The Unique Pharmacometrics of Small Molecule Therapeutic Drug Tracer Imaging for Clinical Oncology


1
Department of Radiology, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA
2
Department of Radiology, Weill Cornell Medical College, New York, NY 10065, USA
*
Author to whom correspondence should be addressed.
Cancers 2020, 12(9), 2712; https://doi.org/10.3390/cancers12092712
Submission received: 27 July 2020
/
Revised: 11 September 2020
/
Accepted: 17 September 2020
/
Published: 22 September 2020
(This article belongs to the Special Issue Cancer Molecular Imaging)
Abstract
:Simple Summary
New clinical radiology scans using trace amounts of therapeutic cancer drugs labeled with radioisotope injected into patients can provide oncologists with fundamentally unique insights about drug delivery to tumors. This new application of radiology aims to improve how cancer drugs are used, towards improving patient outcomes. The article reviews published clinical research in this important new field.
Abstract
Translational development of radiolabeled analogues or isotopologues of small molecule therapeutic drugs as clinical imaging biomarkers for optimizing patient outcomes in targeted cancer therapy aims to address an urgent and recurring clinical need in therapeutic cancer drug development: drug- and target-specific biomarker assays that can optimize patient selection, dosing strategy, and response assessment. Imaging the in vivo tumor pharmacokinetics and biomolecular pharmacodynamics of small molecule cancer drugs offers patient- and tumor-specific data which are not available from other pharmacometric modalities. This review article examines clinical research with a growing pharmacopoeia of investigational small molecule cancer drug tracers.
1. Introduction
Imaging in vivo pharmacokinetics and drug biodistribution, using isotopologues or radiolabeled analogues of chemically synthesized small molecule (<900 Dalton molecular weight) pharmacotherapeutics, administered in microdose (<100 micrograms, with no pharmacologic effects), radiotracer (delivered patient radiation absorbed doses comparable to those from standard radiologic scans) amounts, referred herein simply as drug tracers, is much advocated and increasingly realized in optimizing translational and early clinical phase drug development, with great potential for personalizing cancer care [1,2,3,4,5,6] (Figure 1).
Developing new cancer drugs is a slow, expensive process that usually fails [11,12,13]. The need for new target- and drug-specific pharmacometrics is recurrent and fundamental to the clinical development of new cancer pharmacotherapeutics [14,15]. A growing pharmacopoeia of investigational drug tracers have been clinically tested, with various degrees of qualification [1] as imaging biomarkers (Table 1).
Extensive published research indicates the potentially revolutionary impact of drug tracer imaging as a fundamentally new kind of pharmacometric modality, offering unique, high-impact, patient-, tumor-, and drug-specific in vivo data that can both optimize patient outcomes and economize drug development [56]. National organizations are working to facilitate validation of these and other radiologic biomarkers for the future of patient care [1,3,57,58,59,60,61,62].
The vast majority of published drug tracer research has focused on positron emission tomography (PET) imaging, which can detect picomolar amounts of radiolabeled drug in tissues in vivo [63,64]. In this article, we focus on clinical research with drug tracers developed as drug-specific companion diagnostics for cancer therapy, e.g., 18F-5-fluorouracil, a drug tracer studied for the chemotherapeutic small molecule drug 5-fluorouracil [19]. We discuss the potential of drug tracer imaging to optimize patient selection, dosing strategy, and pharmacometrics in targeted cancer therapy with small molecule drugs in ways that conventional biomarker modalities cannot [14,15,65].
2. What Does Tumor Signal on a Drug Tracer Image Signify?
Medical imaging with drug tracers and other radiotracers typically seeks to detect high tumor signal, relative to background or other reference tissues, commonly termed avidity (Figure 2). Avidity, on imaging, is a net result of biologic factors affecting drug influx, efflux, and retention in tumors [65,66] and of tracer in vivo radiopharmacology (including effective half-life) and imaging technology [67] that affect the detectability of tumor avidity on imagery. Tumor avidity for drug tracers often depends upon strong drug-target binding, i.e., irreversible or slow drug-target dissociation (kOFF). Types of drugs with rapidly reversible drug–receptor binding might not be able to produce a simple tumor avidity “hotspot” signal useful in routine radiology practice as diagnostic or predictive biomarkers, instead requiring more complex image acquisitions and data analyses [63].
Tumor avidity can be a diagnostic imaging biomarker of the molecular target to which the drug tracer binds in vivo. Tumor avidity for the isotopologue I-124 PU-H71 is a well-credentialed imaging biomarker of tumor epichaperome formations [49,68]. Epichaperomes are hyperconnected, integrated functional networks of chaperome units, induced by cellular stress as associated with malignancy [49,68,69,70]. For drugs with multiple molecular targets, tumor avidity might have in vivo specificity as a diagnostic biomarker of one drug target if a particular cancer assayed predominantly expresses one target [9,71].
Absence of tumor avidity (non-avidity) can indicate that the tumor does not express the molecular target or that other in vivo factors prevent the drug tracer from reaching and/or binding to the target [72]. For example, in vivo tumor avidity for F-18 paclitaxel varies inversely with tumor expression of the multidrug resistance (MDR) drug efflux pump; higher tumor F-18 paclitaxel avidity is associated with higher in vivo tumor drug sensitivity [30].
3. Drug Tracer Imaging to Guide Patient Selection
Establishing tumoral target expression is key for rational patient selection for a targeted therapeutic drug. Biopsy-based histologic assay is the conventional gold standard diagnostic biomarker of target expression, but small clinical studies suggest that drug tracer imaging can be a superior predictive biomarker of drug efficacy [30,72].
Our group developed a radiolabeled analog of the therapeutic tyrosine kinase inhibitor dasatinib [9] for PET imaging. Tumor resistance to dasatinib can involve altered tumor pharmacokinetics [73,74,75,76,77,78,79]. Resistant tumors may actively remove dasatinib (e.g., by efflux pumps) [73,74,75] or have mutations in kinase targets that prevent dasatinib binding [73,74,75,80].
Imaging with radiolabeled octreotide fails to detect somatostatin receptor (SSTR) expression in some tumors, but for tumors overexpressing SSTR, on histology, octreotide tracer avidity appears to stratify tumor responses to octreotide therapy [81,82].
The tyrosine kinase inhibitor erlotinib binds to epidermal growth factor receptor (EGFR) exon 19 deletions or exon 21 (L858R) substitution mutations, called sensitizing mutations, affecting the erlotinib binding site. Bahce et al. demonstrated that tumors with sensitizing mutations concentrate [C-11]-erlotinib better than tumors lacking those key mutations—despite equivalent EGFR protein expression—with non-avid tumors progressing more quickly [36].
Tumor avidity for F-18 radiolabeled paclitaxel analogue (FPAC) correlated with in vitro paclitaxel sensitivity in animal models [83] and appeared to correlate with in vivo tumor sensitivity in a small group of human subjects [30,84].
For novel therapeutic drugs that target based upon kinetic selectivity, a simple tissue-based (e.g., immunohistochemistry) target assay might not exist [85]. For example, PU-H71 kinetically selects for epichaperome-integrated HSP90; no epichaperome IHC assay exists and histologic HSP90 expression is not predictive. Tumor avidity for PU-H71 tracer predicts pharmacodynamics and efficacy for PU-H71 [7,8,49,68,69,86] and other small molecule drugs targeting the HSP90 ATPase and interfering with epichaperome formation [8,49].
4. Tumor Pharmacokinetics for Image-Guided Dosing Design
Drug tracer imaging can assay tumor pharmacokinetics, including (1) tumor drug concentrations as a function of time post-drug administration and (2) tumor occupancy by therapeutic drug doses [87]. Tumor pharmacokinetic data from patient imaging can be mathematically modeled for predicting the therapeutic dosage and schedule needed to achieve desired in vivo tumor drug concentration and tumor saturation levels [8,49] (Figure 3). Imaging biodistribution can help to establish correlations with observed organ toxicity that might enable dosing strategies optimized for the therapeutic index [65].
Pioneering clinical research in imaging in vivo tumor pharmacokinetics studied the cytotoxic drug 5-fluorouracil both by PET [88,89,90] and by in vivo nuclear magnetic resonance spectroscopy (NMRS) [18,20,21,91,92,93] and explored the microdose versus therapy dose biodistribution and pharmacokinetics of radiolabeled N-[2-(dimethylamino)ethyl]acridine-4-carboxamide (DACA) [94]. Saleem et al. demonstrated in cancer patients that the prodrug temozolomide undergoes in vivo metabolic conversion to its active form, by studying the PET isotopologue [11C]temozolomide and its in vivo radiometabolites [17]. Saleem and Price [89], analyzing accumulated PET pharmacokinetic data used in previously published studies [16,90,94], evaluated the influence of tumor perfusion on tumor pharmacokinetics.
We and others have explored clinical PET-based approaches to quantifying in vivo tumor drug molar concentrations [8,37,49,95]. Ideally, for tracing pharmacokinetics, drug tracer and (non-labeled) therapeutic drugs are isotopologues, as drug and tracer should then have identical physicochemical characteristics (Figure 1). Microdose (tracer) versus therapy dose in vivo pharmacokinetics, metabolism, and biodistribution can differ significantly, e.g., including potential tumor saturation affecting the accuracy of microdose predictions of tumor molar concentrations achieved by therapy doses. In our trials [7,8,49], we have explored therapy dose (mixing therapeutic drug and tracer together, NCT01393509) and microdose drug tracer PU-H71 imaging (NCT01269593). We validated the quantitative accuracy of non-invasive PET-based measurements of tumor PU-H71 molar concentrations, after administering therapeutic (non-radioactive) PU-H71 dose mixed with drug tracer, using liquid chromatography–tandem mass spectrometry (LC-MS/MS) of PU-H71 concentrations in biopsy specimens (NCT01393509 [8]).
Tumor avidity quantified for derivation of molar concentration must account for any tumor-residualizing radiometabolites. A drug (tracer) might have one or more active metabolites; drug tracer metabolites, produced in vivo, might be radiolabeled or unlabeled. In vivo radiometabolites can be detected by plasma sampling or, for some carbon-11 (C-11) drug tracers, in assayed exhalations [17]. I-124 PU-H71 produces three radiometabolites in vivo but these all spontaneously efflux from the tumor, simplifying tumor analysis. PU-H71 therapeutic drug and its isotopologue are both given intravenously, also simplifying our modeling of drug molar concentrations. Small molecule cancer drugs are often orally administered, with less than 100% bioavailability (compared to the 100% bioavailability of an IV-administered drug). Imaging after oral administration of a drug tracer potentially enables correlation of gastrointestinal drug transit, plasma concentrations, and tumor concentrations.
Conventional selection of a dosing strategy in early-phase therapy trials is based on preclinical PK models to predict sufficient tumor exposure to drug [65]. Our group established a preliminary dose–response relationship for PU-H71 in tumor in vitro cultures, identifying a threshold drug concentration time integral necessary to collapse tumor epichaperomes that correlated with target saturation [49,86]. We next validated the preclinical correlation between tumor pharmacokinetics and tumor pharmacodynamics in vivo, using PET and tissue assays in animal tumor models [86], and now we have explored these PK/PD correlations in cancer patients [7,8,49]. Our clinical PET imaging of tumor kinetics confirmed that tumor Cmax is key, as expected for dose-dependent responses, but our clinical data indicate that in vivo tumor concentrations required for anti-tumor efficacy are an order of magnitude higher than predicted by our prior in vitro data [8]. The PET has helped us to select a phase 2 (NCT03166085) PU-H71 therapeutic dosing strategy that we expect will saturate epichaperome-positive tumors efficaciously [8,49].
Drug tracer imaging enables a non-invasive clinical approach to quantifying in vivo tumor occupancy by therapeutic drug [37,96]. Tumor occupancy or complete saturation by drug uptake are indicated by decreased tumor uptake of drug tracer in the presence of a co-administered therapeutic drug dose, compared to tumor uptake of microdose tracer alone [37,87] (Figure 3). Tumor saturation does not necessarily indicate that all tumor molecular targets are bound by the drug; it can also indicate saturation or alteration of tumor pharmacokinetics [17]. Imaging therapeutic drug inhibition of drug tracer uptake can be used to identify the maximum single therapeutic dose that a tumor can accumulate and retain. Drug tracer imaging can detect whether or not the maximum tolerated dose of a therapeutic drug, determined empirically in phase 1 therapy dose escalation trial, is able to saturate tumors in vivo [16]. Clinical C-11 erlotinib PET imaging demonstrated variable saturation of in vivo tumor EGFR targets in NSCLC patients receiving erlotinib therapeutic doses [37].
Drug tracer imaging potentially could be incorporated into dose escalation therapy trials to detect whether tumor saturation, defined by imaging, occurs prior to reaching the maximum tolerated dose (MTD) level, defined by patient toxicity. Hypothetically, once tumor saturation is achieved, higher therapeutic dose levels might not yield proportional, incremental increases in tumor drug concentrations or responses, despite increasing dose-dependent patient toxicity [73,97,98,99]. Pharmacodynamic studies often reveal a sigmoidal dose-response curve, with “diminishing returns” at higher dose levels [100,101]. If a non-MTD, tumor-saturating dose level, which we call a maximum tumor dose, is detected by imaging, researchers could then pursue identifying the maximum tolerated schedule for that saturating dosage, modeling which steady-state peaks and trough tumor molar concentrations would be obtained [8,49,68]. For example, a lower therapeutic dose of the small molecule tyrosine kinase inhibitor dasatinib, given more frequently, was equally efficacious against leukemia and less toxic to patients, as a less frequent, higher dose strategy [102]. Drug tracer imaging is the only available modality of biomarker assay that enable clinicians to explore this novel dose-learning approach, based upon in vivo tumor pharmacokinetics, in cancer patients.
Drug tracer imaging can aid in the design of combination therapy regimens, in studying drug interactions and pharmacodynamics. Drug tracer imaging revealed an unanticipated drug interaction in an investigational therapy regimen that combined the antiangiogenic therapeutic drug bevacizumab with the cytotoxic drug docetaxel: imaging demonstrated reduced tumor uptake of (C-11) docetaxel isotopologue for four days after bevacizumab administration; these imaging data helped to guide a redesign of this dosing strategy [103]. Saleem et al. [90] studied the effects of a pharmacological intervention (eniluracil modulation) on the in vivo kinetics of the cytotoxic agent 5-fluorouracil in tumors and healthy organs by 5-[18F]-FU drug tracer imaging.
In non-human primates, Kurdzeil et al. [30] used F-18 paclitaxel (FPAC) PET to detect the in vivo effects of tariquidar, an inhibitor of the P-glycoprotein (Pgp) efflux pump that is implicated as a moderator of multidrug resistance (MDR), on paclitaxel kinetics. Tariquidar did not alter FPAC plasma kinetics, but PET successfully detected a change in FPAC biodistribution consistent with Pgp inhibition.
5. How to Make a Drug Tracer
Synthesis of PET or single photon emission computed tomography (SPECT) (i.e., radiolabeled) versions of therapeutic small molecules is usually a more challenging process than, for example, synthesis of radiolabeled versions of therapeutic antibodies. Relatively simple techniques are well-established such that each can radiolabel a wide variety of antibodies; in contrast, synthesizing a radiolabeled version of a drug or tracer that is closely related, structurally, to a drug of interest requires radiochemistry expertise to design a synthetic process that will generate a specific precursor molecule that can be radiolabeled to yield the desired radiotracer. For a drug analogue, when installing a radioisotope, a key design criterion is that the position and type of radioisotope must not significantly compromise the binding affinity of the drug analog to the molecular target(s). Much effort is spent optimizing the synthetic scheme so that radiolabeling can be done at the latest possible synthetic step to minimize potential side products and isotope decay. In general, most PET tracers containing carbon-11, nitrogen-13, fluorine-18, and iodine-124 are synthesized in a two-step method involving radiolabeling of a suitable radioactive precursor followed by deprotection. This is followed by purification, isolation, and formulation in a biocompatible buffer. In considering the position of the radiolabel on the small molecule, the general rule of thumb is that it must be both chemically and metabolically stable. For example, alkyl iodides and benzylic (-CH2) fluorides are highly labile and therefore not common in PET drugs. The available supply and chemical form of the radioisotope dictate the type of chemistry amenable to installing the radiolabel for subsequent in vivo use. Carbon-11 is routinely available as CO2 or CH4 (50–200 GBq) and has to be converted to a suitable reactive form to be installed onto a small organic molecule. Fluorine-18 is commonly available as a non-reactive fluoride (F-) (50–200 GBq) in water and must be completely dried and activated before it is incorporated into an organic molecule using nucleophilic (SN2) displacement or SNAr type reactions. Recent developments in metal-catalyzed aromatic substitution reactions have rapidly expanded the scope of the chemistry with [18F]-F- and have facilitated the production of aromatic- and heterocyclic-fluorinated PET tracers that were previously considered inaccessible with [18F]-F-. Iodine-124 is available as iodide (I-) (0.5–5 GBq) and can be easily installed on a suitable aromatic backbone using direct electrophilic iodination of activated arenes or via a variety of iodo-demetallation reactions on inactivated arene backbones.
In developing drug tracers for human studies, a suitable radiosynthetic method and precursor must facilitate (1) easy, scalable, and reproducible radiochemical yields, (2) automation, (3) easy purification, and (4) formulation in a biocompatible solvent. The radiochemical precursor is a suitable compound whose chemical manipulation with radioactive elements yields the intermediate product that can be converted to the desired final radioactive product or is sometimes the final product itself. The precursor must be reactive enough to facilitate chemical/radiochemical manipulations but stable enough to be vialed and stored for future use (up to two years) without loss of radiochemical yields, isomerization, or chemical transformation that can lead to undesirable products. The precursor and its breakdown products during radiochemical reactions must be easily separable from the final radiochemical compound to ensure that the final product is not only radiochemically pure but also chemically pure. An additional consideration in the precursor design is whether the compound contains toxic elements (e.g., tin, lead, or mercury); a suitable analysis method must be developed to quantify concentrations in the final product. In developing a suitable radiosynthetic method, solvent optimization is essential for each step to address logistical challenges related to automation, purification, and/or formulation due to compatibility, miscibility, corrosiveness, high boiling points, etc. Minimizing the use of HPLC is also advantageous, as is ensuring that separation of the final product can be achieved through physical manipulation.
Once a reproducible synthesis of a candidate drug tracer has been developed, this candidate is subjected to a battery of in vitro and in vivo tests. In vitro testing must demonstrate high affinity binding/avidity of the drug tracer to its target and its ability to be blocked by a suitable competitor, including the drug itself. For example, in designing F-18 SKI-249380, a tracer of the small molecule therapeutic drug dasatinib, we placed a positron-emitting fluorine isotope (F-18) atom to substitute for a hydroxyl group [9,10,71]. Molecular modeling predicted that the substitution had a minimal effect upon drug binding to target kinases, which we confirmed by comparing in vitro pharmacodynamic profiles and potencies and in vivo imaging of competitive inhibition of tracer uptake by a co-administered dasatinib therapeutic dose.
Another important criterion is to ensure that the drug tracer has minimal non-specific binding in the cell or system. This is followed by in vivo testing, in suitable animal models, to demonstrate drug tracer uptake by the tumor or other target-containing tissue(s). This becomes extremely important for privileged organs such as the brain, cerebrospinal fluid (CSF), or tumor, where barriers (e.g., blood–brain barrier, interstitial pressure, low vascularity, etc.) can severely restrict penetration of the drug into the tissue of interest. If the animal imaging studies demonstrate feasibility of using the candidate molecule as a drug tracer, the blood half-life and metabolic profile of the compound is then assessed to demonstrate that the signal from the tissue of interest is arising due to specific binding of the drug tracer to its target rather than merely due to blood pool activity (e.g., high plasma protein binding) or a radioactive metabolic byproduct. For the promising candidate tracer, animal dosimetry studies then measure radioactive dose to organs (mouse); the results are then extrapolated to predict human absorbed and effective doses. To pursue clinical translation to first-in-human studies, further requirements typically include animal toxicology studies, GMP-quality radiochemistry synthesis, radiotracer synthesis validation runs, and institutional and regulatory agency (e.g., FDA) approval of a clinical research protocol embedded within an investigational new drug application package. Radiolabeled small molecule drugs are usually synthesized with a high specific activity, meaning that patients receiving the radiotracer injection, sufficient for imaging of drug biodistribution, often receive only a “microdose” of drug (<100 micrograms). As such, small molecule drug tracers for microdose imaging typically demonstrate no pharmacodynamic effect or pharmacologic toxicity.
6. Does Drug Tracer Imaging Offer Any Advantages over Conventional Tissue-Based PK/PD Biomarkers?
Drug tracer imaging offers potentially revolutionary, multifaceted patient-, tumor-, drug-, and target-specific biomarker data that tissue-based assays cannot, including tumor saturation, drug molar concentrations and kinetics, tumor heterogeneity, and biodistribution data.
Plasma pharmacokinetics, though essential data, often fail to predict tumor pharmacokinetics and response, for which tracer imaging of tumor pharmacokinetics might be a better predictive biomarker [6,8,49,90,98,99,104]. For PU-H71, we found that not only is the maximal tumor concentration achieved important for drug efficacy, but maintaining threshold tumor concentrations for a threshold duration of time was also key [86], whereas plasma pharmacokinetics did not differ significantly between responders and non-responders [8]. For drug administration to specific compartments (e.g., intrathecal, intraperitoneal, intratumoral), plasma pharmacokinetics do not provide a reliable surrogate of drug concentrations and distributions within those spaces and immediately surrounding tissues. The ability to correlate tumor response to tumor dose, not merely to the administered or plasma dose, advances the precision of dose–response analyses from patient- to lesion-level [88,105].
Tumor heterogeneity refers to significant intra- and inter-tumor variations in molecular target expression. Quantifying target expression by imaging of drug tracer uptake throughout a single tumor, as well as in all tumors in a given patient, might achieve stronger correlations with tumor response and insights into mixed tumor responses [72]. Needle biopsy collects biomarker data from only a small sliver of a tumor. Biofluid biomarkers or surrogates, such as circulating tumor cells, offer only a general correlate of systemic tumor burden and biomarker expression.
Drug tracer imaging enables four-dimensional whole-body assessment of drug tracer biodistribution across serial scan time-points. Biodistribution imaging data can reveal unexpected drug tracer accumulation in certain organs that might prompt serum or other assays to monitor for toxicity in that organ during clinical therapy trials, potentially improving patient safety and clarifying drug toxicity profile [106]. Biodistribution might identify compartments of poor drug penetration. PET isotopologues have been used to quantify the blood–brain barrier penetration of small molecule drugs in early-phase development [35,37,40], thereby predicting efficacy against brain metastases and encouraging further testing [34]. Clinical research with a temozolomide drug tracer, when temozolomide was an investigational therapeutic, demonstrated drug uptake in glioma brain tumors with scant drug retention by normal brain, predicting a favorable therapeutic index [17]. Similar clinical data were obtained with a lapatinib drug tracer in breast cancer patients with brain metastases [107,108].
7. Conclusions
Clinical PET assays of tumor targeting by small molecule drugs provide a fundamentally new mode of perception for cancer therapy investigators, enabling clinical testing of new paradigms in targeted drug development. As a pharmacometric modality, clinical PET imaging-based microdose studies, using a trace (<100 micrograms) amount of drug isotopologue or radioanalogue, offer patient-, drug-, and tumor-specific in vivo data that conventional plasma- or biopsy-based pharmacokinetic and biomarker assays cannot.
Author Contributions
Authors M.P.S.D. and N.P. both contributed literature review, text drafting and editing for this monograph. All authors have read and agreed to the published version of the manuscript.
Funding
I-124 PU-H71 research was funded in part through the NIH/NCI Cancer Center Support Grant P30 CA008748, as well as by NIH grants R01 CA172546, P01 CA186866, R56 AG061869, P50 CA192937, R21 CA167803, and R35 CA232130. Samus Therapeutics Inc., the Jane H. Gordon Breast Cancer Research Fund, the Breast Cancer Research Fund, Susan G. Komen for the Cure, and the Experimental Therapeutics Center of MSK also provided support for this research. F-18 SKI249380 research was supported by NIH/NCI grants R21 CA167803, P50 CA086438, R01 CA172546, and R01 CA164490. Memorial Sloan Kettering core facilities are supported by NIH/NCI Cancer Center Support Grant P30 CA008748.
Acknowledgments
The authors gratefully acknowledge manuscript review by Gabriela Chiosis and editorial assistance by Leah Bassity.
Conflicts of Interest
Memorial Sloan Kettering Cancer Center holds the intellectual rights to PU-H71 and [124I]-PU-H71 (PCT/US06/03676, PCT/US2012/045861), which have been licensed to Samus Therapeutics. M.P.S.D. and N.P. have contributed to the patents and have received financial royalties for their IP contributions.
References
- O’Connor, J.P.B.; Aboagye, E.O.; Adams, J.E.; Aerts, H.J.W.L.; Barrington, S.; Beer, A.J.; Boellaard, R.; Bohndiek, S.E.; Brady, M.; Brown, G.; et al. Imaging biomarker roadmap for cancer studies. Nat. Rev. Clin. Oncol. 2017, 14, 169–186. [Google Scholar] [CrossRef] [PubMed]
- Workman, P.; Aboagye, E.O.; Chung, Y.-L.; Griffiths, J.R.; Hart, R.; Leach, M.O.; Maxwell, R.J.; McSheehy, P.M.J.; Price, P.M.; Zweit, J. Minimally invasive pharmacokinetic and pharmacodynamic technologies in hypothesis-testing clinical trials of innovative therapies. J. Natl. Cancer Inst. 2006, 98, 580–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Cancer Institute U.S. National Institutes of Health. A Workshop Regarding What In-Vivo Molecular Imaging Probes Are Needed to Support Future Translational Studies in Cancer Therapeutics. In Strategies for Imaging Priority Targets; NCI: Washington, DC, USA, 2002. Available online: www.cancer.gov (accessed on 1 September 2020).
- Weber, W.A.; Czernin, J.; Phelps, M.E.; Herschman, H.R. Technology Insight: Novel imaging of molecular targets is an emerging area crucial to the development of targeted drugs. Nat. Clin. Pract. Oncol. 2008, 5, 44–54. [Google Scholar] [CrossRef] [Green Version]
- Workman, P.; Burrows, F.; Neckers, L.; Rosen, N. Drugging the cancer chaperone HSP90: Combinatorial therapeutic exploitation of oncogene addiction and tumor stress. Ann. N. Y. Acad. Sci. 2007, 1113, 202–216. [Google Scholar] [CrossRef]
- Seddon, B.M.; Workman, P. The role of functional and molecular imaging in cancer drug discovery and development. Br. J. Radiol. 2003, 76 (Suppl. S2), S128–S138. [Google Scholar] [CrossRef]
- Dunphy, M.P.; Pressl, C.; Pillarsetty, N.; Grkovski, M.; Modi, S.; Jhaveri, K.; Norton, L.; Beattie, B.J.; Zanzonico, P.B.; Zatorska, D.; et al. First-in-Human Trial of Epichaperome-Targeted Positron Emission Tomography in Cancer Patients. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Pillarsetty, N.; Jhaveri, K.; Taldone, T.; Caldas-Lopes, E.; Punzalan, B.; Joshi, S.; Bolaender, A.; Uddin, M.M.; Rodina, A.; Yan, P.; et al. Paradigms for Precision Medicine in Epichaperome Cancer Therapy. Cancer Cell 2019, 36, 559–573. [Google Scholar] [CrossRef]
- Krebs, S.; Veach, D.R.; Carter, L.M.; Grkovski, M.; Fornier, M.; Mauro, M.J.; Voss, M.H.; Danila, D.C.; Burnazi, E.; Null, M.; et al. First-in-Human Trial of Dasatinib-Derivative Tracer for Tumor Kinase-Targeted Positron Emission Tomography. J. Nucl. Med. 2020. [Google Scholar] [CrossRef]
- Veach, D.R.; Namavari, M.; Pillarsetty, N.; Santos, E.B.; Beresten-Kochetkov, T.; Lambek, C.; Punzalan, B.J.; Antczak, C.; Smith-Jones, P.M.; Djaballah, H.; et al. Synthesis and biological evaluation of a fluorine-18 derivative of dasatinib. J. Med. Chem. 2007, 50, 5853–5857. [Google Scholar] [CrossRef]
- Schuhmacher, A.; Gassmann, O.; Hinder, M. Changing R & D models in research-based pharmaceutical companies. J. Transl. Med. 2016, 14, 105. [Google Scholar]
- Wouters, O.J.; McKee, M.; Luyten, J. Estimated research and development investment needed to bring a new medicine to market, 2009–2018. JAMA 2020, 323, 844–853. [Google Scholar] [CrossRef]
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the pharmaceutical industry: New estimates of R & D costs. J. Health Econ. 2016, 47, 20–33. [Google Scholar]
- Peck, C.C.; Cross, J.T. “Getting the dose right”: Facts, a blueprint, and encouragements. Clin. Pharmacol. Ther. 2007, 82, 12–14. [Google Scholar] [CrossRef]
- Powell, J.R.; Gobburu, J.V. Pharmacometrics at FDA: Evolution and impact on decisions. Clin. Pharmacol. Ther. 2007, 82, 97–102. [Google Scholar] [CrossRef]
- Propper, D.; De Bono, J.; Saleem, A.; Ellard, S.; Flanagan, E.; Paul, J.; Ganesan, T.; Talbot, D.; O Aboagye, E.; Price, P.; et al. Use of positron emission tomography in pharmacokinetic studies to investigate therapeutic advantage in a phase I study of 120-hour intravenous infusion XR5000. J. Clin. Oncol. 2003, 21, 203–210. [Google Scholar] [CrossRef]
- Saleem, A.; Brown, G.D.; Brady, F.; Aboagye, E.O.; Osman, S.; Luthra, S.K.; O Ranicar, A.S.; Brock, C.S.; Stevens, M.F.G.; Newlands, E.; et al. Metabolic activation of temozolomide measured in vivo using positron emission tomography. Cancer Res. 2003, 63, 2409–2415. [Google Scholar]
- Presant, C.; Wolf, W.; Waluch, V.; Wiseman, C.; Kennedy, P.; Blayney, D.; Brechner, R.; Kennedy, D. Association of intratumoral pharmacokinetics of fluorouracil with clinical response. Lancet 1994, 343, 1184–1187. [Google Scholar] [CrossRef]
- Kissel, J.; Brix, G.; Bellemann, M.E.; Strauss, L.G. Dimitrakopoulou-Strauss, A.; Port, R.; Haberkorn, U.; Lorenz, W.J. Pharmacokinetic analysis of 5-[18F] fluorouracil tissue concentrations measured with positron emission tomography in patients with liver metastases from colorectal adenocarcinoma. Cancer Res. 1997, 57, 3415–3423. [Google Scholar]
- Presant, C.A.; Wolf, W.; Waluch, V.; Wiseman, C.L.; Weitz, I.; Shani, J. Enhancement of fluorouracil uptake in human colorectal and gastric cancers by interferon or by high-dose Methotrexate: An in vivo human study using noninvasive 19f-magnetic resonance spectroscopy. J. Clin. Oncol. 2000, 18, 255. [Google Scholar] [CrossRef]
- Presant, C.A.; Jacobson, J.; Wolf, W.; Waluch, V.; Weitz, I.C.; Macdonald, J.S. Does leucovorin alter the intratumoral pharmacokinetics of 5-fluorouracil (5-fu)? A southwest oncology group study. Investig. New Drugs 2002, 20, 369–376. [Google Scholar] [CrossRef]
- Levi, J.; Lam, T.; Goth, S.R.; Yaghoubi, S.; Bates, J.; Ren, G.; Jivan, S.; Huynh, T.; Blecha, J.E.; Khattri, R.; et al. Imaging of activated t cells as an early predictor of immune response to anti-PD-1 Therapy. Cancer Res. 2019, 79, 3455–3465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, W.; Le, T.M.; Wei, L.; Poddar, S.; Bazzy, J.; Wang, X.; Uong, N.T.; Abt, E.R.; Capri, J.R.; Austin, W.R.; et al. [18F]CFA as a clinically translatable probe for PET imaging of deoxycytidine kinase activity. Proc. Natl. Acad. Sci. USA 2016, 113, 4027–4032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.M.; Chiu, C.H.; Chen, W.T.; Wu, C.H.; Lin, P.Y.; Huang, Y.Y.; Chen, J.-H.; Tzen, K.-Y.; Shiue, C.-Y.; Lin, W.-J. Evaluation of 5-[(18)F]fluoro-2′-deoxycytidine as a tumor imaging agent: A comparison of [(18)F]FdUrd, [(18)F]FLT and [(18)F]FDG. Appl. Radiat. Isot. 2019, 148, 152–159. [Google Scholar] [CrossRef]
- Young, C.R.; Adler, S.; Eary, J.F.; Lindenberg, M.L.; Jacobs, P.M.; Collins, J.; Kummar, S.; Kurdziel, K.A.; Choyke, P.L.; Mena, E. Biodistribution, tumor detection, and radiation dosimetry of (18)F-5-Fluoro-2′-deoxycytidine with tetrahydrouridine in solid tumors. J. Nucl. Med. 2019, 60, 492–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muthu, P.; Chen, H.X.; Lutz, S. Redesigning human 2′-deoxycytidine kinase enantioselectivity for L-nucleoside analogues as reporters in positron emission tomography. ACS Chem. Biol. 2014, 9, 2326–2333. [Google Scholar] [CrossRef]
- Boothman, D.A.; Briggle, T.V.; Greer, S. Protective, tumor-selective dual pathway activation of 5-fluoro-2′-deoxycytidine provided by tetrahydrouridine in mice bearing mammary adenocarcinoma-755. Cancer Res. 1987, 47, 2344–2353. [Google Scholar]
- Radu, C.G.; Shu, C.J.; Nair-Gill, E.; Shelly, S.M.; Barrio, J.R.; Satyamurthy, N.; Phelps, M.E.; Witte, O.N. Molecular imaging of lymphoid organs and immune activation by positron emission tomography with a new [18F]-labeled 2′-deoxycytidine analog. Nat. Med. 2008, 14, 783–788. [Google Scholar] [CrossRef] [Green Version]
- Kurdziel, K.A.; Kiesewetter, D.O.; Carson, R.E.; Eckelman, W.C.; Herscovitch, P. Biodistribution, radiation dose estimates, and in vivo Pgp modulation studies of 18F-paclitaxel in nonhuman primates. J. Nucl. Med. 2003, 44, 1330–1339. [Google Scholar]
- Kurdziel, K.A.; Kalen, J.D.; Hirsch, J.I.; Wilson, J.D.; Agarwal, R.; Barrett, D.; Bear, H.D.; McCumiskey, J.F. Imaging multidrug resistance with 4-[18F]fluoropaclitaxel. Nucl. Med. Biol. 2007, 34, 823–831. [Google Scholar] [CrossRef]
- Gangloff, A.; Hsueh, W.-A.; Kesner, A.L.; O Kiesewetter, D.; Pio, B.S.; Pegram, M.D.; Beryt, M.; Townsend, A.; Czernin, J.; E Phelps, M.; et al. Estimation of paclitaxel biodistribution and uptake in human-derived xenografts in vivo with (18)F-fluoropaclitaxel. J. Nucl. Med. 2005, 46, 1866–1871. [Google Scholar]
- Kurdziel, K.A.; Kiesewetter, D.O. PET imaging of multidrug resistance in tumors using 18F-fluoropaclitaxel. Curr. Top. Med. Chem. 2010, 10, 1792–1798. [Google Scholar] [CrossRef] [PubMed]
- Fei, X.; Wang, J.-Q.; Miller, K.D.; Sledge, G.W.; Hutchins, G.D.; Zheng, Q.-H. Synthesis of [18F]Xeloda as a novel potential PET radiotracer for imaging enzymes in cancers. Nucl. Med. Biol. 2004, 31, 1033–1041. [Google Scholar] [CrossRef] [PubMed]
- Bauer, T.M.; Shaw, A.T.; Johnson, M.L.; Navarro, A.; Gainor, J.F.; Thurm, H.; Pithavala, Y.K.; Abbattista, A.; Peltz, G.; Felip, E. Brain penetration of lorlatinib: Cumulative incidences of CNS and Non-CNS progression with lorlatinib in patients with previously treated ALK-positive non-small-cell lung cancer. Target. Oncol. 2020, 15, 55–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collier, T.L.; Normandin, M.D.; Stephenson, N.A.; Livni, E.; Liang, S.H.; Wooten, D.W.; Esfahani, S.A.; Stabin, M.G.; Mahmood, U.; Chen, J.; et al. Synthesis and preliminary PET imaging of 11C and 18F isotopologues of the ROS1/ALK inhibitor lorlatinib. Nat. Commun. 2017, 8, 15761. [Google Scholar] [CrossRef]
- Bahce, I.; Smit, E.F.; Lubberink, M.; Van Der Veldt, A.A.M.; Yaqub, M.; Windhorst, A.D.; Schuit, R.C.; Thunnissen, E.; Heideman, D.A.M.; Postmus, P.E.; et al. Development of [(11)C]erlotinib positron emission tomography for in vivo evaluation of EGF receptor mutational status. Clin. Cancer Res. 2013, 19, 183–193. [Google Scholar] [CrossRef] [Green Version]
- Bahce, I.; Yaqub, M.; Errami, H.; Schuit, R.C.; Schober, P.; Thunnissen, E.; Windhorst, A.D.; Lammertsma, A.A.; Smit, E.F.; Hendrikse, N.H. Effects of erlotinib therapy on [11C]erlotinib uptake in EGFR mutated, advanced NSCLC. EJNMMI Res. 2016, 6, 10. [Google Scholar] [CrossRef] [Green Version]
- Slobbe, P.; Windhorst, A.D.; Walsum, M.S.-V.; Smit, E.F.; Niessen, H.G.; Solca, F.; Stehle, G.; Van Dongen, G.A.M.S.; Poot, A.J. A comparative PET imaging study with the reversible and irreversible EGFR tyrosine kinase inhibitors [(11)C]erlotinib and [(18)F]afatinib in lung cancer-bearing mice. EJNMMI Res. 2015, 5, 14. [Google Scholar] [CrossRef] [Green Version]
- Chopra, A.; Shan, L.; Eckelman, W.C.; Leung, K.; Latterner, M.; Bryant, S.H.; Menkens, A. [(11)C]N-(3-Ethynylphenyl)-6,7-bis(2-methoxyethoxy)-4-quinazolinamine. In Molecular Imaging and Contrast Agent Database (MICAD). Mol. Imaging Biol. 2012, 14, 4–13. [Google Scholar] [CrossRef] [Green Version]
- Varrone, A.; Varnäs, K.; Jucaite, A.; Cselényi, Z.; Johnström, P.; Schou, M.; Vazquez-Romero, A.; Moein, M.M.; Halldin, C.; Brown, A.P.; et al. A PET study in healthy subjects of brain exposure of (11)C-labelled osimertinib—A drug intended for treatment of brain metastases in non-small cell lung cancer. J. Cereb. Blood Flow Metab. 2019. [Google Scholar] [CrossRef]
- Huang, S.; Han, Y.; Chen, M.; Hu, K.; Qi, Y.; Sun, P.; Wang, M.; Wu, H.; Li, G.; Wang, Q.; et al. Radiosynthesis and biological evaluation of (18)F-labeled 4-anilinoquinazoline derivative ((18)F-FEA-Erlotinib) as a potential EGFR PET agent. Bioorg. Med. Chem. Lett. 2018, 28, 1143–1148. [Google Scholar] [CrossRef]
- Marijn, V.; Läppchen, T.; Jansen, H.T.; Kivits, S.; Van Driel, A.; Van De Steeg, E.; Van Der Hoorn, J.W.; Sio, C.F.; Steinbach, O.C.; DeGroot, J. PET-CT imaging with [(18)F]-gefitinib to measure Abcb1a/1b (P-gp) and Abcg2 (Bcrp1) mediated drug-drug interactions at the murine blood-brain barrier. Nucl. Med. Biol. 2015, 42, 833–841. [Google Scholar]
- Song, Y.; Xiao, Z.; Wang, K.; Wang, X.; Zhang, C.; Fang, F.; Sun, X.; Shen, B. Development and Evaluation of (18)F-IRS for Molecular Imaging Mutant EGF Receptors in NSCLC. Sci. Rep. 2017, 7, 3121. [Google Scholar] [CrossRef] [PubMed]
- Slobbe, P.; Poot, A.J.; Haumann, R.; Schuit, R.C.; Windhorst, A.D.; Van Dongen, G.A.M.S. Two anti-angiogenic TKI-PET tracers, [(11)C]axitinib and [(11)C]nintedanib: Radiosynthesis, in vivo metabolism and initial biodistribution studies in rodents. Nucl. Med. Biol. 2016, 43, 612–624. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Xie, L.; Zhang, Y.; Fujinaga, M.; Mori, W.; Kurihara, Y.; Yamasaki, T.; Wang, F.; Zhang, M.-R. Pharmacokinetic evaluation of [(11)C]CEP-32496 in nude mice Bearing BRAF(V600E) mutation-induced melanomas. Mol. Imaging 2018, 17, 1536012118795952. [Google Scholar] [CrossRef] [Green Version]
- Saleem, A.; Searle, G.E.; Kenny, L.M.; Huiban, M.; Kozlowski, K.; Waldman, A.; Woodley, L.; Palmieri, C.; Lowdell, C.; Kaneko, T.; et al. Lapatinib access into normal brain and brain metastases in patients with Her-2 overexpressing breast cancer. EJNMMI Res. 2015, 5, 30. [Google Scholar] [CrossRef] [Green Version]
- Poot, A.J.; Van Der Wildt, B.; Walsum, M.S.-V.; Rongen, M.; Schuit, R.C.; Hendrikse, N.H.; Eriksson, J.; Van Dongen, G.A.M.S.; Windhorst, A.D. [11C] Sorafenib: Radiosynthesis and preclinical evaluation in tumor-bearing mice of a new TKI-PET tracer. Nucl. Med. Biol. 2013, 40, 488–497. [Google Scholar] [CrossRef] [Green Version]
- NCT02700763. [18F] Dabrafenib Molecular Imaging in Melanoma Brain Metastasis. Available online: www.clinicaltrials.gov (accessed on 1 September 2020).
- Rodina, A.; Wang, T.; Yan, P.; Gomes, E.D.; Dunphy, M.P.S.; Pillarsetty, N.; Koren, J.I.; Gerecitano, J.F.; Taldone, T.; Zong, H.; et al. The epichaperome is an integrated chaperome network that facilitates tumour survival. Nature 2016, 538, 397–401. [Google Scholar] [CrossRef]
- Di Gialleonardo, V.; Signore, A.; Glaudemans, A.W.; Dierckx, R.A.J.O.; De Vries, E.F. N-(4-18F-fluorobenzoyl)interleukin-2 for PET of human-activated T lymphocytes. J. Nucl. Med. 2012, 53, 679–686. [Google Scholar] [CrossRef] [Green Version]
- Hartimath, S.V.; Manuelli, V.; Zijlma, R.; Signore, A.; Nayak, T.K.; Freimoser-Grundschober, A.; Klein, C.; Dierckx, R.A.; De Vries, E. Pharmacokinetic properties of radiolabeled mutant Interleukin-2v: A PET imaging study. Oncotarget 2018, 9, 7162–7174. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Q.-H.; Fei, X.; DeGrado, T.R.; Wang, J.-Q.; Stone, K.L.; Martinez, T.D.; Gay, D.J.; Baity, W.L.; Mock, B.H.; E Glick-Wilson, B.; et al. Synthesis, biodistribution and micro-PET imaging of a potential cancer biomarker carbon-11 labeled MMP inhibitor (2R)-2-[[4-(6-fluorohex-1-ynyl)phenyl]sulfonylamino]-3-methylbutyric acid [11C]methyl ester. Nucl. Med. Biol. 2003, 30, 753–760. [Google Scholar] [CrossRef]
- Han, N.; Jiang, Y.; Gai, Y.; Liu, Q.; Yuan, L.; Wang, Y.; Li, M.; Zhang, Y.; Lan, X. (11)C-Labeled Pictilisib (GDC-0941) as a molecular tracer targeting phosphatidylinositol 3-Kinase (PI3K) for breast cancer imaging. Contrast Media Mol. Imaging 2019, 2019, 1760184. [Google Scholar] [CrossRef]
- Donnelly, D.J.; Smith, R.A.; Morin, P.; Lipovsek, D.; Gokemeijer, J.; Cohen, D.; Lafont, V.; Tran, T.; Cole, E.L.; Wright, M.; et al. Synthesis and Biologic Evaluation of a Novel (18)F-Labeled Adnectin as a PET Radioligand for Imaging PD-L1 Expression. J. Nucl. Med. 2018, 59, 529–535. [Google Scholar] [CrossRef] [Green Version]
- Makvandi, M.; Pantel, A.; Schwartz, L.E.; Xu, K.; Hsieh, C.-J.; Kim, H.; Li, S.-H.; Doot, R.; Lee, S.; Simpkins, F.; et al. Abstract 3716: Exploring the significance of PARP-1 expression for therapy and clinical PET/CT imaging of PARP-1 in ovarian cancer. Cancer Res. 2017, 77 (Suppl. S13), 3716. [Google Scholar]
- Graham, M.M.; Weber, W.A. Evaluation of the Efficacy of Targeted Imaging Agents. J. Nucl. Med. 2016, 57, 653–659. [Google Scholar] [CrossRef] [Green Version]
- European Imaging Biomarkers Alliance. Available online: http://www.eibir.org/scientific-activities/joint-initiatives/eiball/ (accessed on 1 September 2020).
- FDA Biomarker Qualification Program. Available online: https://www.fda.gov/Drugs/DevelopmentApprovalProcess/DrugDevelopmentToolsQualificationProgram/BiomarkerQualificationProgram/default.htm (accessed on 1 September 2020).
- FDA-NIH Biomarker Working Group. BEST (Biomarkers, EndpointS, and Other Tools) Resource. 2020. Available online: https://www.ncbi.nlm.nih.gov/books/NBK326791/ (accessed on 29 May 2020).
- National Biomarker Development Alliance. Available online: http://nbdabiomarkers.org (accessed on 1 September 2020).
- National Cancer Institute (NCI) Cancer Imaging Program. Available online: https://imaging.cancer.gov/default.htm (accessed on 1 September 2020).
- NCI Clinical Trials Working Group (CTWG) Biomarker Study Evaluation Guidelines. Available online: https://www.cancer.gov/about-nci/organization/ccct/funding/biqsfp/2017-biomarker-study-eval-guide.pdf (accessed on 1 September 2020).
- Bergström, M.; Grahnén, A.; Långström, B. Positron emission tomography microdosing: A new concept with application in tracer and early clinical drug development. Eur. J. Clin. Pharmacol. 2003, 59, 357–366. [Google Scholar] [CrossRef]
- Westerterp, M.; Pruim, J.; Oyen, W.J.; Hoekstra, O.; Paans, A.; Visser, E.; Van Lanschot, J.; Sloof, G.; Boellaard, R. Quantification of FDG PET studies using standardised uptake values in multi-centre trials: Effects of image reconstruction, resolution and ROI definition parameters. Eur. J. Nucl. Med. Mol. Imaging 2007, 34, 392–404. [Google Scholar] [CrossRef]
- Banerji, U.; Workman, P. Critical parameters in targeted drug development: The pharmacological audit trail. Semin. Oncol. 2016, 43, 436–445. [Google Scholar] [CrossRef] [Green Version]
- Jain, R.K. Delivery of molecular and cellular medicine to solid tumors. Adv. Drug Deliv. Rev. 2012, 64, 353–365. [Google Scholar] [CrossRef] [Green Version]
- Badawi, R.D.; Shi, H.; Hu, P.; Chen, S.; Xu, T.; Price, P.M.; Ding, Y.; Spencer, B.A.; Nardo, L.; Liu, W.; et al. First human imaging studies with the EXPLORER total-body pet scanner. J. Nucl. Med. 2019, 60, 299–303. [Google Scholar] [CrossRef]
- Wang, T.; Rodina, A.; Dunphy, M.P.; Corben, A.; Modi, S.; Guzman, M.L.; Gewirth, D.T.; Chiosis, G. Chaperome heterogeneity and its implications for cancer study and treatment. J. Biol. Chem. 2019, 294, 2162–2179. [Google Scholar] [CrossRef] [Green Version]
- Taldone, T.; Wang, T.; Rodina, A.; Pillarsetty, N.V.K.; Digwal, C.S.; Sharma, S.; Yan, P.; Joshi, S.; Pagare, P.P.; Bolaender, A.; et al. A chemical biology approach to the chaperome in cancer-HSP90 and beyond. Cold Spring Harb. Perspect Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Wang, T.; Araujo, T.L.S.; Sharma, S.; Brodsku, J.L.; Chiosis, G. Adapting to stress—Chaperome networks in cancer. Nat. Rev. Cancer 2018, 18, 562–575. [Google Scholar] [CrossRef] [PubMed]
- Dunphy, M.P.; Zanzonico, P.; Veach, D.; Somwar, R.; Pillarsetty, N.; Lewis, J.S.; Larson, S. Dosimetry of (18)F-labeled tyrosine kinase inhibitor SKI-249380, a dasatinib-tracer for PET imaging. Mol. Imaging Biol. 2010. [Google Scholar] [CrossRef] [Green Version]
- Ulaner, G.A.; Hyman, D.M.; Lyashchenko, S.K.; Lewis, J.S.; Carrasquillo, J.A. 89Zr-Trastuzumab PET/CT for Detection of Human Epidermal Growth Factor Receptor 2-Positive Metastases in Patients With Human Epidermal Growth Factor Receptor 2-Negative Primary Breast Cancer. Clin. Nucl. Med. 2017, 42, 912–917. [Google Scholar] [CrossRef]
- Luo, F.R.; Barrett, Y.; Ji, P.; Holly, P.; McCann, E.; Rhyne, P.; Clarke, E.; He, K.; Bleichardt, E.; Blackwood-Chirchir, M. Dasatinib (BMS-354825) pharmacokinetics correlate with pSRC pharmacodynamics in phase I studies of patients with cancer (CA180002, CA180003). J. Clin. Oncol. Meet. Abstr. 2006, 24 (Suppl. S18), 3046. [Google Scholar] [CrossRef]
- Hiwase, D.K.; Saunders, V.; Hewett, D.; Frede, A.; Zrim, S.; Dang, P.; Eadie, L.N.; To, L.B.; Melo, J.; Kumar, S.; et al. Dasatinib cellular uptake and efflux in chronic myeloid leukemia cells: Therapeutic implications. Clin. Cancer Res. 2008, 14, 3881–3888. [Google Scholar] [CrossRef] [Green Version]
- Luo, F.R.; Yang, Z.; Camuso, A.; Smykla, R.; McGlinchey, K.; Fager, K.; Flefleh, C.; Castaneda, S.; Inigo, I.; Kan, D.; et al. Dasatinib (BMS-354825) pharmacokinetics and pharmacodynamic biomarkers in animal models predict optimal clinical exposure. Clin. Cancer Res. 2006, 12, 7180–7186. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Agarwal, S.; Shaik, N.M.; Chen, C.; Yang, Z.; Elmquist, W.F. P-glycoprotein and breast cancer resistance protein influence brain distribution of dasatinib. J. Pharmacol. Exp. Ther. 2009, 330, 956–963. [Google Scholar] [CrossRef] [Green Version]
- Giannoudis, A.; Davies, A.; Lucas, C.M.; Harris, R.J.; Pirmohamed, M.; Clark, R.E. Effective dasatinib uptake may occur without human organic cation transporter 1 (hOCT1): Implications for the treatment of imatinib-resistant chronic myeloid leukemia. Blood 2008, 112, 3348–3354. [Google Scholar] [CrossRef] [Green Version]
- Lagas, J.S.; Van Waterschoot, R.A.; Van Tilburg, V.A.; Hillebrand, M.J.; Lankheet, N.; Rosing, H.; Beijnen, J.H.; Schinkel, A.H. Brain accumulation of dasatinib is restricted by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) and can be enhanced by elacridar treatment. Clin. Cancer Res. 2009, 15, 2344–2351. [Google Scholar] [CrossRef] [Green Version]
- Porkka, K.; Koskenvesa, P.; Lundán, T.; Rimpiläinen, J.; Mustjoki, S.; Smykla, R.; Wild, R.; Luo, R.; Arnan, M.; Brethon, B.; et al. Dasatinib crosses the blood-brain barrier and is an efficient therapy for central nervous system Philadelphia chromosome-positive leukemia. Blood 2008, 112, 1005–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, M.R.; Skaggs, B.J.; Shah, N.P.; Lee, F.Y.; Sawyers, C.L. Comparative analysis of two clinically active BCR-ABL kinase inhibitors reveals the role of conformation-specific binding in resistance. Proc. Natl. Acad. Sci. USA 2005, 102, 3395–3400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamberts, S.; Barker, W.; Reubi, J.-C.; Krenning, E. Somatostatin-receptor imaging in the localization of endocrine tumors. N. Engl. J. Med. 1990, 323, 1246–1249. [Google Scholar] [CrossRef] [PubMed]
- Lamberts, S.W.; Hofland, L.J.; van Koetsveld, P.M.; Reubi, J.C.; Bruining, H.A.; Barker, W.H.; Krenning, E.P. Parallel in vivo and in vitro detection of functional somatostatin receptors in human endocrine pancreatic tumors: Consequences with regard to diagnosis, localization, and therapy. J. Clin. Endocrinol. Metab. 1990, 71, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, W.-A.; Kesner, A.L.; Gangloff, A.; Pegram, M.D.; Beryt, M.; Czernin, J.; Phelps, M.E.; Silverman, D.H.S. Predicting chemotherapy response to paclitaxel with 18F-Fluoropaclitaxel and PET. J. Nucl. Med. 2006, 47, 1995–1999. [Google Scholar]
- Kurdziel, K.A.; Kalen, J.D.; Hirsch, J.I.; Wilson, J.D.; Bear, H.D.; Logan, J.; McCumisky, J.; Moorman-Sykes, K.; Adler, S.; Choyke, P.L. Human dosimetry and preliminary tumor distribution of 18F-fluoropaclitaxel in healthy volunteers and newly diagnosed breast cancer patients using PET/CT. J. Nucl. Med. 2011, 52, 1339–1345. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Ivanov, A.A.; Su, R.; Gonzalez-Pecchi, V.; Qi, Q.; Liu, S.; Webber, P.; McMillan, E.; Rusnak, L.; Pham, C.; et al. The OncoPPi network of cancer-focused protein–protein interactions to inform biological insights and therapeutic strategies. Nat. Commun. 2017, 8, 14356. [Google Scholar] [CrossRef]
- Caldas-Lopes, E.; Cerchietti, L.; Ahn, J.H.; Clement, C.C.; Robles, A.I.; Rodina, A.; Moulick, K.; Taldone, T.; Gozman, A.; Guo, Y.; et al. Hsp90 inhibitor PU-H71, a multimodal inhibitor of malignancy, induces complete responses in triple-negative breast cancer models. Proc. Natl. Acad. Sci. USA 2009, 106, 8368–8373. [Google Scholar] [CrossRef] [Green Version]
- Scheibe, P.O.; Vera, D.R.; Eckelman, W.C. What is to be gained by imaging the same animal before and after treatment? Nucl. Med. Biol. 2005, 32, 727–732. [Google Scholar] [CrossRef]
- Harte, R.J.; Matthews, J.; O’Reilly, S.M.; Tilsley, D.O.; Osman, S.; Brown, G.; Luthra, S.J.; Brady, F.; Jones, T.; Price, P.M. Tumor, normal tissue, and plasma pharmacokinetic studies of fluorouracil biomodulation with N-phosphonacetyl-L-aspartate, folinic acid, and interferon alfa. J. Clin. Oncol. 1999, 17, 1580–1588. [Google Scholar] [CrossRef]
- Saleem, A.; Price, P.M. Early tumor drug pharmacokinetics is influenced by tumor perfusion but not plasma drug exposure. Clin. Cancer Res. 2008, 14, 8184–8190. [Google Scholar] [CrossRef] [Green Version]
- Saleem, A.; Yap, J.; Osman, S.; Brady, F.; Suttle, B.; Lucas, S.V.; Jones, T.; Price, P.M.; Aboagye, E.O.; Yap, J.; et al. Modulation of fluorouracil tissue pharmacokinetics by eniluracil: In-vivo imaging of drug action. Lancet 2000, 355, 2125–2131. [Google Scholar] [CrossRef]
- Evelhoch, J.L. In vivo 19F nuclear magnetic resonance spectroscopy: A potential monitor of 5-fluorouracil pharmacokinetics and metabolism. Invest. New Drugs 1989, 7, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Wolf, W.; Presant, C.A.; Servis, K.L.; El-Tahtawy, A.; Albright, M.J.; Barker, P.B.; Ring, R.; Atkinson, D.; Ong, R.; King, M. Tumor trapping of 5-fluorouracil: In vivo 19F NMR spectroscopic pharmacokinetics in tumor-bearing humans and rabbits. Proc. Natl. Acad. Sci. USA 1990, 87, 492–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Findlay, M.P.N.; Leach, M.O.; Cunningham, D.; Collins, D.J.; Payne, G.S.; Glaholm, J.; Mansi, J.L.; McCready, V.R. The non-invasive monitoring of low dose, infusional 5-fluorouracil and its modulation by interferon-{alpha} using in vivo 19F magnetic resonance spectroscopy in patients with colorectal cancer: A pilot study. Ann. Oncol. 1993, 4, 597–602. [Google Scholar] [CrossRef]
- Saleem, A.; Harte, R.J.; Matthews, J.; Osman, S.; Brady, F.; Luthra, S.K.; Brown, G.D.; Bleehen, N.; Connors, T.; Jones, T.; et al. Pharmacokinetic evaluation of N-[2-(dimethylamino)ethyl]acridine-4-carboxamide in patients by positron emission tomography. J. Clin. Oncol. 2001, 19, 1421–1429. [Google Scholar] [CrossRef]
- Rosso, L.; Brock, C.S.; Gallo, J.M.; Saleem, A.; Price, P.M.; Turkheimer, F.; Aboagye, E.O. A new model for prediction of drug distribution in tumor and normal tissues: Pharmacokinetics of temozolomide in glioma patients. Cancer Res. 2009, 69, 120–127. [Google Scholar] [CrossRef] [Green Version]
- Burvenich, I.; Parakh, S.; Parslow, A.C.; Lee, S.T.; Gan, H.K.; Scott, A.M. Receptor occupancy imaging studies in oncology drug development. AAPS J. 2018, 20, 43. [Google Scholar] [CrossRef]
- Kluger, H.M.; Dudek, A.; McCann, C.; Rink, L.; Ritacco, J.; Adrada, C.; Phouyaphone, N.; Southard, N.; Sznol, M. A phase II trial of dasatinib in advanced melanoma. J. Clin. Oncol. 2009, 27, 9010. [Google Scholar] [CrossRef]
- Brierley, R. 2009 ASCO Annual Meeting. J. Clin. Oncol. 2009, 27, 650. [Google Scholar] [CrossRef]
- Johnson, F.M.; Agrawal, S.; Burris, H.; Rosen, L.; Dhillon, N.; Hong, D.; Blackwood-Chirchir, A.; Luo, F.R.; Sy, O.; Kaul, S.; et al. Phase 1 pharmacokinetic and drug-interaction study of dasatinib in patients with advanced solid tumors. Cancer 2010, 116, 1582–1591. [Google Scholar] [CrossRef]
- Demetri, G.D.; Russo, P.A.; MacPherson, I.; Wang, D.; Morgan, J.A.; Brunton, V.G.; Paliwal, P.; Agrawal, S.; Voi, M.; Evans, T.J. Phase I dose-escalation and pharmacokinetic study of dasatinib in patients with advanced solid tumors. Clin. Cancer Res. 2009, 15, 6232–6240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Tourneau, C.; Lee, J.J.; Siu, L.L. Dose escalation methods in phase I cancer clinical trials. J. Natl. Cancer Inst. 2009, 101, 708–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sessa, C.; Shapiro, G.I.; Bhalla, K.N.; Britten, C.; Jacks, K.S.; Mita, M.; Papadimitrakopoulou, V.; Pluard, T.; Samuel, T.A.; Akimov, M.; et al. First-in-human phase I dose-escalation study of the HSP90 inhibitor AUY922 in patients with advanced solid tumors. Clin. Cancer Res. 2013, 19, 3671–3680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinelli, G.; Soverini, S.; Iacobucci, I.; Baccarani, M. Intermittent targeting as a tool to minimize toxicity of tyrosine kinase inhibitor therapy. Nat. Clin. Pract. Oncol. 2009, 6, 68–69. [Google Scholar] [CrossRef]
- Van Der Veldt, A.A.M.; Lubberink, M.; Bahce, I.; Walraven, M.; De Boer, M.P.; Greuter, H.N.; Hendrikse, N.H.; Eriksson, J.; Windhorst, A.D.; Postmus, P.E.; et al. Rapid decrease in delivery of chemotherapy to tumors after anti-VEGF therapy: Implications for scheduling of anti-angiogenic drugs. Cancer Cell 2012, 21, 82–91. [Google Scholar] [CrossRef] [Green Version]
- Wolf, W.; Presant, C.A. Tumor-based pharmacokinetics has greater significance for anticancer drugs than does blood-based pharmacokinetics. Clin. Phamrmacol. Ther. 2004, 76, 508–509. [Google Scholar] [CrossRef]
- Harte, R.J.O.R.S.; Matthews, J.C.; Babarovic, R.; Brown, G.; Osman, S.; Jones, T.; Price, P.M. Towards chemotherapy dose individualization with tracer derived tissue and tumor pharmacokinetic (pk) parameters. Proc. Am. Meeting ASCO 1995, 14, 1493. [Google Scholar]
- Saleem, A.; Murphy, P.; Plisson, C.; Lahn, M. Why are we failing to implement imaging studies with radiolabelled new molecular entities in early oncology drug development? Sci. World J. 2014, 2014, 269605. [Google Scholar] [CrossRef] [Green Version]
- Saleem, A.; Searle, G.; Kenny, L.M.; Huiban, M.; Waldman, A.; Downie, L.; Lau, M.; Murphy, P.S.; Kozlowski, K.; Lewis, Y.; et al. Brain and tumor penetration of carbon-11–labeled lapatinib ([11C]Lap) in patients (pts) with HER2-overexpressing metastatic breast cancer (MBC). J. Clin. Oncol. 2013, 31 (Suppl. S15), 635. [Google Scholar] [CrossRef]
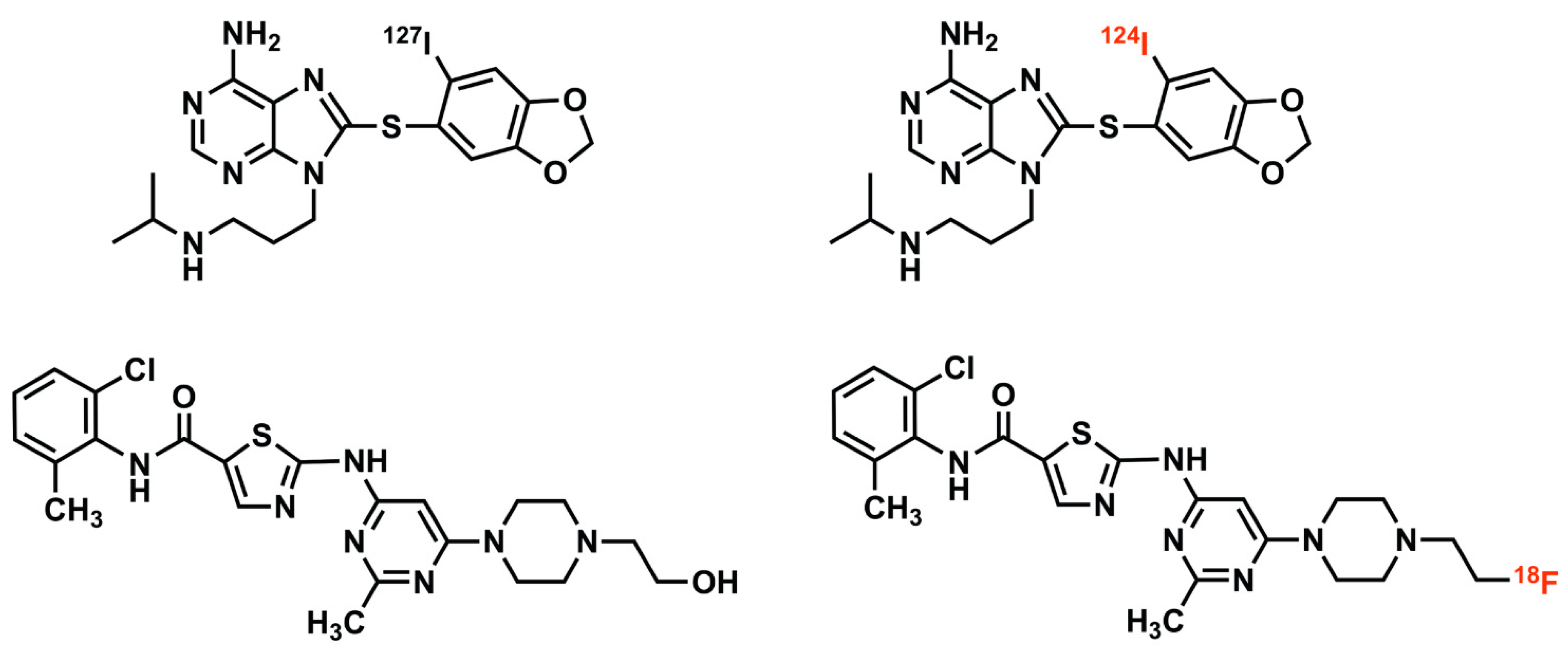
Figure 1.
Drug tracer imaging of small molecule therapeutic compounds employs isotopologues or radiolabeled analogues. For example, I-124 PU-H71 (top right) is an isotopologue of the epichaperome inhibitor therapeutic compound, PU-H71 (top left), identical in molecular structure, differing only in the isotopic form of a constituent atom (stable iodine-127 versus the positron-emitting iodine-124) [7,8]. F-18 SKI-249380 (bottom right) is a radiolabeled analogue of the tyrosine kinase inhibitor therapeutic compound, dasatinib (bottom left). The positron-emitting fluorine-18 atom, in F-18 SKI-249380, replaces a hydroxyl group found in dasatinib; this substitution has minimal pharmacologic effect, enabling F-18 SKI-249380 to be an effective drug tracer of dasatinib [9,10].
Figure 1.
Drug tracer imaging of small molecule therapeutic compounds employs isotopologues or radiolabeled analogues. For example, I-124 PU-H71 (top right) is an isotopologue of the epichaperome inhibitor therapeutic compound, PU-H71 (top left), identical in molecular structure, differing only in the isotopic form of a constituent atom (stable iodine-127 versus the positron-emitting iodine-124) [7,8]. F-18 SKI-249380 (bottom right) is a radiolabeled analogue of the tyrosine kinase inhibitor therapeutic compound, dasatinib (bottom left). The positron-emitting fluorine-18 atom, in F-18 SKI-249380, replaces a hydroxyl group found in dasatinib; this substitution has minimal pharmacologic effect, enabling F-18 SKI-249380 to be an effective drug tracer of dasatinib [9,10].

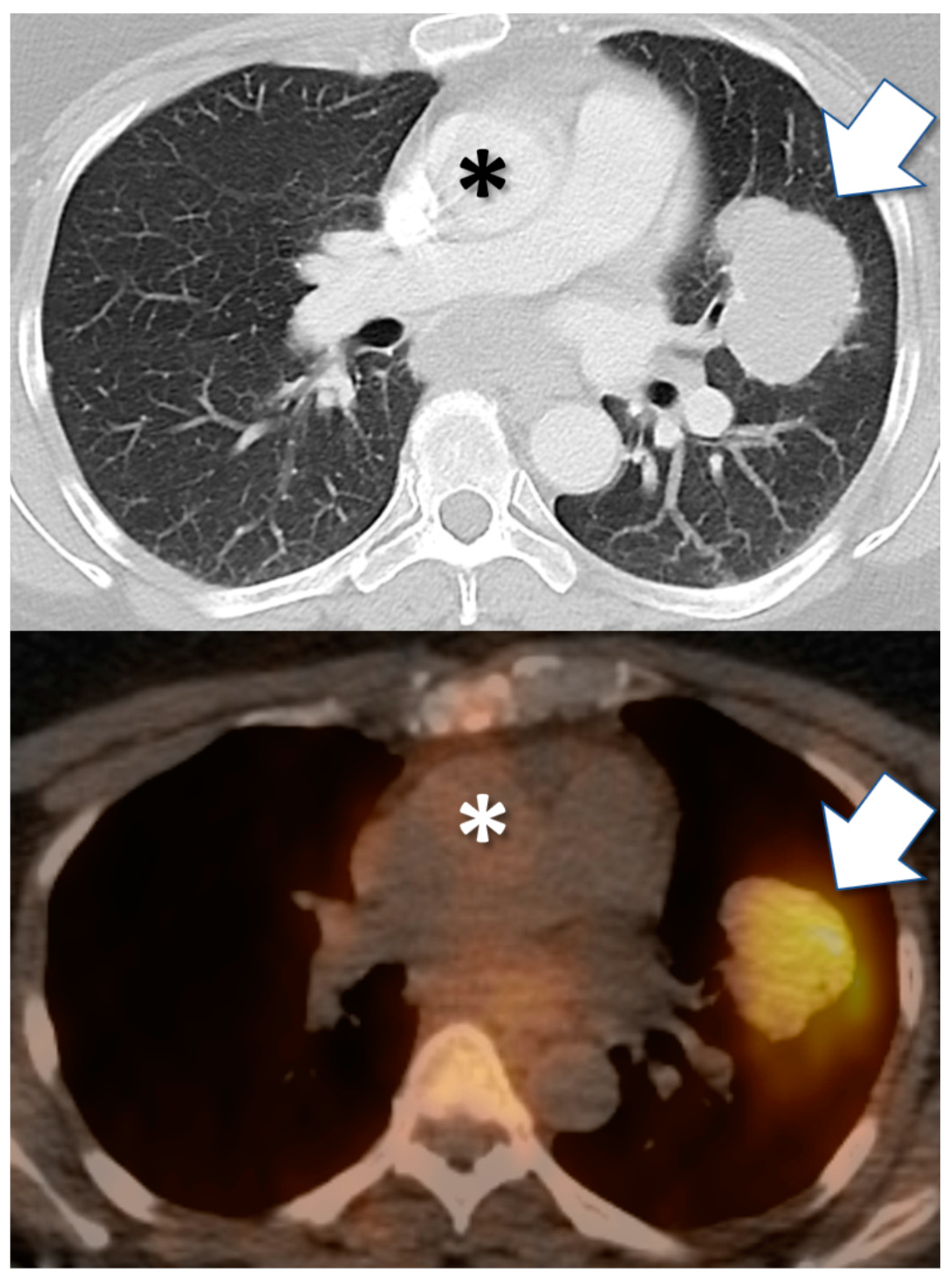
Figure 2.
Drug tracer imaging detects in vivo tumor avidity for therapeutic small molecule compounds. Axial CT (top) and PET/CT fusion (bottom) images from 57-year-old female with clear cell carcinoma of Müllerian origin, including biopsied malignant pulmonary mass (arrow). PET/CT image obtained 24 h after injection with I-124 PU-H71 radiotracer, an isotopologue of the epichaperome inhibitor therapeutic compound PU-H71. PET demonstrates marked retention of drug by tumor, after drug has cleared from blood circulation (e.g., cardiac blood pool, asterisk).
Figure 2.
Drug tracer imaging detects in vivo tumor avidity for therapeutic small molecule compounds. Axial CT (top) and PET/CT fusion (bottom) images from 57-year-old female with clear cell carcinoma of Müllerian origin, including biopsied malignant pulmonary mass (arrow). PET/CT image obtained 24 h after injection with I-124 PU-H71 radiotracer, an isotopologue of the epichaperome inhibitor therapeutic compound PU-H71. PET demonstrates marked retention of drug by tumor, after drug has cleared from blood circulation (e.g., cardiac blood pool, asterisk).

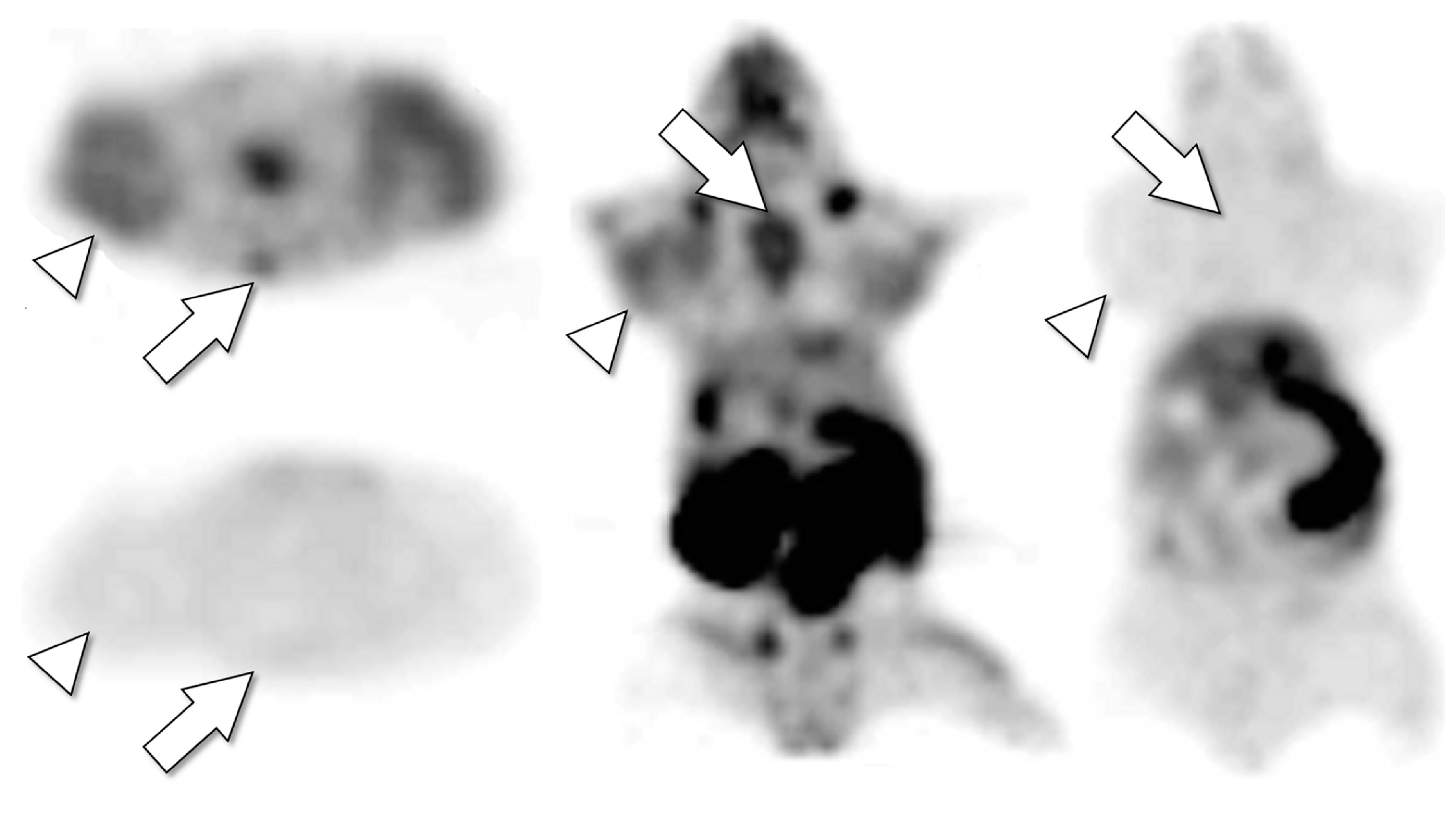
Figure 3.
Drug tracer imaging can detect tumor saturation by therapeutic drug. Axial (left top, left bottom) and coronal (middle, right) PET images of single mouse bearing tumors after IV injection with F-18 SKI249380, radiolabeled analogue of therapeutic tyrosine kinase inhibitor dasatinib. Microdose (radiotracer only) images (left top, middle) detect avid tumors (arrowheads, H1975 lung cancer xenograft) and bone marrow (e.g., sternum, arrows). The next day (after F-18 isotope decay), the mouse was re-imaged after receiving F-18 SKI249380 radiotracer co-injected with therapeutic dose of dasatinib (left bottom, right). The therapeutic dasatinib dose competitively inhibited uptake of the radiotracer in tumors and bone marrow, indicating that the drug and tracer share the same in vivo tumor pharmacokinetic pathways and that these drug-specific tumor pharmacokinetic pathways (cellular kinase targets and/or pre-target uptake and transport mechanisms) were saturated by the single therapeutic dose. The tumor-saturating dose used in the animal model (30 mg/kg) was equivalent (by allometric extrapolation) to a 140 mg human dose, less than the maximum tolerated dose of some solid tumor dasatinib therapy trials [73].
Figure 3.
Drug tracer imaging can detect tumor saturation by therapeutic drug. Axial (left top, left bottom) and coronal (middle, right) PET images of single mouse bearing tumors after IV injection with F-18 SKI249380, radiolabeled analogue of therapeutic tyrosine kinase inhibitor dasatinib. Microdose (radiotracer only) images (left top, middle) detect avid tumors (arrowheads, H1975 lung cancer xenograft) and bone marrow (e.g., sternum, arrows). The next day (after F-18 isotope decay), the mouse was re-imaged after receiving F-18 SKI249380 radiotracer co-injected with therapeutic dose of dasatinib (left bottom, right). The therapeutic dasatinib dose competitively inhibited uptake of the radiotracer in tumors and bone marrow, indicating that the drug and tracer share the same in vivo tumor pharmacokinetic pathways and that these drug-specific tumor pharmacokinetic pathways (cellular kinase targets and/or pre-target uptake and transport mechanisms) were saturated by the single therapeutic dose. The tumor-saturating dose used in the animal model (30 mg/kg) was equivalent (by allometric extrapolation) to a 140 mg human dose, less than the maximum tolerated dose of some solid tumor dasatinib therapy trials [73].


{kind=link}
{kind=link}
{kind=link}
Table 1.
Selected examples of isotopologues and radiolabeled analogues developed as positron emission tomography (PET) imaging tracers of specific therapeutic small molecule cancer drugs, from published peer-reviewed research reports (abstracts available at www.pubmed.gov). Published literature includes numerous other small molecule radiotracers that have been tested as radiologic biomarkers of important oncotherapeutic targets or as companion diagnostics for radionuclide therapy.
Table 1.
Selected examples of isotopologues and radiolabeled analogues developed as positron emission tomography (PET) imaging tracers of specific therapeutic small molecule cancer drugs, from published peer-reviewed research reports (abstracts available at www.pubmed.gov). Published literature includes numerous other small molecule radiotracers that have been tested as radiologic biomarkers of important oncotherapeutic targets or as companion diagnostics for radionuclide therapy.
| Chemotherapy |
|---|
| C-11 N-[(2′-dimethylamino)ethyl]acridine-4-carboxamide (XR5000) [16] C-11 Temozolomide [17] F-18 5-Fluorouracil [18,19,20,21] F-18 AraG [22] F-18 Clofarabine [23] F-18 Deoxycytidine [24,25,26,27,28] F-18 Paclitaxel [29,30,31,32] F-18 Xeloda [33] |
| Anaplastic Lymphoma Kinase (ALK) Inhibitors |
| C-11 Lorlatinib [34,35] F-18 Lorlatinib [35] |
| Epidermal Growth Factor Receptor (EGFR) Tyrosine Kinase Inhibitors |
| C-11 Erlotinib [36,37,38] C-11 Gefitinib [39] C-11 Osimertinib [40] F-18 FEA-Erlotinib [41] F-18 Gefitinib [42] F-18 IRS [43] |
| Other Tyrosine Kinase Inhibitors |
| C-11 Axitinib [44] C-11 CEP-32496 [45] C-11 Lapatinib [46] C-11 Nintedanib [44] C-11 Sorafenib [47] F-18 Afatinib [38] F-18 Dabrafenib [48] F-18 SKI249380 (Dasatinib Analogue) [9] |
| Epichaperome Inhibitor |
| I-124 PU-H71 [7,8,49] |
| IL2-Receptor/Cytokine |
| F-18 IL-2 [50] F-18 FB-IL2v [51] |
| Matrix Metalloproteinase Inhibitor |
| C-11 (2R)-2-[[4-(6-fluorohex-1-ynyl)phenyl]sulfonylamino]-3-methylbutyric acid methyl ester [52] |
| Phosphatidylinositol 3-Kinase (PI3K) Inhibitor |
| C-11 Pictilisib (GDC-0941) [53] |
| PD-L1 |
| F-18 BMS-986192 [54] |
| Poly(Adenosine Diphosphate Ribose) Polymerase (Parp) Inhibitor |
| F-18 Fluorthanatrace [55] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Dunphy, M.P.S.; Pillarsetty, N. The Unique Pharmacometrics of Small Molecule Therapeutic Drug Tracer Imaging for Clinical Oncology. Cancers 2020, 12, 2712. https://doi.org/10.3390/cancers12092712
AMA Style
Dunphy MPS, Pillarsetty N. The Unique Pharmacometrics of Small Molecule Therapeutic Drug Tracer Imaging for Clinical Oncology. Cancers. 2020; 12(9):2712. https://doi.org/10.3390/cancers12092712
Chicago/Turabian StyleDunphy, Mark P. S., and Nagavarakishore Pillarsetty. 2020. "The Unique Pharmacometrics of Small Molecule Therapeutic Drug Tracer Imaging for Clinical Oncology" Cancers 12, no. 9: 2712. https://doi.org/10.3390/cancers12092712
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.