Insight into the Development of PET Radiopharmaceuticals for Oncology

 , , ,
, , ,
Abstract
:1. Introduction
2. Targeting Vectors
2.1. Probes Based on Bioactive Molecules
2.2. Probes Based on Drugs
2.3. Chemical Screens
3. Radiochemistry
3.1. Radiolabeling Strategies
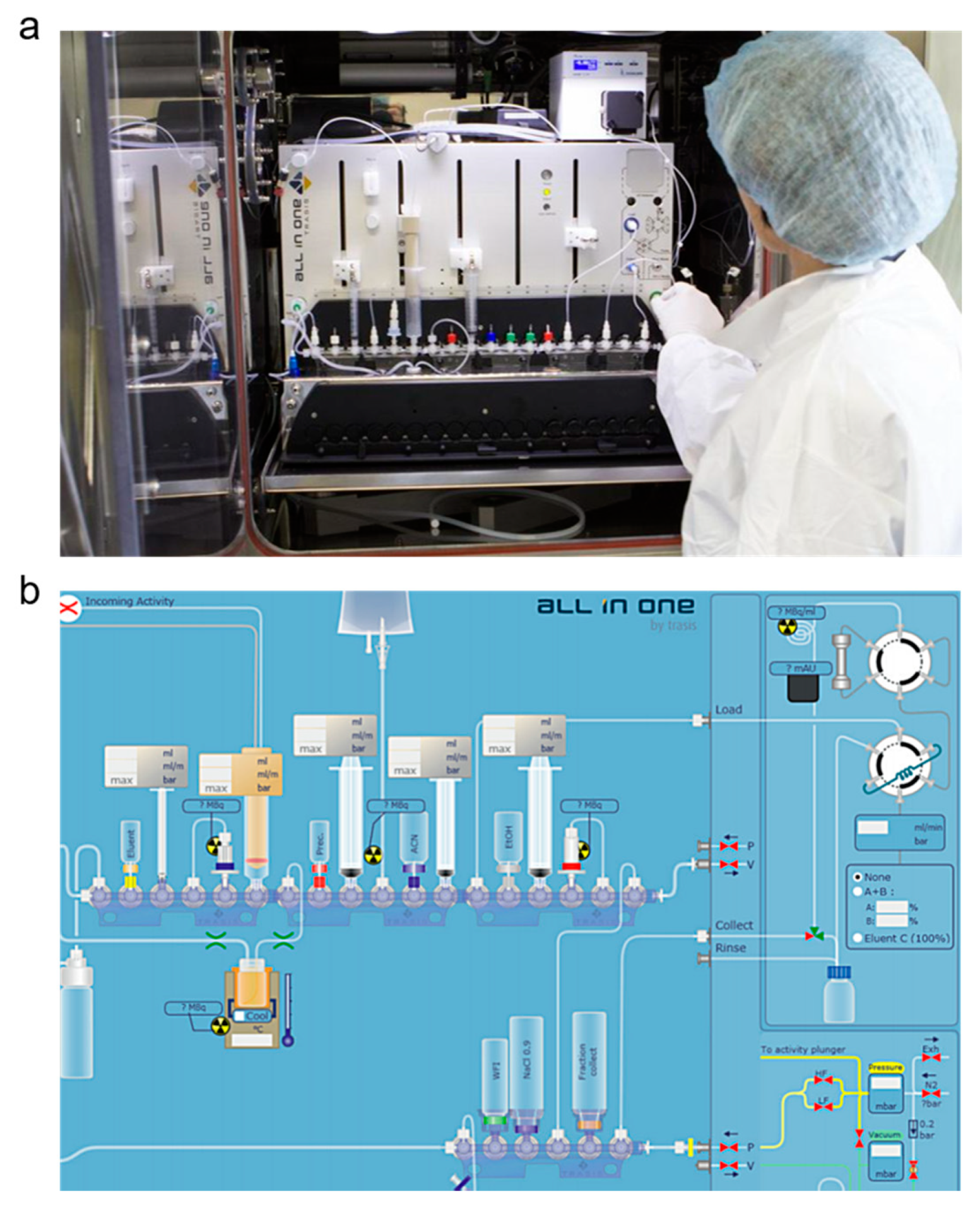
3.2. Automation
3.3. Quality Control
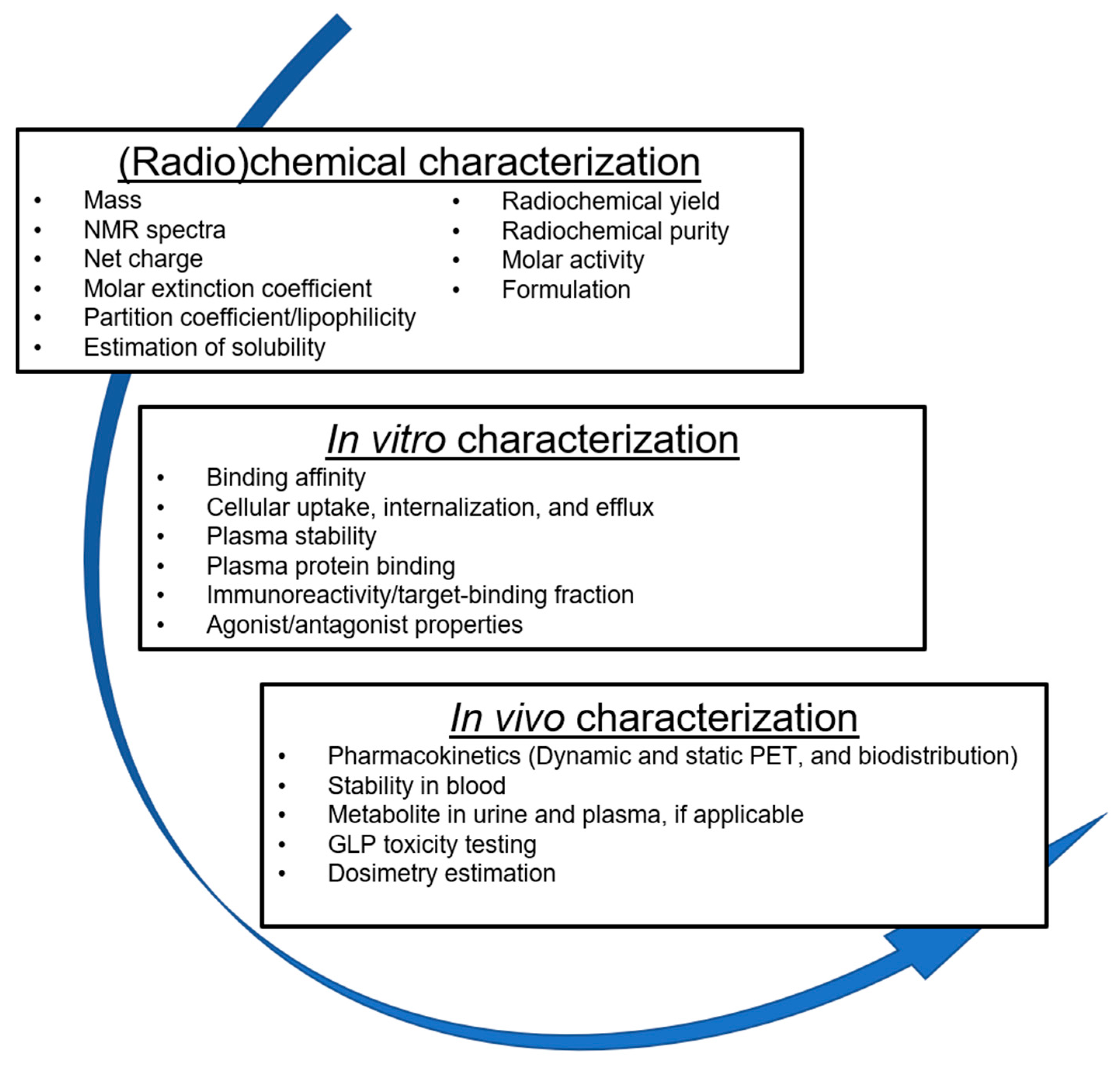
4. Preclinical Experiments
4.1. Binding Affinity
4.2. Internalization and Efflux Assays
4.3. Stability
4.4. Plasma Protein Binding
4.5. Immunoreactivity
4.6. Antagonist and Agonist Assays
4.7. Imaging
4.8. Biodistribution by Dissection
4.9. Specificity
4.10. Time Points
4.11. Dosimetry
4.12. Toxicity
5. Regulatory Considerations
5.1. Nonclinical Evaluation of Radiopharmaceuticals
5.2. Exploratory Approaches for First-in-Human Studies
5.3. Marketing Authorization
6. Perspectives and Summary
Author Contributions
Funding
Conflicts of Interest
References
- Boellaard, R.; Delgado-Bolton, R.; Oyen, W.J.G.; Giammarile, F.; Tatsch, K.; Eschner, W.; Verzijlbergen, F.J.; Barrington, S.F.; Pike, L.C.; Weber, W.A.; et al. FDG PET/CT: EANM procedure guidelines for tumour imaging: version 2.0. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 328–354. [Google Scholar] [CrossRef] [PubMed]
- Cherry, S.; Sorenson, J.; Phelps, M. Physics in Nuclear Medicine, 4th ed.; Saunders, Elsevier Inc.: Philadelphia, PA, USA, 2012; ISBN 9781416051985. [Google Scholar]
- Acton, P.D.; Zhuang, H.; Alavi, A. Quantification in PET. Radiol. Clin. North Am. 2004, 42, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffeth, L.K. Use of Pet/Ct Scanning in Cancer Patients: Technical and Practical Considerations. Baylor Univ. Med. Cent. Proc. 2005, 18, 321–330. [Google Scholar] [CrossRef]
- Lau, J.; Jacobson, O.; Niu, G.; Lin, K.S.; Bénard, F.; Chen, X. Bench to Bedside: Albumin Binders for Improved Cancer Radioligand Therapies. Bioconjug. Chem. 2019, 30, 487–502. [Google Scholar] [CrossRef]
- Chen, K.; Chen, X. Design and Development of Molecular Imaging Probes. Curr. Top. Med. Chem. 2010, 10, 1227–1236. [Google Scholar] [CrossRef]
- Bernard-Gauthier, V.; Bailey, J.J.; Berke, S.; Schirrmacher, R.; Leahy, J.W. Recent advances in the development and application of radiolabeled kinase inhibitors for PET imaging. Molecules 2015, 20, 22000–22027. [Google Scholar] [CrossRef] [Green Version]
- Krebs, S.; Veach, D.R.; Carter, L.M.; Grkovski, M.; Fornier, M.; Mauro, M.J.; Voss, M.H.; Danila, D.C.; Burnazi, E.; Null, M.; et al. First-in-Human Trial of Dasatinib-Derivative Tracer for Tumor Kinase-Targeted Positron Emission Tomography. J. Nucl. Med. 2020. [Google Scholar] [CrossRef]
- Silberstein, E.B.; Ryan, J. Prevalence of adverse reactions in nuclear medicine. Pharmacopeia Committee of the Society of Nuclear Medicine. J. Nucl. Med. 1996, 37, 185–192. [Google Scholar]
- Grewal, R.K.; Ho, A.; Schöder, H. Novel approaches to thyroid cancer treatment and response assessment. Semin. Nucl. Med. 2016, 46, 109–118. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, H.; Takahashi, J.; Fujiwara, T.; Yamaguchi, K.; Abe, Y.; Kubota, K.; Sato, T.; Miyazawa, H.; Hatazawa, J.; Tada, M. High accumulation of 2-deoxy-2-fluorine-18-fluoro-D-galactose by well-differentiated hepatomas of mice and rats. J. Nucl. Med. 1993, 34, 780–786. [Google Scholar] [PubMed]
- Carrasquillo, J.A.; Pandit-Taskar, N.; O’Donoghue, J.A.; Humm, J.L.; Zanzonico, P.; Smith-Jones, P.M.; Divgi, C.R.; Pryma, D.A.; Ruan, S.; Kemeny, N.E.; et al. 124I-huA33 antibody PET of colorectal cancer. J. Nucl. Med. 2011, 52, 1173–1180. [Google Scholar] [CrossRef] [Green Version]
- Grassi, I.; Nanni, C.; Allegri, V.; Morigi, J.J.; Montini, G.C.; Castellucci, P.; Fanti, S. The clinical use of PET with (11)C-acetate. Am. J. Nucl. Med. Mol. Imaging 2012, 2, 33–47. [Google Scholar] [PubMed]
- Chen, W. Clinical applications of PET in brain tumors. J. Nucl. Med. 2007, 48, 1468–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, Y.; Liu, X.; Li, J.; Yao, H.; Yuan, S. Fluorine-18 labeled amino acids for tumor PET/CT imaging. Oncotarget 2017, 8, 60581–60588. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, T.; Kanno, I.; Shishido, F.; Inugami, A.; Higano, S.; Fujita, H.; Murakami, M.; Uemura, K.; Yasui, N.; Mineura, K.; et al. Clinical value of pet with 18F-fluorodeoxyglucose and l-methyl-11C-methionine for diagnosis of recurrent brain tumor and radiation injury. Acta Radiol. 1991, 32, 197–202. [Google Scholar] [CrossRef]
- Verbeek, H.H.G.; Plukker, J.T.M.; Koopmans, K.P.; De Groot, J.W.B.; Hofstra, R.M.W.; Muller Kobold, A.C.; Van Der Horst-Schrivers, A.N.A.; Brouwers, A.H.; Links, T.P. Clinical relevance of 18F-FDG PET and 18F-DOPA PET in recurrent medullary thyroid carcinoma. J. Nucl. Med. 2012, 53, 1863–1871. [Google Scholar] [CrossRef] [Green Version]
- Moller, S.; Law, I.; Munck af Rosenschold, P.; Costa, J.; Poulsen, H.S.; Engelholm, S.A.; Engelholm, S. Prognostic value of 18F-FET PET imaging in re-irradiation of high-grade glioma: Results of a phase I clinical trial. Radiother. Oncol. 2016, 121, 132–137. [Google Scholar] [CrossRef]
- Mittra, E.S.; Koglin, N.; Mosci, C.; Kumar, M.; Hoehne, A.; Keu, K.V.; Iagaru, A.H.; Mueller, A.; Berndt, M.; Bullich, S.; et al. Pilot preclinical and clinical evaluation of (4S)-4-(3-[18F]Fluoropropyl)-L-Glutamate (18F-FSPG) for PET/CT imaging of intracranial malignancies. PLoS ONE 2016, 11, e0148628. [Google Scholar] [CrossRef]
- Dunphy, M.P.S.; Harding, J.J.; Venneti, S.; Zhang, H.; Burnazi, E.M.; Bromberg, J.; Omuro, A.M.; Hsieh, J.J.; Mellinghoff, I.K.; Staton, K.; et al. In vivo PET assay of tumor glutamine flux and metabolism: In-human trial of 18F-(2S,4R)-4-fluoroglutamine. Radiology 2018, 287, 667–675. [Google Scholar] [CrossRef] [Green Version]
- Turkbey, B.; Mena, E.; Shih, J.; Pinto, P.A.; Merino, M.J.; Lindenberg, M.L.; Bernardo, M.; McKinney, Y.L.; Adler, S.; Owenius, R.; et al. Localized prostate cancer detection with 18F FACBC PET/CT: Comparison with MR imaging and histopathologic analysis. Radiology 2014, 270, 849–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savir-Baruch, B.; Schuster, D.M.; Jarkas, N.; Master, V.A.; Nieh, P.T.; Halkar, R.K.; Nye, J.A.; Lewis, M.M.; Crowe, R.J.; Voll, R.J.; et al. Pilot evaluation of anti-1-amino-2-[ 18F] fluorocyclopentane-1- carboxylic acid (anti-2-[ 18F] FACPC) PET-CT in recurrent prostate carcinoma. Mol. Imaging Biol. 2011, 13, 1272–1277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larson, S.M.; Morris, M.; Gunther, I.; Beattie, B.; Humm, J.L.; Akhurst, T.A.; Finn, R.D.; Erdi, Y.; Pentlow, K.; Dyke, J.; et al. Tumor localization of 16β-18F-fluoro-5α- dihydrotestosterone versus 18F-FDG in patients with progressive, metastatic prostate cancer. J. Nucl. Med. 2004, 45, 366–373. [Google Scholar] [PubMed]
- Dehdashti, F.; Picus, J.; Michalski, J.M.; Dence, C.S.; Siegel, B.A.; Katzenellenbogen, J.A.; Welch, M.J. Positron tomographic assessment of androgen receptors in prostatic carcinoma. Eur. J. Nucl. Med. Mol. Imaging 2005, 32, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Oborski, M.J.; Laymon, C.M.; Lieberman, F.S.; Drappatz, J.; Hamilton, R.L.; Mountz, J.M. First use of 18F-labeled ML-10 PET to assess apoptosis change in a newly diagnosed glioblastoma multiforme patient before and early after therapy. Brain Behav. 2014, 4, 312–315. [Google Scholar] [CrossRef]
- Dubash, S.R.; Merchant, S.; Heinzmann, K.; Mauri, F.; Lavdas, I.; Inglese, M.; Kozlowski, K.; Rama, N.; Masrour, N.; Steel, J.F.; et al. Clinical translation of [18F]ICMT-11 for measuring chemotherapy-induced caspase 3/7 activation in breast and lung cancer. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 2285–2299. [Google Scholar] [CrossRef] [Green Version]
- Jadvar, H.; Desai, B.; Conti, P.S. Sodium 18F-fluoride PET/CT of bone, joint, and other disorders. Semin. Nucl. Med. 2015, 45, 58–65. [Google Scholar] [CrossRef] [Green Version]
- Phase 1 Imaging Study of 89Zr-DFO-HuMab-5B1 With HuMab-5B1 - Full Text View - ClinicalTrials. Available online: https://clinicaltrials.gov/ct2/show/NCT02687230 (accessed on 16 April 2020).
- Divgi, C.R.; Uzzo, R.G.; Gatsonis, C.; Bartz, R.; Treutner, S.; Yu, J.Q.; Chen, D.; Carrasquillo, J.A.; Larson, S.; Bevan, P.; et al. Positron emission tomography/computed tomography identification of clear cell renal cell carcinoma: Results from the REDECT trial. J. Clin. Oncol. 2013, 31, 187–194. [Google Scholar] [CrossRef] [Green Version]
- Hekman, M.C.H.; Rijpkema, M.; Aarntzen, E.H.; Mulder, S.F.; Langenhuijsen, J.F.; Oosterwijk, E.; Boerman, O.C.; Oyen, W.J.G.; Mulders, P.F.A. Positron Emission Tomography/Computed Tomography with 89Zr-girentuximab Can Aid in Diagnostic Dilemmas of Clear Cell Renal Cell Carcinoma Suspicion. Eur. Urol. 2018, 74, 257–260. [Google Scholar] [CrossRef]
- Lau, J.; Lin, K.S.; Bénard, F. Past, present, and future: Development of theranostic agents targeting carbonic anhydrase IX. Theranostics 2017, 7, 4322–4339. [Google Scholar] [CrossRef]
- Moek, K.L.; Waaijer, S.J.H.; Kok, I.C.; Suurs, F.V.; Brouwers, A.H.; Menke-Van Der Houven Van Oordt, C.W.; Wind, T.T.; Gietema, J.A.; Schröder, C.P.; Mahesh, S.V.K.; et al. 89Zr-labeled bispecific T-cell engager AMG 211 PET shows AMG 211 accumulation in CD3-rich tissues and clear, heterogeneous tumor uptake. Clin. Cancer Res. 2019, 25, 3517–3527. [Google Scholar] [CrossRef] [PubMed]
- Pandit-Taskar, N.; Postow, M.A.; Hellmann, M.D.; Harding, J.J.; Barker, C.A.; O’Donoghue, J.A.; Ziolkowska, M.; Ruan, S.; Lyashchenko, S.K.; Tsai, F.; et al. First-in-Humans Imaging with 89Zr-Df-IAB22M2C Anti-CD8 Minibody in Patients with Solid Malignancies: Preliminary Pharmacokinetics, Biodistribution, and Lesion Targeting. J. Nucl. Med. 2020, 61, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Jauw, Y.W.S.; Zijlstra, J.M.; de Jong, D.; Vugts, D.J.; Zweegman, S.; Hoekstra, O.S.; van Dongen, G.A.M.S.; Huisman, M.C. Performance of 89Zr-Labeled-Rituximab-PET as an Imaging Biomarker to Assess CD20 Targeting: A Pilot Study in Patients with Relapsed/Refractory Diffuse Large B Cell Lymphoma. PLoS ONE 2017, 12, e0169828. [Google Scholar] [CrossRef] [PubMed]
- Jauw, Y.W.S.; O’Donoghue, J.A.; Zijlstra, J.M.; Hoekstra, O.S.; Willemien Menke-Van Der Houven Van Oordt, C.; Morschhauser, F.; Carrasquillo, J.A.; Zweegman, S.; Pandit-Taskar, N.; Lammertsma, A.A.; et al. 89Zr-immuno-PET: Toward a noninvasive clinical tool to measure target engagement of therapeutic antibodies in vivo. J. Nucl. Med. 2019, 60, 1825–1832. [Google Scholar] [CrossRef]
- Börjesson, P.K.E.; Jauw, Y.W.S.; De Bree, R.; Roos, J.C.; Castelijns, J.A.; Leemans, C.R.; Van Dongen, G.A.M.S.; Boellaard, R. Radiation dosimetry of 89Zr-labeled chimeric monoclonal antibody U36 as used for immuno-PET in head and neck cancer patients. J. Nucl. Med. 2009, 50, 1828–1836. [Google Scholar] [CrossRef] [Green Version]
- Weiss, I.D.; Huff, L.M.; Evbuomwan, M.O.; Xu, X.; Dang, H.D.; Velez, D.S.; Singh, S.P.; Zhang, H.H.; Gardina, P.J.; Lee, J.H.; et al. Screening of cancer tissue arrays identifies CXCR4 on adrenocortical carcinoma: Correlates with expression and quantification on metastases using 64Cu-plerixafor PET. Oncotarget 2017, 8, 73387–73406. [Google Scholar] [CrossRef] [Green Version]
- Lapa, C.; Schreder, M.; Schirbel, A.; Samnick, S.; Kortüm, K.M.; Herrmann, K.; Kropf, S.; Einsele, H.; Buck, A.K.; Wester, H.J.; et al. [68Ga]Pentixafor-PET/CT for imaging of chemokine receptor CXCR4 expression in multiple myeloma - Comparison to [18F]FDG and laboratory values. Theranostics 2017, 7, 205–212. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, M.; Wang, L.; Wang, S.; Kang, F.; Li, G.; Jacobson, O.; Niu, G.; Yang, W.; Wang, J.; et al. Prospective study of 68Ga-NOTA-NFB: Radiation dosimetry in healthy volunteers and first application in glioma patients. Theranostics 2015, 5, 882–889. [Google Scholar] [CrossRef] [Green Version]
- Uptake and Biodistribution of 89Zirconium-labeled Ipilimumab in Ipilimumab Treated Patients With Metastatic Melanoma—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03313323 (accessed on 16 April 2020).
- Bahce, I.; Smit, E.F.; Lubberink, M.; Van Der Veldt, A.A.M.; Yaqub, M.; Windhorst, A.D.; Schuit, R.C.; Thunnissen, E.; Heideman, D.A.M.; Postmus, P.E.; et al. Development of [11C]erlotinib positron emission tomography for in vivo evaluation of EGF receptor mutational status. Clin. Cancer Res. 2013, 19, 183–193. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Li, M.; Li, X.; Meng, X.; Yang, G.; Zhao, S.; Yang, Y.; Ma, L.; Fu, Z.; Yu, J. PET-based biodistribution and radiation dosimetry of epidermal growth factor receptor-selective tracer 11C-PD153035 in humans. J. Nucl. Med. 2009, 50, 303–308. [Google Scholar] [CrossRef] [Green Version]
- van Helden, E.J.; Elias, S.G.; Gerritse, S.L.; van Es, S.C.; Boon, E.; Huisman, M.C.; van Grieken, N.C.T.; Dekker, H.; van Dongen, G.A.M.S.; Vugts, D.J.; et al. [89Zr]Zr-cetuximab PET/CT as biomarker for cetuximab monotherapy in patients with RAS wild-type advanced colorectal cancer. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 849–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindenberg, L.; Adler, S.; Turkbey, I.B.; Mertan, F.; Ton, A.; Do, K.; Kummar, S.; Gonzalez, E.M.; Bhattacharyya, S.; Jacobs, P.M.; et al. Dosimetry and first human experience with 89Zr-panitumumab. Am. J. Nucl. Med. Mol. Imaging 2017, 7, 195–203. [Google Scholar]
- Identifying NSCLC Patients Who Will Benefit from Afatinib Therapy Using [18F]afatinib PET/CT. Available online: http://jnm.snmjournals.org/content/60/supplement_1/145 (accessed on 16 April 2020).
- Velikyan, I.; Schweighöfer, P.; Feldwisch, J.; Seemann, J.; Frejd, F.Y.; Lindman, H.; Sörensen, J. Diagnostic HER2-binding radiopharmaceutical, [68Ga]Ga-ABY-025, for routine clinical use in breast cancer patients. Am. J. Nucl. Med. Mol. Imaging 2019, 9, 12–23. [Google Scholar] [PubMed]
- Keyaerts, M.; Xavier, C.; Heemskerk, J.; Devoogdt, N.; Everaert, H.; Ackaert, C.; Vanhoeij, M.; Duhoux, F.P.; Gevaert, T.; Simon, P.; et al. Phase I study of 68Ga-HER2-Nanobody for PET/CT assessment of HER2 expression in breast carcinoma. J. Nucl. Med. 2016, 57, 27–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulaner, G.A.; Hyman, D.M.; Ross, D.S.; Corben, A.; Chandarlapaty, S.; Goldfarb, S.; McArthur, H.; Erinjeri, J.P.; Solomon, S.B.; Kolb, H.; et al. Detection of HER2-positive metastases in patients with HER2-negative primary breast cancer using 89Zr-trastuzumab PET/CT. J. Nucl. Med. 2016, 57, 1523–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willemien Menke-Van Der Houven Van Oordt, C.; McGeoch, A.; Bergstrom, M.; McSherry, I.; Smith, D.A.; Cleveland, M.; Al-Azzam, W.; Chen, L.; Verheul, H.; Hoekstra, O.S.; et al. Immuno-PET imaging to assess target engagement: Experience from 89Zr-anti-HER3 mAb (GSK2849330) in patients with solid tumors. J. Nucl. Med. 2019, 60, 902–909. [Google Scholar] [CrossRef] [Green Version]
- Bensch, F.; Lamberts, L.E.; Smeenk, M.M.; Jorritsma-Smit, A.; Lub-De Hooge, M.N.; Terwisscha Van Scheltinga, A.G.T.; De Jong, J.R.; Gietema, J.A.; Schröder, C.P.; Thomas, M.; et al. 89Zr-lumretuzumab PET imaging before and during HER3 antibody lumretuzumab treatment in patients with solid tumors. Clin. Cancer Res. 2017, 23, 6128–6137. [Google Scholar] [CrossRef] [Green Version]
- Paquette, M.; Lavallée, É.; Phoenix, S.; Ouellet, R.; Senta, H.; Van Lier, J.E.; Guérin, B.; Lecomte, R.; Turcotte, É.E. Improved estrogen receptor assessment by PET using the novel radiotracer 18 F-4FMFES in estrogen receptor–positive breast cancer patients: An ongoing phase II clinical trial. J. Nucl. Med. 2018, 59, 197–203. [Google Scholar] [CrossRef] [Green Version]
- Lindner, T.; Loktev, A.; Giesel, F.; Kratochwil, C.; Altmann, A.; Haberkorn, U. Targeting of activated fibroblasts for imaging and therapy. EJNMMI Radiopharm. Chem. 2019, 4. [Google Scholar] [CrossRef]
- Kratochwil, C.; Flechsig, P.; Lindner, T.; Abderrahim, L.; Altmann, A.; Mier, W.; Adeberg, S.; Rathke, H.; Röhrich, M.; Winter, H.; et al. 68Ga-FAPI PET/CT: Tracer uptake in 28 different kinds of cancer. J. Nucl. Med. 2019, 60, 801–805. [Google Scholar] [CrossRef] [Green Version]
- Loktev, A.; Lindner, T.; Burger, E.M.; Altmann, A.; Giesel, F.; Kratochwil, C.; Debus, J.; Marmé, F.; Jäger, D.; Mier, W.; et al. Development of fibroblast activation protein-targeted radiotracers with improved tumor retention. J. Nucl. Med. 2019, 60, 1421–1429. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, M.; Frisch, K.; Bender, D.; Keiding, S. The potential use of 2-[18F]fluoro-2-deoxy-D-galactose as a PET/CT tracer for detection of hepatocellular carcinoma. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 1723–1731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baratto, L.; Duan, H.; Maecke, H.R.; Iagaru, A. Imaging the Distribution of Gastrin Releasing Peptide Receptors in Cancer. J. Nucl. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wieser, G.; Mansi, R.; Grosu, A.L.; Schultze-Seemann, W.; Dumont-Walter, R.A.; Meyer, P.T.; Maecke, H.R.; Reubi, J.C.; Weber, W.A. Positron emission tomography (PET) imaging of prostate cancer with a gastrin releasing peptide receptor antagonist - from mice to men. Theranostics 2014, 4, 412–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sah, B.R.; Burger, I.A.; Schibli, R.; Friebe, M.; Dinkelborg, L.; Graham, K.; Borkowski, S.; Bacher-Stier, C.; Valencia, R.; Srinivasan, A.; et al. Dosimetry and First Clinical Evaluation of the New 18F-Radiolabeled Bombesin Analogue BAY 864367 in Patients with Prostate Cancer. J. Nucl. Med. 2015, 56, 372–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minamimoto, R.; Sonni, I.; Hancock, S.; Vasanawala, S.; Loening, A.; Gambhir, S.S.; Iagaru, A. Prospective evaluation of 68 Ga-RM2 PET/MRI in patients with biochemical recurrence of prostate cancer and negative findings on conventional imaging. J. Nucl. Med. 2018, 59, 803–808. [Google Scholar] [CrossRef] [Green Version]
- Maina, T.; Bergsma, H.; Kulkarni, H.R.; Mueller, D.; Charalambidis, D.; Krenning, E.P.; Nock, B.A.; de Jong, M.; Baum, R.P. Preclinical and first clinical experience with the gastrin-releasing peptide receptor-antagonist [68Ga]SB3 and PET/CT. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 964–973. [Google Scholar] [CrossRef]
- Zhang, J.; Niu, G.; Fan, X.; Lang, L.; Hou, G.; Chen, L.; Wu, H.; Zhu, Z.; Li, F.; Chen, X. PET using a GRPR antagonist 68 Ga-RM26 in healthy volunteers and prostate cancer patients. J. Nucl. Med. 2018, 59, 922–928. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Mao, F.; Niu, G.; Peng, L.; Lang, L.; Li, F.; Ying, H.; Wu, H.; Pan, B.; Zhu, Z.; et al. 68Ga-BBN-RGD PET/CT for GRPR and integrin αvβ3 imaging in patients with breast cancer. Theranostics 2018, 8, 1121–1130. [Google Scholar] [CrossRef]
- Zhang, J.; Li, D.; Lang, L.; Zhu, Z.; Wang, L.; Wu, P.; Niu, G.; Li, F.; Chen, X. 68Ga-NOTA-Aca-BBN(7-14) PET/CT in healthy volunteers and glioma patients. J. Nucl. Med. 2016, 57, 9–14. [Google Scholar] [CrossRef] [Green Version]
- Nock, B.A.; Kaloudi, A.; Lymperis, E.; Giarika, A.; Kulkarni, H.R.; Klette, I.; Singh, A.; Krenning, E.P.; de Jong, M.; Maina, T.; et al. Theranostic Perspectives in Prostate Cancer with the Gastrin-Releasing Peptide Receptor Antagonist NeoBOMB1: Preclinical and First Clinical Results. J. Nucl. Med. 2017, 58, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Pan, Q.; Yao, S.; Yu, M.; Wu, W.; Xue, H.; Kiesewetter, D.O.; Zhu, Z.; Li, F.; Zhao, Y.; et al. Glucagon-like peptide-1 receptor PET/CT with 68Ga-NOTA-Exendin-4 for detecting localized insulinoma: A prospective cohort study. J. Nucl. Med. 2016, 57, 715–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrasquillo, J.A.; O’Donoghue, J.A.; Beylergil, V.; Ruan, S.; Pandit-Taskar, N.; Larson, S.M.; Smith-Jones, P.M.; Lyashchenko, S.K.; Ohishi, N.; Ohtomo, T.; et al. I-124 codrituzumab imaging and biodistribution in patients with hepatocellular carcinoma. EJNMMI Res. 2018, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Fleming, I.N.; Manavaki, R.; Blower, P.J.; West, C.; Williams, K.J.; Harris, A.L.; Domarkas, J.; Lord, S.; Baldry, C.; Gilbert, F.J. Imaging tumour hypoxia with positron emission tomography. Br. J. Cancer 2015, 112, 238–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komar, G.; Seppänen, M.; Eskola, O.; Lindholm, P.; Grönroos, T.J.; Forsback, S.; Sipilä, H.; Evans, S.M.; Solin, O.; Minn, H. 18F-EF5: A new PET tracer for imaging hypoxia in head and neck cancer. J. Nucl. Med. 2008, 49, 1944–1951. [Google Scholar] [CrossRef] [Green Version]
- Koh, W.J.; Rasey, J.S.; Evans, M.L.; Grierson, J.R.; Lewellen, T.K.; Graham, M.M.; Krohn, K.A.; Griffin, T.W. Imaging of hypoxia in human tumors with [F-18]fluoromisonidazole. Int. J. Radiat. Oncol. Biol. Phys. 1992, 22, 199–212. [Google Scholar] [CrossRef]
- Savi, A.; Incerti, E.; Fallanca, F.; BettinarDi, V.; Rossetti, F.; Monterisi, C.; Compierchio, A.; Negri, G.; Zannini, P.; Gianolli, L.; et al. First evaluation of PET-based human biodistribution and dosimetry of 18F-FAZA, a tracer for imaging tumor hypoxia. J. Nucl. Med. 2017, 58, 1224–1229. [Google Scholar] [CrossRef] [Green Version]
- Zegers, C.M.L.; van Elmpt, W.; Szardenings, K.; Kolb, H.; Waxman, A.; Subramaniam, R.M.; Moon, D.H.; Brunetti, J.C.; Srinivas, S.M.; Lambin, P.; et al. Repeatability of hypoxia PET imaging using [18F]HX4 in lung and head and neck cancer patients: a prospective multicenter trial. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 1840–1849. [Google Scholar] [CrossRef] [Green Version]
- Lewis, J.S.; Laforest, R.; Dehdashti, F.; Grigsby, P.W.; Welch, M.J.; Siegel, B.A. An imaging comparison of 64Cu-ATSM and 60Cu-ATSM in cancer of the uterine cervix. J. Nucl. Med. 2008, 49, 1177–1182. [Google Scholar] [CrossRef] [Green Version]
- 64Cu-LLP2A for Imaging Multiple Myeloma - Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03804424 (accessed on 16 April 2020).
- Chen, H.; Niu, G.; Wu, H.; Chen, X. Clinical application of radiolabeled RGD peptides for PET imaging of integrin αvβ3. Theranostics 2016, 6, 78–92. [Google Scholar] [CrossRef] [Green Version]
- Beer, A.J.; Haubner, R.; Wolf, I.; Goebel, M.; Luderschmidt, S.; Niemeyer, M.; Grosu, A.-L.; Martinez, M.-J.; Wester, H.J.; Weber, W.A.; et al. PET-based human dosimetry of 18F-galacto-RGD, a new radiotracer for imaging alpha v beta3 expression. J. Nucl. Med. 2006, 47, 763–769. [Google Scholar] [PubMed]
- Chin, F.T.; Shen, B.; Liu, S.; Berganos, R.A.; Chang, E.; Mittra, E.; Chen, X.; Gambhir, S.S. First experience with clinical-grade [ 18F]FPP (RGD) 2: An automated multi-step radiosynthesis for clinical PET studies. Mol. Imaging Biol. 2012, 14, 88–95. [Google Scholar] [CrossRef] [Green Version]
- Doss, M.; Kolb, H.C.; Zhang, J.J.; Bélanger, M.J.; Stubbs, J.B.; Stabin, M.G.; Hostetler, E.D.; Alpaugh, R.K.; Von Mehren, M.; Walsh, J.C.; et al. Biodistribution and radiation dosimetry of the integrin marker 18F-RGD-K5 determined from whole-body PET/CT in monkeys and humans. J. Nucl. Med. 2012, 53, 787–795. [Google Scholar] [CrossRef] [Green Version]
- Mena, E.; Owenius, R.; Turkbey, B.; Sherry, R.; Bratslavsky, G.; Macholl, S.; Miller, M.P.; Somer, E.J.; Lindenberg, L.; Adler, S.; et al. [18F]Fluciclatide in the in vivo evaluation of human melanoma and renal tumors expressing αvβ3 and αvβ5 integrins. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 1879–1888. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.; Guo, N.; Pan, D.; Yu, C.; Weng, Y.; Luo, S.; Ding, H.; Xu, Y.; Wang, L.; Lang, L.; et al. First experience of 18F-alfatide in lung cancer patients using a new lyophilized kit for rapid radiofluorination. J. Nucl. Med. 2013, 54, 691–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.; Pan, D.; Mi, B.; Xu, Y.; Lang, L.; Niu, G.; Yang, M.; Wan, W.; Chen, X. 18 F-Alfatide II PET/CT in healthy human volunteers and patients with brain metastases. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 2021–2028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, K.; Liang, N.; Zhang, J.; Lang, L.; Zhang, W.; Li, S.; Zhao, J.; Niu, G.; Li, F.; Zhu, Z.; et al. 68Ga-NOTA-PRGD2 PET/CT for integrin imaging in patients with lung cancer. J. Nucl. Med. 2015, 56, 1823–1827. [Google Scholar] [CrossRef] [Green Version]
- Hausner, S.H.; Bold, R.J.; Cheuy, L.Y.; Chew, H.K.; Daly, M.E.; Davis, R.A.; Foster, C.C.; Kim, E.J.; Sutcliffe, J.L. Preclinical development and first-in-human imaging of the integrin a v b 6 with [ 18 F]a v b 6 -binding peptide in metastatic carcinoma. Clin. Cancer Res. 2019, 25, 1206–1215. [Google Scholar] [CrossRef] [Green Version]
- Altmann, A.; Sauter, M.; Roesch, S.; Mier, W.; Warta, R.; Debus, J.; Dyckhoff, G.; Herold-Mende, C.; Haberkorn, U. Identification of a novel ITGαvβ6-binding peptide using protein separation and phage display. Clin. Cancer Res. 2017, 23, 4170–4180. [Google Scholar] [CrossRef] [Green Version]
- Kimura, R.H.; Wang, L.; Shen, B.; Huo, L.; Tummers, W.; Filipp, F.V.; Guo, H.H.; Haywood, T.; Abou-Elkacem, L.; Baratto, L.; et al. Evaluation of integrin αvβ6 cystine knot PET tracers to detect cancer and idiopathic pulmonary fibrosis. Nat. Commun. 2019, 10, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Xu, J.; Gonzalez, R.; Lindner, T.; Kratochwil, C.; Miao, Y. 68Ga-DOTA-GGNle-CycMSHhex targets the melanocortin-1 receptor for melanoma imaging. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamberts, L.E.; Menke-Van Der Houven Van Oordt, C.W.; Ter Weele, E.J.; Bensch, F.; Smeenk, M.M.; Voortman, J.; Hoekstra, O.S.; Williams, S.P.; Fine, B.M.; Maslyar, D.; et al. ImmunoPET with anti-mesothelin antibody in patients with pancreatic and ovarian cancer before anti-mesothelin antibody-drug conjugate treatment. Clin. Cancer Res. 2016, 22, 1642–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krolicki, L.; Bruchertseifer, F.; Kunikowska, J.; Koziara, H.; Królicki, B.; Jakuciński, M.; Pawlak, D.; Apostolidis, C.; Mirzadeh, S.; Rola, R.; et al. Prolonged survival in secondary glioblastoma following local injection of targeted alpha therapy with 213Bi-substance P analogue. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 1636–1644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Study of 18F-AlF-NOTA-Neurotensin PET/CT for Imaging Prostate Cancer - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03516045 (accessed on 16 April 2020).
- Picchio, M.; Castellucci, P. Clinical indications of 11C-choline PET/CT in prostate cancer patients with biochemical relapse. Theranostics 2012, 2, 313–317. [Google Scholar] [CrossRef]
- Degrado, T.R.; Coleman, R.E.; Wang, S.; Baldwin, S.W.; Orr, M.D.; Robertson, C.N.; Polascik, T.J.; Price, D.T. Synthesis and evaluation of 18F-labeled choline as an oncologic tracer for positron emission tomography: Initial findings in prostate cancer. Cancer Res. 2001, 61, 110–117. [Google Scholar]
- Schöder, H.M.; Demétrio De Souzaa Franç, P.; Nakajima, R.; Burnazi, E.M.; Roberts, S.; Brand, C.; Grkovski, M.; Mauguen, A.; Dunphy, M.P.; Ghossein, R.; et al. Safety and feasibility of PARP1/2 imaging with 18F-PARPi in patients with head and neck cancer. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef] [Green Version]
- Violet, J.; Sandhu, S.; Iravani, A.; Ferdinandus, J.; Thang, S.P.; Kong, G.; Ravi Kumar, A.; Akhurst, T.; Pattison, D.A.; Beaulieu, A.; et al. Long term follow-up and outcomes of re-treatment in an expanded 50 patient single-center phase II prospective trial of Lutetium-177 ( 177 Lu) PSMA-617 theranostics in metastatic castrate-resistant prostate cancer. J. Nucl. Med. 2019. [Google Scholar] [CrossRef]
- Czarniecki, M.; Mena, E.; Lindenberg, L.; Cacko, M.; Harmon, S.; Radtke, J.P.; Giesel, F.; Turkbey, B.; Choyke, P.L. Keeping up with the prostate-specific membrane antigens (PSMAs): An introduction to a new class of positron emission tomography (PET) imaging agents. Transl. Androl. Urol. 2018, 7, 831–843. [Google Scholar] [CrossRef]
- Rousseau, E.; Wilson, D.; Lacroix-Poisson, F.; Krauze, A.; Chi, K.; Gleave, M.; McKenzie, M.; Tyldesley, S.; Larry Goldenberg, S.; Bénard, F. A prospective study on 18F-DCFPYL PSMA PET/CT imaging in biochemical recurrence of prostate cancer. J. Nucl. Med. 2019, 60, 1587–1593. [Google Scholar] [CrossRef] [Green Version]
- Giesel, F.L.; Knorr, K.; Spohn, F.; Will, L.; Maurer, T.; Flechsig, P.; Neels, O.; Schiller, K.; Amaral, H.; Weber, W.A.; et al. Detection efficacy of 18 F-PSMA-1007 PET/CT in 251 patients with biochemical recurrence of prostate cancer after radical prostatectomy. J. Nucl. Med. 2019, 60, 362–368. [Google Scholar] [CrossRef] [Green Version]
- Pandit-Taskar, N.; O’Donoghue, J.A.; Beylergil, V.; Lyashchenko, S.; Ruan, S.; Solomon, S.B.; Durack, J.C.; Carrasquillo, J.A.; Lefkowitz, R.A.; Gonen, M.; et al. 89Zr-huJ591 immuno-PET imaging in patients with advanced metastatic prostate cancer. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 2093–2105. [Google Scholar] [CrossRef] [Green Version]
- Eiber, M.; Krönke, M.; Wurzer, A.; Ulbrich, L.; Jooß, L.; Maurer, T.; Horn, T.; Schiller, K.; Langbein, T.; Buschner, G.; et al. 18 F-rhPSMA-7 positron emission tomography for the detection of biochemical recurrence of prostate cancer following radical prostatectomy. J. Nucl. Med. 2019. [Google Scholar] [CrossRef]
- Verhoeff, S.R.; van den Heuvel, M.M.; van Herpen, C.M.L.; Piet, B.; Aarntzen, E.H.J.G.; Heskamp, S. Programmed Cell Death-1/Ligand-1 PET Imaging: A Novel Tool to Optimize Immunotherapy? PET Clin. 2020, 15, 35–43. [Google Scholar] [CrossRef] [Green Version]
- Niemeijer, A.N.; Leung, D.; Huisman, M.C.; Bahce, I.; Hoekstra, O.S.; van Dongen, G.A.M.S.; Boellaard, R.; Du, S.; Hayes, W.; Smith, R.; et al. Whole body PD-1 and PD-L1 positron emission tomography in patients with non-small-cell lung cancer. Nat. Commun. 2018, 9, 1–5. [Google Scholar] [CrossRef]
- Huisman, M.; Niemeijer, A.-L.; Windhorst, B.; Schuit, R.; Leung, D.; Hayes, W.; Poot, A.; Bahce, I.; Radonic, T.; Oprea-Lager, D.; et al. Quantification of PD-L1 expression with [ 18 F]BMS-986192 PET/CT in patients with advanced stage non-small-cell lung cancer. J. Nucl. Med. 2020. [Google Scholar] [CrossRef]
- Bensch, F.; van der Veen, E.L.; Lub-de Hooge, M.N.; Jorritsma-Smit, A.; Boellaard, R.; Kok, I.C.; Oosting, S.F.; Schröder, C.P.; Hiltermann, T.J.N.; van der Wekken, A.J.; et al. 89Zr-atezolizumab imaging as a non-invasive approach to assess clinical response to PD-L1 blockade in cancer. Nat. Med. 2018, 24, 1852–1858. [Google Scholar] [CrossRef]
- Carrasquillo, J.A.; Fine, B.M.; Pandit-Taskar, N.; Larson, S.M.; Fleming, S.E.; Fox, J.J.; Cheal, S.M.; O’Donoghue, J.A.; Ruan, S.; Ragupathi, G.; et al. Imaging patients with metastatic castration-resistant prostate cancer using 89Zr-DFO-MSTP2109A anti-STEAP1 antibody. J. Nucl. Med. 2019, 60, 1517–1523. [Google Scholar] [CrossRef] [Green Version]
- Jentzen, W.; Freudenberg, L.; Eising, E.G.; Sonnenschein, W.; Knust, J.; Bockisch, A. Optimized 124I PET dosimetry protocol for radioiodine therapy of differentiated thyroid cancer. J. Nucl. Med. 2008, 49, 1017–1023. [Google Scholar] [CrossRef] [Green Version]
- Hicks, R.J.; Jackson, P.; Kong, G.; Ware, R.E.; Hofman, M.S.; Pattison, D.A.; Akhurst, T.A.; Drummond, E.; Roselt, P.; Callahan, J.; et al. 64Cu-SARTATE PET imaging of patients with neuroendocrine tumors demonstrates high tumor uptake and retention, potentially allowing prospective dosimetry for peptide receptor radionuclide therapy. J. Nucl. Med. 2019, 60, 777–785. [Google Scholar] [CrossRef] [Green Version]
- Nicolas, G.P.; Schreiter, N.; Kaul, F.; Uiters, J.; Bouterfa, H.; Kaufmann, J.; Erlanger, T.E.; Cathomas, R.; Christ, E.; Fani, M.; et al. Sensitivity Comparison of 68Ga-OPS202 and 68Ga-DOTATOC PET/CT in Patients with Gastroenteropancreatic Neuroendocrine Tumors: A Prospective Phase II Imaging Study. J. Nucl. Med. 2018, 59, 915–921. [Google Scholar] [CrossRef] [Green Version]
- Ambrosini, V.; Campana, D.; Bodei, L.; Nanni, C.; Castellucci, P.; Allegri, V.; Montini, G.C.; Tomassetti, P.; Paganelli, G.; Fanti, S. 68Ga-DOTANOC PET/CT clinical impact in patients with neuroendocrine tumors. J. Nucl. Med. 2010, 51, 669–673. [Google Scholar] [CrossRef] [Green Version]
- Krebs, S.; Pandit-Taskar, N.; Reidy, D.; Beattie, B.J.; Lyashchenko, S.K.; Lewis, J.S.; Bodei, L.; Weber, W.A.; O’Donoghue, J.A. Biodistribution and radiation dose estimates for 68 Ga-DOTA-JR11 in patients with metastatic neuroendocrine tumors. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 677–685. [Google Scholar] [CrossRef]
- Shields, A.F.; Grierson, J.R.; Dohmen, B.M.; Machulla, H.J.; Stayanoff, J.C.; Lawhorn-Crews, J.M.; Obradovich, J.E.; Muzik, O.; Mangner, T.J. Imaging proliferation in vivo with [F-18]FLT and positron emission tomography. Nat. Med. 1998, 4, 1334–1336. [Google Scholar] [CrossRef]
- Den Hollander, M.W.; Bensch, F.; Glaudemans, A.W.J.M.; Oude Munnink, T.H.; Enting, R.H.; Den Dunnen, W.F.A.; Heesters, M.A.A.M.; Kruyt, F.A.E.; Lub-De Hooge, M.N.; De Groot, J.C.; et al. TGF-β antibody uptake in recurrent high-grade glioma imaged with 89Zr-fresolimumab PET. J. Nucl. Med. 2015, 56, 1310–1314. [Google Scholar] [CrossRef] [Green Version]
- Gaykema, S.B.M.; Schröder, C.P.; Vitfell-Rasmussen, J.; Chua, S.; Munnink, T.H.O.; Brouwers, A.H.; Bongaerts, A.H.H.; Akimov, M.; Fernandez-Ibarra, C.; Lub-De Hooge, M.N.; et al. 89Zr-trastuzumab and 89Zr-bevacizumab PET to evaluate the effect of the HSP90 inhibitor NVP-AUY922 in metastatic breast cancer patients. Clin. Cancer Res. 2014, 20, 3945–3954. [Google Scholar] [CrossRef] [Green Version]
- Bahce, I.; Huisman, M.C.; Verwer, E.E.; Ooijevaar, R.; Boutkourt, F.; Vugts, D.J.; van Dongen, G.A.M.S.; Boellaard, R.; Smit, E.F. Pilot study of 89Zr-bevacizumab positron emission tomography in patients with advanced non-small cell lung cancer. EJNMMI Res. 2014, 4, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Van Asselt, S.J.; Oosting, S.F.; Brouwers, A.H.; Bongaerts, A.H.H.; De Jong, J.R.; Lub-de Hooge, M.N.; Oude Munnink, T.H.; Fiebrich, H.B.; Sluiter, W.J.; Links, T.P.; et al. Everolimus reduces 89Zr-bevacizumab tumor uptake in patients with neuroendocrine tumors. J. Nucl. Med. 2014, 55, 1087–1092. [Google Scholar] [CrossRef] [Green Version]
- Thorek, D.L.J.; Watson, P.A.; Lee, S.G.; Ku, A.T.; Bournazos, S.; Braun, K.; Kim, K.; Sjostrom, K.; Doran, M.G.; Lamminmaki, U.; et al. Internalization of secreted antigen-targeted antibodies by the neonatal Fc receptor for precision imaging of the androgen receptor axis. Sci. Transl. Med. 2016, 8. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Alavi, A.; Reivich, M. Guest editorial: The conception of FDG-PET imaging. Semin. Nucl. Med. 2002, 32, 2–5. [Google Scholar] [CrossRef] [Green Version]
- Reivich, M.; Kuhl, D.; Wolf, A. The [18F]fluorodeoxyglucose method for the measurement of local cerebral glucose utilization in man. Circ. Res. 1979, 44, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Ido, T.; Wan, C. -N.; Casella, V.; Fowler, J.S.; Wolf, A.P.; Reivich, M.; Kuhl, D.E. Labeled 2-deoxy-D-glucose analogs. 18F-labeled 2-deoxy-2-fluoro-D-glucose, 2-deoxy-2-fluoro-D-mannose and 14C-2-deoxy-2-fluoro-D-glucose. J. Label. Compd. Radiopharm. 1978, 14, 175–183. [Google Scholar] [CrossRef]
- Katsanos, A.H.; Alexiou, G.A.; Fotopoulos, A.D.; Jabbour, P.; Kyritsis, A.P.; Sioka, C. Performance of 18F-FDG, 11C-Methionine, and 18F-FET PET for Glioma Grading: A Meta-analysis. Clin. Nucl. Med. 2019, 44, 864–869. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, H.; Chen, K.; Shao, Y.; Kiesewetter, D.O.; Niu, G.; Chen, X. Boramino acid as a marker for amino acid transporters. Sci. Adv. 2015, 1. [Google Scholar] [CrossRef] [Green Version]
- Nodwell, M.B.; Yang, H.; Čolović, M.; Yuan, Z.; Merkens, H.; Martin, R.E.; Bénard, F.; Schaffer, P.; Britton, R. 18F-Fluorination of Unactivated C-H Bonds in Branched Aliphatic Amino Acids: Direct Synthesis of Oncological Positron Emission Tomography Imaging Agents. J. Am. Chem. Soc. 2017, 139, 3595–3598. [Google Scholar] [CrossRef]
- Nodwell, M.B.; Yang, H.; Merkens, H.; Malik, N.; Čolović, M.; Wagner, B.; Martin, R.E.; Bénard, F.; Schaffer, P.; Britton, R. 18F-branched-chain amino acids: Structure-activity relationships and PET imaging potential. J. Nucl. Med. 2019, 60, 1003–1009. [Google Scholar] [CrossRef] [Green Version]
- Davenport, A.P.; Scully, C.C.G.; de Graaf, C.; Brown, A.J.H.; Maguire, J.J. Advances in therapeutic peptides targeting G protein-coupled receptors. Nat. Rev. Drug Discov. 2020, 1–25. [Google Scholar] [CrossRef]
- Reubi, J.C.; Maecke, H.R. Peptide-based probes for cancer imaging. J. Nucl. Med. 2008, 49, 1735–1738. [Google Scholar] [CrossRef] [Green Version]
- Reubi, J.C. Peptide receptors as molecular targets for cancer diagnosis and therapy. Endocr. Rev. 2003, 24, 389–427. [Google Scholar] [CrossRef] [Green Version]
- Rangger, C.; Haubner, R. Radiolabelled peptides for positron emission tomography and endoradiotherapy in oncology. Pharmaceuticals 2020, 13, 22. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Pan, J.; Lin, K.S.; Dude, I.; Lau, J.; Zeisler, J.; Merkens, H.; Jenni, S.; Guérin, B.; Bénard, F. Targeting the Neuropeptide Y1 Receptor for Cancer Imaging by Positron Emission Tomography Using Novel Truncated Peptides. Mol. Pharm. 2016, 13, 3657–3664. [Google Scholar] [CrossRef]
- Günther, T.; Tulipano, G.; Dournaud, P.; Bousquet, C.; Csaba, Z.; Kreienkamp, H.J.; Lupp, A.; Korbonits, M.; Castaño, J.P.; Wester, H.J.; et al. International union of basic and clinical pharmacology. CV. somatostatin receptors: Structure, function, ligands, and new nomenclature. Pharmacol. Rev. 2018, 70, 763–835. [Google Scholar] [CrossRef] [Green Version]
- Deroose, C.M.; Hindié, E.; Kebebew, E.; Goichot, B.; Pacak, K.; Taïeb, D.; Imperiale, A. Molecular imaging of gastroenteropancreatic neuroendocrine tumors: Current status and future directions. J. Nucl. Med. 2016, 57, 1949–1956. [Google Scholar] [CrossRef] [Green Version]
- Sugae, S.; Suzuki, A.; Takahashi, N.; Minamimoto, R.; Cheng, C.; Theeraladanon, C.; Endo, I.; Togo, S.; Inoue, T.; Shimada, H. Fluorine-18-labeled 5-fluorouracil is a useful radiotracer for differentiation of malignant tumors from inflammatory lesions. Ann. Nucl. Med. 2008, 22, 65–72. [Google Scholar] [CrossRef]
- Jacobson, O.; Weiss, I.D.; Szajek, L.; Farber, J.M.; Kiesewetter, D.O. 64Cu-AMD3100-A novel imaging agent for targeting chemokine receptor CXCR4. Bioorganic Med. Chem. 2009, 17, 1486–1493. [Google Scholar] [CrossRef] [Green Version]
- O’Keefe, D.S.; Bacich, D.J.; Huang, S.S.; Heston, W.D.W. A Perspective on the Evolving Story of PSMA Biology, PSMA-Based Imaging, and Endoradiotherapeutic Strategies. J. Nucl. Med. 2018, 59, 1007–1013. [Google Scholar] [CrossRef]
- Han, M.; Partin, A.W. Current Clinical Applications of the In-capromab Pendetide Scan (ProstaScint(R) Scan, Cyt-356). Rev. Urol. 2001, 3, 165–171. [Google Scholar]
- Kozikowski, A.P.; Nan, F.; Conti, P.; Zhang, J.; Ramadan, E.; Bzdega, T.; Wroblewska, B.; Neale, J.H.; Pshenichkin, S.; Wroblewski, J.T. Design of remarkably simple, yet potent urea-based inhibitors of glutamate carboxypeptidase II (NAALADase). J. Med. Chem. 2001, 44, 298–301. [Google Scholar] [CrossRef]
- Scott, A.M.; Wiseman, G.; Welt, S.; Adjei, A.; Lee, F.T.; Hopkins, W.; Divgi, C.R.; Hanson, L.H.; Mitchell, P.; Gansen, D.N.; et al. A phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin. Cancer Res. 2003, 9, 1639–1647. [Google Scholar]
- Jansen, K.; Heirbaut, L.; Cheng, J.D.; Joossens, J.; Ryabtsova, O.; Cos, P.; Maes, L.; Lambeir, A.M.; De Meester, I.; Augustyns, K.; et al. Selective inhibitors of fibroblast activation protein (FAP) with a (4-quinolinoyl)-glycyl-2-cyanopyrrolidine scaffold. ACS Med. Chem. Lett. 2013, 4, 491–496. [Google Scholar] [CrossRef]
- Lindner, T.; Loktev, A.; Altmann, A.; Giesel, F.; Kratochwil, C.; Debus, J.; Jäger, D.; Mier, W.; Haberkorn, U. Development of quinoline-based theranostic ligands for the targeting of fibroblast activation protein. J. Nucl. Med. 2018, 59, 1415–1422. [Google Scholar] [CrossRef] [Green Version]
- Goldenberg, D.M. Targeted therapy of cancer with radiolabeled antibodies. J. Nucl. Med. 2002, 43, 693–713. [Google Scholar]
- Chiavenna, S.M.; Jaworski, J.P.; Vendrell, A. State of the art in anti-cancer mAbs. J. Biomed. Sci. 2017, 24, 15. [Google Scholar] [CrossRef] [Green Version]
- Ghigna, C.; Valacca, C.; Biamonti, G. Alternative Splicing and Tumor Progression. Curr. Genom. 2008, 9, 556–570. [Google Scholar] [CrossRef] [Green Version]
- Deri, M.A.; Zeglis, B.M.; Francesconi, L.C.; Lewis, J.S. PET imaging with 89Zr: From radiochemistry to the clinic. Nucl. Med. Biol. 2013, 40, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Jauw, Y.W.S.; Menke-van der Houven van Oordt, C.W.; Hoekstra, O.S.; Hendrikse, N.H.; Vugts, D.J.; Zijlstra, J.M.; Huisman, M.C.; van Dongen, G.A.M.S. Immuno-Positron Emission Tomography with Zirconium-89-Labeled Monoclonal Antibodies in Oncology: What Can We Learn from Initial Clinical Trials? Front. Pharmacol. 2016, 7, 131. [Google Scholar] [CrossRef]
- Verma, V.; Sprave, T.; Haque, W.; Simone, C.B.; Chang, J.Y.; Welsh, J.W.; Thomas, C.R. A systematic review of the cost and cost-effectiveness studies of immune checkpoint inhibitors 11 Medical and Health Sciences 1112 Oncology and Carcinogenesis. J. Immunother. Cancer 2018, 6, 128. [Google Scholar] [CrossRef] [Green Version]
- Golebiowski, A.; Klopfenstein, S.R.; Portlock, D.E. Lead compounds discovered from libraries. Curr. Opin. Chem. Biol. 2001, 5, 273–284. [Google Scholar] [CrossRef]
- Comess, K.M.; Schurdak, M.E. Affinity-based screening techniques for enhancing lead discovery. Curr. Opin. Drug Discov. Dev. 2004, 7, 411–416. [Google Scholar]
- Keserü, G.M.; Makara, G.M. The influence of lead discovery strategies on the properties of drug candidates. Nat. Rev. Drug Discov. 2009, 8, 203–212. [Google Scholar] [CrossRef]
- Davies, J.W.; Glick, M.; Jenkins, J.L. Streamlining lead discovery by aligning in silico and high-throughput screening. Curr. Opin. Chem. Biol. 2006, 10, 343–351. [Google Scholar] [CrossRef]
- Shoichet, B.K.; McGovern, S.L.; Wei, B.; Irwin, J.J. Lead discovery using molecular docking. Curr. Opin. Chem. Biol. 2002, 6, 439–446. [Google Scholar] [CrossRef]
- Liu, R.; Li, X.; Lam, K.S. Combinatorial chemistry in drug discovery. Curr. Opin. Chem. Biol. 2017, 38, 117–126. [Google Scholar] [CrossRef]
- Kleiner, R.E.; Dumelin, C.E.; Liu, D.R. Small-molecule discovery from DNA-encoded chemical libraries. Chem. Soc. Rev. 2011, 40, 5707–5717. [Google Scholar] [CrossRef] [Green Version]
- Yuen, L.H.; Franzini, R.M. Achievements, Challenges, and Opportunities in DNA-Encoded Library Research: An Academic Point of View. ChemBioChem 2017, 18, 829–836. [Google Scholar] [CrossRef]
- Mannocci, L.; Leimbacher, M.; Wichert, M.; Scheuermann, J.; Neri, D. 20 years of DNA-encoded chemical libraries. Chem. Commun. 2011, 47, 12747–12753. [Google Scholar] [CrossRef]
- Wichert, M.; Krall, N.; Decurtins, W.; Franzini, R.M.; Pretto, F.; Schneider, P.; Neri, D.; Scheuermann, J. Dual-display of small molecules enables the discovery of ligand pairs and facilitates affinity maturation. Nat. Chem. 2015, 7, 241–249. [Google Scholar] [CrossRef]
- Blind, M.; Blank, M. Aptamer Selection Technology and Recent Advances. Mol. Ther. - Nucleic Acids 2015, 4, e223. [Google Scholar] [CrossRef]
- Ladner, R.C.; Sato, A.K.; Gorzelany, J.; De Souza, M. Phage display-derived peptides as therapeutic alternatives to antibodies. Drug Discov. Today 2004, 9, 525–529. [Google Scholar] [CrossRef]
- Shelat, A.A.; Guy, R.K. Scaffold composition and biological relevance of screening libraries. Nat. Chem. Biol. 2007, 3, 442–446. [Google Scholar] [CrossRef]
- Binz, H.K.; Amstutz, P.; Kohl, A.; Stumpp, M.T.; Briand, C.; Forrer, P.; Grütter, M.G.; Plückthun, A. High-affinity binders selected from designed ankyrin repeat protein libraries. Nat. Biotechnol. 2004, 22, 575–582. [Google Scholar] [CrossRef]
- Heinis, C.; Rutherford, T.; Freund, S.; Winter, G. Phage-encoded combinatorial chemical libraries based on bicyclic peptides. Nat. Chem. Biol. 2009, 5, 502–507. [Google Scholar] [CrossRef]
- Murakami, H.; Ohta, A.; Ashigai, H.; Suga, H. A highly flexible tRNA acylation method for non-natural polypeptide synthesis. Nat. Methods 2006, 3, 357–359. [Google Scholar] [CrossRef]
- Lam, K.S.; Salmon, S.E.; Hersh, E.M.; Hruby, V.J.; Kazmierskit, W.M.; Knappt, R.J. A new type of synthetic peptide library for identifying ligand-binding activity. Nature 1991, 354, 82–84. [Google Scholar] [CrossRef]
- C. Martínez-Ceron, M.; L. Giudicessi, S.; L. Saavedra, S.; M. Gurevich-Messina, J.; Erra-Balsells, R.; Albericio, F.; Cascone, O.; A. Camperi, S. Latest Advances in OBOC Peptide Libraries. Improvements in Screening Strategies and Enlarging the Family From Linear to Cyclic Libraries. Curr. Pharm. Biotechnol. 2016, 17, 449–457. [Google Scholar] [CrossRef]
- Aina, O.H.; Liu, R.; Sutcliffe, J.L.; Marik, J.; Pan, C.X.; Lam, K.S. From combinatorial chemistry to cancer-targeting peptides. Mol. Pharm. 2007, 4, 631–651. [Google Scholar] [CrossRef]
- Song, A.; Zhang, J.; Lebrilla, C.B.; Lam, K.S. A novel and rapid encoding method based on mass spectrometry for “one-bead-one-compound” small molecule combinatorial libraries. J. Am. Chem. Soc. 2003, 125, 6180–6188. [Google Scholar] [CrossRef]
- Joo, S.H.; Xiao, Q.; Ling, Y.; Gopishetty, B.; Pei, D. High-throughput sequence determination of cyclic peptide library members by partial Edman degradation/mass spectrometry. J. Am. Chem. Soc. 2006, 128, 13000–13009. [Google Scholar] [CrossRef]
- Peng, L.; Liu, R.; Marik, J.; Wang, X.; Takada, Y.; Lam, K.S. Combinatorial chemistry identifies high-affinity peptidomimetics against α4β1 integrin for in vivo tumor imaging. Nat. Chem. Biol. 2006, 2, 381–389. [Google Scholar] [CrossRef]
- Komoriya, A.; Green, L.J.; Mervic, M.; Yamada, S.S.; Yamada, K.M.; Humphries, M.J. The minimal essential sequence for a major cell type-specific adhesion site (CS1) within the alternatively spliced type III connecting segment domain of fibronectin is leucine-aspartic acid-valine. J. Biol. Chem. 1991, 266, 15075–15079. [Google Scholar]
- Lin, K.C.; Ateeq, H.S.; Hsiung, S.H.; Chong, L.T.; Zimmerman, C.N.; Castro, A.; Lee, W.C.; Hammond, C.E.; Kalkunte, S.; Chen, L.L.; et al. Selective, tight-binding inhibitors of integrin α4β1 that inhibit allergic airway responses. J. Med. Chem. 1999, 42, 920–934. [Google Scholar] [CrossRef]
- Jiang, M.; Ferdani, R.; Shokeen, M.; Anderson, C.J. Comparison of two cross-bridged macrocyclic chelators for the evaluation of 64Cu-labeled-LLP2A, a peptidomimetic ligand targeting VLA-4-positive tumors. Nucl. Med. Biol. 2013, 40, 245–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaino, W.; Anderson, C.J. PET imaging of very late antigen-4 in melanoma: Comparison of 68Ga- and 64Cu-Labeled NODAGA and CB-TE1A1P-LLP2A conjugates. J. Nucl. Med. 2014, 55, 1856–1863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Minn, I.; Rowe, S.P.; Banerjee, S.R.; Gorin, M.A.; Brummet, M.; Lee, H.S.; Koo, S.M.; Sysa-Shah, P.; Mease, R.C.; et al. Imaging of carbonic anhydrase IX with an 111In-labeled dual-motif inhibitor. Oncotarget 2015, 6, 33733–33742. [Google Scholar] [CrossRef] [Green Version]
- Minn, I.; Koo, S.M.; Lee, H.S.; Brummet, M.; Rowe, S.P.; Gorin, M.A.; Sysa-Shah, P.; Lewis, W.D.; Ahn, H.-H.; Wang, Y.; et al. [64Cu]XYIMSR-06: A dual-motif CAIX ligand for PET imaging of clear cell renal cell carcinoma. Oncotarget 2016, 7, 56471–56479. [Google Scholar] [CrossRef] [Green Version]
- Conti, M.; Eriksson, L. Physics of pure and non-pure positron emitters for PET: a review and a discussion. EJNMMI Phys. 2016, 3, 8. [Google Scholar] [CrossRef] [Green Version]
- Lapi, S.E.; Welch, M.J. A historical perspective on the specific activity of radiopharmaceuticals: What have we learned in the 35 years of the ISRC? Nucl. Med. Biol. 2012, 39, 601–608. [Google Scholar] [CrossRef]
- Sergeev, M.; Lazari, M.; Morgia, F.; Collins, J.; Javed, M.R.; Sergeeva, O.; Jones, J.; Phelps, M.E.; Lee, J.T.; Keng, P.Y.; et al. Performing radiosynthesis in microvolumes to maximize molar activity of tracers for positron emission tomography. Commun. Chem. 2018, 1, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Coenen, H.H.; Gee, A.D.; Adam, M.; Antoni, G.; Cutler, C.S.; Fujibayashi, Y.; Jeong, J.M.; Mach, R.H.; Mindt, T.L.; Pike, V.W.; et al. Consensus nomenclature rules for radiopharmaceutical chemistry — Setting the record straight. Nucl. Med. Biol. 2017, 55, v–xi. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, O.; Chen, X.; Weiss, I.D.; Kiesewetter, D.O.; Farber, J.M. PET of Tumor CXCR4 Expression with 4-18F-T140. J. Nucl. Med. 2010, 51, 1796–1804. [Google Scholar] [CrossRef] [Green Version]
- Dalm, S.U.; Bakker, I.L.; de Blois, E.; Doeswijk, G.N.; Konijnenberg, M.W.; Orlandi, F.; Barbato, D.; Tedesco, M.; Maina, T.; Nock, B.A.; et al. 68 Ga/ 177 Lu-NeoBOMB1, a Novel Radiolabeled GRPR Antagonist for Theranostic Use in Oncology. J. Nucl. Med. 2017, 58, 293–299. [Google Scholar] [CrossRef] [Green Version]
- Nonnekens, J.; Schottelius, M. “Luke! Luke! Don’t! It’s a trap!”—spotlight on bias in animal experiments in nuclear oncology. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 1024–1026. [Google Scholar] [CrossRef] [Green Version]
- Saha, G.B. Production of Radionuclides. In Physics and Radiobiology of Nuclear Medicine; Springer: New York, NY, USA, 2006; pp. 44–55. [Google Scholar]
- Synowiecki, M.A.; Perk, L.R.; Nijsen, J.F.W. Production of novel diagnostic radionuclides in small medical cyclotrons. EJNMMI Radiopharm. Chem. 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Holland, J.P.; Williamson, M.J.; Lewis, J.S. Unconventional Nuclides for Radiopharmaceuticals. Mol. Imaging 2010, 9, 1–20. [Google Scholar] [CrossRef]
- Berger, M.J.; Coursey, J.S.; Zucker, M.A.; Chang, J. Stopping-Power & Range Tables for Electrons, Protons, and Helium Ions. NISTIR 2017, 4999, 1–17. [Google Scholar] [CrossRef]
- Yang, L.; Scott, P.J.H.; Shao, X. [11C]Carbon Dioxide: Starting Point for Labeling PET Radiopharmaceuticals. In Carbon Dioxide Chemistry, Capture and Oil Recovery. In Carbon Dioxide Chemistry, Capture and Oil Recovery; InTech: London, UK, 2018. [Google Scholar]
- Dahl, K.; Halldin, C.; Schou, M. New methodologies for the preparation of carbon-11 labeled radiopharmaceuticals. Clin. Transl. Imaging 2017, 5, 275–289. [Google Scholar] [CrossRef] [Green Version]
- Deng, X.; Rong, J.; Wang, L.; Vasdev, N.; Zhang, L.; Josephson, L.; Liang, S.H. Chemistry for Positron Emission Tomography: Recent Advances in 11 C-, 18 F-, 13 N-, and 15 O-Labeling Reactions. Angew. Chemie - Int. Ed. 2019, 58, 2580–2605. [Google Scholar] [CrossRef]
- Andersson, J.; Truong, P.; Halldin, C. In-target produced [11C]methane: Increased specific radioactivity. Appl. Radiat. Isot. 2009, 67, 106–110. [Google Scholar] [CrossRef]
- Larsen, P.; Ulin, J.; Dahlstrøm, K.; Jensen, M. Synthesis of [11C]iodomethane by iodination of [11C]methane. Appl. Radiat. Isot. 1997, 48, 153–157. [Google Scholar] [CrossRef]
- Preshlock, S.; Tredwell, M.; Gouverneur, V. 18F-Labeling of Arenes and Heteroarenes for Applications in Positron Emission Tomography. Chem. Rev. 2016, 116, 719–766. [Google Scholar] [CrossRef]
- Gouverneur, V. Radiochemistry: Flipping fluoride’s reactivity. Nat. Chem. 2012, 4, 152–154. [Google Scholar] [CrossRef] [PubMed]
- Teare, H.; Robins, E.G.; Årstad, E.; Luthra, S.K.; Gouverneur, V. Synthesis and reactivity of [18F]-N-fluorobenzenesulfonimide. Chem. Commun. 2007, 2330–2332. [Google Scholar] [CrossRef] [PubMed]
- Teare, H.; Robins, E.G.; Kirjavainen, A.; Forsback, S.; Sandford, G.; Solin, O.; Luthra, S.K.; Gouverneur, V. Radiosynthesis and Evaluation of [18F]Selectfluor bis(triflate). Angew. Chemie Int. Ed. 2010, 49, 6821–6824. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Nodwell, M.B.; Yang, H.; Malik, N.; Merkens, H.; Bénard, F.; Martin, R.E.; Schaffer, P.; Britton, R. Site-Selective, Late-Stage C−H 18 F-Fluorination on Unprotected Peptides for Positron Emission Tomography Imaging. Angew. Chemie Int. Ed. 2018, 57, 12733–12736. [Google Scholar] [CrossRef]
- Schirrmacher, R.; Wängler, B.; Bailey, J.; Bernard-Gauthier, V.; Schirrmacher, E.; Wängler, C. Small Prosthetic Groups in 18F-Radiochemistry: Useful Auxiliaries for the Design of 18F-PET Tracers. Semin. Nucl. Med. 2017, 47, 474–492. [Google Scholar] [CrossRef]
- Fani, M.; Maecke, H.R.; Okarvi, S.M. Radiolabeled peptides: Valuable tools for the detection and treatment of cancer. Theranostics 2012, 2, 481–501. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, O.; Kiesewetter, D.O.; Chen, X. Fluorine-18 radiochemistry, labeling strategies and synthetic routes. Bioconjug. Chem. 2015, 26, 1–18. [Google Scholar] [CrossRef]
- Wilson, A.A.; Dannals, R.F.; Ravert, H.T.; Wagner, H.N. Reductive amination of [18F]fluorobenzaldehydes: Radiosyntheses of [2-18F]- and [4-18F]fluorodexetimides. J. Label. Compd. Radiopharm. 1990, 28, 1189–1199. [Google Scholar] [CrossRef]
- Bernard-Gauthier, V.; Bailey, J.J.; Liu, Z.; Wängler, B.; Wängler, C.; Jurkschat, K.; Perrin, D.M.; Schirrmacher, R. From Unorthodox to Established: The Current Status of 18F-Trifluoroborate- And 18F-SiFA-Based Radiopharmaceuticals in PET Nuclear Imaging. Bioconjug. Chem. 2016, 27, 267–279. [Google Scholar] [CrossRef]
- Price, E.W.; Orvig, C. Matching chelators to radiometals for radiopharmaceuticals. Chem. Soc. Rev. 2014, 43, 260–290. [Google Scholar] [CrossRef]
- Notni, J.; Pohle, K.; Wester, H.J. Comparative gallium-68 labeling of TRAP-, NOTA-, and DOTA-peptides: Practical consequences for the future of gallium-68-PET. EJNMMI Res. 2012, 2, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wurzer, A.; DiCarlo, D.; Schmidt, A.; Beck, R.; Eiber, M.; Schwaiger, M.; Wester, H.-J. Radiohybrid ligands: a novel tracer concept exemplified by 18 F- or 68 Ga-labeled rhPSMA-inhibitors. J. Nucl. Med. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roxin, Á.; Zhang, C.; Huh, S.; Lepage, M.; Zhang, Z.; Lin, K.S.; Bénard, F.; Perrin, D.M. A Metal-Free DOTA-Conjugated 18F-Labeled Radiotracer: [18F]DOTA-AMBF3-LLP2A for Imaging VLA-4 Over-Expression in Murine Melanoma with Improved Tumor Uptake and Greatly Enhanced Renal Clearance. Bioconjug. Chem. 2019, 30, 1210–1219. [Google Scholar] [CrossRef] [PubMed]
- Lepage, M.L.; Kuo, H.T.; Roxin, Á.; Huh, S.; Zhang, Z.; Kandasamy, R.; Merkens, H.; Kumlin, J.O.; Limoges, A.; Zeisler, S.K.; et al. Toward 18F-Labeled Theranostics: A Single Agent that Can Be Labeled with 18F, 64Cu, or 177Lu. ChemBioChem 2020, 21, 943–947. [Google Scholar] [CrossRef]
- Vallabhajosula, S. Quality Control of PET Radiopharmaceuticals. In Molecular Imaging; Springer: Berlin/Heidelberg, Germany, 2009; pp. 197–204. [Google Scholar]
- Huang, Y.-Y. An Overview of PET Radiopharmaceuticals in Clinical Use: Regulatory, Quality and Pharmacopeia Monographs of the United States and Europe. In Nuclear Medicine Physics; IntechOpen: London, UK, 2019. [Google Scholar]
- Bigott-Hennkens, H.M.; Dannoon, S.; Lewis, M.R.; Jurisson, S. In vitro receptor binding assays: General methods and considerations. Q. J. Nucl. Med. Mol. Imaging 2008, 52, 245–253. [Google Scholar]
- Hulme, E.C.; Trevethick, M.A. Ligand binding assays at equilibrium: Validation and interpretation. Br. J. Pharmacol. 2010, 161, 1219–1237. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Zeng, X.; Liang, J. Surface plasmon resonance: An introduction to a surface spectroscopy technique. J. Chem. Educ. 2010, 87, 742–746. [Google Scholar] [CrossRef] [Green Version]
- Kumaraswamy, S.; Tobias, R. Label-free kinetic analysis of an antibody-antigen interaction using biolayer interferometry. In Protein-Protein Interactions: Methods and Applications: Second Edition; Springer: New York, NY, USA, 2015; Volume 1278, ISBN 9781493924257. [Google Scholar]
- Duff, M.R.; Grubbs, J.; Howell, E.E. Isothermal titration calorimetry for measuring macromolecule-ligand affinity. J. Vis. Exp. 2011. [Google Scholar] [CrossRef] [Green Version]
- Wienken, C.J.; Baaske, P.; Rothbauer, U.; Braun, D.; Duhr, S. Protein-binding assays in biological liquids using microscale thermophoresis. Nat. Commun. 2010, 1, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Reddy, S.; Robinson, M.K. Immuno-Positron Emission Tomography in Cancer Models. Semin. Nucl. Med. 2010, 40, 182–189. [Google Scholar] [CrossRef] [Green Version]
- Anderson, C.J.; Ferdani, R. Copper-64 radiopharmaceuticals for PET imaging of cancer: Advances in preclinical and clinical research. Cancer Biother. Radiopharm. 2009, 24, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Mansour, N.; Paquette, M.; Ait-Mohand, S.; Dumulon-Perreault, V.; Guérin, B. Evaluation of a novel GRPR antagonist for prostate cancer PET imaging: [64Cu]-DOTHA2-PEG-RM26. Nucl. Med. Biol. 2018, 56, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Luo, G.; Ding, X.; Lu, C. Preclinical experimental models of drug metabolism and disposition in drug discovery and development. Acta Pharm. Sin. B 2012, 2, 549–561. [Google Scholar] [CrossRef] [Green Version]
- Nock, B.A.; Maina, T.; Krenning, E.P.; de Jong, M. “To Serve and Protect”: Enzyme Inhibitors as Radiopeptide Escorts Promote Tumor Targeting. J. Nucl. Med. 2014, 55, 121–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, K.S.; Pan, J.; Amouroux, G.; Turashvili, G.; Mesak, F.; Hundal-Jabal, N.; Pourghiasian, M.; Lau, J.; Jenni, S.; Aparicio, S.; et al. In vivo radioimaging of bradykinin receptor B1, a widely overexpressed molecule in human cancer. Cancer Res. 2015, 75, 387–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lever, S.Z.; Fan, K.H.; Lever, J.R. Tactics for preclinical validation of receptor-binding radiotracers. Nucl. Med. Biol. 2017, 44, 4–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Chen, X. Simple bioconjugate chemistry serves great clinical advances: Albumin as a versatile platform for diagnosis and precision therapy. Chem. Soc. Rev. 2016, 45, 1432–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, C.; Farkas, R.; Borgna, F.; Schmid, R.M.; Benešová, M.; Schibli, R. Synthesis, Radiolabeling, and Characterization of Plasma Protein-Binding Ligands: Potential Tools for Modulation of the Pharmacokinetic Properties of (Radio)Pharmaceuticals. Bioconjug. Chem. 2017, 28, 2372–2383. [Google Scholar] [CrossRef]
- Amini, N.; Nakao, R.; Schou, M.; Halldin, C. Determination of plasma protein binding of positron emission tomography radioligands by high-performance frontal analysis. J. Pharm. Biomed. Anal. 2014, 98, 1401–1443. [Google Scholar] [CrossRef]
- Lindmo, T.; Boven, E.; Cuttitta, F.; Fedorko, J.; Bunn, P.A. Determination of the immunoreactive function of radiolabeled monoclonal antibodies by linear extrapolation to binding at infinite antigen excess. J. Immunol. Methods 1984, 72, 77–89. [Google Scholar] [CrossRef]
- Sharma, S.K.; Lyashchenko, S.K.; Park, H.A.; Pillarsetty, N.; Roux, Y.; Wu, J.; Poty, S.; Tully, K.M.; Poirier, J.T.; Lewis, J.S. A rapid bead-based radioligand binding assay for the determination of target-binding fraction and quality control of radiopharmaceuticals. Nucl. Med. Biol. 2019, 71, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Lambert, D. Drugs and receptors. Contin. Educ. Anaesth. Crit. Care Pain 2004, 4, 181–184. [Google Scholar] [CrossRef]
- Xiao, S.-H.; Reagan, J.; Lee, P.; Fu, A.; Schwandner, R.; Zhao, X.; Knop, J.; Beckmann, H.; Young, S. High Throughput Screening for Orphan and Liganded GPCRs. Comb. Chem. High Throughput Screen. 2008, 11, 195–215. [Google Scholar] [CrossRef] [PubMed]
- Chatziioannou, A.F. Molecular imaging of small animals with dedicated PET tomographs. Eur. J. Nucl. Med. 2002, 29, 98–114. [Google Scholar] [CrossRef] [PubMed]
- Hutchins, G.D.; Miller, M.A.; Soon, V.C.; Receveur, T. Small animal PET imaging. ILAR J. 2008, 49, 54–65. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Zhang, Z.; Merkens, H.; Zeisler, J.; Colpo, N.; Hundal-Jabal, N.; Perrin, D.M.; Lin, K.S.; Bénard, F. 18F-Labeled Cyclized α-Melanocyte-Stimulating Hormone Derivatives for Imaging Human Melanoma Xenograft with Positron Emission Tomography. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef]
- Fueger, B.J.; Czernin, J.; Hildebrandt, I.; Tran, C.; Halpern, B.S.; Stout, D.; Phelps, M.E.; Weber, W.A. Impact of animal handling on the results of 18F-FDG PET studies in mice. J. Nucl. Med. 2006, 47, 999–1006. [Google Scholar]
- Soret, M.; Bacharach, S.L.; Buvat, I. Partial-volume effect in PET tumor imaging. J. Nucl. Med. 2007, 48, 932–945. [Google Scholar] [CrossRef] [Green Version]
- Hildebrandt, I.J.; Su, H.; Weber, W.A. Anesthesia and other considerations for in vivo imaging of small animals. ILAR J. 2008, 49, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Heneweer, C.; Holland, J.P.; Divilov, V.; Carlin, S.; Lewis, J.S. Magnitude of enhanced permeability and retention effect in tumors with different phenotypes: 89Zr-albumin as a model system. J. Nucl. Med. 2011, 52, 625–633. [Google Scholar] [CrossRef] [Green Version]
- Siegel, J.A.; Thomas, S.R.; Stubbs, J.B.; Stabin, M.G.; Hays, M.T.; Koral, K.F.; Robertson, J.S.; Howell, R.W.; Wessels, B.W.; Fisher, D.R.; et al. MIRD pamphlet no. 16: Techniques for quantitative radiopharmaceutical biodistribution data acquisition and analysis for use in human radiation dose estimates. J. Nucl. Med. 1999, 40, 37S–61S. [Google Scholar] [PubMed]
- Bolch, W.E.; Eckerman, K.F.; Sgouros, G.; Thomas, S.R.; Brill, A.B.; Fisher, D.R.; Howell, R.W.; Meredith, R.; Wessels, B.W. MIRD pamphlet No. 21: A generalized schema for radiopharmaceutical dosimetry-standardization of nomenclature. J. Nucl. Med. 2009, 50, 477–484. [Google Scholar] [CrossRef] [Green Version]
- Sparks, R.; Aydogan, B. Comparison of the effectiveness of some common animal data scaling techniques in estimating human radiation dose. Sixth Int. Radiopharm. Dosim. Symp. 1999, 705–716. [Google Scholar]
- Lu, S.; Haskali, M.B.; Ruley, K.M.; Dreyfus, N.J.-F.; DuBois, S.L.; Paul, S.; Liow, J.-S.; Morse, C.L.; Kowalski, A.; Gladding, R.L.; et al. PET ligands [18F]LSN3316612 and [11C]LSN3316612 quantify O -linked-β- N -acetyl-glucosamine hydrolase in the brain. Sci. Transl. Med. 2020, 12, eaau2939. [Google Scholar] [CrossRef] [PubMed]
- FDA ICH M3(R2) Nonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals Q&A. Guideline 2013, 14, 84–86. [CrossRef]
- U.S. Department of Health and Human Services- Food and Drug Administration; Center for Drug Evaluation and Research (CDER) Microdose Radiopharmaceutical Diagnostic Drugs. Nonclinical Study Recommendations Guidance for Industry. Guideline 2018, 1–4. [Google Scholar]
- European Medicines Agency Guideline on the non-clinical requirements for radiopharmaceuticals. Guideline 2018, 44, 1–10.
- Silva-Lima, B.; Carlson, D.; Jones, D.R.; Laurie, D.; Stahl, E.; Maria, V.; Janssens, W.; Robinson, W.T. The European and American use of exploratory approaches for first-in-human studies. Clin. Transl. Sci. 2010, 3, 38–41. [Google Scholar] [CrossRef]
- FDA Guidance for industry, investigators, and reviewers: Exploratory IND studies. Guideline 2006, 25, 167–174. [CrossRef]
- Nimmagadda, S.; Shelake, S.; Pomper, M.G. Preclinical Experimentation in Oncology. In Radiopharmaceutical Chemistry; Springer International Publishing: Cham, Switzerland, 2019; pp. 569–582. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biological Process/Target | Radiopharmaceutical | Vector | Indication | References |
|---|---|---|---|---|
| A33 | [124I]I-huA33 | Antibody | Colorectal cancer | [13] |
| Acetyl-CoA synthetase | [11C]acetate # | Salt | General cancers | [14] |
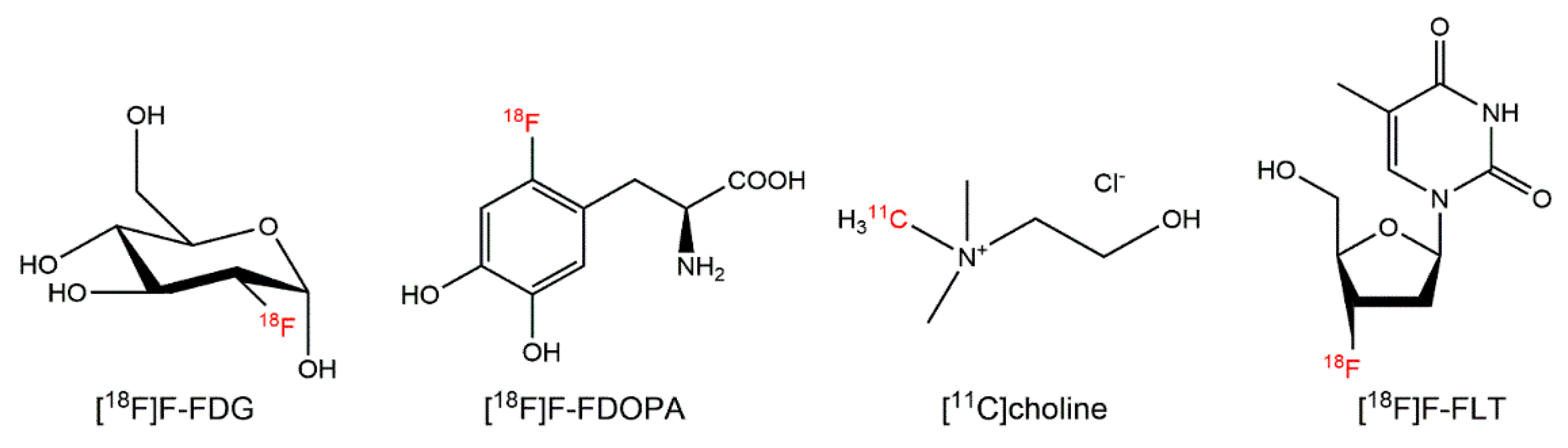
| Amino acid transport | [11C]methionine # [18F]FDOPA # [18F]FET # [18F]FGln [18F]FSPG [18F]FACBC * [18F]FACPC | Amino acid | Glioma, neuroendocrine tumors, prostate cancer | [15,16,17,18,19,20,21,22,23] |
| Androgen receptor (AR) | [18F]FDHT | Hormone | Prostate cancer | [24,25] |
| Apoptosis | [18F]ML-10 [18F]ICMT-11 | Small molecule | Glioblastoma multiforme, breast cancer, lung cancer | [26,27] |
| Bone remodeling | [18F]NaF *,# | Salt | Osseous lesions | [28] |
| CA19.9 | [89Zr]Zr-DFO-HuMab-5B1 | Antibody | Pancreatic cancer and bladder cancer | [29] |
| Carbonic anhydrase 9 (CA-IX) | [124I]I-girentuximab [89Zr]Zr-girentuximab | Antibody | Clear-cell renal cell carcinoma | [30,31,32] |
| Carcinoembryonic antigen (CEA) | [89Zr]Zr-AMG 211 | Bispecific T-cell engager | Gastrointestinal adenocarcinoma | [33] |
| CD8 | [89Zr]Zr-Df-IAB22M2C | Minibody | Melanoma, lung cancer, hepatocarcinoma | [34] |
| CD20 | [89Zr]Zr-rituximab [89Zr]Zr-obinutuzumab | Antibody | B cell lymphoma | [35,36] |
| CD44v6 | [89Zr]Zr-U36 | Antibody | Head and neck cancer | [37] |
| C-X-C chemokine receptor type 4 (CXCR4) | [64Cu]Cu-plerixafor | Small molecule | Hematological and solid malignancies | [38,39,40] |
| [68Ga]Ga-pentixafor [68Ga]Ga-NOTA-NFB | Peptide | |||
| Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) | [89Zr]Zr-ipilimumab | Antibody | Melanoma | [41] |
| Epidermal growth factor receptor (EGFR) | [11C]erlotinib [11C]PD153035 [18F]afatinib | Small molecule | Nonsmall cell lung carcinoma; colorectal cancer | [42,43,44,45,46] |
| [89Zr]Zr-cetuximab [89Zr]Zr-panitumumab | Antibody | |||
| Epidermal growth factor receptor 2 (ERBB2) | [68Ga]Ga-ABY-025 | Affibody | Breast cancer | [47,48,49] |
| [68Ga]Ga-HER2-Nanobody | Nanobody | |||
| [89Zr]Zr-trastuzumab [89Zr]Zr-pertuzumab | Antibody | |||
| Epidermal growth factor receptor 3 (ERBB3) | [89Zr]Zr-GSK2849330 [89Zr]Zr-lumretuzumab | Antibody | Solid malignancies | [50,51] |
| Estrogen receptor (ER) | [18F]FES[18F]4FMFES | Hormone | Breast cancer and gynecologic cancers | [52] |
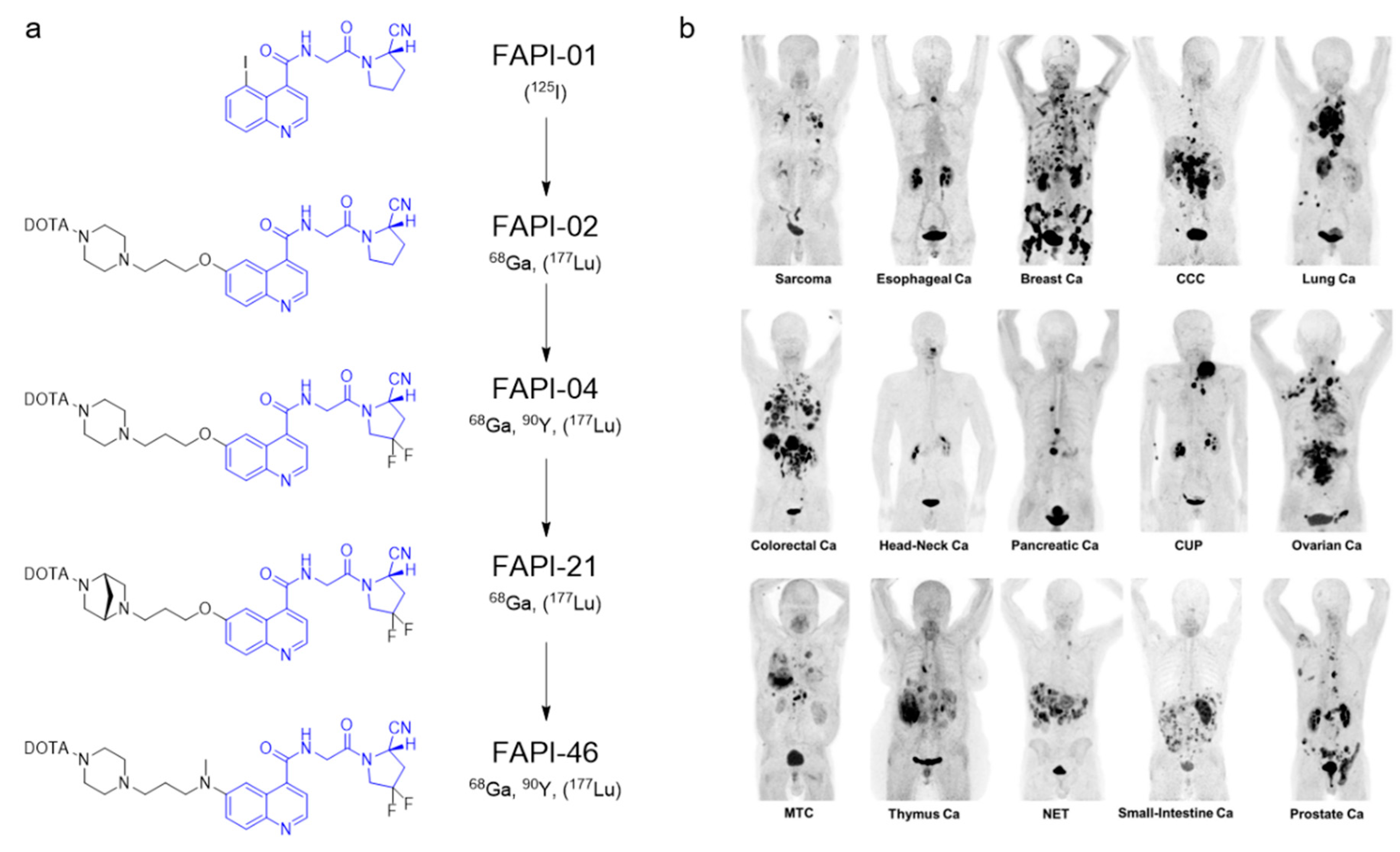
| Fibroblast activation protein α | [68Ga]Ga-FAPI-04 [68Ga]Ga-FAPI-21 [68Ga]Ga-FAPI-46 | Small molecule | Solid malignancies | [53,54,55] |
| Galactose metabolism | [18F]FDGal | Small molecule | Hepatocarcinoma | [56] |
| Gastrin-releasing peptide receptor (GRPR) | [64Cu]Cu-CB-TE2A-AR06 [18F]-BAY 864367 [68Ga]Ga-RM2 [68Ga]Ga-SB3 [68Ga]Ga-RM26 [68Ga]Ga-BBN-RGD [68Ga]Ga-NOTA-Aca-BBN [68Ga]Ga-NeoBOMB1 | Peptide | Prostate cancer, breast cancer, glioma | [57,58,59,60,61,62,63,64,65] |
| Glucagon-like peptide 1 receptor (GLP-1R) | [68Ga]Ga-NOTA-exendin-4 | Peptide | Insulinoma | [66] |
| Glucose metabolism | [18F]FDG *,# | Small molecule | Neoplasm | [1] |
| Glypican 3 | [124I]I-codrituzumab | Antibody | Hepatocarinoma | [67] |
| Hypoxia | [18F]EF5 [18F]FMISO# [18F]FAZA [18F]HX4 [64Cu]Cu-ATSM | Small molecule | Solid malignancies | [68,69,70,71,72,73] |
| Integrin α4β1 | [64Cu]Cu-LLP2A | Peptidomimetic | Multiple myeloma | [74] |
| Integrin αvβ3 | [18F]F-Galacto-RGD [18F]F-FPP(RGD)2 [18F]F-RGD-K5 [18F]F-fluciclatide Al[1 8F]F-alfatide-I Al[18F]F-alfatide-II [68Ga]Ga-NOTA-PRGD2 | Peptide | Solid malignancies | [75,76,77,78,79,80,81,82] |
| Integrin αvβ6 | [18F]F-αvβ6-BP [68Ga]Ga-DOTA-SFITGv6 | Peptide | Head and neck cancer, lung cancer, colorectal cancer, breast cancer, pancreatic cancer | [83,84,85] |
| [18F]FP-R01-MG-F2 [68Ga]Ga-NODAGA-R01-MG | Cystine knot | |||
| Melanocortin-1 receptor (MC1R) | [68Ga]Ga-DOTA-GGNle-CycMSHhex | Peptide | Melanoma | [86] |
| Mesothelin | [89Zr]Zr-MMOT0530A | Antibody | Pancreatic ductal adenocarcinoma and ovarian cancer | [87] |
| Neurokinin 1 receptor (NK1R) | [68Ga]Ga-DOTA-SP | Peptide | Glioma | [88] |
| Neurotensin 1 receptor (NTS1R) | Al[18F]F-NOTA-neurotensin | Peptide | Prostate cancer | [89] |
| Phospholipid synthesis | [11C]choline * [18F]F-choline # | Salt | Prostate cancer | [90,91] |
| Poly(ADP-ribose) polymerase 1 (PARP1) | [18F]PARPi | Small molecule | Head and neck cancer | [92] |
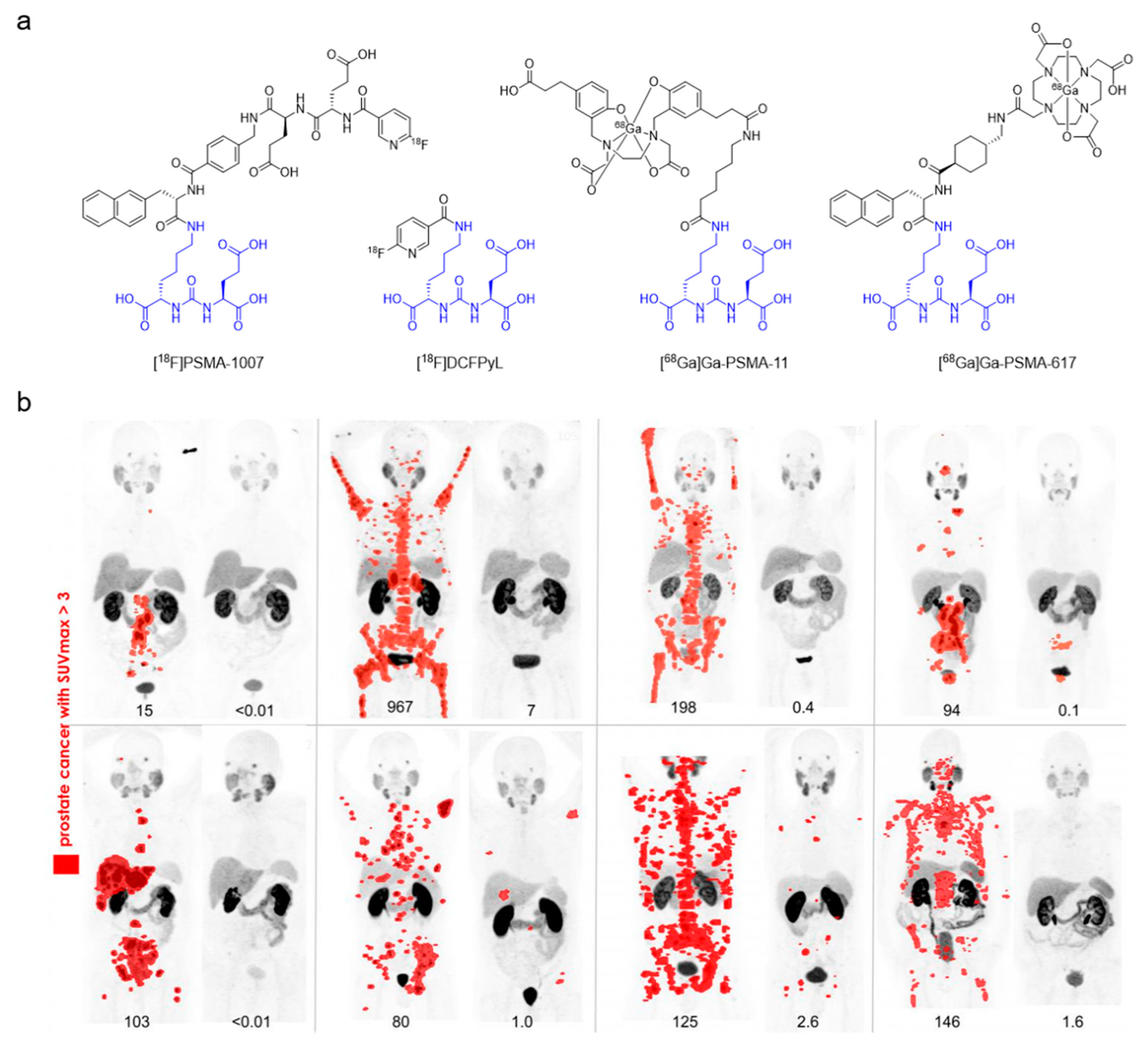
| Prostate-specific membrane antigen (PSMA) | [18F]PSMA-1007 [18F]DCFPyL [18F]DCFBC [18F]rhPSMA-7 [68Ga]Ga-PSMA-11 [68Ga]Ga-PSMA-617 [68Ga]Ga-PSMA-I&T | Peptidomimetic | Prostate cancer | [93,94,95,96,97,98] |
| [89Zr]Zr-HuJ591 | Antibody | |||
| Programmed cell death protein (PD-1) | [89Zr]Zr-durvalumab [89Zr]Zr-nivolumab [89Zr]Zr-pembrolizumab | Antibody | Nonsmall cell lung carcinoma | [99,100] |
| Programmed death-ligand 1 (PD-L1) | [18F]BMS-986192 | Adnectin | Nonsmall cell lung carcinoma, bladder cancer, breast cancer | [100,101,102] |
| [89Zr]Zr-atezolizumab | Antibody | |||
| Six-transmembrane epithelial antigen of prostate-1 (STEAP1) | [89Zr]Zr-DFO-MSTP2109A | Antibody | Prostate cancer | [103] |
| Sodium/iodine transporter | Na[124I]I | Salt | Thyroid cancer | [104] |
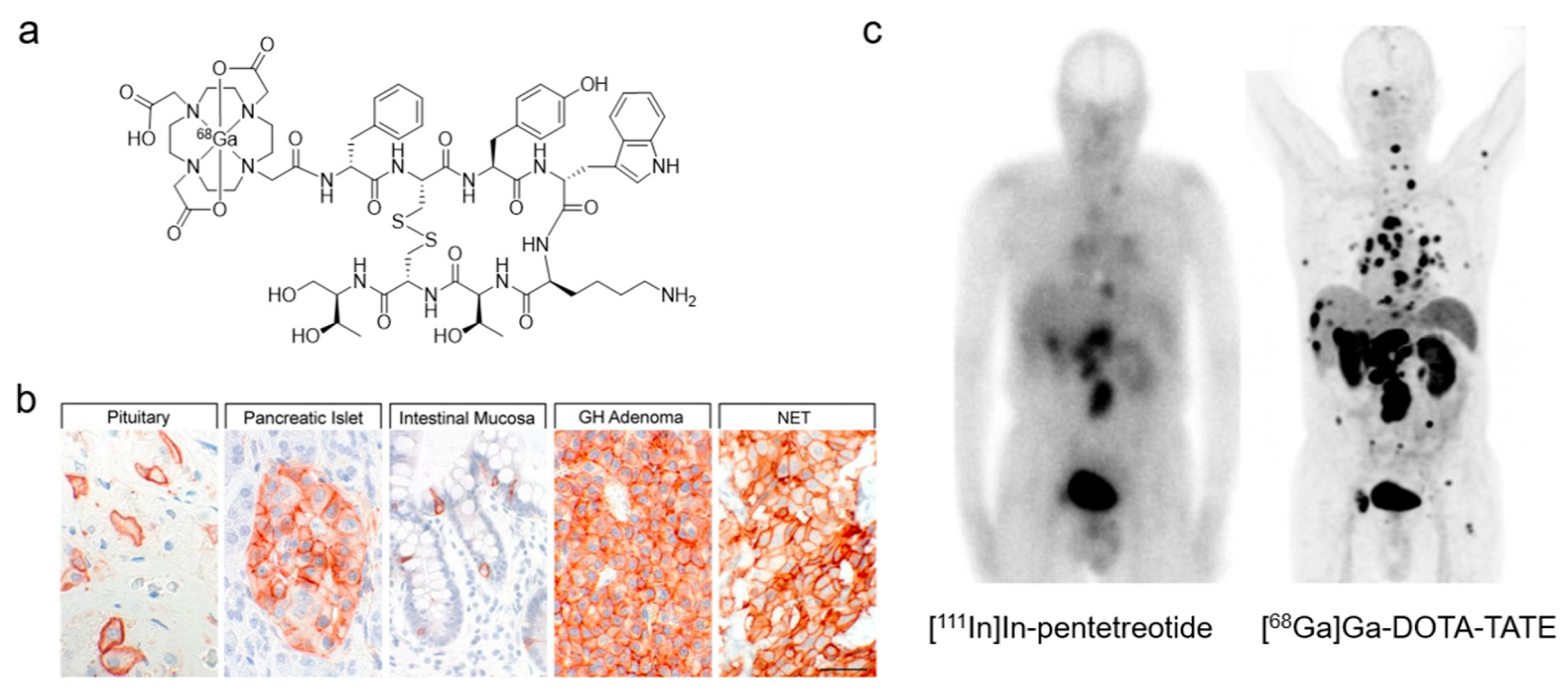
| Somatostatin receptor 2 (SSTR2) | [64Cu]Cu-SARTATE [68Ga]Ga-DOTA-TATE * [68Ga]Ga-DOTA-TOC *,# [68Ga]Ga-DOTA-NOC [68Ga]Ga-NODAGA-JR11 | Peptide | Neuroendocrine tumors | [105,106,107,108] |
| Thymidine kinase (DNA replication) | [18F]FLT # | Nucleoside | Solid malignancies | [109] |
| Transforming growth factor-beta (TGF-β) | [89Zr]Zr-fresolimumab | Antibody | Glioma | [110] |
| Vascular endothelial growth factor receptor (VEGFR) | [89Zr]Zr-bevacizumab | Antibody | Solid malignancies | [111,112,113] |
| Half-Life | Decay Mode | Mean β+ Energy [MeV] | Mean Positron Range in Water [mm] | Production Route | |
|---|---|---|---|---|---|
| 11C | 20.4 min | β+ (99.8%) | 0.386 | 1.2 | 14N(p,α)11C |
| 13N | 10.0 min | β+ (99.8%) | 0.492 | 1.8 | 16O(p,α)13N |
| 15O | 2.0 min | β+ (99.9%) | 0.735 | 3.0 | 15N(p,n)15O |
| 18F | 109.7 min | β+ (96.7%) | 0.250 | 0.6 | 18O(p,n)18F |
| 44Sc | 4.0 h | β+ (94.3%) | 0.632 | 2.4 | 44Ti/44Sc generator |
| 64Cu | 12.7 h | β+ (17.6%) | 0.278 | 0.7 | 64Ni(p,n)64Cu 67Zn(p,α)64Cu |
| 68Ga | 67.7 min | β+ (88.9%) | 0.836 | 3.5 | 68Ge/68Ga generator |
| 82Rb | 1.3 min | β+ (81.8%) β+ (13.1%) | 1.535 1.168 | 7.1 5.0 | 82Sr/82Rb generator |
| 86Y | 14.7 h | β+ (11.9%) β+ (5.6%) β+ (3.6%) | 0.535 0.681 0.883 | 1.9 2.8 3.7 | 86Sr(p,n)86Y |
| 89Zr | 78.4 h | β+ (22.7%) | 0.396 | 1.3 | 89Y(p,n)89Zr |
| 124I | 100.2 h | β+ (11.7%) β+ (10.7%) β+ (0.3%) | 0.687 0.975 0.367 | 2.8 4.4 1.1 | 124Te(p,n)124I |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lau, J.; Rousseau, E.; Kwon, D.; Lin, K.-S.; Bénard, F.; Chen, X. Insight into the Development of PET Radiopharmaceuticals for Oncology. Cancers 2020, 12, 1312. https://doi.org/10.3390/cancers12051312
Lau J, Rousseau E, Kwon D, Lin K-S, Bénard F, Chen X. Insight into the Development of PET Radiopharmaceuticals for Oncology. Cancers. 2020; 12(5):1312. https://doi.org/10.3390/cancers12051312
Chicago/Turabian StyleLau, Joseph, Etienne Rousseau, Daniel Kwon, Kuo-Shyan Lin, François Bénard, and Xiaoyuan Chen. 2020. "Insight into the Development of PET Radiopharmaceuticals for Oncology" Cancers 12, no. 5: 1312. https://doi.org/10.3390/cancers12051312