
Techniques for the Detection of Sickle Cell Disease: A Review



Abstract
:1. Introduction
2. Clinical Picture of the Inherited Hemoglobin Disorders
3. Techniques and Assays to Diagnose and Monitor SCD
4. Current Techniques to Diagnose and Monitor SCD
4.1. Complete Blood Cell Count
4.2. Peripheral Blood Smear
4.3. Solubility Sickling Test
4.4. Hemoglobin Electrophoresis
4.5. Isoelectric Focusing
4.6. High Performance Liquid Chromatography
4.7. Genetic Test
4.7.1. Polymerase Chain Reaction (PCR)-Based Techniques
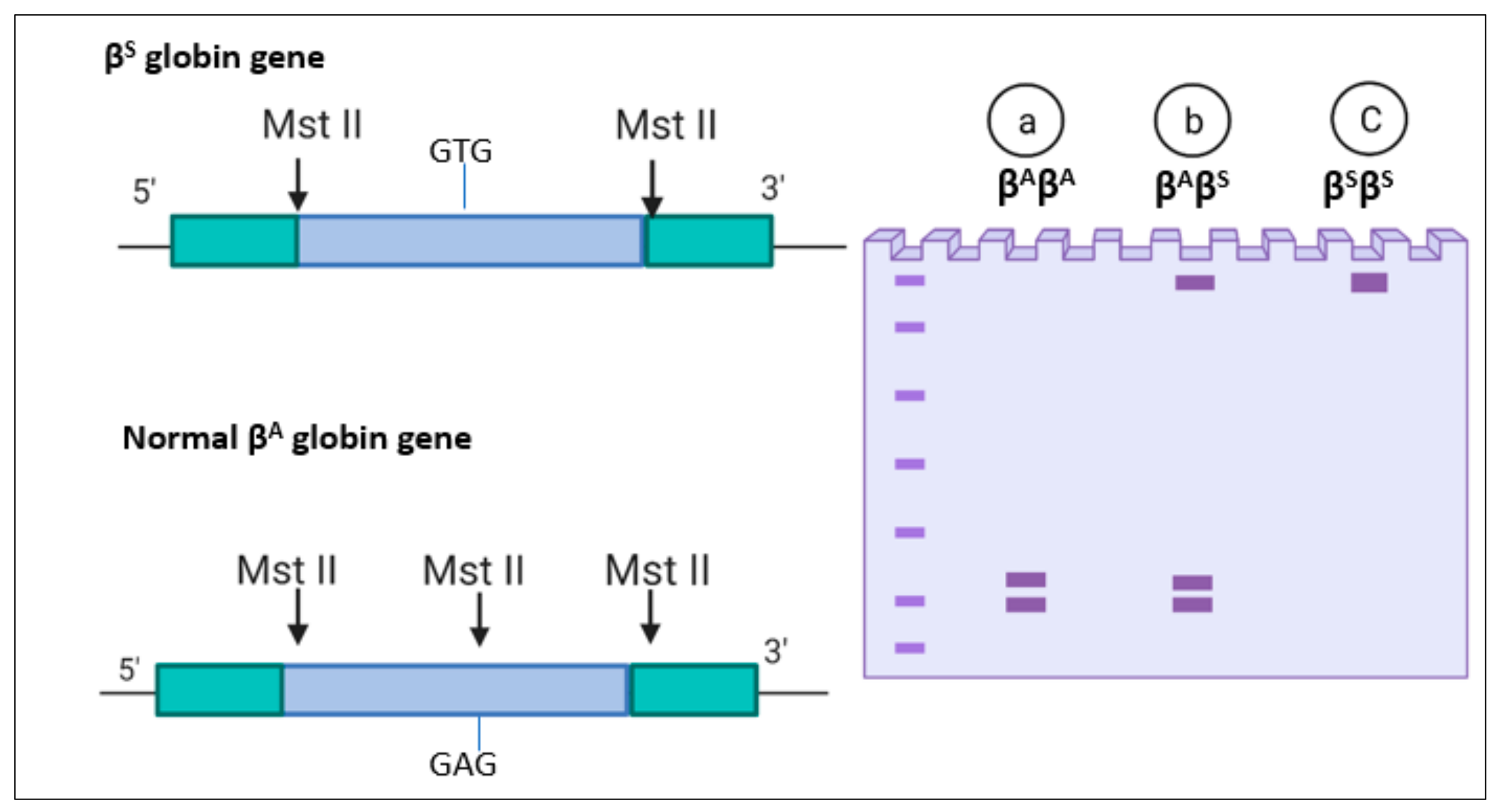
4.7.2. Restriction Fragment Length Polymorphism
4.7.3. DNA Microarrays and Sequencing Techniques
5. Innovative Techniques for the Diagnosis and Monitoring of SCD
5.1. Image Processing Techniques
5.2. Emerging Flow Cytometry
5.3. Mechanical Differentiation of Sickle Cells
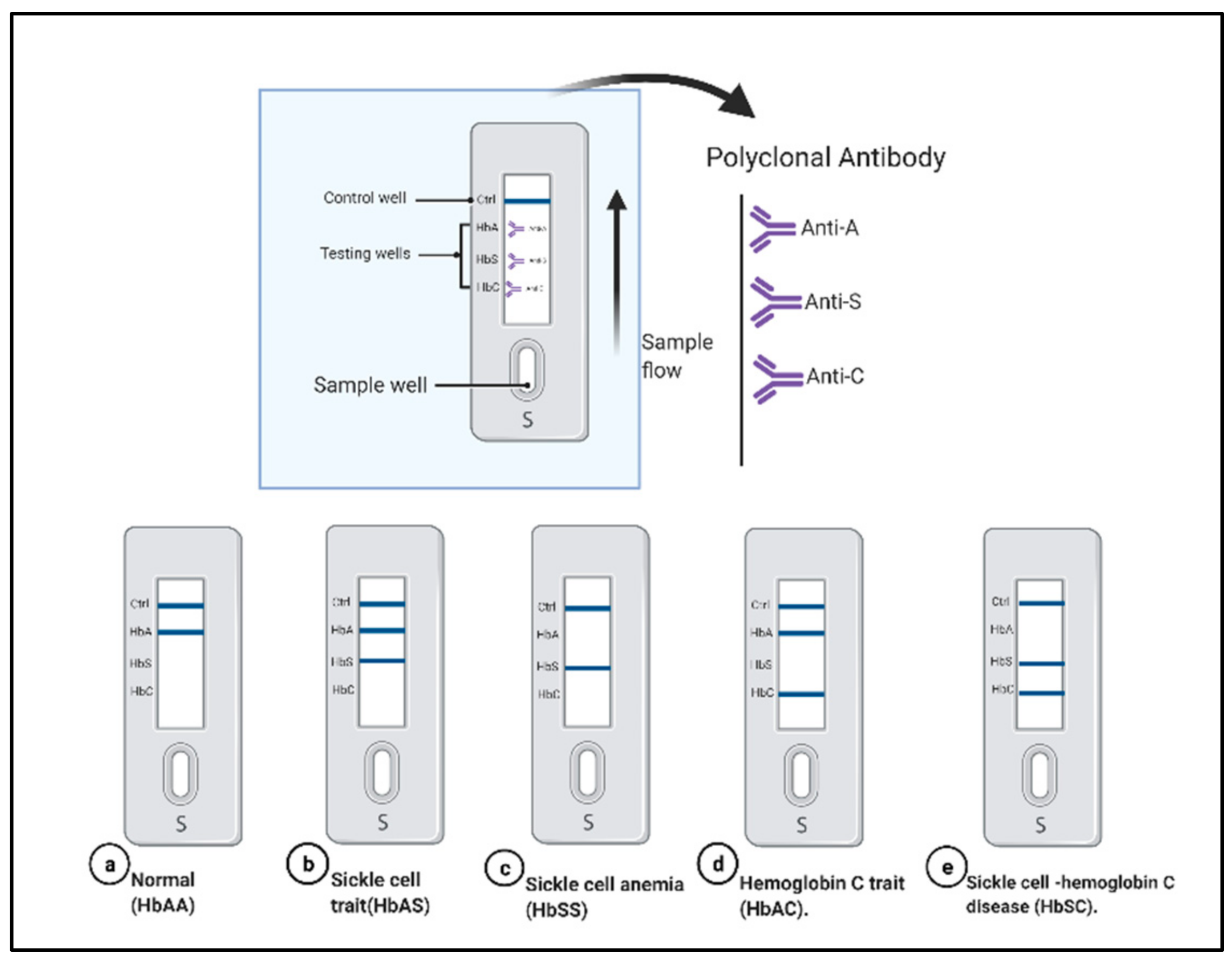
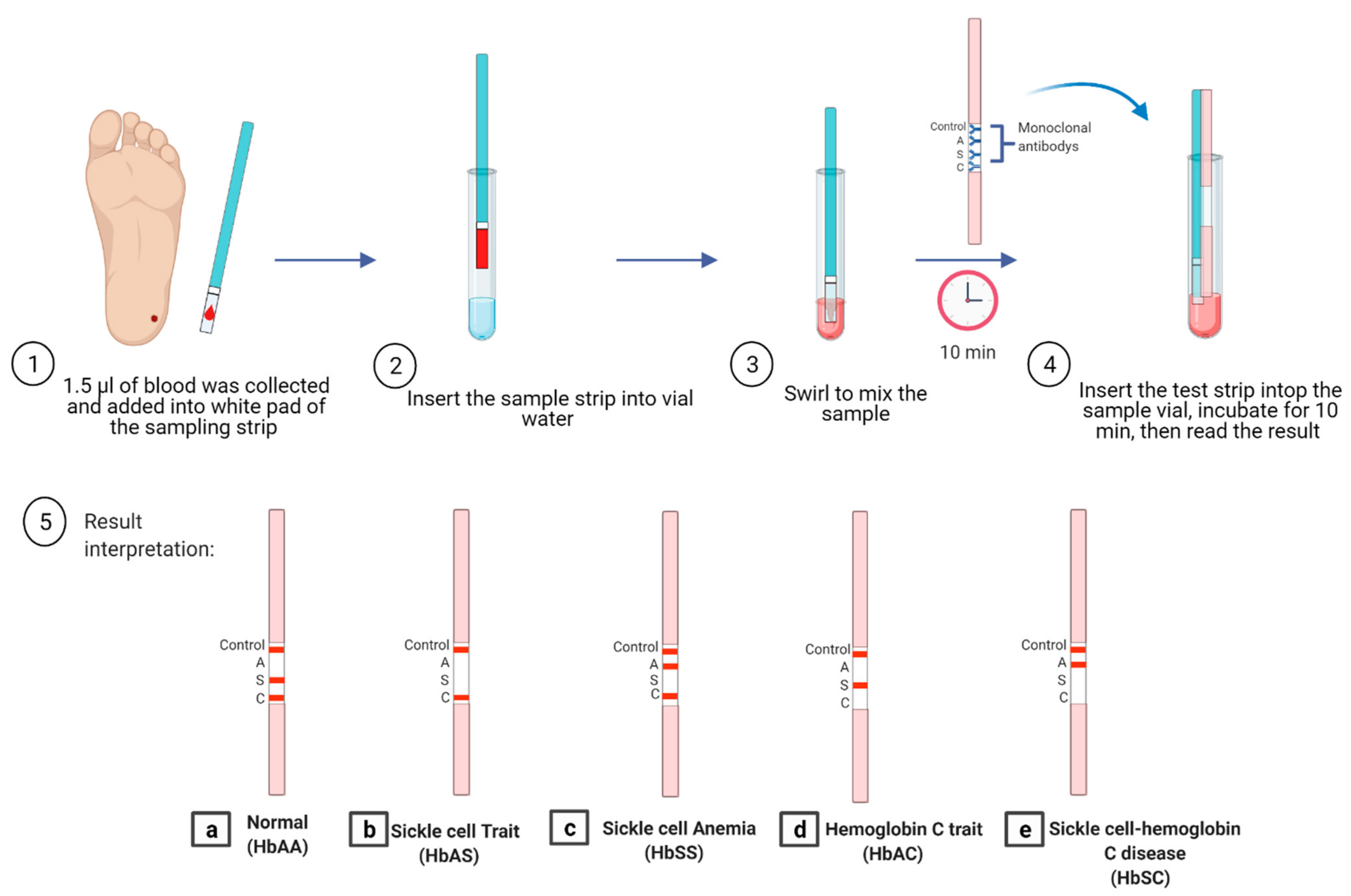
5.4. Lateral Flow Immunoassay
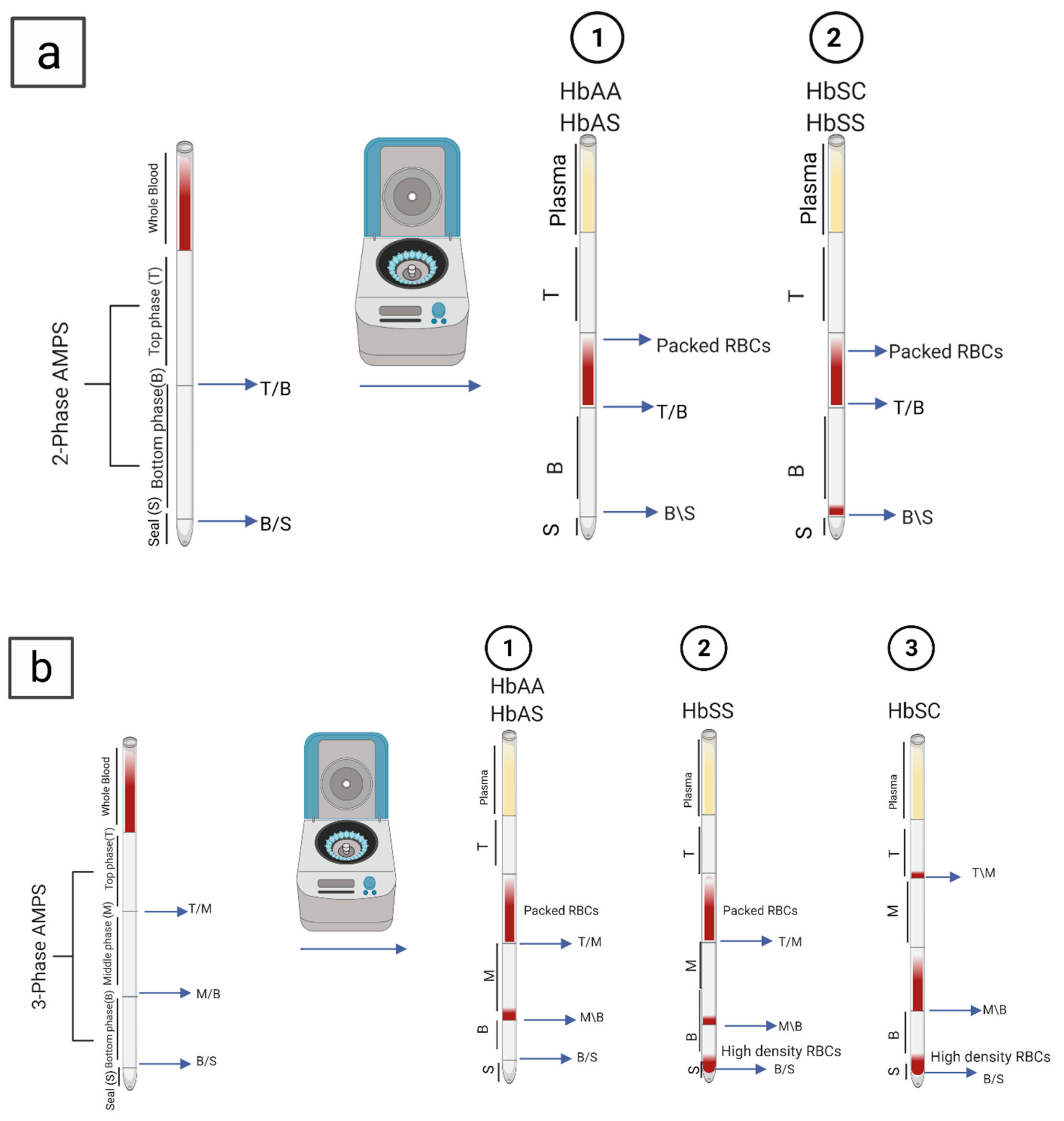
5.5. Density-Based Separation
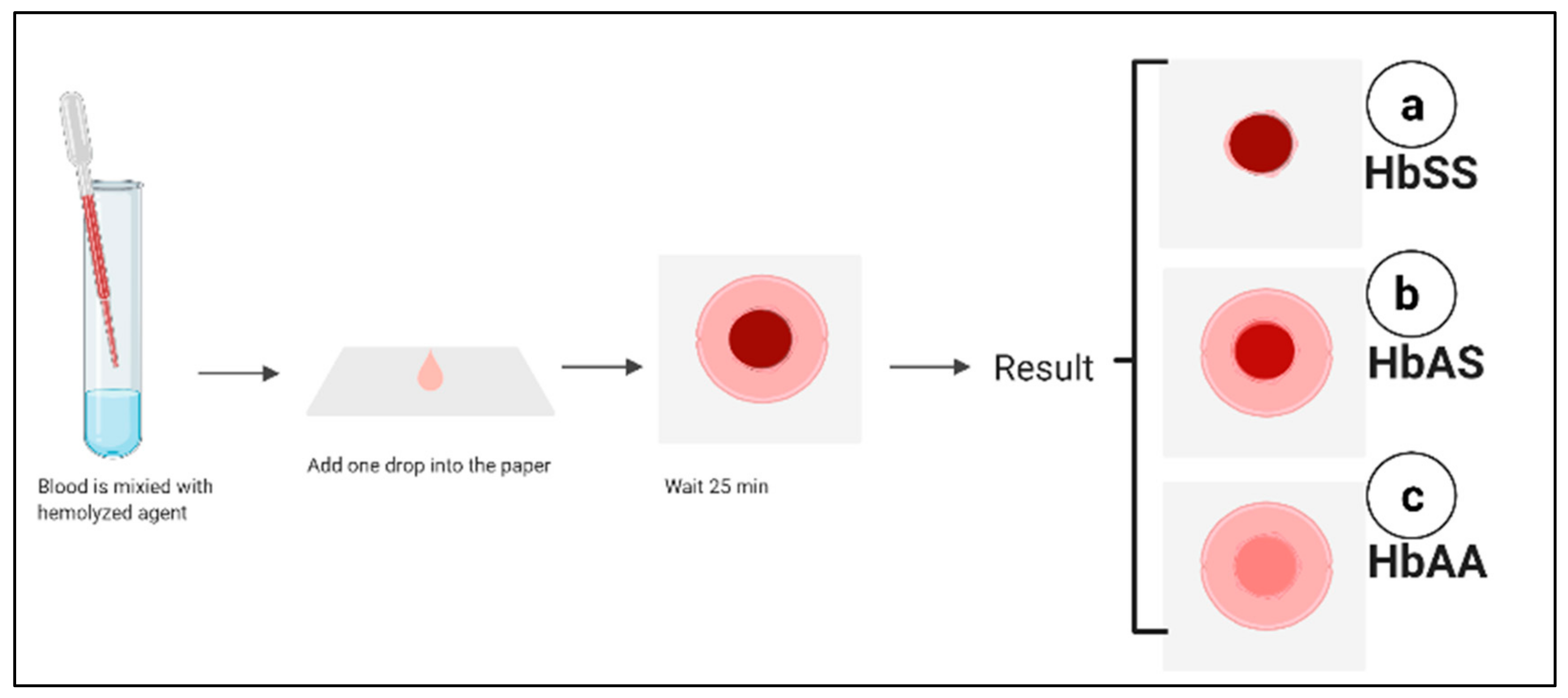
5.6. Paper-Based Hemoglobin Solubility Test
5.7. Sensors Based Techniques
5.7.1. Fluorescence Based Optofluidic Resonator
5.7.2. Sensors Based on Electrical Impedance Signal
5.7.3. Quartz Crystal Microbalance (QCM)
5.7.4. Genosensors
5.8. The Pyrosequencing Technique
6. Conclusions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Technique | Sensitivity | Specificity | Accuracy | Advantages | Disadvantage | Result | Ref. |
|---|---|---|---|---|---|---|---|
| Peripheral blood smear (PBF) | 35.0%. | 96.7% | 90.5% | Simple preparation, inexpensive, Turnaround time (TAT) is 44 min | Dependence on the pathologist’s skills, does not differentiate between different types of SCD | Detect sickle cells | [22] |
| Solubility and Sickling | Sickling: 65.0% Solubility: 45.0%. | Sickling: 95.6% Solubility: 90.0%. | Sickling: 92.5% Solubility: 85.5%. | Easy, inexpensive, fast, affordable, TAT 38 min for sickling, TAT for solubility 70 min | Testing newborns shows false-negative result, does not differentiate between SCD types | Detect the sickling event. | [27] |
| Capillary electrophoresis | Not reported | Not reported | Not reported | Reliable, ability to distinguish most types of sickle cell disease including heterozygous. | Expensive, requires skilled technicians | identify and quantify HbF, Hb A, Hb A2, Hb S, Hb C, Hb Barts and other | [36] |
| Isoelectric focusing (IEF) | Not reported | Not reported | Not reported | Detect HbS and HbA easily in a high concentration of HbF, Hb D-Punjab easily separated from HbS, need small volume of the sample, able to use dried blood spot, TAT is 45 min. | Expensive, requires highly trained staff to interpret the results. | Hb A, Hb F, Hb C, Hb S, Hb E and Hb O Arab | [38] |
| High-performance liquid chromatography (HPLC) | Not reported | Not reported | Not reported | Reliable, ability to distinguish most types of sickle cell disease including heterozygous, fully automated | Misdiagnoses the new variants that mimic HbS, Expensive and needs trained personnel, not practical in limited resources areas | Detect Hb F, Hb A2, Hb S, Hb C, Hb Barts, and other Hb variants. | [18,44] |
| Amplification-refractory mutation system (ARMS) polymerase chain reaction (PCR) for prenatal analysis | 75% | Not reported | Not reported | Simple, can be used for prenatal diagnosis | Low sensitivity, maternal cell DNA contamination | βSβS βAβS βAβA | [49] |
| Allele-Specific Recombinase Polymerase Amplification | 100% | βA: 94.7% βS:97.1% | <95% | Affordable, rapid (less than 30 min), low-cost, accurate | This test is difficult to design, missing some single nucleotide polymorphisms (SNPs), costly and laborious assay | βA βS | [100] [101] |
| Emerging technologies | |||||||
| Image processing technique | 96.55% | Not reported | 95% | Automated method to detect sickle cells, minimize the error of dependence on the naked eye | Cannot distinguish between different types of SCD, cannot be used to determine the severity of the disease, affected by different conditions that can affect the red blood cells (RBCs) number as in blood transfusion, expensive, needs special equipment such as camera connected to microscope | Detect Sickling RBCs | [66] |
| Propose deep learning models | Not reported | Not reported | 99.54% | Indicate the sickle RBCs automatically in one shot, minimize the error of dependence on the naked eye | Cannot distinguish between different types of SCD, cannot be used to determine the severity of the disease, affected by different conditions that can affect the RBCs number as in blood transfusion, needs special equipment such as camera connected to microscope., time consuming, ignore other cells which leads to false diagnosis | Detect Sickling RBCs | [67] |
| Smartphone microchip with microscope and machine learning algorithms | Not reported | Not reported | Not reported | Can be used as point of care (POC) to monitor the diseases severity, reduce the cost | Test is based on the morphology of the RBCs, cannot distinguish between different types of SCD, affected by different conditions that can affects the RBCs morphology | Detect Sickling RBCs | [68] |
| Electrical impedance microflow cytometry | 91% | 86% | Not reported | Used to monitor the sickling events accurately | Does not differentiate between different type of SCD, need to be validated | Electrical impedance of the sickle cells Detect Sickling RBCs | [19] [73] |
| Imaging flow cytometry | Not reported | Not reported | Not reported | Robust test, can be automated to correlate the percentage of HbF and the percentage of sickled cells, biomarker of disease severity | Effected by agents that reduce polymerization of HbS, laborious | Used to quantify sickled cells | [71] |
| Optical tweezer to capture red blood | Not reported | Not reported | Not reported | Can be a monitor test, simple | Cannot indicate the severity of the disease in heterozygous states | Measuring red blood cell elasticity | [102] |
| Photoacoustic Flow cytometry | Not reported | Not reported | Not reported | Simple, low-cost, uses cellphone-like camera. | It is not clear if it can be used to monitor the disease severity, cannot distinguish between sickle cells trait and sickle cell disease | Determine the RBCs Sickling | [72] |
| lateral flow Immunoassay sickle SCAN | 90% | 100% | 98% | Simple, rapid | Relies on polyclonal antibody, more expensive, low specificity and cross reactivity, qualitative test, the intensity of band shows inconsistency, does not identify hemoglobin F, limit of detection of Hb A is 2% | Identify HbC and HbS | [103] |
| lateral flow Immunoassay HemoTypeSC | 93.4% | 99.9% | 99.1% | Cost-effective, rapid, POC | Cannot detect all hemoglobin variants, does not differentiate between HbSS and sick-le-β0-thalassemia, misinterpretation of the result in cases with recent blood transfusion | HbAA, HbAS, HbAC, HbSC, and HbCC | [80,81] |
| HemeChipMicro-elecrophoresis assay | 100% | HbSS 98.7% Other type 100% | 100% | Reliable. POC, inexpensive, simple | Interpretation requires skills, the need for web-based image for automated results | SCD-SS, SCD-SC, and SCD Trait Hb E Disease | [90] |
| SCD-AMPS 2-phase | 90% | 97% | 77% | Inexpensive, simple POC | Interpretation is difficult, less reliable, affected by different conditions that decrease the number of dense cells, may not be appropriate for neonatal screening, low sensitivity and specificity | Identifies Hb S and Hb A | [18,84,85] |
| SCD-AMPS 3-phase | 91% | 88% | 69% | Identifies Hb S, Hb A and Hb C | [18,84,85] | ||
| Paper-based hemoglobin solubility test | 94.2% | 97.7% | 96.9% | Simple, rapid, inexpensive POC, does not need trained personal | Difficult to distinguish HbAS (trait) from HbSC, humidity can affect the test result, low sensitivity and specificity | Diagnosis of HbSS | [89] |
| Quartz crystal microbalance (QCM) sensor | Not reported | Not reported | Not reported | Reliable, simple, POC, low-cost | Not a diagnostic test | Determine RBC’s elasticity | [95] |
| Electrochemical genosensor | 1.23 × 105 ohmLmmol-1 cm-2 | Not reported | Not reported | Simple, low cost, POC | Determination of SCA trait only | Detect βAβS | [96] |
| Surface plasmon resonance-based biosensor | Not reported | Not reported | Not reported | Simple, rapid | Needs PCR product, needs to be validated | Detect βSβS | [98] |
| The Pyrosequencing technique (PyS) | 98.2% | Not reported | Sickle cell anemia 98.7% sickle cell- hemoglobin C disease with 98.7%, and the heterozygous with 92.2%, | Diagnose heterozygous SCD, simple, fast, low cost, suitable for large scale | Misclassification, false negativity, depends on primer design | Detect βSβS, Sβ0 thalassemia, Sβ+ thalassemia, and aickle-hemoglobin C | [99] |
Author Contributions
Funding
Conflicts of Interest
References
- Kato, G.J.; Piel, F.B.; Reid, C.D.; Gaston, M.H.; Ohene-frempong, K.; Krishnamurti, L.; Smith, W.R.; Panepinto, J.A.; Weatherall, D.J.; Costa, F.F.; et al. Sickle cell disease. Nat. Rev. Dis. Primers 2018, 4, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagel, R.L.; Johnson, J.; Bookchin, R.M.; Garel, M.C.; Rosa, J.; Schiliro, G.; Castro, O. Beta-chain contact sites in the haemoglobin S polymer. Nature 1980, 283, 832–834. [Google Scholar] [CrossRef]
- Steinberg, M.H. Pathophysiology of sickle cell disease. Baillieres. Clin. Haematol. 1998, 11, 163–184. [Google Scholar] [CrossRef]
- Edelstein, S.J.; Telford, J.N.; Crepeau, R.H. Structure of fibers of sickle cell hemoglobin. Proc. Natl. Acad. Sci. USA 1973, 70, 1104–1107. [Google Scholar] [CrossRef] [Green Version]
- Benesch, R.E.; Kwong, S.; Benesch, R. The effects of α chain mutations cis and trans to the β6 mutation on the polymerization of sickle cell haemoglobin. Nature 1982, 299, 231. [Google Scholar] [CrossRef]
- Pagnier, J.; Mears, J.G.; Dunda-Belkhodja, O.; Schaefer-Rego, K.E.; Beldjord, C.; Nagel, R.L.; Labie, D. Evidence for the multicentric origin of the sickle cell hemoglobin gene in Africa. Proc. Natl. Acad. Sci. USA 1984, 81, 1771–1773. [Google Scholar] [CrossRef] [Green Version]
- Lipowsky, H.H.; Williams, M.E. Shear rate dependency of red cell sequestration in skin capillaries in sickle cell disease and its variation with vasoocclusive crisis. Microcirculation 1997, 4, 289–301. [Google Scholar] [CrossRef]
- Piel, F.B.; Patil, A.P.; Howes, R.E.; Nyangiri, O.A.; Peter W Gething, M.D.; William H Temperley, T.N.W.; Weatherall, D.J.; Hay, S.I. Global epidemiology of sickle haemoglobin in neonates: A contemporary geostatistical model-based map and population estimates. Lancet 2013, 381, 142–151. [Google Scholar] [CrossRef] [Green Version]
- Mulumba, L.L.; Wilson, L. Sickle cell disease among children in Africa: An integrative literature review and global recommendations Lucky. Int. J. Afr. Nurs. Sci. 2015, 3, 56–64. [Google Scholar] [CrossRef] [Green Version]
- Lubeck, D.; Agodoa, I.; Bhakta, N.; Danese, M.; Pappu, K.; Howard, R.; Gleeson, M.; Halperin, M.; Lanzkron, S. Estimated Life Expectancy and Income of Patients With Sickle Cell Disease Compared with Those Without Sickle Cell Disease. JAMA Netw. Open 2019, 2, e1915374. [Google Scholar] [CrossRef]
- Wajcman, H.; Moradkhani, K. Abnormal haemoglobins: Detection & characterization. Indian J. Med. Res. 2011, 134, 538–546. [Google Scholar] [PubMed]
- Da Fonseca, S.F.; Amorim, T.; Purificação, A.; Goncalves, M.D.S.; Boasorte, N.C.A. Hemoglobin A2 values in sickle cell disease patients quantified by high performance liquid chromatography and the influence of alpha thalassemia. Rev. Bras. Hematol. Hemoter. 2015, 37, 296–301. [Google Scholar] [CrossRef] [Green Version]
- Vincent, O.; Oluwaseyi, B.; James, B.; Saidat, L. Coinheritance of B-Thalassemia and Sickle Cell Anaemia in Southwestern Nigeria. Ethiop. J. Health Sci. 2016, 26, 517–522. [Google Scholar] [CrossRef] [Green Version]
- Da Guarda, C.C.; Yahouédéhou, S.C.M.A.; Santiago, R.P.; Neres, J.S.D.S.; Fernandes, C.F.D.L.; Aleluia, M.M.; Figueiredo, C.V.B.; Fiuza, L.M.; Carvalho, S.P.; De Oliveira, R.M.; et al. Sickle cell disease: A distinction of two most frequent genotypes (HbSS and HbSC). PLoS ONE 2020, 15, e0228399. [Google Scholar] [CrossRef]
- Figueiredo, M.S. The importance of hemoglobin A 2 determination. Rev. Bras. Hematol. Hemoter. 2015, 37, 287–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagel, R.L.; Fabry, M.E.; Steinberg, M.H. The paradox of hemoglobin SC disease. Blood Rev. 2003, 17, 167–178. [Google Scholar] [CrossRef]
- Quinn, C.T. Minireview: Clinical severity in sickle cell disease: The challenges of definition and prognostication. Exp. Biol. Med. 2016, 241, 679–688. [Google Scholar] [CrossRef]
- Alapan, Y.; Fraiwan, A.; Kucukal, E.; Hasan, M.N.; Ung, R.; Kim, M.; Odame, I.; Little, J.A.; Gurkan, U.A. Emerging point-of-care technologies for sickle cell disease screening and monitoring. Expert Rev. Med. Devices 2016, 13, 1073–1093. [Google Scholar] [CrossRef] [Green Version]
- Ilyas, S.; Simonson, A.E.; Asghar, W. Emerging point-of-care technologies for sickle cell disease diagnostics. Clin. Chim. Acta 2020, 501, 85–91. [Google Scholar] [CrossRef]
- Greene, D.N.; Vaugn, C.P.; Crews, B.O.; Agarwal, A.M. Advances in detection of hemoglobinopathies. Clin. Chim. Acta 2014, 439, 50–57. [Google Scholar] [CrossRef]
- Nwogoh, B.; Transfusion, B.; State, E.; Transfusion, B.; State, R. The Peripheral Blood Film. Peripher. Blood Film 1974, 12, 71–79. [Google Scholar] [CrossRef]
- De Haan, K.; Ceylan Koydemir, H.; Rivenson, Y.; Tseng, D.; Van Dyne, E.; Bakic, L.; Karinca, D.; Liang, K.; Ilango, M.; Gumustekin, E.; et al. Automated screening of sickle cells using a smartphone-based microscope and deep learning. NPJ Digit. Med. 2020, 3. [Google Scholar] [CrossRef] [PubMed]
- Acharya, V.; Prakasha, K. Computer Aided Technique to Separate the Red Blood Cells, Categorize them and Diagnose Sickle Cell Anemia. J. Eng. Sci. Technol. Rev. 2019, 12, 67–80. [Google Scholar] [CrossRef]
- Diggs, L.W.; Naumann, H.N.; Diggs, B.M. A Solubility Test for Sickle Cell-Hemoglobin. Lab. Med. 1975, 6, 38–41. [Google Scholar] [CrossRef] [Green Version]
- Tubman, V.N.; Field, J.J. Sickle solubility test to screen for sickle cell trait: What’s the harm? Hematol. Am. Soc. Hematol. Educ. Progr. 2015, 2015, 433–435. [Google Scholar] [CrossRef]
- Elasbali, A.M.; Alalem, A.M.; Alshammari, E.M.A.; Khan, S.; Adnan, M.; Haque, S. Prevalence of HbS Gene in Marzouk Region of Southern Libya. Egypt. Acad. J. Biol. Sci. C Physiol. Mol. Biol. 2015, 7, 27–37. [Google Scholar] [CrossRef]
- Okwi, A.L.; Byarugaba, W.; Parkes, A.T.R. of S. and S.T. and P.; Ocaido, M. The Reliability of Sickling and Solubility Tests and Peripheral Blood Film Method for Sickle Cell Disease Screening at District Health Centers in Uganda A. Clin. Mother Child Heal. 2010, 7, 1–5. [Google Scholar] [CrossRef]
- Kotila, T. Guidelines for teh diagnosis of the haemoglobinopathies in Nigeria. Ann. Ibadan Postgrad. Med. 2011, 8, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Derbigny, W.A. Cellulose acetate electrophoresis of hemoglobin. Methods Mol. Biol. 2019, 1855, 81–85. [Google Scholar] [CrossRef]
- Rentapalli, B.; Gade, J.; MD, D.; Ganji, S.B.; Kiranmai, P.; Revupalli, S.; Kavali, S.; Ramtenki, A. Agar Gel Alkaline Electrophoresis As A Cost Effective Screening Modality For Abnormal Hemoglobins. IOSR J. Dent. Med. Sci. 2017, 16, 39–43. [Google Scholar] [CrossRef]
- Khosa, S.M.; Usman, M.; Moinuddin, M.; Mehmood, H.O.; Qamar, K. Comparative analysis of cellulose acetate hemoglobin electrophoresis and high performance liquid chromatography for quantitative determination of hemoglobin A2. Blood Res. 2015, 50, 46–50. [Google Scholar] [CrossRef] [Green Version]
- Hughes, J. Comparison of Electrophoresis on Citrate Agar, Cellulose Acetate, or Starch for Hemoglobin Identification system. Clin. Chem. 1975, 1076, 1072–1076. [Google Scholar]
- Wild, B.J.; Bain, B.J. Detection and quantitation of normal and variant haemoglobins: An analytical review. Ann. Clin. Biochem. 2004, 41, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Hicks, E.J.; Criep, J.A.; Nordschow, C.D. Citrate Agar Gel Electrophoresis: Application to Detection of Hemoglobinopathies. Lab. Med. 1974, 5, 43–46. [Google Scholar] [CrossRef]
- Chen, F.T.A.; Liu, C.M.; Hsieh, Y.Z.; Sternberg, J.C. Capillary electrophoresis—A new clinical tool. Clin. Chem. 1991, 37, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Cotton, F.; Wolff, F.; Gulbis, B. Automated Capillary Electrophoresis in the Screening for Hemoglobinopathies. Methods Mol. Biol. 2013, 984, 139–151. [Google Scholar] [CrossRef]
- Reddy, M.N.; Franciosi, R.A. Rapid quantitation of hemoglobin S by isoelectric focusing. Ann. Clin. Lab. Sci. 1994, 24, 401–406. [Google Scholar] [PubMed]
- Frömmel, C. Newborn Screening for Sickle Cell Disease and Other Hemoglobinopathies: A Short Review on Classical Laboratory Methods-Isoelectric Focusing, HPLC, and Capillary Electrophoresis. Int. J. neonatal Screen. 2018, 4, 39. [Google Scholar] [CrossRef] [Green Version]
- McGann, P.T.; Hoppe, C. The pressing need for point-of-care diagnostics for sickle cell disease: A review of current and future technologies. Blood Cells Mol. Dis. 2017, 67, 104–113. [Google Scholar] [CrossRef]
- Ou, C.; Rognerud, C.L. Diagnosis of hemoglobinopathies: Electrophoresis vs. HPLC. Clin. Chim. Acta 2001, 313, 187–194. [Google Scholar] [CrossRef]
- Pant, L.; Kalita, D.; Singh, S.; Kudesia, M.; Mendiratta, S.; Mittal, M.; Mathur, A. Detection of Abnormal Hemoglobin Variants by HPLC Method: Common Problems with Suggested Solutions. Int. Sch. Res. Not. 2014, 2014, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, S.I.; Haga, J.A.; Castleberry, S.M.; Hall, R.B.; Thompson, W.C. Validation of an automated HPLC method for quantification of hemoglobin S. Clin. Chem. 1997, 43, 1667–1669. [Google Scholar] [CrossRef]
- Gupta, P.K.; Kumar, H.; Kumar, S.; Jaiprakash, M. Cation exchange high performance liquid chromatography for diagnosis of haemoglobinopathies. Med. J. Armed Forces India 2009, 65, 33–37. [Google Scholar] [CrossRef] [Green Version]
- Nair, S. Potential Pithfalls in Using HPLC and its Interpretation in Diagnosing HbS. J. Rare Dis. Res. Treat. 2018, 3, 9–12. [Google Scholar] [CrossRef]
- Clark, B.E.; Thein, S.L. Molecular diagnosis of haemoglobin disorders. Clin. Lab. Haematol. 2004, 26, 159–176. [Google Scholar] [CrossRef]
- Waterfall, C.M.; Cobb, B.D. Single tube genotyping of sickle cell anaemia using PCR-based SNP analysis. Nucleic Acids Res. 2001, 29. [Google Scholar] [CrossRef] [Green Version]
- Yue, L.; Lin, M.; Chen, J.T.; Zhan, X.F.; Zhong, D.S.; Monte-Nguba, S.M.; Liu, P.F.; Pan, X.F.; Huang, J.H.; Wang, X.; et al. Rapid screening for sickle cell disease by polymerase chain reaction-high resolution melting analysis. Mol. Med. Rep. 2014, 9, 2479–2484. [Google Scholar] [CrossRef] [Green Version]
- Toye, E.T.; van Marle, G.; Hutchins, W.; Abgabiaje, O.; Okpuzor, J. Single tube allele specific PCR: A low cost technique for molecular screening of sickle cell anaemia in Nigeria. Afr. Health Sci. 2018, 18, 995–1002. [Google Scholar] [CrossRef]
- Newton, C.R.; Graham, A.; Heptinstall, L.E.; Powell, S.J.; Summers, C.; Kalsheker, N.; Smith, J.C.; Markham, A.F. Analysis of any point mutation in DNA. The amplification refractory mutation system (ARMS). Nucleic Acids Res. 1989, 17, 2503–2516. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.J.; Shrivastava, A.C.; Shrikhande, A. V Prenatal diagnosis of sickle cell disease by the technique of PCR. Indian J. Hematol. Blood Transfus. 2015, 31, 233–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.Y.; Ugozzoli, L.; Pal, B.K.; Wallace, R.B. Allele-specific enzymatic amplification of β-globin genomic DNA for diagnosis of sickle cell anemia. Proc. Natl. Acad. Sci. USA 1989, 86, 2757–2760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, J.T.; Milner, P.F.; Summer, M.E.; Nallaseth, F.S.; Fadel, H.E.; Reindollar, R.H.; McDonough, P.G. Use of restriction endonucleases for mapping the allele for β(s)-globin. Proc. Natl. Acad. Sci. USA 1982, 79, 3628–3631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geever, R.F.; Wilson, L.B.; Nallaseth, F.S.; Milner, P.F.; Bittner, M. Direct identification of sickle cell anemia by blot hybridization. Proc. Natl. Acad. Sci. USA 1981, 78, 5081–5085. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, G.R. A Simplified and Cheapest Method for the Diagnosis of Sickle Cell using Whole Blood PCR and RFLP in Nepal. Tribhuvan Univ. J. 2016, 30, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Bumgarner, R. Overview of dna microarrays: Types, applications, and their future. Curr. Protoc. Mol. Biol. 2013, 6137, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Fertrin, K.Y.; Costa, F.F. Genomic polymorphisms in sickle cell disease: Implications for clinical diversity and treatment. Expert Rev. Hematol. 2010, 3, 443–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben Hamda, C.; Sangeda, R.; Mwita, L.; Meintjes, A.; Nkya, S.; Panji, S.; Mulder, N.; Guizani-Tabbane, L.; Benkahla, A.; Makani, J.; et al. A common molecular signature of patients with sickle cell disease revealed by microarray meta-analysis and a genome-wide association study. PLoS ONE 2018, 13, e0199461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suwinski, P.; Ong, C.; Ling, M.H.T.; Poh, Y.M.; Khan, A.M.; Ong, H.S. Advancing Personalized Medicine Through the Application of Whole Exome Sequencing and Big Data Analytics. Front. Genet. 2019, 10, 49. [Google Scholar] [CrossRef] [Green Version]
- Rampersaud, E.; Palmer, L.E.; Hankins, J.S.; Sheehan, V.A.; Bi, W.; Mulder, H.; Kang, G.; Estepp, J.H.; Wang, S.; Thrasher, A.; et al. Precision Medicine for Sickle Cell Disease through Whole Genome Sequencing. Blood 2018, 132, 3641. [Google Scholar] [CrossRef]
- Flanagan, J.M.; Sheehan, V.; Linder, H.; Howard, T.A.; Wang, Y.-D.; Hoppe, C.C.; Aygun, B.; Adams, R.J.; Neale, G.A.; Ware, R.E. Genetic mapping and exome sequencing identify 2 mutations associated with stroke protection in pediatric patients with sickle cell anemia. Blood 2013, 121, 3237–3245. [Google Scholar] [CrossRef] [Green Version]
- Sheehan, V.A.; Crosby, J.R.; Sabo, A.; Mortier, N.A.; Howard, T.A.; Muzny, D.M.; Dugan-perez, S.; Aygun, B.; Nottage, K.A.; Boerwinkle, E.; et al. Whole Exome Sequencing Identifies Novel Genes for Fetal Hemoglobin Response to Hydroxyurea in Children with Sickle Cell Anemia. PLoS ONE 2014, 9, e110740. [Google Scholar] [CrossRef]
- Mwesigw, S.; Moulds, J.M.; Chen, A.; Flanagan, J.; Sheehan, V.A.; George, A.; Hanchard, N.A. Whole-exome sequencing of sickle cell disease patients with hyperhemolysis syndrome suggests a role for rare variation in disease predisposition. Transfusion 2018, 58, 726–735. [Google Scholar] [CrossRef]
- Simoneaux, R. Whole Genome Sequencing of Pediatric Sickle Cell Disease Patients. Oncol. Times 2019, 41, 11–12. [Google Scholar] [CrossRef]
- Abdulraheemfadhel, M.; Humaidi, A.J.; Razzaqoleiwi, S. processing-based diagnosis of sickle cell anemia in erythrocyteImages. 2017 Annu. Conf. New Trends Inf. Commun. Technol. Appl. IRAQ NTICT 2017 2017, 203–207. [Google Scholar] [CrossRef]
- Veluchamy, M.; Perumal, K.; Ponuchamy, T. Feature extraction and classification of blood cells using artificial neural network. Am. J. Appl. Sci. 2012, 9, 615–619. [Google Scholar] [CrossRef] [Green Version]
- Chy, T.S.; Rahaman, M.A. Automatic Sickle Cell Anemia Detection Using Image Processing Technique. Proceedings of 2018 International Conference on Advancement in Electrical and Electronic Engineering (ICAEEE), Gazipur, Bangladesh, 22–24 November 2019; pp. 1–4. [Google Scholar] [CrossRef]
- Alzubaidi, L.; Fadhel, M.A.; Al-shamma, O.; Zhang, J.; Duan, Y. Deep learning models for classification of red blood cells in microscopy images to aid in sickle cell anemia diagnosis. Electron 2020, 9, 427. [Google Scholar] [CrossRef] [Green Version]
- Ilyas, S.; Sher, M.; Du, E.; Asghar, W. Smartphone-based sickle cell disease detection and monitoring for point-of-care settings. Biosens. Bioelectron. 2020, 165. [Google Scholar] [CrossRef]
- Samsel, L.; McCoy, J.P., Jr. Imaging Flow Cytometry for the Study of Erythroid Cell Biology and Pathology. J. Immunol. Methods 2016, 59. [Google Scholar] [CrossRef] [Green Version]
- Van Beers, E.J.; Samsel, L.; Mendelsohn, L.; Saiyed, R.; Fertrin, K.Y.; Brantner, C.A.; Daniels, M.P.; Nichols, J.; Mccoy, J.P.; Kato, G.J. Imaging flow cytometry for automated detection of hypoxia-induced erythrocyte shape change in sickle cell disease. Am. J. Hematol. 2014, 89, 598–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fertrin, K.Y.; Samsel, L.; van Beers, E.J.; Mendelsohn, L.; Kato, G.J.; McCoy, J.P. Sickle Cell Imaging Flow Cytometry Assay (SIFCA). Imaging Flow Cytometry 2016, 279–292. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.; Nedosekin, D.A.; Menyaev, Y.A.; Sarimollaoglu, M.; Proskurnin, M.A.; Zharov, V.P. Photoacoustic Flow Cytometry for Single Sickle Cell Detection In Vitro and In Vivo. Anal. Cell. Pathol. 2016, 2016, 2642361. [Google Scholar] [CrossRef] [Green Version]
- Liua, J.; Qianga, Y.; Alvarezb, O.; Dua, E. Electrical impedance microflow cytometry with oxygen control for detection of sickle cells. Sens. Actuators B Chem. 2018, 255, 2392–2398. [Google Scholar] [CrossRef]
- Brandao, M.M.; Fontes, A.; Barjas-Castro, M.L.; Barbosa, L.C.; Costa, F.F.; Cesar, C.L.; Saad, S.T.O. Optical tweezers for measuring red blood cell elasticity: Application to the study of drug response in sickle cell disease. Eur. J. Haematol. 2003, 70, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Qiang, Y.; Liu, J.; Dao, M.; Suresh, S.; Du, E. Mechanical fatigue of human red blood cells. Proc. Natl. Acad. Sci. USA 2019, 116, 19828–19834. [Google Scholar] [CrossRef] [Green Version]
- Du, E.; Diez-Silva, M.; Kato, G.J.; Dao, M.; Suresh, S. Kinetics of sickle cell biorheology and implications for painful vasoocclusive crisis. Proc. Natl. Acad. Sci. USA 2015, 112, 1422–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javidi, B.; Markman, A.; Rawat, S.; O’Connor, T.; Anand, A.; Andemariam, B. Sickle cell disease diagnosis based on spatiotemporal cell dynamics analysis using 3D printed shearing digital holographic microscopy. Opt. Express 2018, 26, 13614. [Google Scholar] [CrossRef]
- Kanter, J.; Telen, M.J.; Hoppe, C.; Roberts, C.L.; Kim, J.S.; Yang, X. Validation of a novel point of care testing device for sickle cell disease. BMC Med. 2015, 13, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mcgann, P.T.; Schaefer, B.A.; Paniagua, M.; Howard, T.A.; Ware, R.E. Characteristics of a rapid, point-of-care lateral flow immunoassay for the diagnosis of sickle cell disease. Am. J. Hematol. 2016, 91, 205–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinn, C.T.; Paniagua, M.C.; DiNello, R.K.; Panchal, A.; Geisberg, M. A rapid, inexpensive and disposable point-of-care blood test for sickle cell disease using novel, highly specific monoclonal antibodies. Br. J. Haematol. 2016, 175, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Nnodu, O.; Isa, H.; Nwegbu, M.; Ohiaeri, C.; Adegoke, S.; Chianumba, R.; Ugwu, N.; Brown, B.; Olaniyi, J.; Okocha, E.; et al. HemoTypeSC, a low-cost point-of-care testing device for sickle cell disease: Promises and challenges. Blood Cells Mol. Dis. 2019, 78, 22–28. [Google Scholar] [CrossRef]
- Nankanja, R.; Kadhumbula, S.; Tagoola, A.; Geisberg, M.; Serrao, E.; Balyegyusa, S. HemoTypeSC Demonstrates >99% Field Accuracy in a Sickle Cell Disease Screening Initiative in Children of Southeastern Uganda. Am. J. Hematol. 2019, 94, E164–E166. [Google Scholar] [CrossRef] [Green Version]
- Kakou Danho, J.B.; Atiméré, Y.N.; Koné, D.; Yéo, D.D.; Couitchéré, L. Feasibility Study of the “HemoTypeSC” Test for the Rapid Screening of Sickle Cell Disease in Côte D’Ivoire. Adv. Hematol. 2021, 2021, 8862039. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.A.; Patton, M.R.; Hennek, J.W.; Lee, S.Y.R.; D’alesio-Spina, G.; Yang, X.; Kanter, J.; Shevkoplyas, S.S.; Brugnara, C.; Whitesides, G.M. Density-based separation in multiphase systems provides a simple method to identify sickle cell disease. Proc. Natl. Acad. Sci. USA 2014, 111, 14864–14869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.A.; Chunda-Liyoka, C.; Hennek, J.W.; Mantina, H.; Lee, S.Y.R.; Patton, M.R.; Sambo, P.; Sinyangwe, S.; Kankasa, C.; Chintu, C.; et al. Evaluation of a density-based rapid diagnostic test for sickle cell disease in a clinical setting in Zambia. PLoS ONE 2014, 9, e114540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knowlton, S.M.; Sencan, I.; Aytar, Y.; Khoory, J.; Heeney, M.M.; Ghiran, I.C.; Tasoglu, S. Sickle cell detection using a smartphone. Sci. Rep. 2015, 5, 15022. [Google Scholar] [CrossRef] [PubMed]
- Piety, N.Z.; Yang, X.; Lezzar, D.; George, A.; Shevkoplyas, S.S. A rapid paper-based test for quantifying sickle hemoglobin in blood samples from patients with sickle cell disease. Am. J. Hematol. 2015, 90, 478–482. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Kanter, J.; Piety, N.Z.; Benton, M.S.; Vignesa, S.M.; Shevkoplyas, S. A simple, rapid, low-cost diagnostic test for sickle cell disease. Lab Chip 2013, 13, 1464–1467. [Google Scholar] [CrossRef]
- Piety, N.Z.; Yang, X.; Kanter, J.; Vignes, S.M.; George, A.; Shevkoplyas, S.S. Validation of a low-cost paper-based screening test for sickle cell anemia. PLoS ONE 2016, 11, e0144901. [Google Scholar] [CrossRef] [Green Version]
- Hasan, M.N.; Fraiwan, A.; An, R.; Alapan, Y.; Ung, R.; Akkus, A.; Xu, J.Z.; Rezac, A.J.; Kocmich, N.J.; Creary, M.S.; et al. Paper-based microchip electrophoresis for point-of-care hemoglobin testing. Analyst 2020, 145, 2525–2542. [Google Scholar] [CrossRef] [Green Version]
- Dai, H.; Yin, C.; Ye, X.; Jiang, B.; Ran, M.; Cao, Z. A possible pathogenetic factor of sickle-cell disease based on fluorescent analysis via an optofluidic resonator. Sci. Rep. 2017, 7, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Qiang, Y.; Du, E. Measurement of Electrical Properties of Sickle Cells From Electrical Impedance of Cell Suspension. Am. Soc. Mech. 2017. [Google Scholar] [CrossRef]
- Liu, J.; Qiang, Y.; Alvarez, O.; Du, E. Electrical Impedance Characterization of Erythrocyte Response to Cyclic Hypoxia in Sickle Cell Disease. ACS Sens. 2019, 4, 1783–1790. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, R.; Chen, F.; Jiang, T.; Wang, H. QCM-based aptamer selection and detection of Salmonella Typhimurium. Food Chem. 2016. [Google Scholar] [CrossRef] [PubMed]
- Efremov, V.; Lakshmanan, R.S.; Byrne, B.; Killard, A.J. Simple and convenient measurement of RBC deformability using QCM integrated with a novel model of cell viscoelasticity. Sens. Actuators B Chem. 2018, 266, 472–476. [Google Scholar] [CrossRef]
- Brazaca, L.C.; Bramorski, C.B.; Cancino-bernardi, J.; Janegitz, B.C. A Genosensor for Sickle Cell Anemia Trait Determination. Electroanalysis 2017, 29, 773–777. [Google Scholar] [CrossRef]
- Kumar, M.; Guo, Y.; Zhang, P. Highly sensitive and selective oligonucleotide sensor for sickle cell disease gene using photon upconverting nanoparticles. Biosens. Bioelectron. 2010, 24, 1522–1526. [Google Scholar] [CrossRef] [Green Version]
- Breveglieri, G.; D’Aversa, E.; Cosenza, L.C.; Boutou, E.; Balassopoulou, A.; Voskaridou, E.; Gambari, R.; Borgatti, M. Detection of the sickle hemoglobin allele using a surface plasmon resonance based biosensor. Sensors Actuators B Chem. 2019, 296, 126604. [Google Scholar] [CrossRef]
- De Martino, C.C.; Alencar, C.S.; Loureiro, P.; de Freitas Carneiro-Proietti, A.B.; de Alvarenga Máximo, C.; Mota, R.A.; Rodrigues, D.O.W.; Junior, N.G.; Kelly, S.; Sabino, E.C. Use of an automated pyrosequencing technique for confirmation of sickle cell disease. PLoS ONE 2019, 14, e0216020. [Google Scholar] [CrossRef] [Green Version]
- Natoli, M.E.; Chang, M.M.; Kundrod, K.A.; Coole, J.B.; Airewele, G.E.; Tubman, V.N.; Richards-Kortum, R.R. Allele-Specific Recombinase Polymerase Amplification to Detect Sickle Cell Disease in Low-Resource Settings. Anal. Chem. 2021. [Google Scholar] [CrossRef] [PubMed]
- Voisey, J.; Morris, C.P. SNP technologies for drug discovery: A current review. Curr. Drug Discov. Technol. 2008, 5, 230–235. [Google Scholar] [CrossRef]
- Zhu, R.; Avsievich, T. Optical Tweezers in Studies of Red Blood Cells. Cells 2020, 9, 545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bond, M.; Hunt, B.; Flynn, B.; Huhtinen, P.; Ware, R.; Richards-kortum, R. Towards a point-of-care strip test to diagnose sickle cell anemia. PLoS ONE 2017, 12, e0177732. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arishi, W.A.; Alhadrami, H.A.; Zourob, M. Techniques for the Detection of Sickle Cell Disease: A Review. Micromachines 2021, 12, 519. https://doi.org/10.3390/mi12050519
Arishi WA, Alhadrami HA, Zourob M. Techniques for the Detection of Sickle Cell Disease: A Review. Micromachines. 2021; 12(5):519. https://doi.org/10.3390/mi12050519
Chicago/Turabian StyleArishi, Wjdan A., Hani A. Alhadrami, and Mohammed Zourob. 2021. "Techniques for the Detection of Sickle Cell Disease: A Review" Micromachines 12, no. 5: 519. https://doi.org/10.3390/mi12050519