
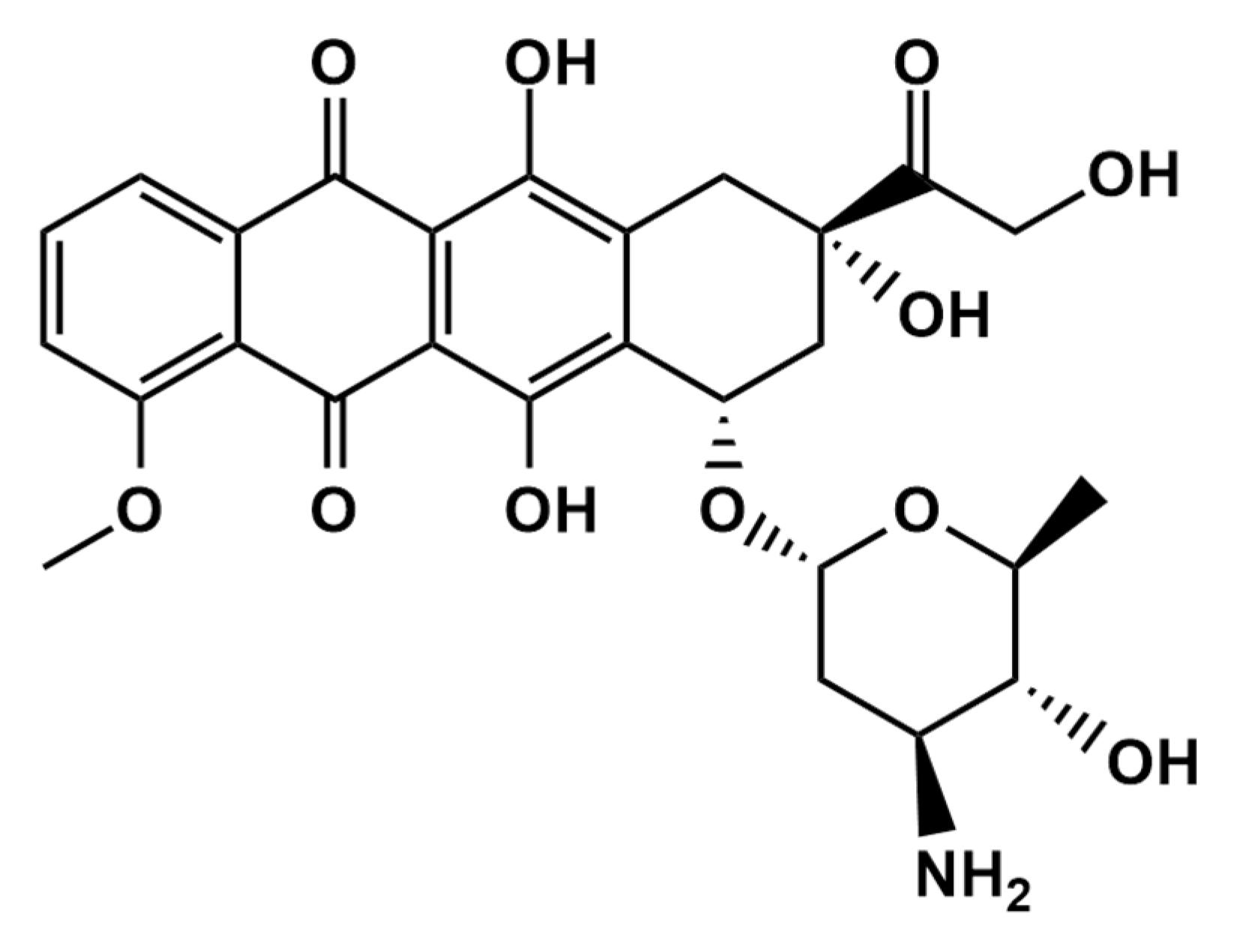
Sensitive and Selective Electrochemical Detection of Epirubicin as Anticancer Drug Based on Nickel Ferrite Decorated with Gold Nanoparticles

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Instrumentation
2.3. Synthesis of NiFe2O4
2.4. Synthesis of Au@NiFe2O4
2.5. Preparation of Au@NiFe2O4/SPE
2.6. Preparation of Real Samples
3. Results
3.1. Characterizations of Au@NiFe2O4 Nanocomposite
3.2. Electrochemical Performance
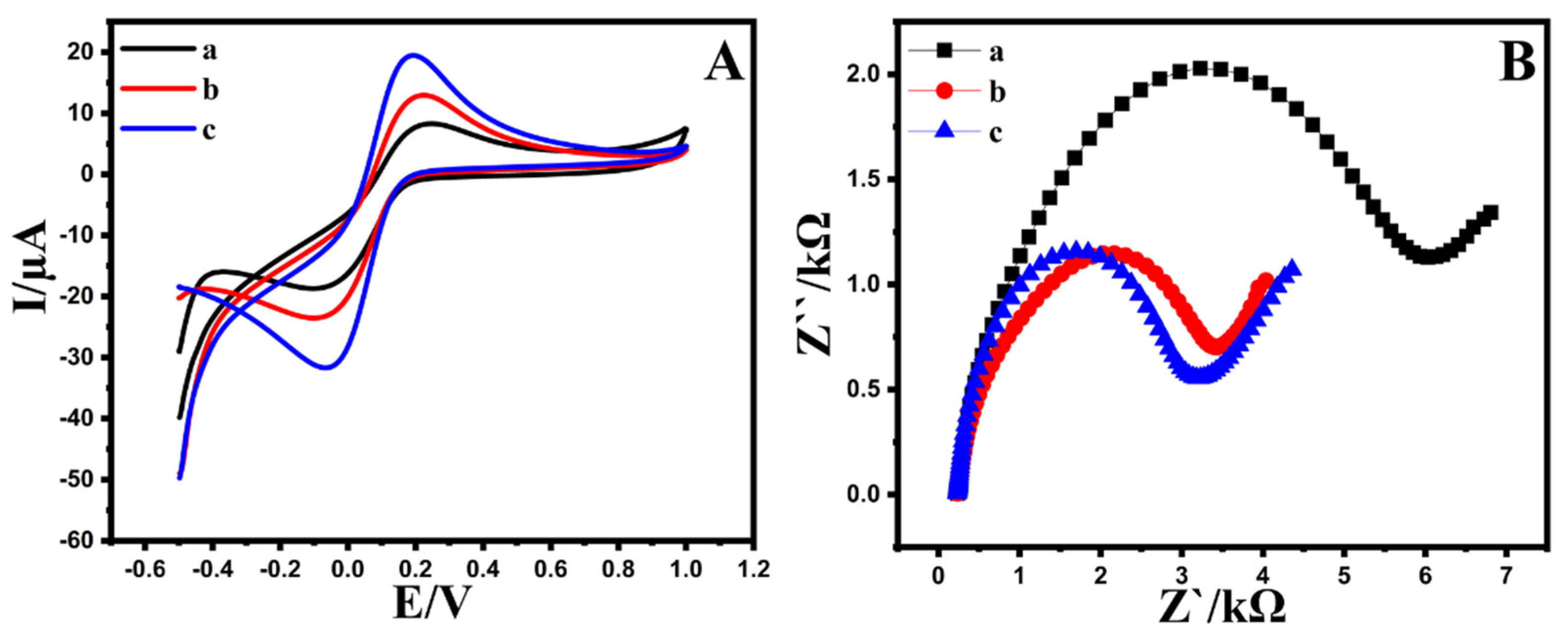
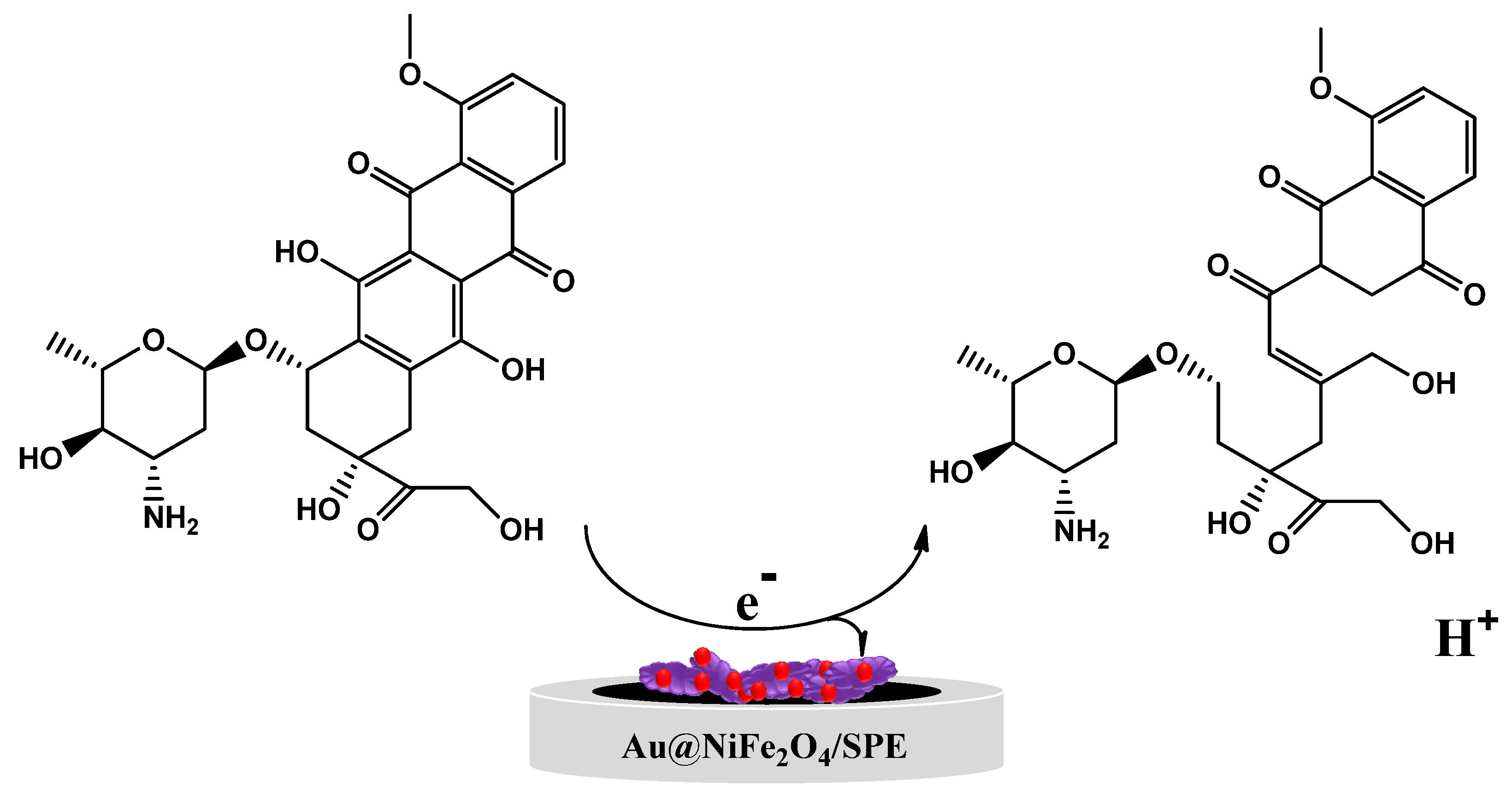
3.2.1. Influence of Modifier on the Electrochemical Oxidation of EPR
3.2.2. Heterogeneous Electron Transfer Rate Constant (K°)
3.3. Optimization of Conditions for Developing Sensitive and Selective Au@NiFe2O4/SPE
3.3.1. Effect of Physical and Chemical Properties
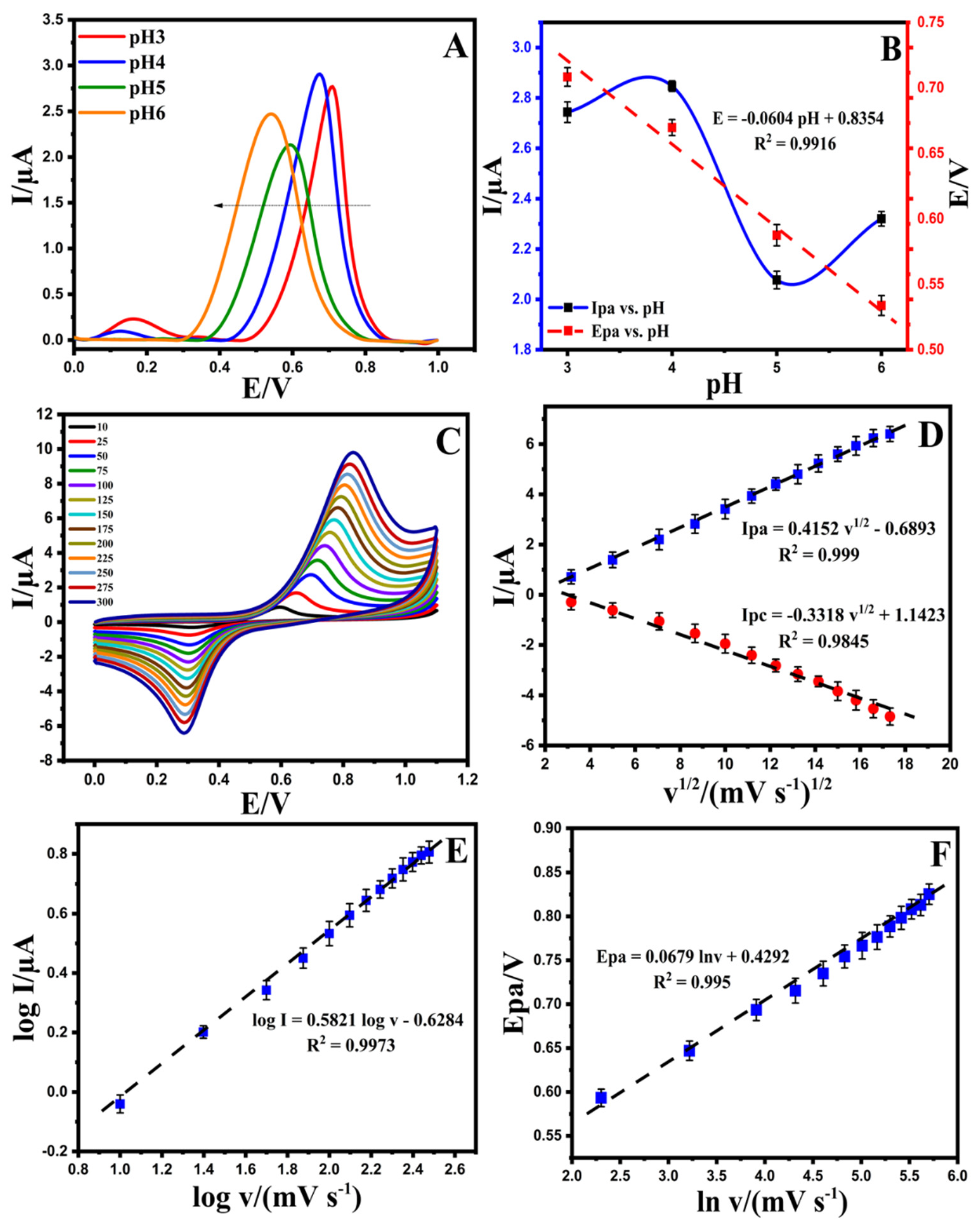
3.3.2. The Effect of pH
3.3.3. The Effect of Scan Rate
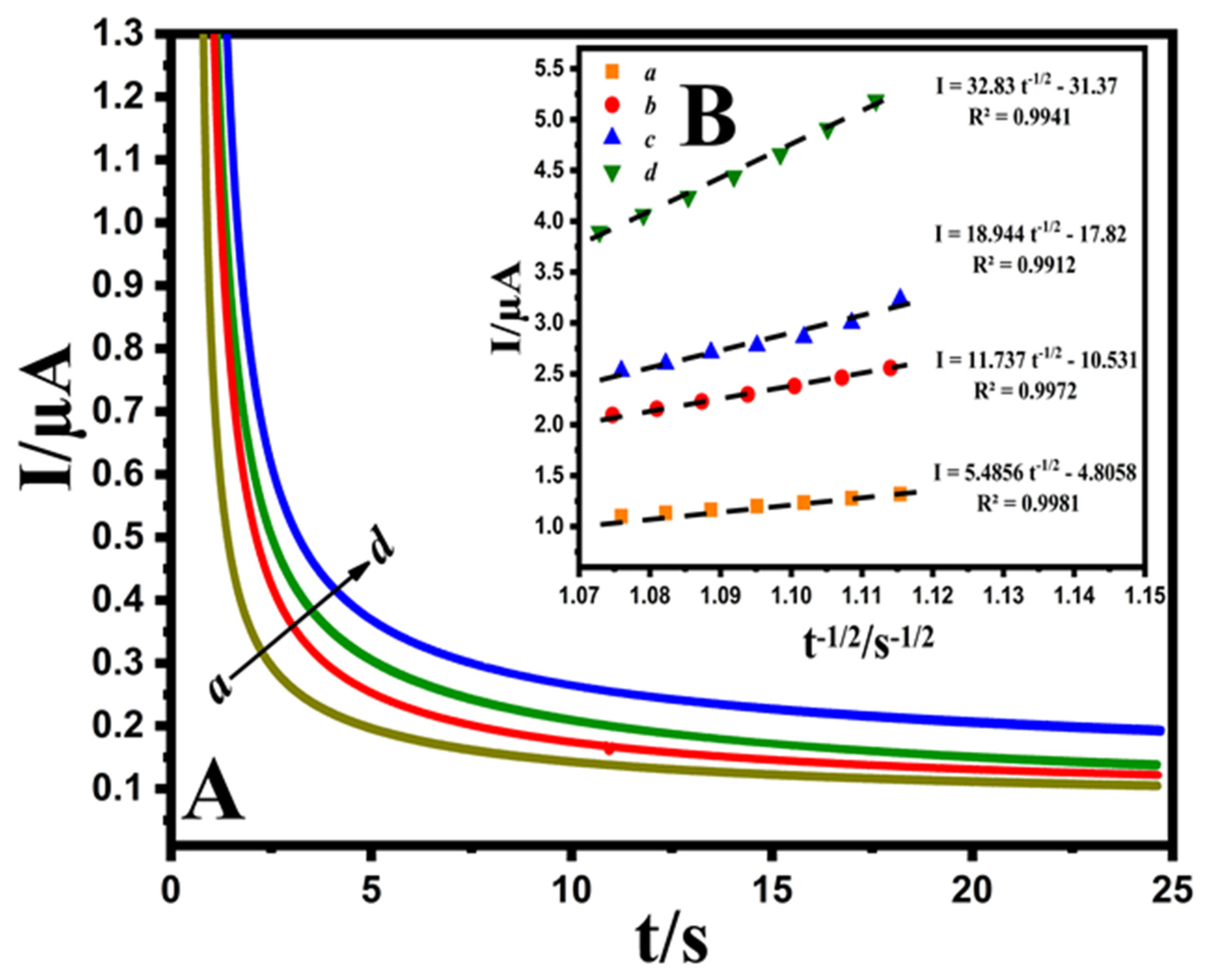
3.3.4. Chronoamperometric Study
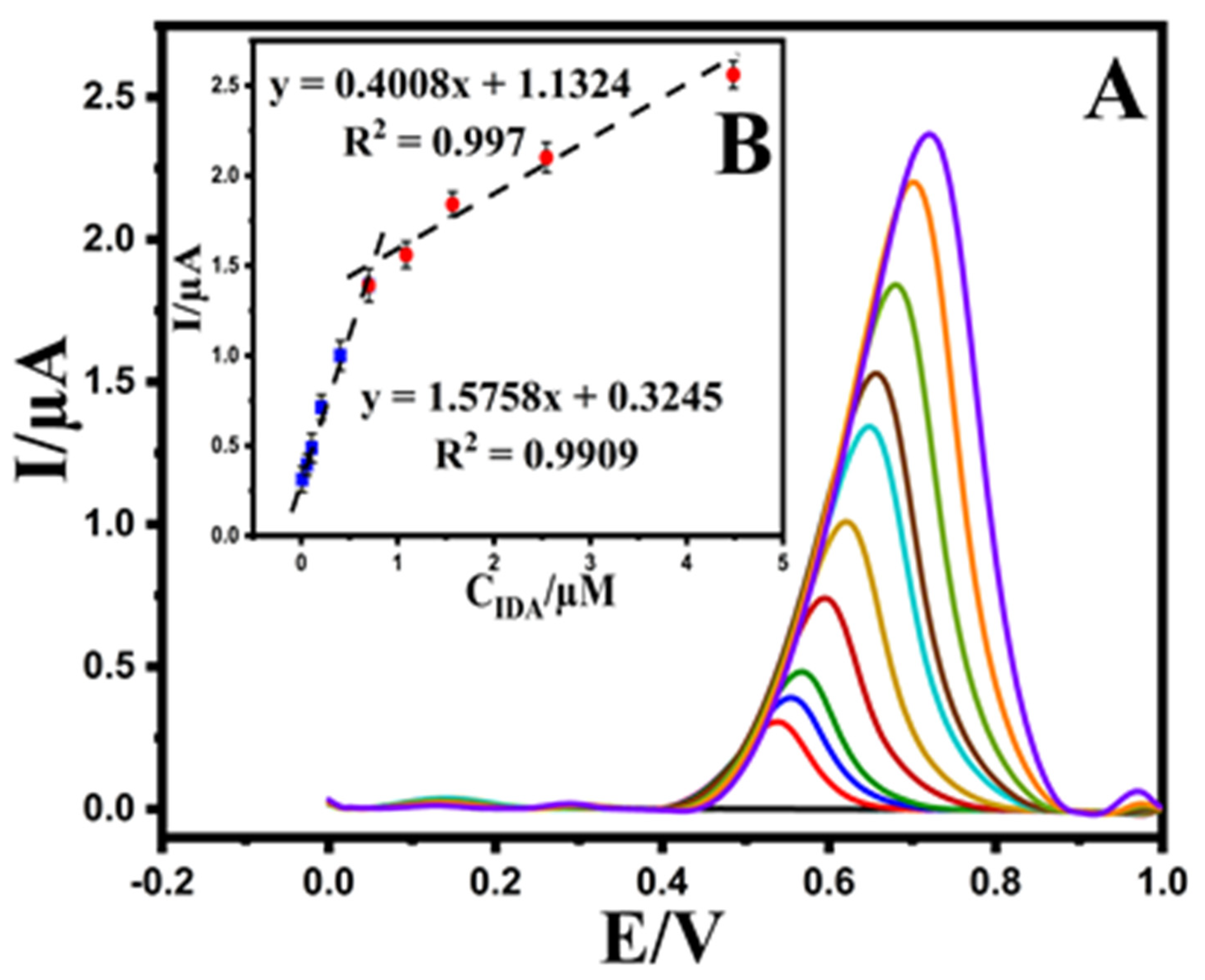
3.4. Determination of EPR at the Au@NiFe2O4/SPE
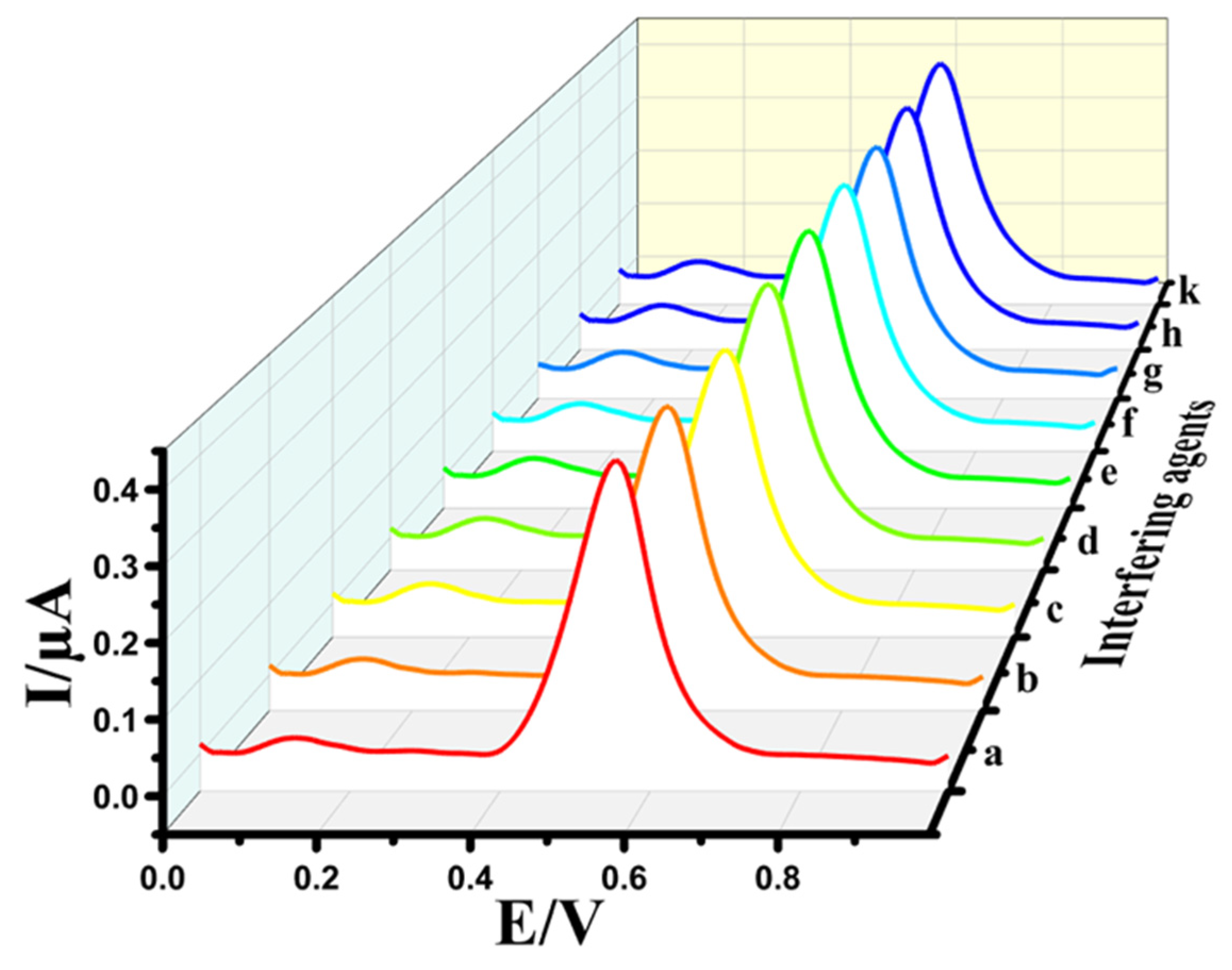
3.5. Selectivity of Au@NiFe2O4/SPE
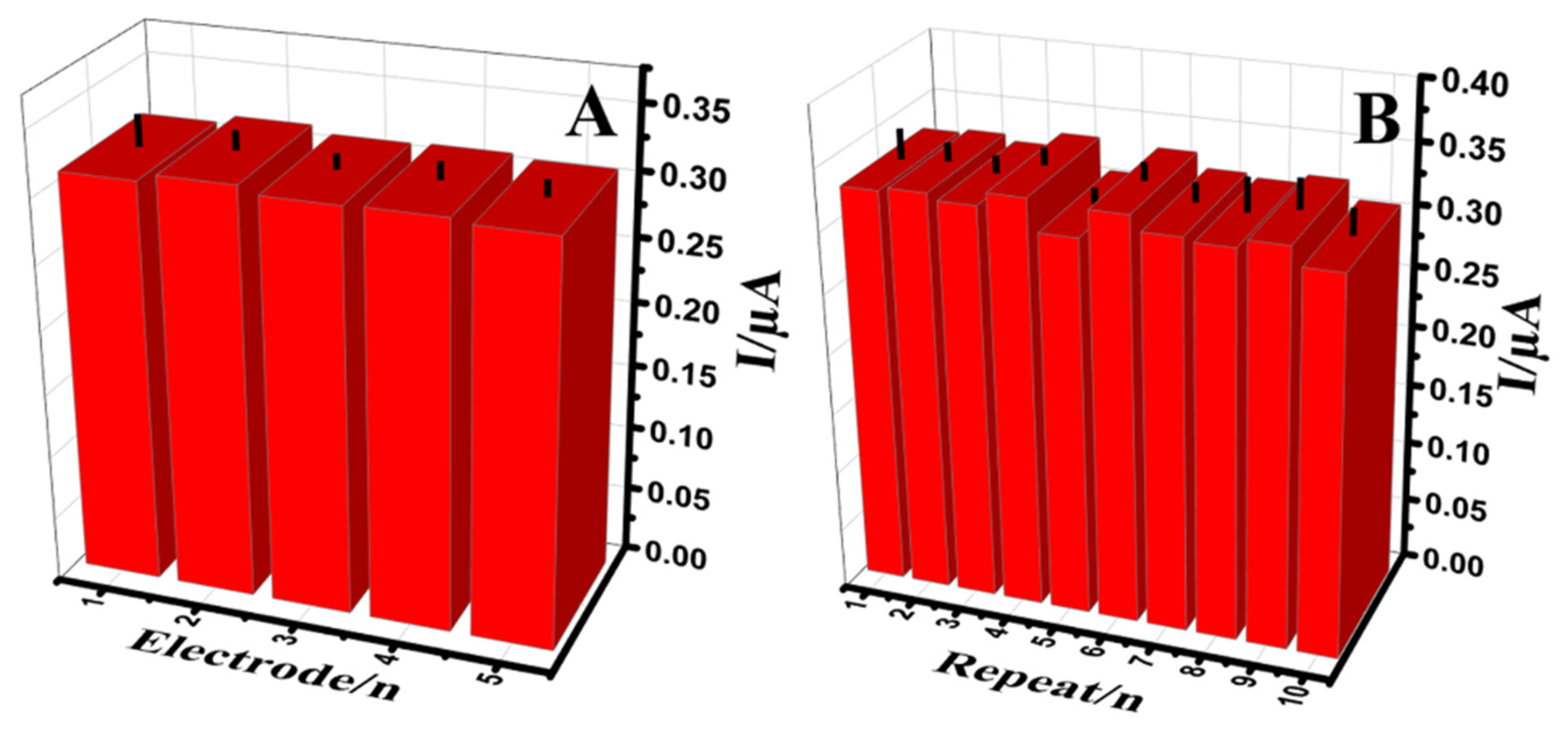
3.6. Reproducibility, Repeatability, Stability and Reusability of the Au@NiFe2O4/SPE
3.7. Real Samples Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Eksborg, S.; Hardell, L.; Bengtsson, N.O.; Sjodin, M.; Elfsson, B. Epirubicin as a Single Agent Therapy for the Treatment of Breast-Cancer—A Pharmacokinetic and Clinical-Study. Med. Oncol. Tumor Pharmacother. 1992, 9, 75–80. [Google Scholar] [CrossRef]
- Munster, P.; Marchion, D.; Bicaku, E.; Schmitt, M.; Lee, J.H.; DeConti, R.; Simon, G.; Fishman, M.; Minton, S.; Garrett, C.; et al. Phase I trial of histone deacetylase inhibition by valproic acid followed by the topoisomerase II inhibitor epirubicin in advanced solid tumors: A clinical and translational study. J. Clin. Oncol. 2007, 25, 1979–1985. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, J.; Retz, M.; Weining, C.; Albers, P.; Frohneberg, D.F.; Becker, T.; Funke, P.J.; Walz, P.; Langbein, S.; Schiller, M.; et al. Adjuvant systemic chemotherapy with cisplatin plus methotrexate (CM) versus methotrexate, vinblastine, epirubicin, and cisplatin (MVEC) for locally advanced bladder cancer: Results of a randomized, multicenter phase III study in Germany. J. Urol. 2003, 169, 337. [Google Scholar]
- Kuroda, M.; Kotake, T.; Akaza, H.; Hinotsu, S.; Kakizoe, T.; Grp, The Japanese Urothelial Cancer Research Group. Efficacy of dose-intensified MEC (methotrexate, epirubicin and cisplatin) chemotherapy for advanced urothelial carcinoma: A prospective randomized trial comparing MEC and M-VAC (methotrexate, vinblastine, doxorubicin and cisplatin). Jpn. J. Clin. Oncol. 1998, 28, 497–501. [Google Scholar] [CrossRef] [Green Version]
- Hajian, R.; Ekhlasi, E.; Daneshvar, R. Spectroscopic and Electrochemical Studies on the Interaction of Epirubicin with Fish Sperm DNA. E-J. Chem. 2012, 9, 1587–1598. [Google Scholar] [CrossRef] [Green Version]
- Charak, S.; Jangir, D.K.; Tyagi, G.; Mehrotra, R. Interaction studies of Epirubicin with DNA using spectroscopic techniques. J. Mol. Struct. 2011, 1000, 150–154. [Google Scholar] [CrossRef]
- Dodde, W.I.; Maring, J.G.; Hendriks, G.; Wachters, F.M.; Groen, H.J.; de Vries, E.G.; Uges, D.R. Determination of epirubicin and its metabolite epirubicinol in saliva and plasma by HPLC. Ther. Drug Monit. 2003, 25, 433–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fogli, S.; Danesi, R.; Innocenti, F.; Di Paolo, A.; Bocci, G.; Barbara, C.; Del Tacca, M. An improved HPLC method for therapeutic drug monitoring of daunorubicin, idarubicin, doxorubicin, epirubicin, and their 13-dihydro metabolites in human plasma. Ther. Drug Monit. 1999, 21, 367–375. [Google Scholar] [CrossRef]
- Gopinath, P.; Veluswami, S.; Thangarajan, R.; Gopisetty, G. RP-HPLC-UV Method for Estimation of Fluorouracil-Epirubicin-Cyclophosphamide and Their Metabolite Mixtures in Human Plasma (Matrix). J. Chromatogr. Sci. 2018, 56, 488–497. [Google Scholar] [CrossRef] [PubMed]
- Camaggi, C.M.; Comparsi, R.; Strocchi, E.; Testoni, F.; Pannuti, F. Hplc Analysis of Doxorubicin, Epirubicin and Fluorescent Metabolites in Biological-Fluids. Cancer Chemother. Pharmacol. 1988, 21, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Tariq, M.; Thomas, S.; Singh, A.; Talegaonkar, S. Developed and validated stability indicating HPLC method for the determination of epirubicin in bulk drug, marketed injection and polymeric nanoparticles. Braz. J. Pharm. Sci. 2018, 54. [Google Scholar] [CrossRef] [Green Version]
- Duffy, P.M.; Hayes, M.C.; Cooper, A.; Smart, C.J. Determination and reversal of resistance to epirubicin intravesical chemotherapy. A flow cytometric model. Br. J. Urol. 1996, 77, 819–823. [Google Scholar] [CrossRef]
- El-Kimary, E.I.; El-Yazbi, A.F. An eco-friendly stability-indicating spectrofluorimetric method for the determination of two anticancer stereoisomer drugs in their pharmaceutical preparations following micellar enhancement: Application to kinetic degradation studies. Spectrochim. Acta A 2016, 163, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Greco, F.; Arif, I.; Botting, R.; Fante, C.; Quintieri, L.; Clementi, C.; Schiavon, O.; Pasut, G. Polysialic acid as a drug carrier: Evaluation of a new polysialic acid-epirubicin conjugate and its comparison against established drug carriers. Polym. Chem. 2013, 4, 1600–1609. [Google Scholar] [CrossRef]
- Sottani, C.; Leoni, E.; Porro, B.; Montagna, B.; Amatu, A.; Sottotetti, F.; Quaretti, P.; Poggi, G.; Minoia, C. Validation of an LC-MS/MS method for the determination of epirubicin in human serum of patients undergoing Drug Eluting Microsphere-Transarterial Chemoembolization (DEM-TACE). J. Chromatogr. B. 2009, 877, 3543–3548. [Google Scholar] [CrossRef] [PubMed]
- Fotoohi, K.; Skarby, T.; Soderhall, S.; Peterson, C.; Albertioni, F. Interference of 7-hydroxymethotrexate with the determination of methotrexate in plasma samples from children with acute lymphoblastic leukemia employing routine clinical assays. J. Chromatogr. B. 2005, 817, 139–144. [Google Scholar] [CrossRef]
- Liu, X.J.; Liu, J.Z.; Huang, Y.Y.; Zhao, R.; Liu, G.Q.; Chen, Y. Determination of methotrexate in human serum by high-performance liquid chromatography combined with pseudo template molecularly iniprinted polymer. J. Chromatogr. A 2009, 1216, 7533–7538. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wu, Y.J.; Liu, J.X.; Ye, B.X. DNA Langmuir-Blodgett modified glassy carbon electrode as voltammetric sensor for determinate of methotrexate. Electrochim. Acta 2009, 54, 1408–1413. [Google Scholar] [CrossRef]
- Gao, L.; Wu, Y.J.; Liu, J.X.; Ye, B.X. Anodic voltammetric behaviors of methotrexate at a glassy carbon electrode and its determination in spiked human urine. J. Electroanal. Chem. 2007, 610, 131–136. [Google Scholar] [CrossRef]
- Guo, Y.J.; Chen, Y.H.; Zhao, Q.; Shuang, S.M.; Dong, C. Electrochemical Sensor for Ultrasensitive Determination of Doxorubicin and Methotrexate Based on Cyclodextrin-Graphene Hybrid Nanosheets. Electroanalysis 2011, 23, 2400–2407. [Google Scholar] [CrossRef]
- Karimi-Maleh, H.; Karimi, F.; Fu, L.; Sanati, A.L.; Alizadeh, M.; Karaman, C.; Orooji, Y. Cyanazine herbicide monitoring as a hazardous substance by a DNA nanostructure biosensor. J. Hazard. Mater. 2022, 423, 127058. [Google Scholar] [CrossRef]
- Karimi-Maleh, H.; Yola, M.L.; Atar, N.; Orooji, Y.; Karimi, F.; Kumar, P.S.; Rouhi, J.; Baghayeri, M. A novel detection method for organophosphorus insecticide fenamiphos: Molecularly imprinted electrochemical sensor based on core-shell Co3O4@MOF-74 nanocomposite. J. Colloid Interface Sci. 2021, 592, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Karimi-Maleh, H.; Orooji, Y.; Karimi, F.; Alizadeh, M.; Baghayeri, M.; Rouhi, J.; Tajik, S.; Beitollahi, H.D.; Agarwal, S.; Gupta, V.K.; et al. A critical review on the use of potentiometric based biosensors for biomarkers detection. Biosens. Bioelectron. 2021, 184, 113252. [Google Scholar] [CrossRef] [PubMed]
- Karimi-Maleh, H.; Alizadeh, M.; Orooji, Y.; Karimi, F.; Baghayeri, M.; Rouhi, J.; Tajik, S.; Beitollahi, H.; Agarwal, S.; Gupta, V.K.; et al. Guanine-Based DNA Biosensor Amplified with Pt/SWCNTs Nanocomposite as Analytical Tool for Nanomolar Determination of Daunorubicin as an Anticancer Drug: A Docking/Experimental Investigation. Ind. Eng. Chem. Res. 2021, 60, 816–823. [Google Scholar] [CrossRef]
- Medetalibeyoglu, H.; Beytur, M.; Manap, S.; Karaman, C.; Kardas, F.; Akyildirim, O.; Kotan, G.; Yuksek, H.; Atar, N.; Yola, M.L. Molecular Imprinted Sensor Including Au Nanoparticles/Polyoxometalate/Two-Dimensional Hexagonal Boron Nitride Nanocomposite for Diazinon Recognition. ECS J. Solid State Sci. Technol. 2020, 9, 101006. [Google Scholar] [CrossRef]
- Boke, C.P.; Karaman, O.; Medetalibeyoglu, H.; Karaman, C.; Atar, N.; Yola, M.L. A new approach for electrochemical detection of organochlorine compound lindane: Development of molecular imprinting polymer with polyoxometalate/carbon nitride nanotubes composite and validation. Microchem. J. 2020, 157, 105012. [Google Scholar] [CrossRef]
- Karimi-Maleh, H.; Keyvanfard, M.; Alizad, K.; Fouladgar, M.; Beitollahi, H.; Mokhtari, A.; Gholami-Orimi, F. Voltammetric Determination of N-Actylcysteine Using Modified Multiwall Carbon Nanotubes Paste Electrode. Int. J. Electrochem. Sci. 2011, 6, 6141–6150. [Google Scholar]
- Ensafi, A.A.; Dadkhah-Tehrani, S.; Karimi-Maleh, H. A Voltammetric Sensor for the Simultaneous Determination of L-Cysteine and Tryptophan Using a p-Aminophenol-Multiwall Carbon Nanotube Paste Electrode. Anal. Sci. 2011, 27, 409–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ensafi, A.A.; Karimi-Maleh, H.; Mallakpour, S. N-(3,4-Dihydroxyphenethyl)-3,5-dinitrobenzamide-Modified Multiwall Carbon Nanotubes Paste Electrode as a Novel Sensor for Simultaneous Determination of Penicillamine, Uric acid, and Tryptophan. Electroanalysis 2011, 23, 1478–1487. [Google Scholar] [CrossRef]
- Raoof, J.B.; Ojani, R.; Karimi-Maleh, H. Electrocatalytic oxidation of glutathione at carbon paste electrode modified with 2,7-bis (ferrocenyl ethyl) fluoren-9-one: Application as a voltammetric sensor. J. Appl. Electrochem. 2009, 39, 1169–1175. [Google Scholar] [CrossRef]
- Karaman, C.; Karaman, O.; Yola, B.B.; Ulker, İ.; Atar, N.; Yola, M.L. A novel electrochemical Aflatoxin B1 immunosensor based on gold nanoparticles decorated porous graphene nanoribbon and Ag nanocubes incorporated MoS2 nanosheets. New J. Chem. 2021, 45, 11222–11233. [Google Scholar] [CrossRef]
- Ozcan, N.; Karaman, C.; Atar, N.; Karaman, O.; Yola, M.L. A Novel Molecularly Imprinting Biosensor Including Graphene Quantum Dots/Multi-Walled Carbon Nanotubes Composite for Interleukin-6 Detection and Electrochemical Biosensor Validation. ECS J. Solid State Sci. Technol. 2020, 9, 121010. [Google Scholar] [CrossRef]
- Huang, L.; Chen, D.C.; Ding, Y.; Feng, S.; Wang, Z.L.; Liu, M.L. Nickel-Cobalt Hydroxide Nanosheets Coated on NiCo2O4 Nanowires Grown on Carbon Fiber Paper for High-Performance Pseudocapacitors. Nano Lett. 2013, 13, 3135–3139. [Google Scholar] [CrossRef]
- Yang, W.L.; Gao, Z.; Ma, J.; Zhang, X.M.; Wang, J.; Liu, J.Y. Hierarchical NiCo2O4@NiO core-shell hetero-structured nanowire arrays on carbon cloth for a high-performance flexible all-solid-state electrochemical capacitor. J. Mater. Chem. A 2014, 2, 1448–1457. [Google Scholar] [CrossRef]
- Al Sharabati, M.; Abokwiek, R.; Al-Othman, A.; Tawalbeh, M.; Karaman, C.; Orooji, Y.; Karimi, F. Biodegradable polymers and their nano-composites for the removal of endocrine-disrupting chemicals (EDCs) from wastewater: A review. Environ. Res. 2021, 202, 111694. [Google Scholar] [CrossRef]
- Karimi, F.; Ayati, A.; Tanhaei, B.; Sanati, A.L.; Afshar, S.; Kardan, A.; Dabirifar, Z.; Karaman, C. Removal of metal ions using a new magnetic chitosan nano-bio-adsorbent; A powerful approach in water treatment. Environ. Res. 2022, 203, 111753. [Google Scholar] [CrossRef] [PubMed]
- Karaman, O.; Ozdogan, H.; Uncu, V.A.; Karaman, C.; Tamar, A.G. Investigation of the effects of different composite materials on neutron contamination caused by medical LINAC. Kerntechnik 2020, 85, 401–407. [Google Scholar] [CrossRef]
- Akca, A.; Karaman, O.; Karaman, C.; Atar, N.; Yola, M.L. A comparative study of CO catalytic oxidation on the single vacancy and di-vacancy graphene supported single-atom iridium catalysts: A DFT analysis. Surf. Interfaces 2021, 25, 101293. [Google Scholar] [CrossRef]
- Karimi-Maleh, H.; Ayati, A.; Davoodi, R.; Tanhaei, B.; Karimi, F.; Malekmohammadi, S.; Orooji, Y.; Fu, L.; Sillanpaa, M. Recent advances in using of chitosan-based adsorbents for removal of pharmaceutical contaminants: A review. J. Clean. Prod. 2021, 291, 125880. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, X.; Li, Y.; Liu, Z.T.; Hao, Z.P. Synthesis of graphene-NiFe2O4 nanocomposites and their electrochemical capacitive behavior. J. Mater. Chem. A 2013, 1, 6393–6399. [Google Scholar] [CrossRef]
- Venkatachalam, V.; Jayavel, R. Novel Synthesis of Ni-Ferrite (NiFe2O4) Electrode Material for Supercapacitor Applications. AIP Conf. Proc. 2015, 1665, 140016. [Google Scholar] [CrossRef]
- Anwar, S.; Muthu, K.S.; Ganesh, V.; Lakshminarasimhan, N. A Comparative Study of Electrochemical Capacitive Behavior of NiFe2O4 Synthesized by Different Routes. J. Electrochem. Soc. 2011, 158, A976–A981. [Google Scholar] [CrossRef]
- Ensafi, A.A.; Jafari-Asl, M.; Rezaei, B.; Allafchian, A.R. Simultaneous determination of guanine and adenine in DNA based on NiFe2O4 magnetic nanoparticles decorated MWCNTs as a novel electrochemical sensor using adsorptive stripping voltammetry. Sens. Actuators B Chem. 2013, 177, 634–642. [Google Scholar] [CrossRef]
- Chen, H.; Yan, J.Q.; Wu, H.; Zhang, Y.X.; Liu, S.Z. One-pot fabrication of NiFe2O4 nanoparticles on alpha-Ni(OH)(2) nanosheet for enhanced water oxidation. J. Power Sources 2016, 324, 499–508. [Google Scholar] [CrossRef]
- Tarkistani, M.A.M.; Komalla, V.; Kayser, V. Recent Advances in the Use of Iron-Gold Hybrid Nanoparticles for Biomedical Applications. Nanomaterials 2021, 11, 1227. [Google Scholar] [CrossRef]
- Dawson, K.; Baudequin, M.; O’Riordan, A. Single on-chip gold nanowires for electrochemical biosensing of glucose. Analyst 2011, 136, 4507–4513. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.J.; Zhang, R.; Dong, C.; Cheng, F.Q.; Guo, Y.J. Sensitive electrochemical sensor for nitrite ions based on rose-like AuNPs/MoS2/graphene composite. Biosens. Bioelectron. 2019, 142, 111529. [Google Scholar] [CrossRef] [PubMed]
- Er, E.; Erk, N. Construction of a sensitive electrochemical sensor based on 1T-MoS2 nanosheets decorated with shape-controlled gold nanostructures for the voltammetric determination of doxorubicin. Microchim. Acta 2020, 187, 1–9. [Google Scholar] [CrossRef]
- Prakash, S.; Chakrabarty, T.; Singh, A.K.; Shahi, V.K. Polymer thin films embedded with metal nanoparticles for electrochemical biosensors applications. Biosens. Bioelectron. 2013, 41, 43–53. [Google Scholar] [CrossRef]
- Yola, M.L.; Atar, N. Development of cardiac troponin-I biosensor based on boron nitride quantum dots including molecularly imprinted polymer. Biosens. Bioelectron. 2019, 126, 418–424. [Google Scholar] [CrossRef]
- Yang, B.; Wang, C.; Xiao, R.; Yu, H.Y.; Huang, C.Q.; Wang, J.X.; Xu, J.L.; Liu, H.M.; Xia, F.; Xiao, J.Z. High NH3 selectivity of NiFe2O4 sensing electrode for potentiometric sensor at elevated temperature. Anal. Chim. Acta 2019, 1089, 165–173. [Google Scholar] [CrossRef]
- Karaman, C.; Karaman, O.; Atar, N.; Yola, M.L. Sustainable electrode material for high-energy supercapacitor: Biomass-derived graphene-like porous carbon with three-dimensional hierarchically ordered ion highways. Phys. Chem. Chem. Phys. 2021, 23, 12807–12821. [Google Scholar] [CrossRef] [PubMed]
- Pawar, R.C.; Kang, S.; Ahn, S.H.; Lee, C.S. Gold nanoparticle modified graphitic carbon nitride/multi-walled carbon nanotube (g-C3N4/CNTs/Au) hybrid photocatalysts for effective water splitting and degradation. RSC Adv. 2015, 5, 24281–24292. [Google Scholar] [CrossRef]
- Amulya, M.A.S.; Nagaswarupa, H.P.; Kumar, M.R.A.; Ravikumar, C.R.; Prashantha, S.C.; Kusuma, K.B. Sonochemical synthesis of NiFe2O4 nanoparticles: Characterization and their photocatalytic and electrochemical applications. Appl. Surf. Sci. Adv. 2020, 1, 100023. [Google Scholar] [CrossRef]
- Naidu, T.M.; Narayana, P.L. Synthesis and Characterization of Fe-TiO2 and NiFe2O4 Nanoparticles and Its Thermal Properties. J. Nanosci. Technol. 2019, 5, 769–772. [Google Scholar] [CrossRef]
- Deeth, R.J.; Anastasi, A.; Diedrich, C.; Randell, K. Molecular modelling for transition metal complexes: Dealing with d-electron effects. Coord. Chem. Rev. 2009, 253, 795–816. [Google Scholar] [CrossRef]
- Ceylan, A.; Ozcan, S.; Ni, C.; Shah, S.I. Solid state reaction synthesis of NiFe2O4 nanoparticles. J. Magn. Magn. Mater. 2008, 320, 857–863. [Google Scholar] [CrossRef]
- Jandaghi, N.; Jahani, S.; Foroughi, M.M.; Kazemipour, M.; Ansari, M. Cerium-doped flower-shaped ZnO nano-crystallites as a sensing component for simultaneous electrochemical determination of epirubicin and methotrexate. Microchim. Acta 2020, 187, 24. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.T.; Xie, J.M.; Tao, L.; Tian, H.; Wang, S.; Ding, H. Simultaneous electrochemical determination of epirubicin and methotrexate in human blood using a disposable electrode modified with nano-Au/MWNTs-ZnO composites. Sens. Actuators B Chem. 2014, 204, 360–367. [Google Scholar] [CrossRef]
- Zhang, H.J. Fabrication of a single-walled carbon nanotube-modified glassy carbon electrode and its application in the electrochemical determination of epirubicin. J. Nanoparticle Res. 2004, 6, 665–669. [Google Scholar] [CrossRef]
- Karimi, F.; Shojaei, A.F.; Tabatabaeian, K.; Shakeri, S. CoFe2O4 nanoparticle/ionic liquid modified carbon paste electrode as an amplified sensor for epirubicin analysis as an anticancer drug. J. Mol. Liq. 2017, 242, 685–689. [Google Scholar] [CrossRef]
- Bardajee, G.R.; Sharifi, M.; Mahmoodian, H. Novel CMC-CdTe/ZnS QDs Nanosensor for the Detection of Anticancer Drug Epirubicin. J. Fluoresc. 2021, 31, 651–658. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metrics | Au@NiFe2O4/SPE |
|---|---|
| response variability (RSD, %) | 3.56 |
| sensitivity (μA μmol L−1) | 1.5758 ± 0.14 a |
| linear dynamic range (μM) | 0.01–0.7 and 0.7–3.6 |
| correlation coefficient | 0.9909 and 0.997 |
| limit of detection (LOD, nM) | 5.32 |
| Method | Modified Electrode | LOD (μM) | LWR a (μM) | Ref. |
|---|---|---|---|---|
| DPV b | Ce-ZnO/GCE c | 0.0023 | 0.01–600 | [58] |
| SWV d | Au/MWNTs-ZnO/SPE | 0.0025 | 0.005–0.2 | [59] |
| LSV e | SWNT–DCP/SPE f | 0.02 | 0.05–10 | [60] |
| SWV | CoFe2O4/1,3-DPIBr g/CPE | 0.01 | 0.04–450 | [61] |
| fluorescence | CMC-CdTe/ZnS QDs | 0.04 | - | [62] |
| HPLC | - | 0.0149 | 1.87–187 | [11] |
| LC–MS/MS | - | 1.87 | 5.6–374.5 | [15] |
| DPV | Au@NiFe2O4/SPE | 0.0053 | 0.01–3.6 | Our work |
| Sample | Spiked (µM) | Found (µM) a | RSD (%) | Recovery (%) |
|---|---|---|---|---|
| Human plasma | 0.4 | 0.39 ± 0.01 | 2.14 | 97.5 |
| 0.6 | 0.6 ± 0.05 | 2.84 | 100.1 | |
| 0.8 | 0.79 ± 0.04 | 3.52 | 98.8 | |
| 1.0 | 1.0 ± 0.01 | 2.98 | 100.0 | |
| Urine | 0.4 | 0.41 ± 0.01 | 3.15 | 102.5 |
| 0.6 | 0.59 ± 0.06 | 2.89 | 98.4 | |
| 0.8 | 0.80 ± 0.03 | 4.11 | 100.0 | |
| 1.0 | 1.03 ± 0.03 | 2.59 | 103.0 | |
| Injection | - | 1.15 ± 0.14 | - | - |
| 0.4 | 1.51 ± 0.08 | 3.7 | 102.6 | |
| 0.6 | 1.78 ± 0.09 | 4.27 | 105.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehmandoust, M.; Erk, N.; Karaman, C.; Karimi, F.; Salmanpour, S. Sensitive and Selective Electrochemical Detection of Epirubicin as Anticancer Drug Based on Nickel Ferrite Decorated with Gold Nanoparticles. Micromachines 2021, 12, 1334. https://doi.org/10.3390/mi12111334
Mehmandoust M, Erk N, Karaman C, Karimi F, Salmanpour S. Sensitive and Selective Electrochemical Detection of Epirubicin as Anticancer Drug Based on Nickel Ferrite Decorated with Gold Nanoparticles. Micromachines. 2021; 12(11):1334. https://doi.org/10.3390/mi12111334
Chicago/Turabian StyleMehmandoust, Mohammad, Nevin Erk, Ceren Karaman, Fatemeh Karimi, and Sadegh Salmanpour. 2021. "Sensitive and Selective Electrochemical Detection of Epirubicin as Anticancer Drug Based on Nickel Ferrite Decorated with Gold Nanoparticles" Micromachines 12, no. 11: 1334. https://doi.org/10.3390/mi12111334