial. Jpn. J. Clin. Oncol 1992, 22, 172–176. [Google Scholar] [PubMed]
- Cunningham, D.; Starling, N.; Rao, S.; Iveson, T.; Nicolson, M.; Coxon, F.; Middleton, G.; Daniel, F.; Oates, J.; Norman, A.R.; et al. Capecitabine and oxaliplatin for advanced esophagogastric cancer. N. Engl. J. Med. 2008, 358, 36–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Ryu, J.; Gandara, D.; Bold, R.J.; Urayama, S.; Tanaka, M.; Goldberg, Z.; Follette, D.; Narayan, S.; Lau, D. A phase II study of paclitaxel, carboplatin, and radiation with or without surgery for esophageal cancer. J. Thorac. Oncol. 2007, 2, 153–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.Y.; Do, Y.R.; Park, K.U.; Kim, M.K.; Lee, K.H.; Bae, S.H.; Ryoo, H.M.; Baek, J.H.; Song, H.S. A multi-center phase II study of docetaxel plus cisplatin as first-line therapy in patients with metastatic squamous cell esophageal cancer. Cancer Chemother. Pharmacol. 2010, 66, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, G.K.; Yanala, U.; Ravipati, A.; Follet, M.; Vijayakumar, M.; Are, C. Global trends in esophageal cancer. J. Surg. Oncol. 2017, 115, 564–579. [Google Scholar] [CrossRef] [Green Version]
- Siewert, J.R.; Stein, H.J.; Feith, M.; Bruecher, B.L.; Bartels, H.; Fink, U. Histologic tumor type is an independent prognostic parameter in esophageal cancer: Lessons from more than 1000 consecutive resections at a single center in the Western world. Ann. Surg. 2001, 234, 360–367; discussion 368–369. [Google Scholar] [CrossRef]
- Amos, L.A.; Lowe, J. How Taxol stabilises microtubule structure. Chem. Biol. 1999, 6, R65–R69. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Martin, A.; Alba, E.; Ciruelos, E.; Cortes, J.; Llombart, A.; Lluch, A.; Andres, R.; Alvarez, I.; Aramendia, J.M.; de la Pena, F.A.; et al. Nab-Paclitaxel in Metastatic Breast Cancer: Defining the Best Patient Profile. Curr. Cancer Drug Targets 2016, 16, 415–428. [Google Scholar] [CrossRef]
- Gradishar, W.J.; Tjulandin, S.; Davidson, N.; Shaw, H.; Desai, N.; Bhar, P.; Hawkins, M.; O'Shaughnessy, J. Phase III trial of nanoparticle albumin-bound paclitaxel compared with polyethylated castor oil-based paclitaxel in women with breast cancer. J. Clin. Oncol. 2005, 23, 7794–7803. [Google Scholar] [CrossRef] [PubMed]
- Socinski, M.A.; Bondarenko, I.; Karaseva, N.A.; Makhson, A.M.; Vynnychenko, I.; Okamoto, I.; Hon, J.K.; Hirsh, V.; Bhar, P.; Zhang, H.; et al. Weekly nab-paclitaxel in combination with carboplatin versus solvent-based paclitaxel plus carboplatin as first-line therapy in patients with advanced non-small-cell lung cancer: Final results of a phase III trial. J. Clin. Oncol. 2012, 30, 2055–2062. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, D.; El-Maraghi, R.H.; Hammel, P.; Heinemann, V.; Kunzmann, V.; Sastre, J.; Scheithauer, W.; Siena, S.; Tabernero, J.; Teixeira, L.; et al. Nab-Paclitaxel plus gemcitabine for metastatic pancreatic cancer: Long-term survival from a phase III trial. J. Natl. Cancer Inst. 2015, 107, 2. [Google Scholar] [CrossRef] [PubMed]
- Loong, H.H.; Chan, A.C.; Wong, A.C. Evolving Evidence of the Efficacy and Safety of nab-Paclitaxel in the Treatment of Cancers with Squamous Histologies. J. Cancer 2016, 7, 268–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Qin, R.; Wang, Z.K.; Dai, G.H. Nanoparticle albumin-bound paclitaxel combined with cisplatin as the first-line treatment for metastatic esophageal squamous cell carcinoma. Onco Targets Ther. 2013, 6, 585–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Jiang, Y.; Zhou, X.; Chen, Q.; Huang, Z.; Xu, Y.; Gong, L.; Yu, H.; Yang, H.; Liu, J.; et al. Phase II study of neoadjuvant therapy with nab-paclitaxel and cisplatin followed by surgery in patients with locally advanced esophageal squamous cell carcinoma. Oncotarget 2016, 7, 50624–50634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngoi, N.Y.L.; Choong, C.; Lee, J.; Bellot, G.; Wong, A.L.A.; Goh, B.C.; Pervaiz, S. Targeting Mitochondrial Apoptosis to Overcome Treatment Resistance in Cancer. Cancers 2020, 12, 574. [Google Scholar] [CrossRef] [Green Version]
- Wertz, I.E.; Kusam, S.; Lam, C.; Okamoto, T.; Sandoval, W.; Anderson, D.J.; Helgason, E.; Ernst, J.A.; Eby, M.; Liu, J.; et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 2011, 471, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Bolomsky, A.; Vogler, M.; Kose, M.C.; Heckman, C.A.; Ehx, G.; Ludwig, H.; Caers, J. MCL-1 inhibitors, fast-lane development of a new class of anti-cancer agents. J. Hematol. Oncol. 2020, 13, 173. [Google Scholar] [CrossRef]
- Perciavalle, R.M.; Stewart, D.P.; Koss, B.; Lynch, J.; Milasta, S.; Bathina, M.; Temirov, J.; Cleland, M.M.; Pelletier, S.; Schuetz, J.D.; et al. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat. Cell Biol. 2012, 14, 575–583. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.M.; Giltnane, J.M.; Balko, J.M.; Schwarz, L.J.; Guerrero-Zotano, A.L.; Hutchinson, K.E.; Nixon, M.J.; Estrada, M.V.; Sanchez, V.; Sanders, M.E.; et al. MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell Metab. 2017, 26, 633–647.e7. [Google Scholar] [CrossRef] [Green Version]
- Mojsa, B.; Lassot, I.; Desagher, S. Mcl-1 ubiquitination: Unique regulation of an essential survival protein. Cells 2014, 3, 418–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y. Assembly and structure of protein phosphatase 2A. Sci. China Ser. C Life Sci. 2009, 52, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Chen, Y.; Zhang, P.; Jeffrey, P.D.; Shi, Y. Structure of a protein phosphatase 2A holoenzyme: Insights into B55-mediated Tau dephosphorylation. Mol. Cell 2008, 31, 873–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seshacharyulu, P.; Pandey, P.; Datta, K.; Batra, S.K. Phosphatase: PP2A structural importance, regulation and its aberrant expression in cancer. Cancer Lett. 2013, 335, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Nifoussi, S.K.; Ratcliffe, N.R.; Ornstein, D.L.; Kasof, G.; Strack, S.; Craig, R.W. Inhibition of protein phosphatase 2A (PP2A) prevents Mcl-1 protein dephosphorylation at the Thr-163/Ser-159 phosphodegron, dramatically reducing expression in Mcl-1-amplified lymphoma cells. J. Biol. Chem. 2014, 289, 21950–21959. [Google Scholar] [CrossRef] [Green Version]
- Hong, C.S.; Ho, W.; Zhang, C.; Yang, C.; Elder, J.B.; Zhuang, Z. LB100, a small molecule inhibitor of PP2A with potent chemo- and radio-sensitizing potential. Cancer Biol. Ther. 2015, 16, 821–833. [Google Scholar] [CrossRef] [Green Version]
- Chung, V.; Mansfield, A.S.; Braiteh, F.; Richards, D.; Durivage, H.; Ungerleider, R.S.; Johnson, F.; Kovach, J.S. Safety, Tolerability, and Preliminary Activity of LB-100, an Inhibitor of Protein Phosphatase 2A, in Patients with Relapsed Solid Tumors: An Open-Label, Dose Escalation, First-in-Human, Phase I Trial. Clin. Cancer Res. 2017, 23, 3277–3284. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, Q.; Wang, H.; Jiang, D.; Xu, C.; Cui, J.; Zhang, Q.; Wang, H.; Huang, J.; Su, J.; Wu, G.S.; et al. Pharmacological Inhibition of PP2A Overcomes Nab-Paclitaxel Resistance by Downregulating MCL1 in Esophageal Squamous Cell Carcinoma (ESCC). Cancers 2021, 13, 4766. https://doi.org/10.3390/cancers13194766
Song Q, Wang H, Jiang D, Xu C, Cui J, Zhang Q, Wang H, Huang J, Su J, Wu GS, et al. Pharmacological Inhibition of PP2A Overcomes Nab-Paclitaxel Resistance by Downregulating MCL1 in Esophageal Squamous Cell Carcinoma (ESCC). Cancers. 2021; 13(19):4766. https://doi.org/10.3390/cancers13194766
Chicago/Turabian StyleSong, Qi, Herui Wang, Dongxian Jiang, Chen Xu, Jing Cui, Qi Zhang, Haixing Wang, Jie Huang, Jieakesu Su, Gen Sheng Wu, and et al. 2021. "Pharmacological Inhibition of PP2A Overcomes Nab-Paclitaxel Resistance by Downregulating MCL1 in Esophageal Squamous Cell Carcinoma (ESCC)" Cancers 13, no. 19: 4766. https://doi.org/10.3390/cancers13194766