
EGFR in Cancer: Signaling Mechanisms, Drugs, and Acquired Resistance


Abstract
:Simple Summary
Abstract
1. Introduction
2. Physiological and Mutational Activation of EGFR
2.1. Ligand-Induced Receptor Activation
2.2. Mutation-Induced Receptor Activation
2.2.1. Extracellular Domain Mutations and Deletions
2.2.2. Mutations within the Intracellular and Kinase Domains of EGFR
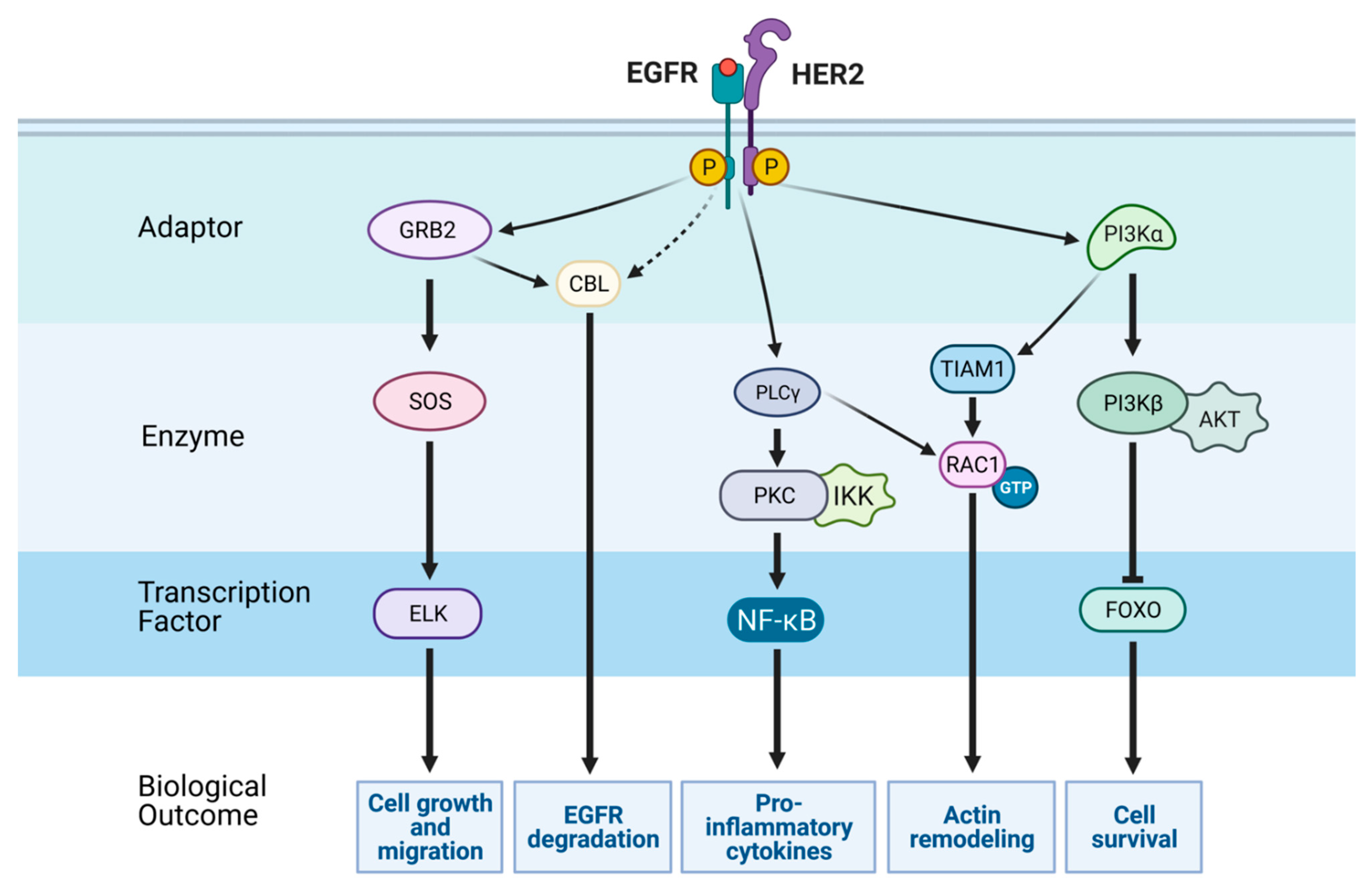
3. The Major Signaling Pathways Downstream of EGFR and Regulation of Transcription
4. Crosstalk between EGFR and Other Signaling Pathways
5. Roles for Non-Coding RNAs in EGFR Signaling
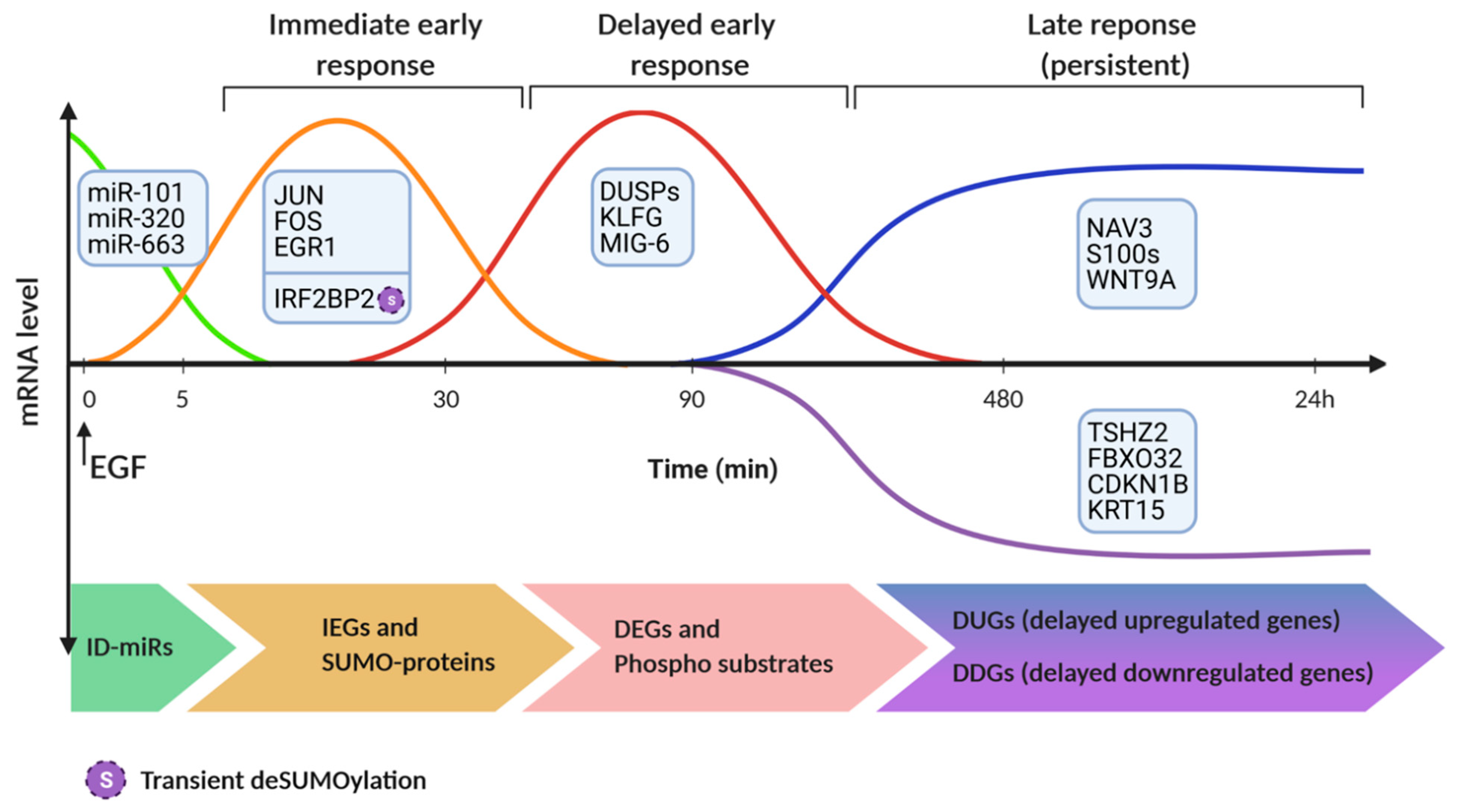
6. Proteomic and Epigenetic Analyses of EGF-Induced Molecular Switches
7. Roles for EGFR in Metastasis
8. Patient Resistance to Anti-Cancer Drugs Targeting EGFR
8.1. Resistance to EGFR-Specific TKIs
8.2. Resistance to EGFR mAbs
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Savage, C.R., Jr.; Cohen, S. Epidermal growth factor and a new derivative. Rapid isolation procedures and biological and chemical characterization. J. Biol. Chem. 1972, 247, 7609–7611. [Google Scholar] [CrossRef]
- Cohen, S.; Ushiro, H.; Stoscheck, C.; Chinkers, M. A native 170,000 epidermal growth factor receptor-kinase complex from shed plasma membrane vesicles. J. Biol. Chem. 1982, 257, 1523–1531. [Google Scholar] [CrossRef]
- Downward, J.; Yarden, Y.; Mayes, E.; Scrace, G.; Totty, N.; Stockwell, P.; Ullrich, A.; Schlessinger, J.; Waterfield, M.D. Close similarity of epidermal growth factor receptor and v-erb-B oncogene protein sequences. Nature 1984, 307, 521–527. [Google Scholar] [CrossRef]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Paez, J.G.; Janne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [Green Version]
- Pao, W.; Miller, V.; Zakowski, M.; Doherty, J.; Politi, K.; Sarkaria, I.; Singh, B.; Heelan, R.; Rusch, V.; Fulton, L.; et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. USA 2004, 101, 13306–13311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell. Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef]
- Arteaga, C.L.; Engelman, J.A. ERBB receptors: From oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell 2014, 25, 282–303. [Google Scholar] [CrossRef] [Green Version]
- Lemmon, M.A.; Schlessinger, J.; Ferguson, K.M. The EGFR family: Not so prototypical receptor tyrosine kinases. Cold Spring Harb. Perspect. Biol. 2014, 6, a020768. [Google Scholar] [CrossRef]
- Kovacs, E.; Zorn, J.A.; Huang, Y.; Barros, T.; Kuriyan, J. A Structural Perspective on the Regulation of the Epidermal Growth Factor Receptor. Annu. Rev. Biochem. 2015, 84, 739–764. [Google Scholar] [CrossRef] [Green Version]
- Waterman, H.; Katz, M.; Rubin, C.; Shtiegman, K.; Lavi, S.; Elson, A.; Jovin, T.; Yarden, Y. A mutant EGF-receptor defective in ubiquitylation and endocytosis unveils a role for Grb2 in negative signaling. EMBO J. 2002, 21, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Shtiegman, K.; Kochupurakkal, B.S.; Zwang, Y.; Pines, G.; Starr, A.; Vexler, A.; Citri, A.; Katz, M.; Lavi, S.; Ben-Basat, Y.; et al. Defective ubiquitinylation of EGFR mutants of lung cancer confers prolonged signaling. Oncogene 2007, 26, 6968–6978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosesson, Y.; Mills, G.B.; Yarden, Y. Derailed endocytosis: An emerging feature of cancer. Nat. Rev. Cancer 2008, 8, 835–850. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Bhargava, R.; Gerald, W.L.; Li, A.R.; Pan, Q.; Lal, P.; Ladanyi, M.; Chen, B. EGFR gene amplification in breast cancer: Correlation with epidermal growth factor receptor mRNA and protein expression and HER-2 status and absence of EGFR-activating mutations. Mod. Pathol. 2005, 18, 1027–1033. [Google Scholar] [CrossRef] [PubMed]
- Ooi, A.; Takehana, T.; Li, X.; Suzuki, S.; Kunitomo, K.; Iino, H.; Fujii, H.; Takeda, Y.; Dobashi, Y. Protein overexpression and gene amplification of HER-2 and EGFR in colorectal cancers: An immunohistochemical and fluorescent in situ hybridization study. Mod. Pathol. 2004, 17, 895–904. [Google Scholar] [CrossRef]
- Hirsch, F.R.; Varella-Garcia, M.; Cappuzzo, F. Predictive value of EGFR and HER2 overexpression in advanced non-small-cell lung cancer. Oncogene 2009, 28 (Suppl. S1), S32–S37. [Google Scholar] [CrossRef] [Green Version]
- Batra, S.K.; Castelino-Prabhu, S.; Wikstrand, C.J.; Zhu, X.; Humphrey, P.A.; Friedman, H.S.; Bigner, D.D. Epidermal growth factor ligand-independent, unregulated, cell-transforming potential of a naturally occurring human mutant EGFRvIII gene. Cell Growth Differ. 1995, 6, 1251–1259. [Google Scholar]
- Moscatello, D.K.; Holgado-Madruga, M.; Emlet, D.R.; Montgomery, R.B.; Wong, A.J. Constitutive activation of phosphatidylinositol 3-kinase by a naturally occurring mutant epidermal growth factor receptor. J. Biol. Chem. 1998, 273, 200–206. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.H.; Mukasa, A.; Bonavia, R.; Flynn, R.A.; Brewer, Z.E.; Cavenee, W.K.; Furnari, F.B.; White, F.M. Quantitative analysis of EGFRvIII cellular signaling networks reveals a combinatorial therapeutic strategy for glioblastoma. Proc. Natl. Acad. Sci. USA 2007, 104, 12867–12872. [Google Scholar] [CrossRef] [Green Version]
- Sugawa, N.; Ekstrand, A.J.; James, C.D.; Collins, V.P. Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc. Natl. Acad. Sci. USA 1990, 87, 8602–8606. [Google Scholar] [CrossRef] [Green Version]
- Wong, A.J.; Ruppert, J.M.; Bigner, S.H.; Grzeschik, C.H.; Humphrey, P.A.; Bigner, D.S.; Vogelstein, B. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc. Natl. Acad. Sci. USA 1992, 89, 2965–2969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekstrand, A.J.; Sugawa, N.; James, C.D.; Collins, V.P. Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N- and/or C-terminal tails. Proc. Natl. Acad. Sci. USA 1992, 89, 4309–4313. [Google Scholar] [CrossRef] [Green Version]
- Banisadr, A.; Eick, M.; Beri, P.; Parisian, A.D.; Yeoman, B.; Placone, J.K.; Engler, A.J.; Furnari, F. EGFRvIII uses intrinsic and extrinsic mechanisms to reduce glioma adhesion and increase migration. J. Cell Sci. 2020, 133, jcs247189. [Google Scholar] [CrossRef]
- Frederick, L.; Wang, X.Y.; Eley, G.; James, C.D. Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res. 2000, 60, 1383–1387. [Google Scholar] [PubMed]
- Wang, Z.; Longo, P.A.; Tarrant, M.K.; Kim, K.; Head, S.; Leahy, D.J.; Cole, P.A. Mechanistic insights into the activation of oncogenic forms of EGF receptor. Nat. Struct. Mol. Biol. 2011, 18, 1388–1393. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.L.; Yuan, J.Q.; Wang, K.F.; Fu, X.H.; Han, X.R.; Threapleton, D.; Yang, Z.Y.; Mao, C.; Tang, J.L. The prevalence of EGFR mutation in patients with non-small cell lung cancer: A systematic review and meta-analysis. Oncotarget 2016, 7, 78985–78993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cappuzzo, F.; Hirsch, F.R.; Rossi, E.; Bartolini, S.; Ceresoli, G.L.; Bemis, L.; Haney, J.; Witta, S.; Danenberg, K.; Domenichini, I.; et al. Epidermal growth factor receptor gene and protein and gefitinib sensitivity in non-small-cell lung cancer. J. Natl. Cancer Inst. 2005, 97, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Greulich, H.; Chen, T.H.; Feng, W.; Janne, P.A.; Alvarez, J.V.; Zappaterra, M.; Bulmer, S.E.; Frank, D.A.; Hahn, W.C.; Sellers, W.R.; et al. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med. 2005, 2, e313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Politi, K.; Zakowski, M.F.; Fan, P.D.; Schonfeld, E.A.; Pao, W.; Varmus, H.E. Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Genes Dev. 2006, 20, 1496–1510. [Google Scholar] [CrossRef] [Green Version]
- Stewart, E.L.; Tan, S.Z.; Liu, G.; Tsao, M.S. Known and putative mechanisms of resistance to EGFR targeted therapies in NSCLC patients with EGFR mutations—A review. Transl. Lung Cancer Res. 2015, 4, 67–81. [Google Scholar] [PubMed]
- Yasuda, H.; Park, E.; Yun, C.H.; Sng, N.J.; Lucena-Araujo, A.R.; Yeo, W.L.; Huberman, M.S.; Cohen, D.W.; Nakayama, S.; Ishioka, K.; et al. Structural, biochemical, and clinical characterization of epidermal growth factor receptor (EGFR) exon 20 insertion mutations in lung cancer. Sci. Transl. Med. 2013, 5, 216ra177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mok, T.S.; Wu, Y.L.; Thongprasert, S.; Yang, C.H.; Chu, D.T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef]
- Mitsudomi, T.; Morita, S.; Yatabe, Y.; Negoro, S.; Okamoto, I.; Tsurutani, J.; Seto, T.; Satouchi, M.; Tada, H.; Hirashima, T.; et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): An open label, randomised phase 3 trial. Lancet Oncol. 2010, 11, 121–128. [Google Scholar] [CrossRef]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef]
- Zhou, C.; Wu, Y.L.; Chen, G.; Feng, J.; Liu, X.Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011, 12, 735–742. [Google Scholar] [CrossRef]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2, e73. [Google Scholar] [CrossRef] [Green Version]
- Konieczkowski, D.J.; Johannessen, C.M.; Garraway, L.A. A Convergence-Based Framework for Cancer Drug Resistance. Cancer Cell 2018, 33, 801–815. [Google Scholar] [CrossRef] [Green Version]
- Mancini, M.; Yarden, Y. Mutational and network level mechanisms underlying resistance to anti-cancer kinase inhibitors. Semin. Cell Dev. Biol. 2016, 50, 164–176. [Google Scholar] [CrossRef]
- Tumbrink, H.L.; Heimsoeth, A.; Sos, M.L. The next tier of EGFR resistance mutations in lung cancer. Oncogene 2021, 40, 1–11. [Google Scholar] [CrossRef]
- Niederst, M.J.; Hu, H.; Mulvey, H.E.; Lockerman, E.L.; Garcia, A.R.; Piotrowska, Z.; Sequist, L.V.; Engelman, J.A. The allelic context of the C797S mutation acquired upon treatment with third generation EGFR inhibitors impacts sensitivity to subsequent treatment strategies. Clin. Cancer Res. 2015, 21, 3924–3933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thress, K.S.; Paweletz, C.P.; Felip, E.; Cho, B.C.; Stetson, D.; Dougherty, B.; Lai, Z.; Markovets, A.; Vivancos, A.; Kuang, Y.; et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat. Med. 2015, 21, 560–562. [Google Scholar] [CrossRef] [Green Version]
- Russo, M.; Crisafulli, G.; Sogari, A.; Reilly, N.M.; Arena, S.; Lamba, S.; Bartolini, A.; Amodio, V.; Magri, A.; Novara, L.; et al. Adaptive mutability of colorectal cancers in response to targeted therapies. Science 2019, 366, 1473–1480. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Kim, Y.; Park, I.; Cho, J.Y.; Lee, K.A. Detection of EGFR-SEPT14 fusion in cell-free DNA of a patient with advanced gastric cancer: A case report. World J. Clin. Cases 2021, 9, 2884–2889. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, H.B.; Chen, X.; Yang, X.; Ye, Y.; Bekaii-Saab, T.; Zheng, Y.; Zhang, Y. A Rare EGFR-SEPT14 Fusion in a Patient with Colorectal Adenocarcinoma Responding to Erlotinib. Oncologist 2020, 25, 203–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Z.; Brown, B.P.; Kim, S.; Ferguson, D.; Pavlick, D.C.; Jayakumaran, G.; Benayed, R.; Gallant, J.N.; Zhang, Y.K.; Yan, Y.; et al. Structure-function analysis of oncogenic EGFR Kinase Domain Duplication reveals insights into activation and a potential approach for therapeutic targeting. Nat. Commun. 2021, 12, 1382. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.-M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Takezawa, K.; Pirazzoli, V.; Arcila, M.E.; Nebhan, C.A.; Song, X.; de Stanchina, E.; Ohashi, K.; Janjigian, Y.Y.; Spitzler, P.J.; Melnick, M.A.; et al. HER2 amplification: A potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov. 2012, 2, 922–933. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; LaFramboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 2012, 44, 852–860. [Google Scholar] [CrossRef]
- Mueller, K.L.; Madden, J.M.; Zoratti, G.L.; Kuperwasser, C.; List, K.; Boerner, J.L. Fibroblast-secreted hepatocyte growth factor mediates epidermal growth factor receptor tyrosine kinase inhibitor resistance in triple-negative breast cancers through paracrine activation of Met. Breast Cancer Res. 2012, 14, R104. [Google Scholar] [CrossRef] [Green Version]
- Yano, S.; Wang, W.; Li, Q.; Matsumoto, K.; Sakurama, H.; Nakamura, T.; Ogino, H.; Kakiuchi, S.; Hanibuchi, M.; Nishioka, Y.; et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations. Cancer Res. 2008, 68, 9479–9487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pao, W.; Wang, T.Y.; Riely, G.J.; Miller, V.A.; Pan, Q.; Ladanyi, M.; Zakowski, M.F.; Heelan, R.T.; Kris, M.G.; Varmus, H.E. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005, 2, e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohashi, K.; Sequist, L.V.; Arcila, M.E.; Moran, T.; Chmielecki, J.; Lin, Y.L.; Pan, Y.; Wang, L.; de Stanchina, E.; Shien, K.; et al. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc. Natl. Acad. Sci. USA 2012, 109, E2127–E2133. [Google Scholar] [CrossRef] [Green Version]
- Jakobsen, K.R.; Demuth, C.; Sorensen, B.S.; Nielsen, A.L. The role of epithelial to mesenchymal transition in resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Transl. Lung Cancer Res. 2016, 5, 172–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Planchard, D.; Loriot, Y.; André, F.; Gobert, A.; Auger, N.; Lacroix, L.; Soria, J.C. EGFR-independent mechanisms of acquired resistance to AZD9291 in EGFR T790M-positive NSCLC patients. Ann. Oncol. 2015, 26, 2073–2078. [Google Scholar] [CrossRef]
- Eberlein, C.A.; Stetson, D.; Markovets, A.A.; Al-Kadhimi, K.J.; Lai, Z.; Fisher, P.R.; Meador, C.B.; Spitzler, P.; Ichihara, E.; Ross, S.J.; et al. Acquired Resistance to the Mutant-Selective EGFR Inhibitor AZD9291 Is Associated with Increased Dependence on RAS Signaling in Preclinical Models. Cancer Res. 2015, 75, 2489–2500. [Google Scholar] [CrossRef] [Green Version]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef] [Green Version]
- Ramalingam, S.S.; Yang, J.C.; Lee, C.K.; Kurata, T.; Kim, D.W.; John, T.; Nogami, N.; Ohe, Y.; Mann, H.; Rukazenkov, Y.; et al. Osimertinib As First-Line Treatment of EGFR Mutation-Positive Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 841–849. [Google Scholar] [CrossRef] [Green Version]
- Lievre, A.; Bachet, J.B.; Le Corre, D.; Boige, V.; Landi, B.; Emile, J.F.; Cote, J.F.; Tomasic, G.; Penna, C.; Ducreux, M.; et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006, 66, 3992–3995. [Google Scholar] [CrossRef] [Green Version]
- Di Fiore, F.; Blanchard, F.; Charbonnier, F.; Le Pessot, F.; Lamy, A.; Galais, M.P.; Bastit, L.; Killian, A.; Sesboue, R.; Tuech, J.J.; et al. Clinical relevance of KRAS mutation detection in metastatic colorectal cancer treated by Cetuximab plus chemotherapy. Br. J. Cancer 2007, 96, 1166–1169. [Google Scholar] [CrossRef] [Green Version]
- Bardelli, A.; Corso, S.; Bertotti, A.; Hobor, S.; Valtorta, E.; Siravegna, G.; Sartore-Bianchi, A.; Scala, E.; Cassingena, A.; Zecchin, D.; et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013, 3, 658–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertotti, A.; Migliardi, G.; Galimi, F.; Sassi, F.; Torti, D.; Isella, C.; Cora, D.; Di Nicolantonio, F.; Buscarino, M.; Petti, C.; et al. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011, 1, 508–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yonesaka, K.; Zejnullahu, K.; Okamoto, I.; Satoh, T.; Cappuzzo, F.; Souglakos, J.; Ercan, D.; Rogers, A.; Roncalli, M.; Takeda, M.; et al. Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci. Transl. Med. 2011, 3, 99ra86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montagut, C.; Dalmases, A.; Bellosillo, B.; Crespo, M.; Pairet, S.; Iglesias, M.; Salido, M.; Gallen, M.; Marsters, S.; Tsai, S.P.; et al. Identification of a mutation in the extracellular domain of the Epidermal Growth Factor Receptor conferring cetuximab resistance in colorectal cancer. Nat. Med. 2012, 18, 221–223. [Google Scholar] [CrossRef]
- Aird, K.M.; Ghanayem, R.B.; Peplinski, S.; Lyerly, H.K.; Devi, G.R. X-linked inhibitor of apoptosis protein inhibits apoptosis in inflammatory breast cancer cells with acquired resistance to an ErbB1/2 tyrosine kinase inhibitor. Mol. Cancer Ther. 2010, 9, 1432–1442. [Google Scholar] [CrossRef] [Green Version]
- Martin, A.P.; Miller, A.; Emad, L.; Rahmani, M.; Walker, T.; Mitchell, C.; Hagan, M.P.; Park, M.A.; Yacoub, A.; Fisher, P.B.; et al. Lapatinib resistance in HCT116 cells is mediated by elevated MCL-1 expression and decreased BAK activation and not by ERBB receptor kinase mutation. Mol. Pharmacol. 2008, 74, 807–822. [Google Scholar] [CrossRef] [Green Version]
- Garrett, J.T.; Olivares, M.G.; Rinehart, C.; Granja-Ingram, N.D.; Sanchez, V.; Chakrabarty, A.; Dave, B.; Cook, R.S.; Pao, W.; McKinely, E.; et al. Transcriptional and posttranslational up-regulation of HER3 (ErbB3) compensates for inhibition of the HER2 tyrosine kinase. Proc. Natl. Acad. Sci. USA 2011, 108, 5021–5026. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.T.; Kim, H.; Liska, D.; Gao, S.; Christensen, J.G.; Weiser, M.R. MET activation mediates resistance to lapatinib inhibition of HER2-amplified gastric cancer cells. Mol. Cancer Ther. 2012, 11, 660–669. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Greger, J.; Shi, H.; Liu, Y.; Greshock, J.; Annan, R.; Halsey, W.; Sathe, G.M.; Martin, A.M.; Gilmer, T.M. Novel mechanism of lapatinib resistance in HER2-positive breast tumor cells: Activation of AXL. Cancer Res. 2009, 69, 6871–6878. [Google Scholar] [CrossRef] [Green Version]
- Giuliano, M.; Hu, H.; Wang, Y.C.; Fu, X.; Nardone, A.; Herrera, S.; Mao, S.; Contreras, A.; Gutierrez, C.; Wang, T.; et al. Upregulation of ER Signaling as an Adaptive Mechanism of Cell Survival in HER2-Positive Breast Tumors Treated with Anti-HER2 Therapy. Clin. Cancer Res. 2015, 21, 3995–4003. [Google Scholar] [CrossRef] [Green Version]
- Brady, S.W.; Zhang, J.; Tsai, M.H.; Yu, D. PI3K-independent mTOR activation promotes lapatinib resistance and IAP expression that can be effectively reversed by mTOR and Hsp90 inhibition. Cancer Biol. Ther. 2015, 16, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Eichhorn, P.J.; Gili, M.; Scaltriti, M.; Serra, V.; Guzman, M.; Nijkamp, W.; Beijersbergen, R.L.; Valero, V.; Seoane, J.; Bernards, R.; et al. Phosphatidylinositol 3-kinase hyperactivation results in lapatinib resistance that is reversed by the mTOR/phosphatidylinositol 3-kinase inhibitor NVP-BEZ235. Cancer Res. 2008, 68, 9221–9230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trowe, T.; Boukouvala, S.; Calkins, K.; Cutler, R.E., Jr.; Fong, R.; Funke, R.; Gendreau, S.B.; Kim, Y.D.; Miller, N.; Woolfrey, J.R.; et al. EXEL-7647 inhibits mutant forms of ErbB2 associated with lapatinib resistance and neoplastic transformation. Clin. Cancer Res. 2008, 14, 2465–2475. [Google Scholar] [CrossRef] [Green Version]
- Hanker, A.B.; Brewer, M.R.; Sheehan, J.H.; Koch, J.P.; Sliwoski, G.R.; Nagy, R.; Lanman, R.; Berger, M.F.; Hyman, D.M.; Solit, D.B.; et al. An Acquired HER2(T798I) Gatekeeper Mutation Induces Resistance to Neratinib in a Patient with HER2 Mutant-Driven Breast Cancer. Cancer Discov. 2017, 7, 575–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scaltriti, M.; Rojo, F.; Ocana, A.; Anido, J.; Guzman, M.; Cortes, J.; Di Cosimo, S.; Matias-Guiu, X.; Ramon y Cajal, S.; Arribas, J.; et al. Expression of p95HER2, a truncated form of the HER2 receptor, and response to anti-HER2 therapies in breast cancer. J. Natl. Cancer Inst. 2007, 99, 628–638. [Google Scholar] [CrossRef] [Green Version]
- Nagy, P.; Friedlander, E.; Tanner, M.; Kapanen, A.I.; Carraway, K.L.; Isola, J.; Jovin, T.M. Decreased accessibility and lack of activation of ErbB2 in JIMT-1, a herceptin-resistant, MUC4-expressing breast cancer cell line. Cancer Res. 2005, 65, 473–482. [Google Scholar]
- Berns, K.; Horlings, H.M.; Hennessy, B.T.; Madiredjo, M.; Hijmans, E.M.; Beelen, K.; Linn, S.C.; Gonzalez-Angulo, A.M.; Stemke-Hale, K.; Hauptmann, M.; et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell 2007, 12, 395–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagata, Y.; Lan, K.H.; Zhou, X.; Tan, M.; Esteva, F.J.; Sahin, A.A.; Klos, K.S.; Li, P.; Monia, B.P.; Nguyen, N.T.; et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 2004, 6, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Hanker, A.B.; Pfefferle, A.D.; Balko, J.M.; Kuba, M.G.; Young, C.D.; Sánchez, V.; Sutton, C.R.; Cheng, H.; Perou, C.M.; Zhao, J.J.; et al. Mutant PIK3CA accelerates HER2-driven transgenic mammary tumors and induces resistance to combinations of anti-HER2 therapies. Proc. Natl. Acad. Sci. USA 2013, 110, 14372–14377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Zi, X.; Zhao, Y.; Mascarenhas, D.; Pollak, M. Insulin-like growth factor-I receptor signaling and resistance to trastuzumab (Herceptin). J. Natl. Cancer Inst. 2001, 93, 1852–1857. [Google Scholar] [CrossRef] [Green Version]
- Shattuck, D.L.; Miller, J.K.; Carraway, K.L., 3rd; Sweeney, C. Met receptor contributes to trastuzumab resistance of Her2-overexpressing breast cancer cells. Cancer Res. 2008, 68, 1471–1477. [Google Scholar] [CrossRef] [Green Version]
- Gallardo, A.; Lerma, E.; Escuin, D.; Tibau, A.; Muñoz, J.; Ojeda, B.; Barnadas, A.; Adrover, E.; Sánchez-Tejada, L.; Giner, D.; et al. Increased signalling of EGFR and IGF1R, and deregulation of PTEN/PI3K/Akt pathway are related with trastuzumab resistance in HER2 breast carcinomas. Br. J. Cancer 2012, 106, 1367–1373. [Google Scholar] [CrossRef] [Green Version]
- Musolino, A.; Naldi, N.; Bortesi, B.; Pezzuolo, D.; Capelletti, M.; Missale, G.; Laccabue, D.; Zerbini, A.; Camisa, R.; Bisagni, G.; et al. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J. Clin. Oncol. 2008, 26, 1789–1796. [Google Scholar] [CrossRef] [PubMed]
- Bon, G.; Pizzuti, L.; Laquintana, V.; Loria, R.; Porru, M.; Marchiò, C.; Krasniqi, E.; Barba, M.; Maugeri-Saccà, M.; Gamucci, T.; et al. Loss of HER2 and decreased T-DM1 efficacy in HER2 positive advanced breast cancer treated with dual HER2 blockade: The SePHER Study. J. Exp. Clin. Cancer Res. 2020, 39, 279. [Google Scholar] [CrossRef]
- Hynes, N.E.; MacDonald, G. ErbB receptors and signaling pathways in cancer. Curr. Opin. Cell Biol. 2009, 21, 177–184. [Google Scholar] [CrossRef]
- Shaul, Y.D.; Seger, R. The MEK/ERK cascade: From signaling specificity to diverse functions. Biochim. Biophys. Acta 2007, 1773, 1213–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katz, M.; Amit, I.; Yarden, Y. Regulation of MAPKs by growth factors and receptor tyrosine kinases. Biochim. Biophys. Acta 2007, 1773, 1161–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef] [Green Version]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef]
- Tarcic, G.; Avraham, R.; Pines, G.; Amit, I.; Shay, T.; Lu, Y.; Zwang, Y.; Katz, M.; Ben-Chetrit, N.; Jacob-Hirsch, J.; et al. EGR1 and the ERK-ERF axis drive mammary cell migration in response to EGF. FASEB J. 2012, 26, 1582–1592. [Google Scholar] [CrossRef] [Green Version]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [Green Version]
- Köstler, W.J.; Zeisel, A.; Körner, C.; Tsai, J.M.; Jacob-Hirsch, J.; Ben-Chetrit, N.; Sharma, K.; Cohen-Dvashi, H.; Yitzhaky, A.; Lader, E.; et al. Epidermal growth-factor—Induced transcript isoform variation drives mammary cell migration. PLoS ONE 2013, 8, e80566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchwalter, G.; Gross, C.; Wasylyk, B. Ets ternary complex transcription factors. Gene 2004, 324, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Ginty, D.D.; Greenberg, M.E. Coupling of the RAS-MAPK pathway to gene activation by RSK2, a growth factor-regulated CREB kinase. Science 1996, 273, 959–963. [Google Scholar] [CrossRef] [PubMed]
- Amit, I.; Citri, A.; Shay, T.; Lu, Y.; Katz, M.; Zhang, F.; Tarcic, G.; Siwak, D.; Lahad, J.; Jacob-Hirsch, J.; et al. A module of negative feedback regulators defines growth factor signaling. Nat. Genet. 2007, 39, 503–512. [Google Scholar] [CrossRef]
- Tullai, J.W.; Schaffer, M.E.; Mullenbrock, S.; Sholder, G.; Kasif, S.; Cooper, G.M. Immediate-early and delayed primary response genes are distinct in function and genomic architecture. J. Biol. Chem. 2007, 282, 23981–23995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avraham, R.; Sas-Chen, A.; Manor, O.; Steinfeld, I.; Shalgi, R.; Tarcic, G.; Bossel, N.; Zeisel, A.; Amit, I.; Zwang, Y.; et al. EGF decreases the abundance of microRNAs that restrain oncogenic transcription factors. Sci. Signal. 2010, 3, ra43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kedmi, M.; Ben-Chetrit, N.; Korner, C.; Mancini, M.; Ben-Moshe, N.B.; Lauriola, M.; Lavi, S.; Biagioni, F.; Carvalho, S.; Cohen-Dvashi, H.; et al. EGF induces microRNAs that target suppressors of cell migration: miR-15b targets MTSS1 in breast cancer. Sci. Signal. 2015, 8, ra29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avraham, R.; Yarden, Y. Feedback regulation of EGFR signalling: Decision making by early and delayed loops. Nat. Rev. Mol. Cell Biol. 2011, 12, 104–117. [Google Scholar] [CrossRef]
- Cohen-Dvashi, H.; Ben-Chetrit, N.; Russell, R.; Carvalho, S.; Lauriola, M.; Nisani, S.; Mancini, M.; Nataraj, N.; Kedmi, M.; Roth, L.; et al. Navigator-3, a modulator of cell migration, may act as a suppressor of breast cancer progression. EMBO Mol. Med. 2015, 7, 299–314. [Google Scholar] [CrossRef] [Green Version]
- Albrecht, M.; Stichel, D.; Muller, B.; Merkle, R.; Sticht, C.; Gretz, N.; Klingmuller, U.; Breuhahn, K.; Matthaus, F. TTCA: An R package for the identification of differentially expressed genes in time course microarray data. BMC Bioinform. 2017, 18, 33. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Grassi, M.L.; Palma, C.S.; Thome, C.H.; Lanfredi, G.P.; Poersch, A.; Faca, V.M. Proteomic analysis of ovarian cancer cells during epithelial-mesenchymal transition (EMT) induced by epidermal growth factor (EGF) reveals mechanisms of cell cycle control. J. Proteom. 2017, 151, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Zinatizadeh, M.R.; Miri, S.R.; Zarandi, P.K.; Chalbatani, G.M.; Raposo, C.; Mirzaei, H.R.; Akbari, M.E.; Mahmoodzadeh, H. The Hippo Tumor Suppressor Pathway (YAP/TAZ/TEAD/MST/LATS) and EGFR-RAS-RAF-MEK in cancer metastasis. Genes Dis. 2021, 8, 48–60. [Google Scholar] [CrossRef]
- He, C.; Mao, D.; Hua, G.; Lv, X.; Chen, X.; Angeletti, P.C.; Dong, J.; Remmenga, S.W.; Rodabaugh, K.J.; Zhou, J.; et al. The Hippo/YAP pathway interacts with EGFR signaling and HPV oncoproteins to regulate cervical cancer progression. EMBO Mol. Med. 2015, 7, 1426–1449. [Google Scholar] [CrossRef]
- Shostak, K.; Chariot, A. EGFR and NF-kappaB: Partners in cancer. Trends Mol. Med. 2015, 21, 385–393. [Google Scholar] [CrossRef]
- Pan, D.; Jiang, C.; Ma, Z.; Blonska, M.; You, M.J.; Lin, X. MALT1 is required for EGFR-induced NF-kappaB activation and contributes to EGFR-driven lung cancer progression. Oncogene 2016, 35, 919–928. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Cai, J.; Wang, Q.; Wang, Y.; Liu, M.; Yang, J.; Zhou, J.; Kang, C.; Li, M.; Jiang, C. Long Noncoding RNA NEAT1, Regulated by the EGFR Pathway, Contributes to Glioblastoma Progression Through the WNT/beta-Catenin Pathway by Scaffolding EZH2. Clin. Cancer Res. 2018, 24, 684–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangelberger, D.; Kern, D.; Loipetzberger, A.; Eberl, M.; Aberger, F. Cooperative Hedgehog-EGFR signaling. Front. Biosci. (Landmark Ed.) 2012, 17, 90–99. [Google Scholar] [CrossRef] [Green Version]
- Doheny, D.; Manore, S.G.; Wong, G.L.; Lo, H.W. Hedgehog Signaling and Truncated GLI1 in Cancer. Cells 2020, 9, 2114. [Google Scholar] [CrossRef] [PubMed]
- Eberl, M.; Klingler, S.; Mangelberger, D.; Loipetzberger, A.; Damhofer, H.; Zoidl, K.; Schnidar, H.; Hache, H.; Bauer, H.C.; Solca, F.; et al. Hedgehog-EGFR cooperation response genes determine the oncogenic phenotype of basal cell carcinoma and tumour-initiating pancreatic cancer cells. EMBO Mol. Med. 2012, 4, 218–233. [Google Scholar] [CrossRef] [PubMed]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012, 22, 1760–1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sas-Chen, A.; Aure, M.R.; Leibovich, L.; Carvalho, S.; Enuka, Y.; Korner, C.; Polycarpou-Schwarz, M.; Lavi, S.; Nevo, N.; Kuznetsov, Y.; et al. LIMT is a novel metastasis inhibiting lncRNA suppressed by EGF and downregulated in aggressive breast cancer. EMBO Mol. Med. 2016, 8, 1052–1064. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, J.; You, Z.; Yin, Y.; Liu, L.; Kang, Y.; Li, S.; Ning, S.; Li, H.; Gong, Y.; et al. LncRNA TINCR favors tumorigenesis via STAT3-TINCR-EGFR-feedback loop by recruiting DNMT1 and acting as a competing endogenous RNA in human breast cancer. Cell Death Dis. 2021, 12, 83. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Wang, Q.; Wang, Y.; Huang, K.; Yang, C.; Li, Y.; Yi, K.; Kang, C. EGFR/c-myc axis regulates TGFbeta/Hippo/Notch pathway via epigenetic silencing miR-524 in gliomas. Cancer Lett. 2017, 406, 12–21. [Google Scholar] [CrossRef]
- Liang, Y.; Xu, X.; Wang, T.; Li, Y.; You, W.; Fu, J.; Liu, Y.; Jin, S.; Ji, Q.; Zhao, W.; et al. The EGFR/miR-338-3p/EYA2 axis controls breast tumor growth and lung metastasis. Cell Death Dis. 2017, 8, e2928. [Google Scholar] [CrossRef] [Green Version]
- Verduci, L.; Strano, S.; Yarden, Y.; Blandino, G. The circRNA-microRNA code: Emerging implications for cancer diagnosis and treatment. Mol. Oncol. 2019, 13, 669–680. [Google Scholar] [CrossRef] [Green Version]
- Enuka, Y.; Lauriola, M.; Feldman, M.E.; Sas-Chen, A.; Ulitsky, I.; Yarden, Y. Circular RNAs are long-lived and display only minimal early alterations in response to a growth factor. Nucleic Acids Res. 2016, 44, 1370–1383. [Google Scholar] [CrossRef]
- Gao, X.; Xia, X.; Li, F.; Zhang, M.; Zhou, H.; Wu, X.; Zhong, J.; Zhao, Z.; Zhao, K.; Liu, D.; et al. Circular RNA-encoded oncogenic E-cadherin variant promotes glioblastoma tumorigenicity through activation of EGFR-STAT3 signalling. Nat. Cell Biol. 2021, 23, 278–291. [Google Scholar] [CrossRef]
- Barysch, S.V.; Stankovic-Valentin, N.; Miedema, T.; Karaca, S.; Doppel, J.; Nait Achour, T.; Vasudeva, A.; Wolf, L.; Sticht, C.; Urlaub, H.; et al. Transient deSUMOylation of IRF2BP proteins controls early transcription in EGFR signaling. EMBO Rep. 2021, 22, e49651. [Google Scholar] [CrossRef]
- Roth, L.; Srivastava, S.; Lindzen, M.; Sas-Chen, A.; Sheffer, M.; Lauriola, M.; Enuka, Y.; Noronha, A.; Mancini, M.; Lavi, S.; et al. SILAC identifies LAD1 as a filamin-binding regulator of actin dynamics in response to EGF and a marker of aggressive breast tumors. Sci. Signal. 2018, 11, eaan0949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nava, M.; Dutta, P.; Zemke, N.R.; Farias-Eisner, R.; Vadgama, J.V.; Wu, Y. Transcriptomic and ChIP-sequence interrogation of EGFR signaling in HER2+ breast cancer cells reveals a dynamic chromatin landscape and S100 genes as targets. BMC Med. Genom. 2019, 12, 32. [Google Scholar] [CrossRef]
- Enuka, Y.; Feldman, M.E.; Chowdhury, A.; Srivastava, S.; Lindzen, M.; Sas-Chen, A.; Massart, R.; Cheishvili, D.; Suderman, M.J.; Zaltsman, Y.; et al. Epigenetic mechanisms underlie the crosstalk between growth factors and a steroid hormone. Nucleic Acids Res. 2017, 45, 12681–12699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganesh, K.; Massague, J. Targeting metastatic cancer. Nat. Med. 2021, 27, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Witsch, E.; Sela, M.; Yarden, Y. Roles for growth factors in cancer progression. Physiology 2010, 25, 85–101. [Google Scholar] [CrossRef] [Green Version]
- Biber, G.; Ben-Shmuel, A.; Sabag, B.; Barda-Saad, M. Actin regulators in cancer progression and metastases: From structure and function to cytoskeletal dynamics. Int. Rev. Cell Mol. Biol. 2020, 356, 131–196. [Google Scholar]
- Nobes, C.D.; Hall, A. Rho GTPases control polarity, protrusion, and adhesion during cell movement. J. Cell Biol. 1999, 144, 1235–1244. [Google Scholar] [CrossRef] [Green Version]
- Etienne-Manneville, S.; Hall, A. Cdc42 regulates GSK-3beta and adenomatous polyposis coli to control cell polarity. Nature 2003, 421, 753–756. [Google Scholar] [CrossRef]
- Petrie, R.J.; Doyle, A.D.; Yamada, K.M. Random versus directionally persistent cell migration. Nat. Rev. Mol. Cell Biol. 2009, 10, 538–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pegtel, D.M.; Ellenbroek, S.I.; Mertens, A.E.; van der Kammen, R.A.; de Rooij, J.; Collard, J.G. The Par-Tiam1 complex controls persistent migration by stabilizing microtubule-dependent front-rear polarity. Curr. Biol. 2007, 17, 1623–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankov, R.; Endo, Y.; Even-Ram, S.; Araki, M.; Clark, K.; Cukierman, E.; Matsumoto, K.; Yamada, K.M. A Rac switch regulates random versus directionally persistent cell migration. J. Cell Biol. 2005, 170, 793–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asokan, S.B.; Johnson, H.E.; Rahman, A.; King, S.J.; Rotty, J.D.; Lebedeva, I.P.; Haugh, J.M.; Bear, J.E. Mesenchymal chemotaxis requires selective inactivation of myosin II at the leading edge via a noncanonical PLCgamma/PKCalpha pathway. Dev. Cell 2014, 31, 747–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Wang, Q.; Wang, Y.; Chen, X.; Wang, Z. PLC-gamma1 and Rac1 coregulate EGF-induced cytoskeleton remodeling and cell migration. Mol. Endocrinol. 2009, 23, 901–913. [Google Scholar] [CrossRef] [Green Version]
- Kazanietz, M.G.; Barrio-Real, L.; Casado-Medrano, V.; Baker, M.J.; Lopez-Haber, C. The P-Rex1/Rac signaling pathway as a point of convergence for HER/ErbB receptor and GPCR responses. Small GTPases 2018, 9, 297–303. [Google Scholar] [CrossRef]
- Tavares, S.; Vieira, A.F.; Taubenberger, A.V.; Araujo, M.; Martins, N.P.; Bras-Pereira, C.; Polonia, A.; Herbig, M.; Barreto, C.; Otto, O.; et al. Actin stress fiber organization promotes cell stiffening and proliferation of pre-invasive breast cancer cells. Nat. Commun. 2017, 8, 15237. [Google Scholar] [CrossRef]
- Philippar, U.; Roussos, E.T.; Oser, M.; Yamaguchi, H.; Kim, H.D.; Giampieri, S.; Wang, Y.; Goswami, S.; Wyckoff, J.B.; Lauffenburger, D.A.; et al. A Mena invasion isoform potentiates EGF-induced carcinoma cell invasion and metastasis. Dev. Cell 2008, 15, 813–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckert, M.A.; Lwin, T.M.; Chang, A.T.; Kim, J.; Danis, E.; Ohno-Machado, L.; Yang, J. Twist1-induced invadopodia formation promotes tumor metastasis. Cancer Cell 2011, 19, 372–386. [Google Scholar] [CrossRef] [Green Version]
- Ben-Chetrit, N.; Chetrit, D.; Russell, R.; Korner, C.; Mancini, M.; Abdul-Hai, A.; Itkin, T.; Carvalho, S.; Cohen-Dvashi, H.; Koestler, W.J.; et al. Synaptojanin 2 is a druggable mediator of metastasis and the gene is overexpressed and amplified in breast cancer. Sci. Signal. 2015, 8, ra7. [Google Scholar] [CrossRef] [Green Version]
- Gewinner, C.; Wang, Z.C.; Richardson, A.; Teruya-Feldstein, J.; Etemadmoghadam, D.; Bowtell, D.; Barretina, J.; Lin, W.M.; Rameh, L.; Salmena, L.; et al. Evidence that inositol polyphosphate 4-phosphatase type II is a tumor suppressor that inhibits PI3K signaling. Cancer Cell 2009, 16, 115–125. [Google Scholar] [CrossRef] [Green Version]
- Sundqvist, A.; Vasilaki, E.; Voytyuk, O.; Bai, Y.; Morikawa, M.; Moustakas, A.; Miyazono, K.; Heldin, C.H.; Ten Dijke, P.; van Dam, H. TGFβ and EGF signaling orchestrates the AP-1- and p63 transcriptional regulation of breast cancer invasiveness. Oncogene 2020, 39, 4436–4449. [Google Scholar] [CrossRef] [PubMed]
- Wrenn, E.D.; Yamamoto, A.; Moore, B.M.; Huang, Y.; McBirney, M.; Thomas, A.J.; Greenwood, E.; Rabena, Y.F.; Rahbar, H.; Partridge, S.C.; et al. Regulation of Collective Metastasis by Nanolumenal Signaling. Cell 2020, 183, 395–410.e19. [Google Scholar] [CrossRef] [PubMed]
- Day, K.C.; Lorenzatti Hiles, G.; Kozminsky, M.; Dawsey, S.J.; Paul, A.; Broses, L.J.; Shah, R.; Kunja, L.P.; Hall, C.; Palanisamy, N.; et al. HER2 and EGFR Overexpression Support Metastatic Progression of Prostate Cancer to Bone. Cancer Res. 2017, 77, 74–85. [Google Scholar] [CrossRef] [Green Version]
- Lau, T.S.; Chan, L.K.; Wong, E.C.; Hui, C.W.; Sneddon, K.; Cheung, T.H.; Yim, S.F.; Lee, J.H.; Yeung, C.S.; Chung, T.K.; et al. A loop of cancer-stroma-cancer interaction promotes peritoneal metastasis of ovarian cancer via TNFα-TGFα-EGFR. Oncogene 2017, 36, 3576–3587. [Google Scholar] [CrossRef]
- Wyckoff, J.; Wang, W.; Lin, E.Y.; Wang, Y.; Pixley, F.; Stanley, E.R.; Graf, T.; Pollard, J.W.; Segall, J.; Condeelis, J. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 2004, 64, 7022–7029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarden, Y.; Pines, G. The ERBB network: At last, cancer therapy meets systems biology. Nat. Rev. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Balaban, N.Q.; Merrin, J.; Chait, R.; Kowalik, L.; Leibler, S. Bacterial persistence as a phenotypic switch. Science 2004, 305, 1622–1625. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hata, A.N.; Niederst, M.J.; Archibald, H.L.; Gomez-Caraballo, M.; Siddiqui, F.M.; Mulvey, H.E.; Maruvka, Y.E.; Ji, F.; Bhang, H.-e.C.; Krishnamurthy Radhakrishna, V.; et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med. 2016, 22, 262–269. [Google Scholar] [CrossRef]
- Yun, C.H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janne, P.A.; Yang, J.C.; Kim, D.W.; Planchard, D.; Ohe, Y.; Ramalingam, S.S.; Ahn, M.J.; Kim, S.W.; Su, W.C.; Horn, L.; et al. Azd9291 in egfr inhibitor-resistant non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.; Wu, Y.-L.; Ahn, M.-J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.M.E.; et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soria, J.-C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef]
- Sforza, V.; Martinelli, E.; Ciardiello, F.; Gambardella, V.; Napolitano, S.; Martini, G.; Della Corte, C.; Cardone, C.; Ferrara, M.L.; Reginelli, A.; et al. Mechanisms of resistance to anti-epidermal growth factor receptor inhibitors in metastatic colorectal cancer. World J. Gastroenterol. 2016, 22, 6345–6361. [Google Scholar] [CrossRef]
- Bardelli, A.; Siena, S. Molecular mechanisms of resistance to cetuximab and panitumumab in colorectal cancer. J. Clin. Oncol. 2010, 28, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Leto, S.M.; Trusolino, L. Primary and acquired resistance to EGFR-targeted therapies in colorectal cancer: Impact on future treatment strategies. J. Mol. Med. 2014, 92, 709–722. [Google Scholar] [CrossRef] [Green Version]
- Lievre, A.; Ouine, B.; Canet, J.; Cartier, A.; Amar, Y.; Cacheux, W.; Mariani, O.; Guimbaud, R.; Selves, J.; Lecomte, T.; et al. Protein biomarkers predictive for response to anti-EGFR treatment in RAS wild-type metastatic colorectal carcinoma. Br. J. Cancer 2017, 117, 1819–1827. [Google Scholar] [CrossRef] [Green Version]
- Gerlinger, M.; McGranahan, N.; Dewhurst, S.M.; Burrell, R.A.; Tomlinson, I.; Swanton, C. Cancer: Evolution within a lifetime. Annu. Rev. Genet. 2014, 48, 215–236. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Receptor | Class | Name of the Drug | Trade Name | Year of Approval | Company | Indication | Main Resistance Mechanisms |
|---|---|---|---|---|---|---|---|
| EGFR | TKI | Gefitinib | Iressa | 2003 | AstraZeneca | NSCLC | Resistance to first and second generation EGFR-TKIs: T790M mutation [37], amplification of MET [47] or HER2 [48], overexpression of AXL [49] or HGF [50,51], mutations in downstream molecules [52,53], phenotypic transformation [54]. Resistance to 2nd-line osimertinib: C797S mutation [42], MET or HER2 amplification [55], activation of downstream signaling pathways [56], phenotypic transformation [57]. Resistance to 1st-line osimertinib: MET amplification, C797X mutations, EGFR WT or HER2 amplification, mutations in downstream molecules [58]. |
| Erlotinib | Tarceva | 2004 | Genentech/OSI | NSCLC/Pancreatic cancer | |||
| Afatinib # | Gilotrif | 2013 | Boehringer Ingelheim | NSCLC | |||
| Dacomitinib # | Vizimpro | 2018 | Pfizer Inc. | NSCLC | |||
| Osimertinib | Tagrisso | 2015 | AstraZeneca | NSCLC | |||
| Vandetanib * | Caprelsa | 2011 | Genzyme Corp | Medullary thyroid cancer | |||
| mAb | Cetuximab | Erbitux | 2004 | Imclone/Bristol-Meyers Squib | CCR, head and neck cancer | Resistance to EGFR mAbs: Genomic alterations in downstream molecules [59,60], activation of bypass receptors (e.g., MET, HER2) [61,62,63], mutations in the extracellular domain of EGFR (e.g., S492R) [64]. | |
| Panitumumab | Vectibix | 2006 | Amgen | CCR | |||
| Necitumumab | Portrazza | 2015 | Eli Lilly Co | NSCLC | |||
| HER2 | TKI | Lapatinib | Tykerb | 2007 | GSK | Breast cancer | Resistance to HER2 TKIs: Inhibition of apoptotic pathways [65,66], activation of bypass receptors (i.e., HER3, MET, AXL, ER) [67,68,69,70], activation of downstream signaling pathways [71,72], T798I mutation [73,74]. |
| Neratinib # | Nerlynx | 2017 | Puma Biotech | Breast cancer | |||
| Tucatinib | Tukysa | 2020 | Seagen | Breast cancer | |||
| mAb/ADC | Trastuzumab | Herceptin | 1998 | Genentech | Breast cancer, gastric or GEJ adenocarcinoma | Resistance to HER2 mAbs: Expression of a truncated form of HER2 (p95HER2) [75], decreased mAb binding to the receptor due to overexpression of Mucin 4 (epitope masking) [76], activation of downstream signaling pathways [77,78,79], activation of bypass receptors [80,81,82], impaired ADCC (FcγRIII 158 V/F or 158 F/F polymorphisms) [83], loss of HER2 [84]. | |
| Pertuzumab | Perjeta | 2012 | Genentech | Breast cancer | |||
| Trastuzumab emtansine | Kadcyla | 2013 | Genentech | Breast cancer | |||
| Trastuzumab deruxtecan | Enhertu | 2019 | Daiichi Sankyo | Breast cancer, gastric or GEJ adenocarcinoma | |||
| Margetuximab | Margenza | 2020 | Macrogenics Inc | Breast cancer |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uribe, M.L.; Marrocco, I.; Yarden, Y. EGFR in Cancer: Signaling Mechanisms, Drugs, and Acquired Resistance. Cancers 2021, 13, 2748. https://doi.org/10.3390/cancers13112748
Uribe ML, Marrocco I, Yarden Y. EGFR in Cancer: Signaling Mechanisms, Drugs, and Acquired Resistance. Cancers. 2021; 13(11):2748. https://doi.org/10.3390/cancers13112748
Chicago/Turabian StyleUribe, Mary Luz, Ilaria Marrocco, and Yosef Yarden. 2021. "EGFR in Cancer: Signaling Mechanisms, Drugs, and Acquired Resistance" Cancers 13, no. 11: 2748. https://doi.org/10.3390/cancers13112748