
LightSpot®-FL-1 Fluorescent Probe: An Innovative Tool for Cancer Drug Resistance Analysis by Direct Detection and Quantification of the P-glycoprotein (P-gp) on Monolayer Culture and Spheroid Triple Negative Breast Cancer Models

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Experimental Section
2.1. LightSpot®-FL-1 Chemical Synthesis Achievement
2.2. Monolayer Cell Culture and Spheroid Formation
2.3. Lightspot®-Fl-1 Cellular Uptake and Outtake Analysis by Flow Cytometry
2.4. LightSpot®-FL-1 Fluorescent Staining on Monolayer and Suspension Cell Cultures
2.5. LightSpot®-FL-1 and Anti-P-gp Co-staining Analysis on Monolayer and Suspension Cell Cultures
2.6. LightSpot®-FL-1 Fluorescent Staining on SUM1315 Spheroids
2.7. P-gp Expression Level Measurement in SUM1315 Monolayer Cell Line and in Spheroid Models after Olaparib Treatment
2.8. Statistical Analysis
3. Results
3.1. Synthesis and Characterization of the LightSpot®-FL-1 Probe
3.2. LightSpot®-FL-1 Fluorescent Probe Selectively Targets P-gp in the DU4475 and SUM1315 TNBC Cell Line
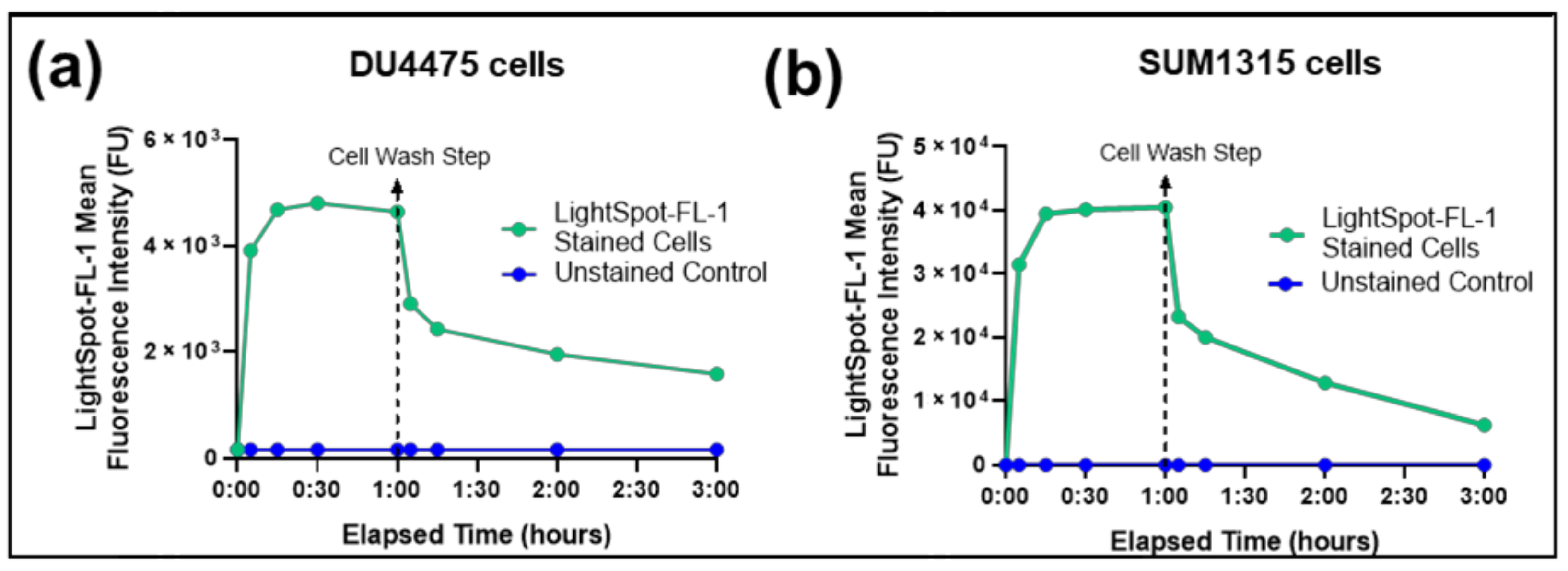
3.2.1. LightSpot®-FL-1 Kinetic Uptake by Flow Cytometry
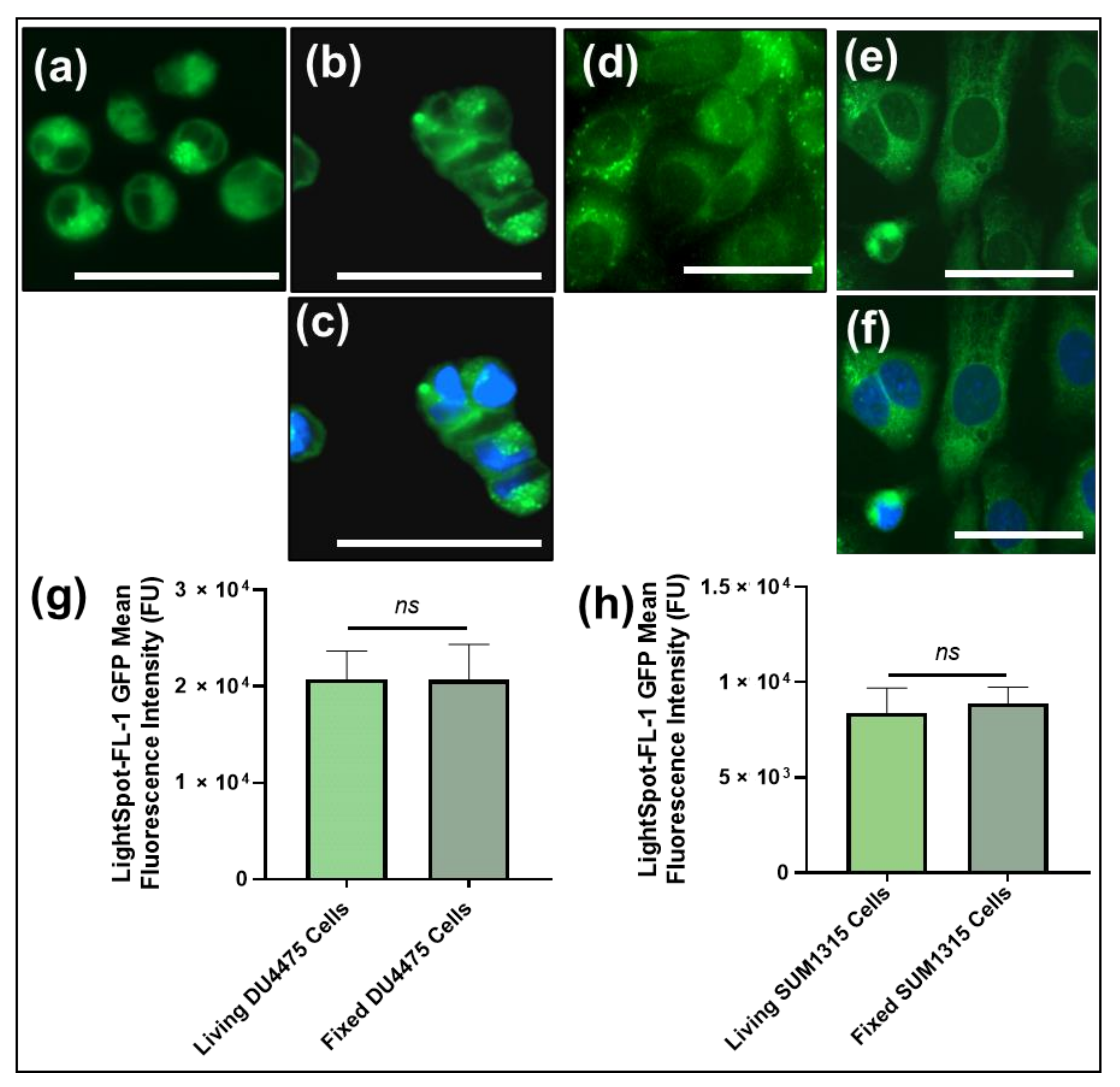
Lightspot®-FL-1 Staining Intensity Comparison between Living or Fixed Cells by Fluorescence Imaging
P-gp Co-Staining with LightSpot-FL-1 and Immunostaining
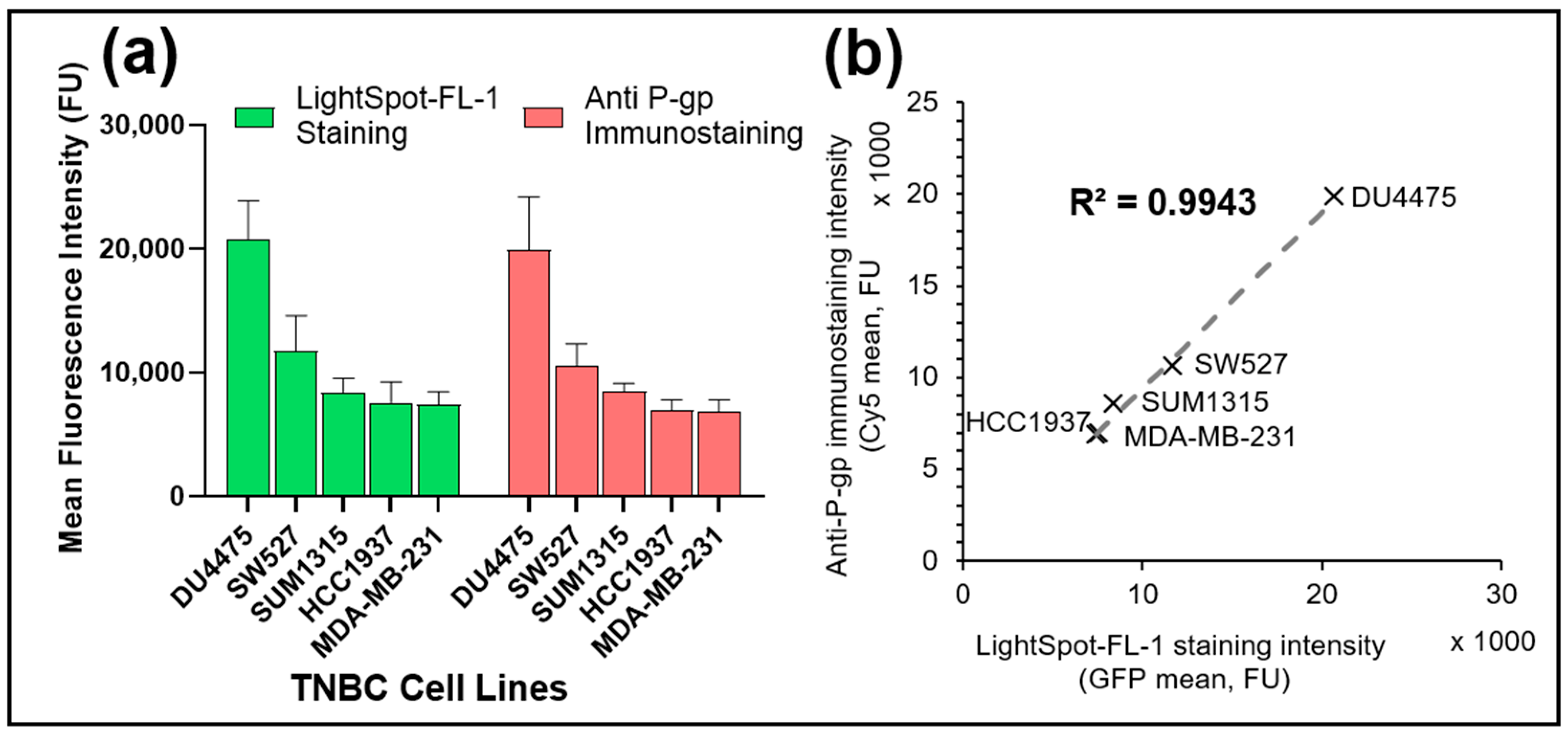
3.3. LightSpot®-FL-1 Allows Sensitive Detection and Quantification of Basal P-gp Expression Level in Various TNBC Cell Lines
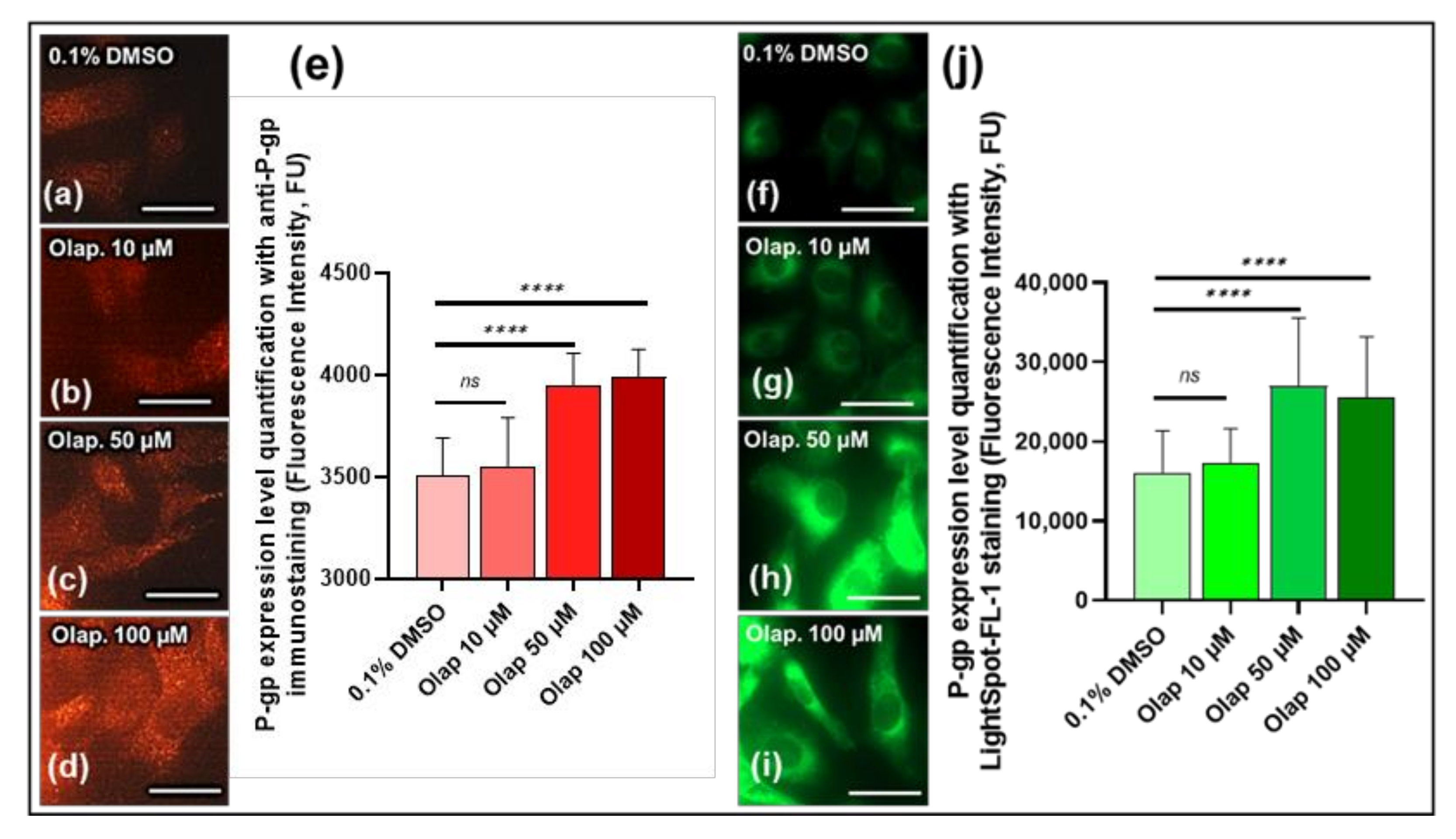
3.4. LightSpot®-FL-1 Detects and Quantifies Olaparib-induced P-gp expression Level on SUM1315 Monolayer Cultures
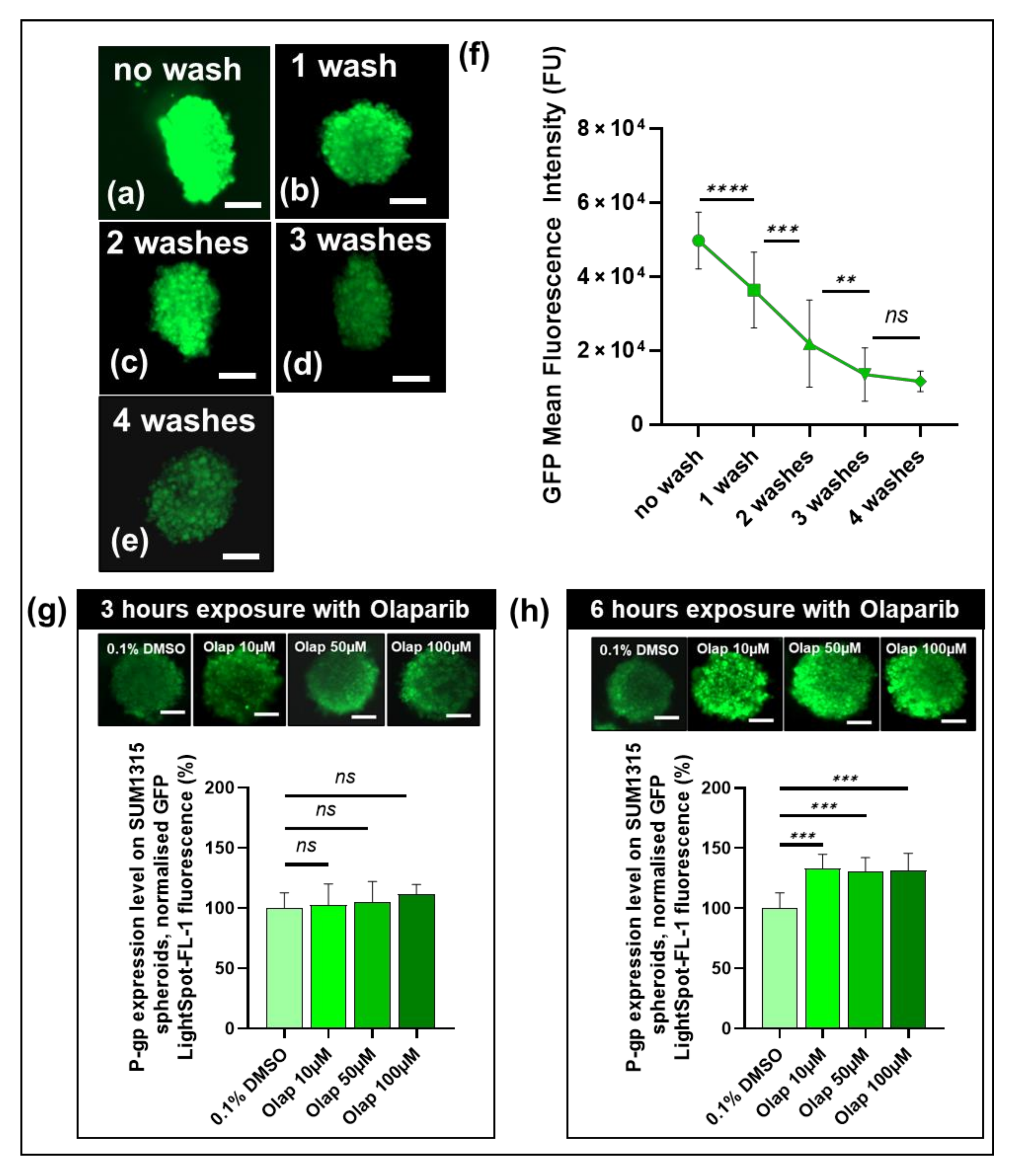
3.5. The Cell-permeant LightSpot®-FL-1 Detects and Quantifies Basal and Induced P-gp Expression in SUM1315 Spheroid Cell Mass Models
4. Discussion
5. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Juliano, R.L.; Ling, V.A. Surface Glycoprotein Modulating Drug Permeability in Chinese Hamster Ovary Cell Mutants. Biochim. Biophys. Acta. 1976, 455, 152–162. [Google Scholar] [CrossRef]
- Holland, I.B.; Cole, S.P.C.; Kuchler, K.; Higgins, C.F. Chapter 1—Phylogenetic and Functional Classification of ABC (ATP-Binding Cassette) Systems. In ABC Proteins: From Bacteria to Man; Academic Press Inc., Elsevier: Amsterdam, The Netherland, 2003; pp. 3–35. [Google Scholar]
- Bamdad, M.; Brousseau, P.; Denizeau, F. Identification of a Multidrug Resistance-like System in Tetrahymena Pyriformis: Evidence for a New Detoxication Mechanism in Freshwater Ciliates. FEBS Lett. 1999, 456, 389–393. [Google Scholar] [CrossRef] [Green Version]
- Bamdad, M.; Reader, S.; Grolière, C.A.; Bohatier, J.; Denizeau, F. Uptake and Efflux of Polycyclic Aromatic Hydrocarbons by Tetrahymena Pyriformis: Evidence for a Resistance Mechanism. Cytometry 1997, 28, 170–175. [Google Scholar] [CrossRef]
- Vache, C.; Camares, O.; Cardoso-Ferreira, M.-C.; Dastugue, B.; Creveaux, I.; Vaury, C.; Bamdad, M. A Potential Genomic Biomarker for the Detection of Polycyclic Aromatic Hydrocarbon Pollutants: Multidrug Resistance Gene 49 in Drosophila Melanogaster. Environ. Toxicol. Chem. 2007, 26, 1418–1424. [Google Scholar] [CrossRef]
- Valton, E.; Wawrzyniak, I.; Amblard, C.; Combourieu, B.; Bayle, M.-L.; Desmolles, F.; Kwiatkowski, F.; Penault-Llorca, F.; Bamdad, M. P-Gp Expression Levels in the Erythrocytes of Brown Trout: A New Tool for Aquatic Sentinel Biomarker Development. Biomarkers 2017, 22, 566–574. [Google Scholar] [CrossRef]
- Gottesman, M.M. Mechanisms of Cancer Drug Resistance. Annu. Rev. Med. 2002, 53, 615–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the Role of ABC Transporters in Multidrug-Resistant Cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef]
- Theodoulou, F.L.; Kerr, I.D. ABC Transporter Research: Going Strong 40 Years On. Biochem. Soc. Trans. 2015, 43, 1033–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silverman, J.A. Multidrug-Resistance Transporters. Pharm. Biotechnol. 1999, 12, 353–386. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.-C. The Molecular Mechanisms of Chemoresistance in Cancers. Oncotarget 2017, 8, 59950–59964. [Google Scholar] [CrossRef] [Green Version]
- Sarkadi, B.; Homolya, L.; Szakács, G.; Váradi, A. Human Multidrug Resistance ABCB and ABCG Transporters: Participation in a Chemoimmunity Defense System. Physiol. Rev. 2006, 86, 1179–1236. [Google Scholar] [CrossRef]
- Leonard, G.D.; Fojo, T.; Bates, S.E. The Role of ABC Transporters in Clinical Practice. Oncologist 2003, 8, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, D.; Martin, P.; Busschots, S.; Thery, J.; O’Leary, J.J.; Hennessy, B.T.; Stordal, B. PARP Inhibitors as P-Glyoprotein Substrates. J. Pharm. Sci. 2014, 103, 1913–1920. [Google Scholar] [CrossRef]
- Azzariti, A.; Porcelli, L.; Simone, G.M.; Quatrale, A.E.; Colabufo, N.A.; Berardi, F.; Perrone, R.; Zucchetti, M.; D’Incalci, M.; Xu, J.M.; et al. Tyrosine Kinase Inhibitors and Multidrug Resistance Proteins: Interactions and Biological Consequences. Cancer Chemoth. Pharm. 2010, 65, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Tomás, R. Multidrug Resistance: Retrospect and Prospects in Anti-Cancer Drug Treatment. Curr. Med. Chem. 2006, 13, 1859–1876. [Google Scholar] [CrossRef]
- Nedeljković, M.; Damjanović, A. Mechanisms of Chemotherapy Resistance in Triple-Negative Breast Cancer-How We Can Rise to the Challenge. Cells 2019, 8, 957. [Google Scholar] [CrossRef] [Green Version]
- Yamada, A.; Ishikawa, T.; Ota, I.; Kimura, M.; Shimizu, D.; Tanabe, M.; Chishima, T.; Sasaki, T.; Ichikawa, Y.; Morita, S.; et al. High Expression of ATP-Binding Cassette Transporter ABCC11 in Breast Tumors Is Associated with Aggressive Subtypes and Low Disease-Free Survival. Breast Cancer Res. Treat. 2013, 137, 773–782. [Google Scholar] [CrossRef]
- Linn, S.C.; Giaccone, G.; van Diest, P.J.; Blokhuis, W.M.D.; van der Valk, P.; van Kalken, C.K.; Kuiper, C.M.; Pinedo, H.M.; Baak, J.P.A. Prognostic Relevance of P-Glycoprotein Expression in Breast Cancer. Ann. Oncol. 1995, 6, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.; Tiriveedhi, V. Perplexing Role of P-Glycoprotein in Tumor Microenvironment. Front. Oncol. 2020, 10, 265. [Google Scholar] [CrossRef] [PubMed]
- Baekelandt, M.M.; Holm, R.; Nesland, J.M.; Tropé, C.G.; Kristensen, G.B. P-Glycoprotein Expression Is a Marker for Chemotherapy Resistance and Prognosis in Advanced Ovarian Cancer. Anticancer Res. 2000, 20, 1061–1067. [Google Scholar]
- Verrelle, P.; Frédéric, M.; Fonck, Y.; Feillel, V.; Dionet, C.; Kwiatkowski, F.; Plagne, R.; Chassagne, J. Clinical Relevance of Immunohistochemical Detection of Multidrug Resistance P-Glycoprotein in Breast Carcinoma. J. Natl. Cancer Inst. 1991, 83, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Irvin, W.J.; Carey, L.A. What Is Triple-Negative Breast Cancer? Eur. J. Cancer 2008, 44, 2799–2805. [Google Scholar] [CrossRef]
- Prat, A.; Pineda, E.; Adamo, B.; Galván, P.; Fernández, A.; Gaba, L.; Díez, M.; Viladot, M.; Arance, A.; Muñoz, M. Clinical Implications of the Intrinsic Molecular Subtypes of Breast Cancer. Breast 2015, 24 (Suppl. 2), S26–S35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, L.; Duan, J.-J.; Bian, X.-W.; Yu, S.-C. Triple-Negative Breast Cancer Molecular Subtyping and Treatment Progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef]
- Liedtke, C.; Mazouni, C.; Hess, K.R.; André, F.; Tordai, A.; Mejia, J.A.; Symmans, W.F.; Gonzalez-Angulo, A.M.; Hennessy, B.; Green, M.; et al. Response to Neoadjuvant Therapy and Long-Term Survival in Patients with Triple-Negative Breast Cancer. J. Clin. Oncol. 2008, 26, 1275–1281. [Google Scholar] [CrossRef]
- Carey, L.A.; Dees, E.C.; Sawyer, L.; Gatti, L.; Moore, D.T.; Collichio, F.; Ollila, D.W.; Sartor, C.I.; Graham, M.L.; Perou, C.M. The Triple Negative Paradox: Primary Tumor Chemosensitivity of Breast Cancer Subtypes. Clin. Cancer. Res. 2007, 13, 2329–2334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dufour, R.; Daumar, P.; Mounetou, E.; Aubel, C.; Kwiatkowski, F.; Abrial, C.; Vatoux, C.; Penault-Llorca, F.; Bamdad, M. BCRP and P-Gp Relay Overexpression in Triple Negative Basal-like Breast Cancer Cell Line: A Prospective Role in Resistance to Olaparib. Sci. Rep. 2015, 5, 12670. [Google Scholar] [CrossRef] [PubMed]
- Dubois, C.; Dufour, R.; Daumar, P.; Aubel, C.; Szczepaniak, C.; Blavignac, C.; Mounetou, E.; Penault-Llorca, F.; Bamdad, M. Development and Cytotoxic Response of Two Proliferative MDA-MB-231 and Non-Proliferative SUM1315 Three-Dimensional Cell Culture Models of Triple-Negative Basal-like Breast Cancer Cell Lines. Oncotarget 2017, 8, 95316–95331. [Google Scholar] [CrossRef] [PubMed]
- Dubois, C.; Daumar, P.; Aubel, C.; Gauthier, J.; Vidalinc, B.; Mounetou, E.; Penault-Llorca, F.; Bamdad, M. The New Synthetic Serum-Free Medium OptiPASS Promotes High Proliferation and Drug Efficacy Prediction on Spheroids from MDA-MB-231 and SUM1315 Triple-Negative Breast Cancer Cell Lines. J. Clin. Med. Res. 2019, 8, 397. [Google Scholar] [CrossRef] [Green Version]
- Dubois, C.; Martin, F.; Hassel, C.; Magnier, F.; Daumar, P.; Aubel, C.; Guerder, S.; Mounetou, E.; Penault-Lorca, F.; Bamdad, M. Low-Dose and Long-Term Olaparib Treatment Sensitizes MDA-MB-231 and SUM1315 Triple-Negative Breast Cancers Spheroids to Fractioned Radiotherapy. J. Clin. Med. 2019, 9, 64. [Google Scholar] [CrossRef] [Green Version]
- Goisnard, A.; Dubois, C.; Daumar, P.; Aubel, C.; Depresle, M.; Gauthier, J.; Vidalinc, B.; Penault-Llorca, F.; Mounetou, E.; Bamdad, M. The New Serum-Free OptiPASS® Medium in Cold and Oxygen-Free Conditions: An Innovative Conservation Method for the Preservation of MDA-MB-231 Triple Negative Breast Cancer Spheroids. Cancers 2021, 13, 1945. [Google Scholar] [CrossRef] [PubMed]
- Booij, T.H.; Price, L.S.; Danen, E.H.J. 3D Cell-Based Assays for Drug Screens: Challenges in Imaging, Image Analysis, and High-Content Analysis. SLAS Discov. 2019, 24, 615–627. [Google Scholar] [CrossRef] [Green Version]
- Bamdad, M.; Daumar, P.; Dubois, C.; Gauthier, J.; Goisnard, A.; Mounetou, E.; Penault-Llorca, F.; Vidalinc, B. LightSpot Patent: Method for Detecting, Localising and Quantifying MDR Proteins. International Patent Application WO 2021/064191 A1, 8 April 2021. [Google Scholar]
- Bush, T.L.; Payton, M.; Heller, S.; Chung, G.; Hanestad, K.; Rottman, J.B.; Loberg, R.; Friberg, G.; Kendall, R.L.; Saffran, D.; et al. AMG 900, a Small-Molecule Inhibitor of Aurora Kinases, Potentiates the Activity of Microtubule-Targeting Agents in Human Metastatic Breast Cancer Models. Mol. Cancer Ther. 2013, 12, 2356–2366. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Jaitak, V. Natural Products as Multidrug Resistance Modulators in Cancer. Eur. J. Med. Chem. 2019, 176, 268–291. [Google Scholar] [CrossRef]
- Kartal-Yandim, M.; Adan-Gokbulut, A.; Baran, Y. Molecular Mechanisms of Drug Resistance and Its Reversal in Cancer. Crit. Rev. Biotechnol. 2016, 36, 716–726. [Google Scholar] [CrossRef] [Green Version]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-Negative Breast Cancer: Clinical Features and Patterns of Recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- André, F.; Zielinski, C.C. Optimal Strategies for the Treatment of Metastatic Triple-Negative Breast Cancer with Currently Approved Agents. Ann. Oncol. 2012, 23, vi46–vi51. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Fatayer, H.; Hanby, A.M.; Horgan, K.; Perry, S.L.; Valleley, E.M.A.; Verghese, E.T.; Williams, B.J.; Thorne, J.L.; Hughes, T.A. Neoadjuvant Chemotherapy Induces Expression Levels of Breast Cancer Resistance Protein That Predict Disease-Free Survival in Breast Cancer. PLoS ONE 2013, 8, e62766. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhang, W.; Jin, Y.; Wang, H.; Gu, F.; Zhou, J.; Lao, Z.; Xu, Z.; Tang, F.; Zou, L.; et al. Evaluating the Response of Neoadjuvant Chemotherapy for Treatment of Breast Cancer: Are Tumor Biomarkers and Dynamic Contrast Enhanced MR Images Useful Predictive Tools? J. Thorac. Dis. 2014, 6, 785–794. [Google Scholar] [CrossRef]
- Rudas, M.; Filipits, M.; Taucher, S.; Stranzl, T.; Steger, G.G.; Jakesz, R.; Pirker, R.; Pohl, G. Expression of MRP1, LRP and Pgp in Breast Carcinoma Patients Treated with Preoperative Chemotherapy. Breast Cancer Res. Treat. 2003, 81, 149–157. [Google Scholar] [CrossRef]
- Saaby, L.; Brodin, B. A Critical View on In Vitro Analysis of P-Glycoprotein (P-Gp) Transport Kinetics. J. Pharm. Sci. 2017, 106, 2257–2264. [Google Scholar] [CrossRef] [Green Version]
- Jouan, E.; Le Vée, M.; Mayati, A.; Denizot, C.; Parmentier, Y.; Fardel, O. Evaluation of P-Glycoprotein Inhibitory Potential Using a Rhodamine 123 Accumulation. Pharmaceutics 2016, 8, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamrang, Z.; Arthanari, Y.; Clarke, D.; Pluen, A. Quantitative Assessment of P-Glycoprotein Expression and Function Using Confocal Image Analysis. Microsc. Microanal. 2014, 20, 1329–1339. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Arias, I.M. Intracellular Trafficking of P-Glycoprotein. Int. J. Biochem. Cell Biol. 2012, 44, 461–464. [Google Scholar] [CrossRef] [Green Version]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomal Sequestration of Hydrophobic Weak Base Chemotherapeutics Triggers Lysosomal Biogenesis and Lysosome-Dependent Cancer Multidrug Resistance. Oncotarget 2015, 6, 1143–1156. [Google Scholar] [CrossRef] [Green Version]
- Garland, M.; Yim, J.J.; Bogyo, M. A Bright Future for Precision Medicine: Advances in Fluorescent Chemical Probe Design and Their Clinical Application. Cell Chem. Biol. 2016, 23, 122–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnaud, O.; Koubeissi, A.; Ettouati, L.; Terreux, R.; Alamé, G.; Grenot, C.; Dumontet, C.; Di Pietro, A.; Paris, J.; Falson, P. Potent and Fully Noncompetitive Peptidomimetic Inhibitor of Multidrug Resistance P-Glycoprotein. J. Med. Chem. 2010, 53, 6720–6729. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, T.; Sahni, S.; Sharp, D.M.; Arvind, A.; Jansson, P.J.; Richardson, D.R. P-Glycoprotein Mediates Drug Resistance via a Novel Mechanism Involving Lysosomal Sequestration. J. Biol. Chem. 2013, 288, 31761–31771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hraběta, J.; Belhajová, M.; Šubrtová, H.; Merlos Rodrigo, M.A.; Heger, Z.; Eckschlager, T. Drug Sequestration in Lysosomes as One of the Mechanisms of Chemoresistance of Cancer Cells and the Possibilities of Its Inhibition. Int. J. Mol. Sci. 2020, 21, 392. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goisnard, A.; Daumar, P.; Dubois, C.; Aubel, C.; Roux, M.; Depresle, M.; Gauthier, J.; Vidalinc, B.; Penault-Llorca, F.; Mounetou, E.; et al. LightSpot®-FL-1 Fluorescent Probe: An Innovative Tool for Cancer Drug Resistance Analysis by Direct Detection and Quantification of the P-glycoprotein (P-gp) on Monolayer Culture and Spheroid Triple Negative Breast Cancer Models. Cancers 2021, 13, 4050. https://doi.org/10.3390/cancers13164050
Goisnard A, Daumar P, Dubois C, Aubel C, Roux M, Depresle M, Gauthier J, Vidalinc B, Penault-Llorca F, Mounetou E, et al. LightSpot®-FL-1 Fluorescent Probe: An Innovative Tool for Cancer Drug Resistance Analysis by Direct Detection and Quantification of the P-glycoprotein (P-gp) on Monolayer Culture and Spheroid Triple Negative Breast Cancer Models. Cancers. 2021; 13(16):4050. https://doi.org/10.3390/cancers13164050
Chicago/Turabian StyleGoisnard, Antoine, Pierre Daumar, Clémence Dubois, Corinne Aubel, Manon Roux, Marie Depresle, Jean Gauthier, Bernard Vidalinc, Frédérique Penault-Llorca, Emmanuelle Mounetou, and et al. 2021. "LightSpot®-FL-1 Fluorescent Probe: An Innovative Tool for Cancer Drug Resistance Analysis by Direct Detection and Quantification of the P-glycoprotein (P-gp) on Monolayer Culture and Spheroid Triple Negative Breast Cancer Models" Cancers 13, no. 16: 4050. https://doi.org/10.3390/cancers13164050