

White Adipose Tissue Dysfunction: Pathophysiology and Emergent Measurements

Abstract
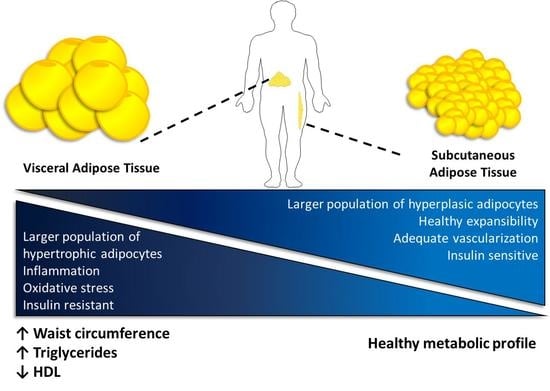
:
1. Introduction
2. Adipose Tissue Composition and Function
3. Adipose Tissue Dysfunction
3.1. Role of ECM Remodeling in Adipose Tissue Dysfunction
3.2. Inflammation as a Key Component of AT Dysfunction and Metabolic Impairments
3.3. Adipose Tissue Dysfunction and Metabolic Disturbances
4. Current and Emergent Parameters for the Assessment of Adipose Tissue Dysfunction
4.1. Current Parameters Associated to Obesity
4.2. Parameters Associated to AT Dysfunction
4.2.1. Visceral Adiposity Index (VAI)
4.2.2. Hypertriglyceridemic Waist (HTGW)
4.2.3. Metabolic Flexibility
4.2.4. Biomarkers
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hafekost, K.; Lawrence, D.; Mitrou, F.; O’Sullivan, T.A.; Zubrick, S.R. Tackling overweight and obesity: Does the public health message match the science? BMC Med. 2013, 11, 41. [Google Scholar] [CrossRef] [Green Version]
- The GBD 2015 Obesity Collaborators. Health Effects of Overweight and Obesity in 195 Countries over 25 Years. N. Engl. J. Med. 2017, 377, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Burhans, M.S.; Hagman, D.K.; Kuzma, J.N.; Schmidt, K.A.; Kratz, M. Contribution of Adipose Tissue Inflammation to the Development of Type 2 Diabetes Mellitus. Compr. Physiol. 2018, 9, 1–58. [Google Scholar] [CrossRef] [PubMed]
- Esser, N.; Legrand-Poels, S.; Piette, J.; Scheen, A.J.; Paquot, N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res. Clin. Pract. 2014, 105, 141–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Oria, R.; Genchi, V.A.; Caccioppoli, C.; Calderoni, I.; Marrano, N.; Biondi, G.; Borrelli, A.; Di Gioia, L.; Giorgino, F.; Laviola, L. Impact of Dysfunctional Adipose Tissue Depots on the Cardiovascular System. Int. J. Mol. Sci. 2022, 23, 14296. [Google Scholar] [CrossRef] [PubMed]
- Muscogiuri, G.; Pugliese, G.; Laudisio, D.; Castellucci, B.; Barrea, L.; Savastano, S.; Colao, A. The impact of obesity on immune response to infection: Plausible mechanisms and outcomes. Obes. Rev. 2021, 22, e13216. [Google Scholar] [CrossRef] [PubMed]
- Stefan, N.; Birkenfeld, A.L.; Schulze, M.B.; Ludwig, D.S. Obesity and impaired metabolic health in patients with COVID-19. Nat. Rev. Endocrinol. 2020, 16, 341–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, E.D.; Spiegelman, B.M. What we talk about when we talk about fat. Cell 2014, 156, 20–44. [Google Scholar] [CrossRef] [Green Version]
- Engin, A.B. Adipocyte-Macrophage Cross-Talk in Obesity. Adv. Exp. Med. Biol. 2017, 960, 327–343. [Google Scholar] [CrossRef]
- Bluher, M. Adipose tissue dysfunction contributes to obesity related metabolic diseases. Best Pract. Res. Clin. Endocrinol. Metab. 2013, 27, 163–177. [Google Scholar] [CrossRef]
- Longo, M.; Zatterale, F.; Naderi, J.; Parrillo, L.; Formisano, P.; Raciti, G.A.; Beguinot, F.; Miele, C. Adipose Tissue Dysfunction as Determinant of Obesity-Associated Metabolic Complications. Int. J. Mol. Sci. 2019, 20, 2358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tchkonia, T.; Thomou, T.; Zhu, Y.; Karagiannides, I.; Pothoulakis, C.; Jensen, M.D.; Kirkland, J.L. Mechanisms and metabolic implications of regional differences among fat depots. Cell Metab. 2013, 17, 644–656. [Google Scholar] [CrossRef] [Green Version]
- Arner, P.; Ryden, M. Human white adipose tissue: A highly dynamic metabolic organ. J. Intern. Med. 2022, 291, 611–621. [Google Scholar] [CrossRef]
- Gaborit, B.; Sengenes, C.; Ancel, P.; Jacquier, A.; Dutour, A. Role of Epicardial Adipose Tissue in Health and Disease: A Matter of Fat? Compr. Physiol. 2017, 7, 1051–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grigoras, A.; Balan, R.A.; Caruntu, I.D.; Giusca, S.E.; Lozneanu, L.; Avadanei, R.E.; Rusu, A.; Riscanu, L.A.; Amalinei, C. Perirenal Adipose Tissue-Current Knowledge and Future Opportunities. J. Clin. Med. 2021, 10, 1291. [Google Scholar] [CrossRef]
- Schafer, K.; Drosos, I.; Konstantinides, S. Perivascular adipose tissue: Epiphenomenon or local risk factor? Int. J. Obes. 2017, 41, 1311–1323. [Google Scholar] [CrossRef]
- Berry, R.; Rodeheffer, M.S. Characterization of the adipocyte cellular lineage in vivo. Nat. Cell Biol. 2013, 15, 302–308. [Google Scholar] [CrossRef]
- Wang, Q.A.; Tao, C.; Gupta, R.K.; Scherer, P.E. Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat. Med. 2013, 19, 1338–1344. [Google Scholar] [CrossRef]
- McLaughlin, T.; Craig, C.; Liu, L.F.; Perelman, D.; Allister, C.; Spielman, D.; Cushman, S.W. Adipose Cell Size and Regional Fat Deposition as Predictors of Metabolic Response to Overfeeding in Insulin-Resistant and Insulin-Sensitive Humans. Diabetes 2016, 65, 1245–1254. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.M.; Lun, M.; Wang, M.; Senyo, S.E.; Guillermier, C.; Patwari, P.; Steinhauser, M.L. Loss of white adipose hyperplastic potential is associated with enhanced susceptibility to insulin resistance. Cell Metab. 2014, 20, 1049–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y. Angiogenesis modulates adipogenesis and obesity. J. Clin. Investig. 2007, 117, 2362–2368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Ojeda, F.J.; Mendez-Gutierrez, A.; Aguilera, C.M.; Plaza-Diaz, J. Extracellular Matrix Remodeling of Adipose Tissue in Obesity and Metabolic Diseases. Int. J. Mol. Sci. 2019, 20, 4888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fasshauer, M.; Bluher, M. Adipokines in health and disease. Trends Pharmacol. Sci. 2015, 36, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Kirichenko, T.V.; Markina, Y.V.; Bogatyreva, A.I.; Tolstik, T.V.; Varaeva, Y.R.; Starodubova, A.V. The Role of Adipokines in Inflammatory Mechanisms of Obesity. Int. J. Mol. Sci. 2022, 23, 14982. [Google Scholar] [CrossRef]
- Arner, E.; Mejhert, N.; Kulyte, A.; Balwierz, P.J.; Pachkov, M.; Cormont, M.; Lorente-Cebrian, S.; Ehrlund, A.; Laurencikiene, J.; Heden, P.; et al. Adipose tissue microRNAs as regulators of CCL2 production in human obesity. Diabetes 2012, 61, 1986–1993. [Google Scholar] [CrossRef] [Green Version]
- Kappeler, L. Role of Adipose Tissue microRNAs in the Onset of Metabolic Diseases and Implications in the Context of the DOHaD. Cells 2022, 11, 3711. [Google Scholar] [CrossRef]
- Yao, J.; Wu, D.; Qiu, Y. Adipose tissue macrophage in obesity-associated metabolic diseases. Front. Immunol. 2022, 13, 977485. [Google Scholar] [CrossRef]
- Li, C.; Xu, M.M.; Wang, K.; Adler, A.J.; Vella, A.T.; Zhou, B. Macrophage polarization and meta-inflammation. Transl. Res. 2018, 191, 29–44. [Google Scholar] [CrossRef]
- Ruggiero, A.D.; Key, C.C.; Kavanagh, K. Adipose Tissue Macrophage Polarization in Healthy and Unhealthy Obesity. Front. Nutr. 2021, 8, 625331. [Google Scholar] [CrossRef]
- Lee, M.J.; Wu, Y.; Fried, S.K. Adipose tissue heterogeneity: Implication of depot differences in adipose tissue for obesity complications. Mol. Asp. Med. 2013, 34, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Leibel, R.L.; Rosenbaum, M.; Hirsch, J. Changes in energy expenditure resulting from altered body weight. N. Engl. J. Med. 1995, 332, 621–628. [Google Scholar] [CrossRef]
- Schlogl, M.; Piaggi, P.; Pannacciuli, N.; Bonfiglio, S.M.; Krakoff, J.; Thearle, M.S. Energy Expenditure Responses to Fasting and Overfeeding Identify Phenotypes Associated with Weight Change. Diabetes 2015, 64, 3680–3689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuthbertson, D.J.; Steele, T.; Wilding, J.P.; Halford, J.C.; Harrold, J.A.; Hamer, M.; Karpe, F. What have human experimental overfeeding studies taught us about adipose tissue expansion and susceptibility to obesity and metabolic complications? Int. J. Obes. 2017, 41, 853–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iacobini, C.; Pugliese, G.; Blasetti Fantauzzi, C.; Federici, M.; Menini, S. Metabolically healthy versus metabolically unhealthy obesity. Metabolism 2019, 92, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Ryden, M.; Andersson, D.P.; Bergstrom, I.B.; Arner, P. Adipose tissue and metabolic alterations: Regional differences in fat cell size and number matter, but differently: A cross-sectional study. J. Clin. Endocrinol. Metab. 2014, 99, E1870–E1876. [Google Scholar] [CrossRef] [Green Version]
- White, U.; Beyl, R.A.; Ravussin, E. A higher proportion of small adipocytes is associated with increased visceral and ectopic lipid accumulation during weight gain in response to overfeeding in men. Int. J. Obes. 2022, 46, 1560–1563. [Google Scholar] [CrossRef]
- Kawai, T.; Autieri, M.V.; Scalia, R. Adipose tissue inflammation and metabolic dysfunction in obesity. Am. J. Physiol.-Cell Physiol. 2021, 320, C375–C391. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation, metaflammation and immunometabolic disorders. Nature 2017, 542, 177–185. [Google Scholar] [CrossRef]
- Carobbio, S.; Pellegrinelli, V.; Vidal-Puig, A. Adipose Tissue Function and Expandability as Determinants of Lipotoxicity and the Metabolic Syndrome. Adv. Exp. Med. Biol. 2017, 960, 161–196. [Google Scholar] [CrossRef]
- Taylor, R.; Holman, R.R. Normal weight individuals who develop type 2 diabetes: The personal fat threshold. Clin. Sci. 2015, 128, 405–410. [Google Scholar] [CrossRef] [Green Version]
- Johannsen, D.L.; Tchoukalova, Y.; Tam, C.S.; Covington, J.D.; Xie, W.; Schwarz, J.M.; Bajpeyi, S.; Ravussin, E. Effect of 8 weeks of overfeeding on ectopic fat deposition and insulin sensitivity: Testing the “adipose tissue expandability” hypothesis. Diabetes Care 2014, 37, 2789–2797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tchoukalova, Y.D.; Votruba, S.B.; Tchkonia, T.; Giorgadze, N.; Kirkland, J.L.; Jensen, M.D. Regional differences in cellular mechanisms of adipose tissue gain with overfeeding. Proc. Natl. Acad. Sci. USA 2010, 107, 18226–18231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corvera, S.; Solivan-Rivera, J.; Yang Loureiro, Z. Angiogenesis in adipose tissue and obesity. Angiogenesis 2022, 25, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Crewe, C.; An, Y.A.; Scherer, P.E. The ominous triad of adipose tissue dysfunction: Inflammation, fibrosis and impaired angiogenesis. J. Clin. Investig. 2017, 127, 74–82. [Google Scholar] [CrossRef] [Green Version]
- Orsso, C.E.; Colin-Ramirez, E.; Field, C.J.; Madsen, K.L.; Prado, C.M.; Haqq, A.M. Adipose Tissue Development and Expansion from the Womb to Adolescence: An Overview. Nutrients 2020, 12, 2735. [Google Scholar] [CrossRef]
- Blaak, E.E.; van Baak, M.A.; Kemerink, G.J.; Pakbiers, M.T.; Heidendal, G.A.; Saris, W.H. Beta-adrenergic stimulation and abdominal subcutaneous fat blood flow in lean, obese, and reduced-obese subjects. Metabolism 1995, 44, 183–187. [Google Scholar] [CrossRef]
- Goossens, G.H.; Bizzarri, A.; Venteclef, N.; Essers, Y.; Cleutjens, J.P.; Konings, E.; Jocken, J.W.; Cajlakovic, M.; Ribitsch, V.; Clement, K.; et al. Increased adipose tissue oxygen tension in obese compared with lean men is accompanied by insulin resistance, impaired adipose tissue capillarization and inflammation. Circulation 2011, 124, 67–76. [Google Scholar] [CrossRef] [Green Version]
- Trayhurn, P. Hypoxia and adipose tissue function and dysfunction in obesity. Physiol. Rev. 2013, 93, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Fujisaka, S.; Usui, I.; Ikutani, M.; Aminuddin, A.; Takikawa, A.; Tsuneyama, K.; Mahmood, A.; Goda, N.; Nagai, Y.; Takatsu, K.; et al. Adipose tissue hypoxia induces inflammatory M1 polarity of macrophages in an HIF-1alpha-dependent and HIF-1alpha-independent manner in obese mice. Diabetologia 2013, 56, 1403–1412. [Google Scholar] [CrossRef] [Green Version]
- Babapoor-Farrokhran, S.; Gill, D.; Alzubi, J.; Mainigi, S.K. Atrial fibrillation: The role of hypoxia-inducible factor-1-regulated cytokines. Mol. Cell. Biochem. 2021, 476, 2283–2293. [Google Scholar] [CrossRef]
- Darby, I.A.; Hewitson, T.D. Hypoxia in tissue repair and fibrosis. Cell Tissue Res. 2016, 365, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.C.; Lee, J. Cellular and molecular players in adipose tissue inflammation in the development of obesity-induced insulin resistance. Biochim. Biophys. Acta 2014, 1842, 446–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rausch, M.E.; Weisberg, S.; Vardhana, P.; Tortoriello, D.V. Obesity in C57BL/6J mice is characterized by adipose tissue hypoxia and cytotoxic T-cell infiltration. Int. J. Obes. 2008, 32, 451–463. [Google Scholar] [CrossRef] [Green Version]
- Henegar, C.; Tordjman, J.; Achard, V.; Lacasa, D.; Cremer, I.; Guerre-Millo, M.; Poitou, C.; Basdevant, A.; Stich, V.; Viguerie, N.; et al. Adipose tissue transcriptomic signature highlights the pathological relevance of extracellular matrix in human obesity. Genome Biol. 2008, 9, R14. [Google Scholar] [CrossRef]
- Johnston, E.K.; Abbott, R.D. Adipose Tissue Paracrine-, Autocrine-, and Matrix-Dependent Signaling during the Development and Progression of Obesity. Cells 2023, 12, 407. [Google Scholar] [CrossRef]
- Khan, T.; Muise, E.S.; Iyengar, P.; Wang, Z.V.; Chandalia, M.; Abate, N.; Zhang, B.B.; Bonaldo, P.; Chua, S.; Scherer, P.E. Metabolic dysregulation and adipose tissue fibrosis: Role of collagen VI. Mol. Cell. Biol. 2009, 29, 1575–1591. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Santibanez, G.; Lumeng, C.N. Macrophages and the regulation of adipose tissue remodeling. Annu. Rev. Nutr. 2014, 34, 57–76. [Google Scholar] [CrossRef]
- Boutens, L.; Stienstra, R. Adipose tissue macrophages: Going off track during obesity. Diabetologia 2016, 59, 879–894. [Google Scholar] [CrossRef] [Green Version]
- Liang, W.; Qi, Y.; Yi, H.; Mao, C.; Meng, Q.; Wang, H.; Zheng, C. The Roles of Adipose Tissue Macrophages in Human Disease. Front. Immunol. 2022, 13, 908749. [Google Scholar] [CrossRef]
- Hammarstedt, A.; Gogg, S.; Hedjazifar, S.; Nerstedt, A.; Smith, U. Impaired Adipogenesis and Dysfunctional Adipose Tissue in Human Hypertrophic Obesity. Physiol. Rev. 2018, 98, 1911–1941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, N.; Liang, H.; Zen, K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front. Immunol. 2014, 5, 614. [Google Scholar] [CrossRef] [Green Version]
- Castoldi, A.; Naffah de Souza, C.; Camara, N.O.; Moraes-Vieira, P.M. The Macrophage Switch in Obesity Development. Front. Immunol. 2015, 6, 637. [Google Scholar] [CrossRef] [Green Version]
- Cinti, S.; Mitchell, G.; Barbatelli, G.; Murano, I.; Ceresi, E.; Faloia, E.; Wang, S.; Fortier, M.; Greenberg, A.S.; Obin, M.S. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J. Lipid Res. 2005, 46, 2347–2355. [Google Scholar] [CrossRef] [Green Version]
- Murano, I.; Barbatelli, G.; Parisani, V.; Latini, C.; Muzzonigro, G.; Castellucci, M.; Cinti, S. Dead adipocytes, detected as crown-like structures, are prevalent in visceral fat depots of genetically obese mice. J. Lipid Res. 2008, 49, 1562–1568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Investig. 2006, 116, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; Hunter, D.; Huber, R.; Lemieux, J.; Slaymaker, S.; Vaddi, K.; Charo, I.; Leibel, R.L.; Ferrante, A.W., Jr. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J. Clin. Investig. 2006, 116, 115–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Zhao, J.; Meng, H.; Zhang, X. Adipose Tissue-Resident Immune Cells in Obesity and Type 2 Diabetes. Front. Immunol. 2019, 10, 1173. [Google Scholar] [CrossRef]
- Keophiphath, M.; Achard, V.; Henegar, C.; Rouault, C.; Clement, K.; Lacasa, D. Macrophage-secreted factors promote a profibrotic phenotype in human preadipocytes. Mol. Endocrinol. 2009, 23, 11–24. [Google Scholar] [CrossRef] [Green Version]
- Sorisky, A.; Molgat, A.S.; Gagnon, A. Macrophage-induced adipose tissue dysfunction and the preadipocyte: Should I stay (and differentiate) or should I go? Adv. Nutr. 2013, 4, 67–75. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; He, J.; Wang, H.; Zhu, D.; Bi, Y. Adipose Morphology: A Critical Factor in Regulation of Human Metabolic Diseases and Adipose Tissue Dysfunction. Obes. Surg. 2020, 30, 5086–5100. [Google Scholar] [CrossRef]
- Chung, S.; Lapoint, K.; Martinez, K.; Kennedy, A.; Boysen Sandberg, M.; McIntosh, M.K. Preadipocytes mediate lipopolysaccharide-induced inflammation and insulin resistance in primary cultures of newly differentiated human adipocytes. Endocrinology 2006, 147, 5340–5351. [Google Scholar] [CrossRef]
- Lee, Y.S.; Li, P.; Huh, J.Y.; Hwang, I.J.; Lu, M.; Kim, J.I.; Ham, M.; Talukdar, S.; Chen, A.; Lu, W.J.; et al. Inflammation is necessary for long-term but not short-term high-fat diet-induced insulin resistance. Diabetes 2011, 60, 2474–2483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kern, P.A.; Di Gregorio, G.B.; Lu, T.; Rassouli, N.; Ranganathan, G. Adiponectin expression from human adipose tissue: Relation to obesity, insulin resistance, and tumor necrosis factor-alpha expression. Diabetes 2003, 52, 1779–1785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gariballa, S.; Alkaabi, J.; Yasin, J.; Al Essa, A. Total adiponectin in overweight and obese subjects and its response to visceral fat loss. BMC Endocr. Disord. 2019, 19, 55. [Google Scholar] [CrossRef]
- Reilly, S.M.; Saltiel, A.R. Adapting to obesity with adipose tissue inflammation. Nat. Rev. Endocrinol. 2017, 13, 633–643. [Google Scholar] [CrossRef]
- Hernandez, E.D.; Lee, S.J.; Kim, J.Y.; Duran, A.; Linares, J.F.; Yajima, T.; Muller, T.D.; Tschop, M.H.; Smith, S.R.; Diaz-Meco, M.T.; et al. A macrophage NBR1-MEKK3 complex triggers JNK-mediated adipose tissue inflammation in obesity. Cell Metab. 2014, 20, 499–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zatterale, F.; Longo, M.; Naderi, J.; Raciti, G.A.; Desiderio, A.; Miele, C.; Beguinot, F. Chronic Adipose Tissue Inflammation Linking Obesity to Insulin Resistance and Type 2 Diabetes. Front. Physiol. 2019, 10, 1607. [Google Scholar] [CrossRef]
- Bai, Y.; Sun, Q. Macrophage recruitment in obese adipose tissue. Obes. Rev. 2015, 16, 127–136. [Google Scholar] [CrossRef] [Green Version]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Reynolds, C.M.; McGillicuddy, F.C.; Harford, K.A.; Finucane, O.M.; Mills, K.H.; Roche, H.M. Dietary saturated fatty acids prime the NLRP3 inflammasome via TLR4 in dendritic cells-implications for diet-induced insulin resistance. Mol. Nutr. Food Res. 2012, 56, 1212–1222. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Tate, M.; Mathew, G.; Vince, J.E.; Ritchie, R.H.; de Haan, J.B. Oxidative Stress and NLRP3-Inflammasome Activity as Significant Drivers of Diabetic Cardiovascular Complications: Therapeutic Implications. Front. Physiol. 2018, 9, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legrand-Poels, S.; Esser, N.; L’Homme, L.; Scheen, A.; Paquot, N.; Piette, J. Free fatty acids as modulators of the NLRP3 inflammasome in obesity/type 2 diabetes. Biochem. Pharmacol. 2014, 92, 131–141. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esser, N.; L’Homme, L.; De Roover, A.; Kohnen, L.; Scheen, A.J.; Moutschen, M.; Piette, J.; Legrand-Poels, S.; Paquot, N. Obesity phenotype is related to NLRP3 inflammasome activity and immunological profile of visceral adipose tissue. Diabetologia 2013, 56, 2487–2497. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.K.; Cheung, S.W.; Cheng, K.K. NLRP3 Inflammasome Activation in Adipose Tissues and Its Implications on Metabolic Diseases. Int. J. Mol. Sci. 2020, 21, 4184. [Google Scholar] [CrossRef]
- Vandanmagsar, B.; Youm, Y.H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef]
- Unamuno, X.; Gomez-Ambrosi, J.; Ramirez, B.; Rodriguez, A.; Becerril, S.; Valenti, V.; Moncada, R.; Silva, C.; Salvador, J.; Fruhbeck, G.; et al. NLRP3 inflammasome blockade reduces adipose tissue inflammation and extracellular matrix remodeling. Cell. Mol. Immunol. 2021, 18, 1045–1057. [Google Scholar] [CrossRef]
- Jiang, N.; Li, Y.; Shu, T.; Wang, J. Cytokines and inflammation in adipogenesis: An updated review. Front. Med. 2019, 13, 314–329. [Google Scholar] [CrossRef]
- Bourlier, V.; Sengenes, C.; Zakaroff-Girard, A.; Decaunes, P.; Wdziekonski, B.; Galitzky, J.; Villageois, P.; Esteve, D.; Chiotasso, P.; Dani, C.; et al. TGFbeta family members are key mediators in the induction of myofibroblast phenotype of human adipose tissue progenitor cells by macrophages. PLoS ONE 2012, 7, e31274. [Google Scholar] [CrossRef]
- Tanaka, M.; Ikeda, K.; Suganami, T.; Komiya, C.; Ochi, K.; Shirakawa, I.; Hamaguchi, M.; Nishimura, S.; Manabe, I.; Matsuda, T.; et al. Macrophage-inducible C-type lectin underlies obesity-induced adipose tissue fibrosis. Nat. Commun. 2014, 5, 4982. [Google Scholar] [CrossRef] [Green Version]
- Rabhi, N.; Desevin, K.; Belkina, A.C.; Tilston-Lunel, A.; Varelas, X.; Layne, M.D.; Farmer, S.R. Obesity-induced senescent macrophages activate a fibrotic transcriptional program in adipocyte progenitors. Life Sci. Alliance 2022, 5, e202101286. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, C.Y.; Jang, J.E.; Lee, S.E.; Koh, E.H.; Lee, K.U. Mitochondrial Dysfunction in Adipocytes as a Primary Cause of Adipose Tissue Inflammation. Diabetes Metab. J. 2019, 43, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ojeda, F.J.; Ozla, J.; Gil, Á.; Concepción, M.A. Chapter 1—Oxidative Stress and Inflammation in Obesity and Metabolic Syndrome. In Obesity; del Moral, A.M., María, C., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 1–15. [Google Scholar] [CrossRef]
- Xu, L.; Yan, X.; Zhao, Y.; Wang, J.; Liu, B.; Yu, S.; Fu, J.; Liu, Y.; Su, J. Macrophage Polarization Mediated by Mitochondrial Dysfunction Induces Adipose Tissue Inflammation in Obesity. Int. J. Mol. Sci. 2022, 23, 9252. [Google Scholar] [CrossRef] [PubMed]
- Masschelin, P.M.; Cox, A.R.; Chernis, N.; Hartig, S.M. The Impact of Oxidative Stress on Adipose Tissue Energy Balance. Front. Physiol. 2019, 10, 1638. [Google Scholar] [CrossRef]
- Manna, P.; Jain, S.K. Obesity, Oxidative Stress, Adipose Tissue Dysfunction and the Associated Health Risks: Causes and Therapeutic Strategies. Metab. Syndr. Relat. Disord. 2015, 13, 423–444. [Google Scholar] [CrossRef] [Green Version]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Park, S.Y.; Choi, C.S. Insulin Resistance: From Mechanisms to Therapeutic Strategies. Diabetes Metab. J. 2022, 46, 15–37. [Google Scholar] [CrossRef]
- Khalid, M.; Alkaabi, J.; Khan, M.A.B.; Adem, A. Insulin Signal Transduction Perturbations in Insulin Resistance. Int. J. Mol. Sci. 2021, 22, 8590. [Google Scholar] [CrossRef]
- Glass, C.K.; Olefsky, J.M. Inflammation and lipid signaling in the etiology of insulin resistance. Cell Metab. 2012, 15, 635–645. [Google Scholar] [CrossRef] [Green Version]
- Lauterbach, M.A.; Wunderlich, F.T. Macrophage function in obesity-induced inflammation and insulin resistance. Pflügers Arch.-Eur. J. Physiol. 2017, 469, 385–396. [Google Scholar] [CrossRef] [Green Version]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yazici, D.; Sezer, H. Insulin Resistance, Obesity and Lipotoxicity. Adv. Exp. Med. Biol. 2017, 960, 277–304. [Google Scholar] [CrossRef] [PubMed]
- Engin, A.B. What Is Lipotoxicity? Adv. Exp. Med. Biol. 2017, 960, 197–220. [Google Scholar] [CrossRef]
- Nielsen, S.; Guo, Z.; Johnson, C.M.; Hensrud, D.D.; Jensen, M.D. Splanchnic lipolysis in human obesity. J. Clin. Investig. 2004, 113, 1582–1588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sears, B.; Perry, M. The role of fatty acids in insulin resistance. Lipids Health Dis. 2015, 14, 121. [Google Scholar] [CrossRef] [Green Version]
- Sobczak, A.I.S.; Blindauer, C.A.; Stewart, A.J. Changes in Plasma Free Fatty Acids Associated with Type-2 Diabetes. Nutrients 2019, 11, 2022. [Google Scholar] [CrossRef] [Green Version]
- Oh, Y.S.; Bae, G.D.; Baek, D.J.; Park, E.Y.; Jun, H.S. Fatty Acid-Induced Lipotoxicity in Pancreatic Beta-Cells During Development of Type 2 Diabetes. Front. Endocrinol. 2018, 9, 384. [Google Scholar] [CrossRef]
- Tumova, J.; Andel, M.; Trnka, J. Excess of free fatty acids as a cause of metabolic dysfunction in skeletal muscle. Physiol. Res. 2016, 65, 193–207. [Google Scholar] [CrossRef]
- Nishi, H.; Higashihara, T.; Inagi, R. Lipotoxicity in Kidney, Heart, and Skeletal Muscle Dysfunction. Nutrients 2019, 11, 1664. [Google Scholar] [CrossRef] [Green Version]
- Lessard, J.; Laforest, S.; Pelletier, M.; Leboeuf, M.; Blackburn, L.; Tchernof, A. Low abdominal subcutaneous preadipocyte adipogenesis is associated with visceral obesity, visceral adipocyte hypertrophy, and a dysmetabolic state. Adipocyte 2014, 3, 197–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veilleux, A.; Caron-Jobin, M.; Noel, S.; Laberge, P.Y.; Tchernof, A. Visceral adipocyte hypertrophy is associated with dyslipidemia independent of body composition and fat distribution in women. Diabetes 2011, 60, 1504–1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, X.; Peng, D. The exchangeable apolipoproteins in lipid metabolism and obesity. Clin. Chim. Acta 2020, 503, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Jung, U.J.; Choi, M.S. Obesity and its metabolic complications: The role of adipokines and the relationship between obesity, inflammation, insulin resistance, dyslipidemia and nonalcoholic fatty liver disease. Int. J. Mol. Sci. 2014, 15, 6184–6223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashid, S.; Barrett, P.H.; Uffelman, K.D.; Watanabe, T.; Adeli, K.; Lewis, G.F. Lipolytically modified triglyceride-enriched HDLs are rapidly cleared from the circulation. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 483–487. [Google Scholar] [CrossRef] [Green Version]
- Rashid, S.; Trinh, D.K.; Uffelman, K.D.; Cohn, J.S.; Rader, D.J.; Lewis, G.F. Expression of human hepatic lipase in the rabbit model preferentially enhances the clearance of triglyceride-enriched versus native high-density lipoprotein apolipoprotein A-I. Circulation 2003, 107, 3066–3072. [Google Scholar] [CrossRef] [Green Version]
- Stadler, J.T.; Marsche, G. Obesity-Related Changes in High-Density Lipoprotein Metabolism and Function. Int. J. Mol. Sci. 2020, 21, 8985. [Google Scholar] [CrossRef]
- Zhong, G.C.; Huang, S.Q.; Peng, Y.; Wan, L.; Wu, Y.Q.; Hu, T.Y.; Hu, J.J.; Hao, F.B. HDL-C is associated with mortality from all causes, cardiovascular disease and cancer in a J-shaped dose-response fashion: A pooled analysis of 37 prospective cohort studies. Eur. J. Prev. Cardiol. 2020, 27, 1187–1203. [Google Scholar] [CrossRef] [Green Version]
- Neeland, I.J.; Turer, A.T.; Ayers, C.R.; Powell-Wiley, T.M.; Vega, G.L.; Farzaneh-Far, R.; Grundy, S.M.; Khera, A.; McGuire, D.K.; de Lemos, J.A. Dysfunctional adiposity and the risk of prediabetes and type 2 diabetes in obese adults. JAMA 2012, 308, 1150–1159. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.H.; Ha, K.H.; Kim, D.J. Visceral Fat Mass Has Stronger Associations with Diabetes and Prediabetes than Other Anthropometric Obesity Indicators among Korean Adults. Yonsei Med. J. 2016, 57, 674–680. [Google Scholar] [CrossRef] [PubMed]
- Piche, M.E.; Tchernof, A.; Despres, J.P. Obesity Phenotypes, Diabetes, and Cardiovascular Diseases. Circ. Res. 2020, 126, 1477–1500. [Google Scholar] [CrossRef] [PubMed]
- Wildman, R.P.; Muntner, P.; Reynolds, K.; McGinn, A.P.; Rajpathak, S.; Wylie-Rosett, J.; Sowers, M.R. The obese without cardiometabolic risk factor clustering and the normal weight with cardiometabolic risk factor clustering: Prevalence and correlates of 2 phenotypes among the US population (NHANES 1999–2004). Arch. Intern. Med. 2008, 168, 1617–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Zhuang, R.; Luo, X.; Yin, L.; Pang, C.; Feng, T.; You, H.; Zhai, Y.; Ren, Y.; Zhang, L.; et al. Prevalence of Metabolically Healthy Obese and Metabolically Obese but Normal Weight in Adults Worldwide: A Meta-Analysis. Horm. Metab. Res. 2015, 47, 839–845. [Google Scholar] [CrossRef]
- Bluher, M. Metabolically Healthy Obesity. Endocr. Rev. 2020, 41, bnaa004. [Google Scholar] [CrossRef] [Green Version]
- Tsatsoulis, A.; Paschou, S.A. Metabolically Healthy Obesity: Criteria, Epidemiology, Controversies, and Consequences. Curr. Obes. Rep. 2020, 9, 109–120. [Google Scholar] [CrossRef]
- Executive summary of the clinical guidelines on the identification, evaluation, and treatment of overweight and obesity in adults. Arch. Intern. Med. 1998, 158, 1855–1867. [CrossRef]
- Alberti, K.G.; Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z.; Cleeman, J.I.; Donato, K.A.; Fruchart, J.C.; James, W.P.; Loria, C.M.; Smith, S.C., Jr.; et al. Harmonizing the metabolic syndrome: A joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009, 120, 1640–1645. [Google Scholar] [CrossRef] [Green Version]
- Ross, R.; Neeland, I.J.; Yamashita, S.; Shai, I.; Seidell, J.; Magni, P.; Santos, R.D.; Arsenault, B.; Cuevas, A.; Hu, F.B.; et al. Waist circumference as a vital sign in clinical practice: A Consensus Statement from the IAS and ICCR Working Group on Visceral Obesity. Nat. Rev. Endocrinol. 2020, 16, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Amato, M.C.; Giordano, C.; Pitrone, M.; Galluzzo, A. Cut-off points of the visceral adiposity index (VAI) identifying a visceral adipose dysfunction associated with cardiometabolic risk in a Caucasian Sicilian population. Lipids Health Dis. 2011, 10, 183. [Google Scholar] [CrossRef] [Green Version]
- Blackburn, P.; Lemieux, I.; Lamarche, B.; Bergeron, J.; Perron, P.; Tremblay, G.; Gaudet, D.; Despres, J.P. Hypertriglyceridemic waist: A simple clinical phenotype associated with coronary artery disease in women. Metabolism 2012, 61, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.E.; Mandarino, L.J. Fuel selection in human skeletal muscle in insulin resistance: A reexamination. Diabetes 2000, 49, 677–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, M.J.; Braun, W.; Enderle, J.; Bosy-Westphal, A. Beyond BMI: Conceptual Issues Related to Overweight and Obese Patients. Obes. Facts 2016, 9, 193–205. [Google Scholar] [CrossRef]
- Neeland, I.J.; Ross, R.; Despres, J.P.; Matsuzawa, Y.; Yamashita, S.; Shai, I.; Seidell, J.; Magni, P.; Santos, R.D.; Arsenault, B.; et al. Visceral and ectopic fat, atherosclerosis, and cardiometabolic disease: A position statement. Lancet Diabetes Endocrinol. 2019, 7, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Amato, M.C.; Giordano, C.; Galia, M.; Criscimanna, A.; Vitabile, S.; Midiri, M.; Galluzzo, A.; AlkaMeSy Study Group. Visceral Adiposity Index: A reliable indicator of visceral fat function associated with cardiometabolic risk. Diabetes Care 2010, 33, 920–922. [Google Scholar] [CrossRef] [Green Version]
- Amato, M.C.; Giordano, C. Visceral adiposity index: An indicator of adipose tissue dysfunction. Int. J. Endocrinol. 2014, 2014, 730827. [Google Scholar] [CrossRef] [Green Version]
- Lemieux, I.; Poirier, P.; Bergeron, J.; Almeras, N.; Lamarche, B.; Cantin, B.; Dagenais, G.R.; Despres, J.P. Hypertriglyceridemic waist: A useful screening phenotype in preventive cardiology? Can. J. Cardiol. 2007, 23, 23B–31B. [Google Scholar] [CrossRef] [Green Version]
- Sam, S.; Haffner, S.; Davidson, M.H.; D’Agostino, R.B., Sr.; Feinstein, S.; Kondos, G.; Perez, A.; Mazzone, T. Hypertriglyceridemic waist phenotype predicts increased visceral fat in subjects with type 2 diabetes. Diabetes Care 2009, 32, 1916–1920. [Google Scholar] [CrossRef] [Green Version]
- Cunha de Oliveira, C.; Carneiro Roriz, A.K.; Eickemberg, M.; Barreto Medeiros, J.M.; Barbosa Ramos, L. Hypertriglyceridemic waist phenotype: Association with metabolic disorders and visceral fat in adults. Nutr. Hosp. 2014, 30, 25–31. [Google Scholar] [CrossRef]
- LeBlanc, S.; Coulombe, F.; Bertrand, O.F.; Bibeau, K.; Pibarot, P.; Marette, A.; Almeras, N.; Lemieux, I.; Despres, J.P.; Larose, E. Hypertriglyceridemic Waist: A Simple Marker of High-Risk Atherosclerosis Features Associated with Excess Visceral Adiposity/Ectopic Fat. J. Am. Heart Assoc. 2018, 7, e008139. [Google Scholar] [CrossRef] [Green Version]
- Arsenault, B.J.; Lemieux, I.; Despres, J.P.; Wareham, N.J.; Kastelein, J.J.; Khaw, K.T.; Boekholdt, S.M. The hypertriglyceridemic-waist phenotype and the risk of coronary artery disease: Results from the EPIC-Norfolk prospective population study. CMAJ 2010, 182, 1427–1432. [Google Scholar] [CrossRef] [Green Version]
- Minambres, I.; Sanchez-Hernandez, J.; Cuixart, G.; Sanchez-Pinto, A.; Sarroca, J.; Perez, A. Characterization of the hypertriglyceridemic waist phenotype in patients with type 2 diabetes mellitus in Spain: An epidemiological study. Rev. Clin. Esp. 2021, 221, 576–581. [Google Scholar] [CrossRef]
- Zheng, X.; Ren, X.; Jiang, M.; Han, L. Association between hypertriglyceridemic-waist phenotype and cardiovascular disease: A cohort study and meta-analysis. Front. Cardiovasc. Med. 2022, 9, 940168. [Google Scholar] [CrossRef] [PubMed]
- Borges, L.D.; Comini, L.O.; de Oliveira, L.C.; Dias, H.H.; Ferreira, E.S.; Batistelli, C.R.S.; da Costa, G.D.; Moreira, T.R.; da Silva, R.G.; Cotta, R.M.M. Hypertriglyceridemic waist phenotype and associated factors in individuals with arterial hypertension and/or diabetes mellitus. J. Nutr. Sci. 2021, 10, e74. [Google Scholar] [CrossRef] [PubMed]
- Cai, R.; Zhou, J.; Bai, L.; Dong, Y.; Ding, W. Hypertriglyceridemic-waist phenotype is strongly associated with cardiovascular risk factor clustering in Chinese adolescents. Sci. Rep. 2022, 12, 15464. [Google Scholar] [CrossRef] [PubMed]
- Goodpaster, B.H.; Sparks, L.M. Metabolic Flexibility in Health and Disease. Cell Metab. 2017, 25, 1027–1036. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.L.; Soeters, M.R.; Wust, R.C.I.; Houtkooper, R.H. Metabolic Flexibility as an Adaptation to Energy Resources and Requirements in Health and Disease. Endocr. Rev. 2018, 39, 489–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galgani, J.E.; Moro, C.; Ravussin, E. Metabolic flexibility and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1009–E1017. [Google Scholar] [CrossRef] [Green Version]
- Yu, E.A.; Le, N.A.; Stein, A.D. Measuring Postprandial Metabolic Flexibility to Assess Metabolic Health and Disease. J. Nutr. 2021, 151, 3284–3291. [Google Scholar] [CrossRef]
- Branson, R.D.; Johannigman, J.A. The measurement of energy expenditure. Nutr. Clin. Pract. 2004, 19, 622–636. [Google Scholar] [CrossRef]
- Patel, H.; Kerndt, C.C.; Bhardwaj, A. Physiology, Respiratory Quotient; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Sparks, L.M.; Ukropcova, B.; Smith, J.; Pasarica, M.; Hymel, D.; Xie, H.; Bray, G.A.; Miles, J.M.; Smith, S.R. Relation of adipose tissue to metabolic flexibility. Diabetes Res. Clin. Pract. 2009, 83, 32–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheja, L.; Heeren, J. The endocrine function of adipose tissues in health and cardiometabolic disease. Nat. Rev. Endocrinol. 2019, 15, 507–524. [Google Scholar] [CrossRef] [PubMed]
- Okada-Iwabu, M.; Yamauchi, T.; Iwabu, M.; Honma, T.; Hamagami, K.; Matsuda, K.; Yamaguchi, M.; Tanabe, H.; Kimura-Someya, T.; Shirouzu, M.; et al. A small-molecule AdipoR agonist for type 2 diabetes and short life in obesity. Nature 2013, 503, 493–499. [Google Scholar] [CrossRef]
- Choi, H.M.; Doss, H.M.; Kim, K.S. Multifaceted Physiological Roles of Adiponectin in Inflammation and Diseases. Int. J. Mol. Sci. 2020, 21, 1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiernan, K.; MacIver, N.J. The Role of the Adipokine Leptin in Immune Cell Function in Health and Disease. Front. Immunol. 2020, 11, 622468. [Google Scholar] [CrossRef] [PubMed]
- Zuo, H.; Shi, Z.; Yuan, B.; Dai, Y.; Wu, G.; Hussain, A. Association between serum leptin concentrations and insulin resistance: A population-based study from China. PLoS ONE 2013, 8, e54615. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Lohakare, A.C. Association of Leptin and Carotid Intima-Media Thickness in Overweight and Obese Individuals: A Cross-Sectional Study. J. Assoc. Physicians India 2020, 68, 19–23. [Google Scholar] [PubMed]
- Castela, I.; Morais, J.; Barreiros-Mota, I.; Silvestre, M.P.; Marques, C.; Rodrigues, C.; Ismael, S.; Araujo, J.R.; Angelo-Dias, M.; Martins, C.; et al. Decreased adiponectin/leptin ratio relates to insulin resistance in adults with obesity. Am. J. Physiol. Endocrinol. Metab. 2023, 324, E115–E119. [Google Scholar] [CrossRef]
- Inoue, M.; Maehata, E.; Yano, M.; Taniyama, M.; Suzuki, S. Correlation between the adiponectin-leptin ratio and parameters of insulin resistance in patients with type 2 diabetes. Metabolism 2005, 54, 281–286. [Google Scholar] [CrossRef]
- Lee, K.W.; Shin, D. Prospective Associations of Serum Adiponectin, Leptin, and Leptin-Adiponectin Ratio with Incidence of Metabolic Syndrome: The Korean Genome and Epidemiology Study. Int. J. Environ. Res. Public Health 2020, 17, 3287. [Google Scholar] [CrossRef]

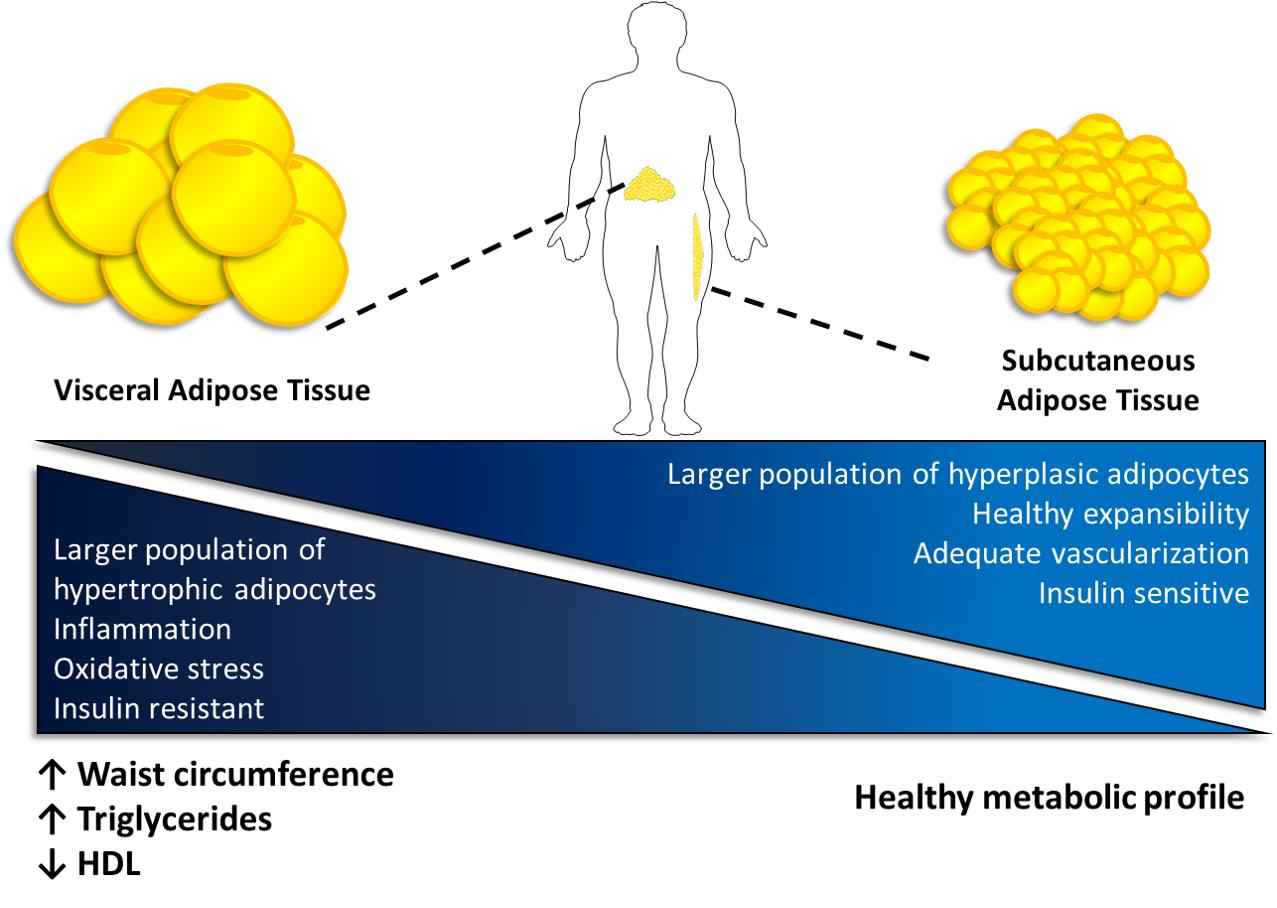{kind=link}
| Measurements Associated to Obesity | Description | Advantages | Disadvantages |
|---|---|---|---|
| BMI [128] | Weight divided by height squared. Depending on its value, classifies the nutritional status of adults as “underweight”, “normal”, “overweight” and “obese”. | Simple and quick to assess in clinical practice. | Does not discriminate body composition or AT dysfunction. |
| Waist circumference [128,129] | Assesses fat accumulation at the abdominal level. | Simple and quick to assess in clinical practice. | Does not distinguish between VAT and SAT. There is no consensus on the measurement technique. |
| Metabolic Syndrome [129] | A cluster of risk factors for cardiovascular disease and type 2 diabetes mellitus. Is diagnosed when at least three out of five metabolic alterations are present: abdominal obesity measured through waist circumference, hypertriglyceridemia, hypertension, low levels of HDL, and/or high fasting glycemia. | Well documented in the medical literature. | Diagnosis of metabolic syndrome may be too late to evaluate the susceptibility to develop metabolic alterations. |
| Waist circumference thresholds within BMI categories [130] | Evaluates risk of future coronary events through waist circumference thresholds stratified by BMI. | Simple and quick to assess in clinical practice. It could improve cardiometabolic risk management in adults with excess adiposity. | It needs to be validated in different populations and ethnic groups. |
| Measurements associated with adipose dysfunction | |||
| VAI [131] | Mathematical model that estimates visceral functionality based on BMI, waist circumference, triglycerides, and HDL levels. | Inversely related to insulin sensitivity and independently associated with cardiometabolic risk. | It may not be useful in the morbid obesity population (BMI > 40 kg /m2). It needs to be validated in non-Caucasian populations. |
| HTGW [132] | Simultaneous presence of an increased waist circumference and elevated fasting triglyceride concentrations. | Associated with the content of VAT, it could screen those individuals susceptible to developing metabolic alterations more quickly. | It needs to be validated in different populations and ethnic groups. |
| Metabolic flexibility [133] | Evaluates the ability of an organism to adapt substrate oxidation to substrate availability, measured through the change in respiratory quotient under metabolic challenges such as overnight fasting or hyperinsulinemic–euglycemic clamp. | It could improve the early detection of subjects susceptible to developing metabolic disorders | Studies are needed to validate the impact of VAT dysfunction on metabolic flexibility |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santillana, N.; Astudillo-Guerrero, C.; D’Espessailles, A.; Cruz, G. White Adipose Tissue Dysfunction: Pathophysiology and Emergent Measurements. Nutrients 2023, 15, 1722. https://doi.org/10.3390/nu15071722
Santillana N, Astudillo-Guerrero C, D’Espessailles A, Cruz G. White Adipose Tissue Dysfunction: Pathophysiology and Emergent Measurements. Nutrients. 2023; 15(7):1722. https://doi.org/10.3390/nu15071722
Chicago/Turabian StyleSantillana, Natalia, Camila Astudillo-Guerrero, Amanda D’Espessailles, and Gonzalo Cruz. 2023. "White Adipose Tissue Dysfunction: Pathophysiology and Emergent Measurements" Nutrients 15, no. 7: 1722. https://doi.org/10.3390/nu15071722