

The Protective Effect of 11-Keto-β-Boswellic Acid against Diabetic Cardiomyopathy in Rats Entails Activation of AMPK
 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Chemicals and Drugs
2.3. STZ-Induced T1DM
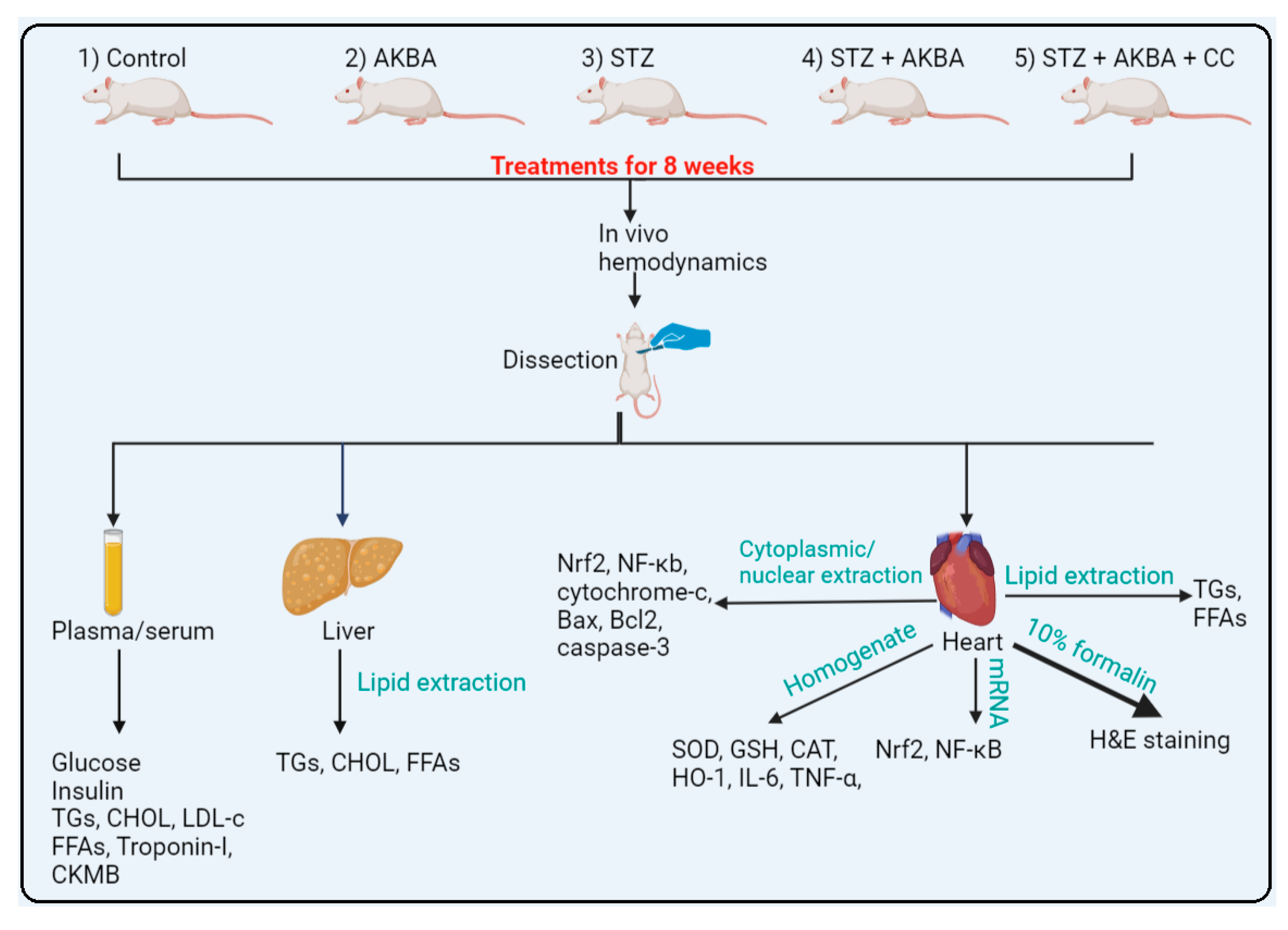
2.4. Experimental Design
2.5. Regimen and Dose Selections
2.6. Collection of Plasma and Biochemical Measurement
2.7. Measurement of Cardiac Function
2.8. LV Collection and Processing
2.9. Serum and LV Lipid Measurement
2.10. Analyses in LV Tissue
2.11. Measurement of the Mitochondria Permeability Transition Pore Potential (mtPTP) Opening
2.12. Real-Time PCR
2.13. Histopathological Evaluation
2.14. Statistical Analysis
3. Results
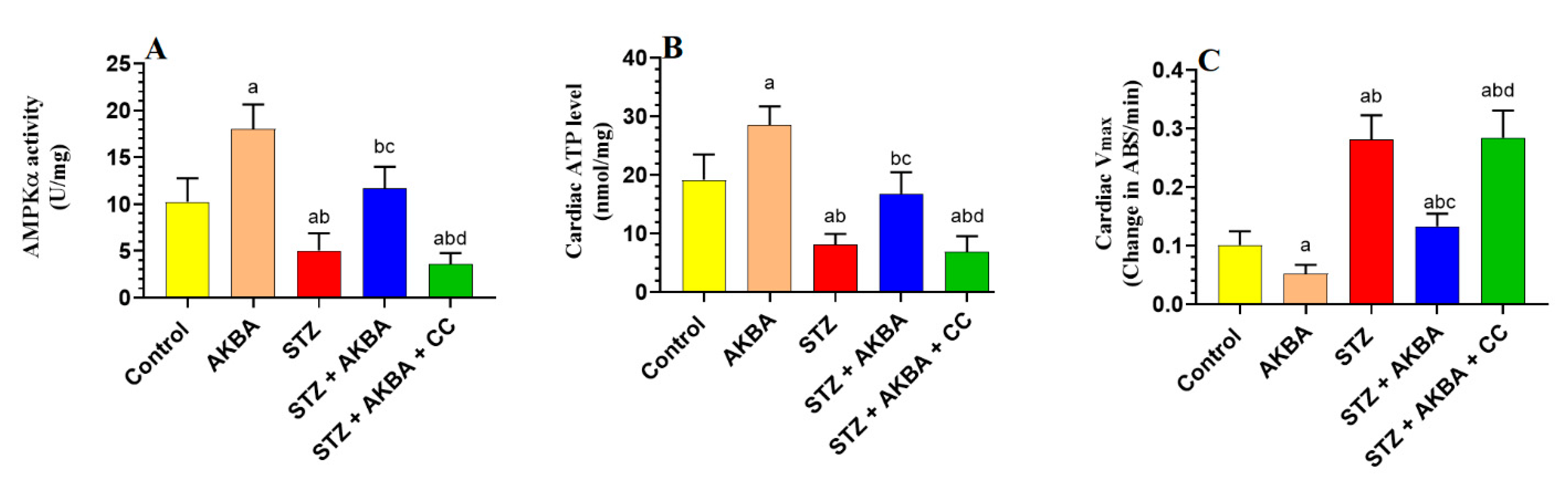
3.1. AKBA Activated AMPK, Improved Mitochondrial Function, and Increased ATP in the LVs of STZ-Diabetic Hearts
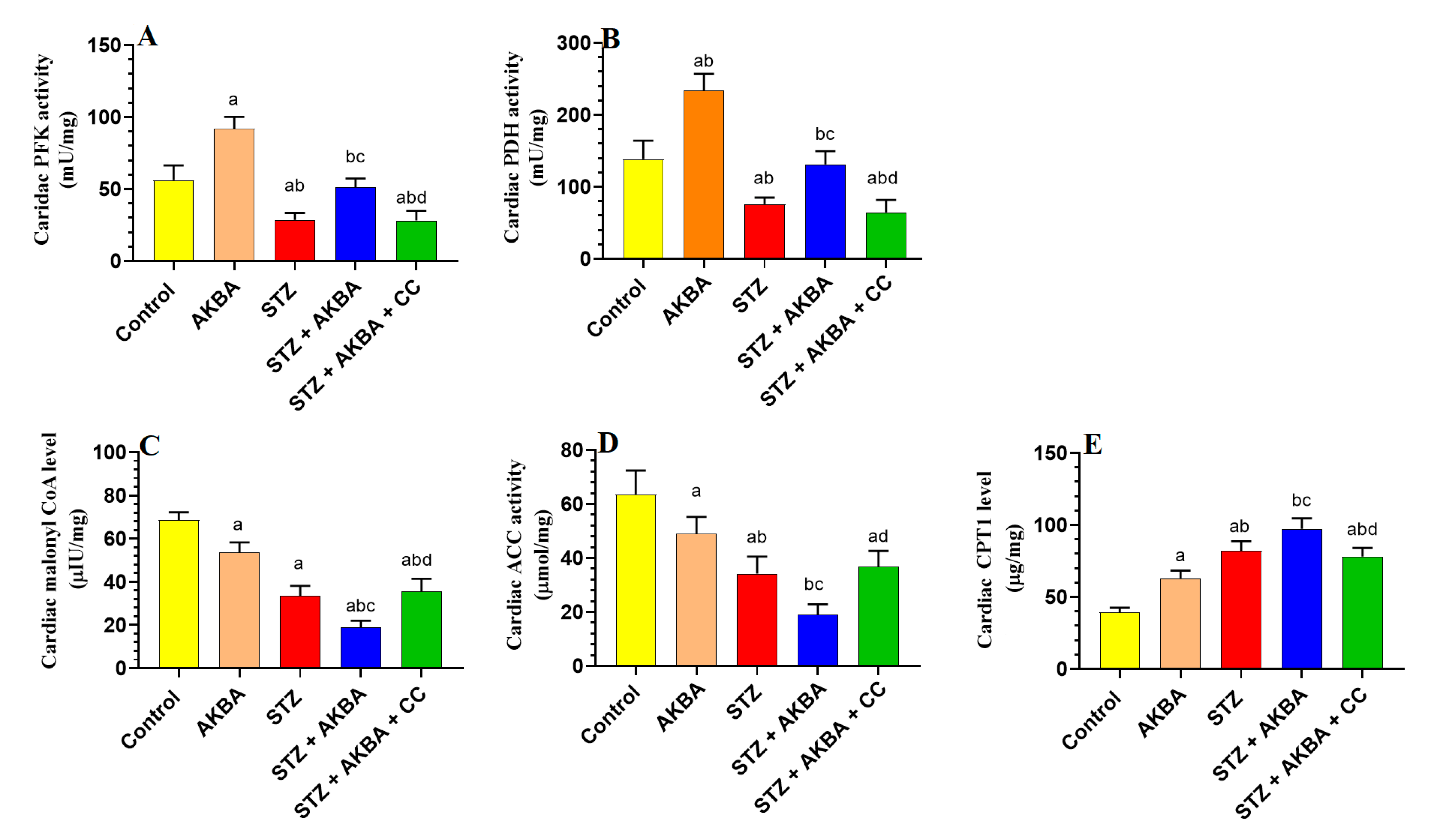
3.2. AKBA Stimulated Glycolysis and FA Oxidation in the LVs of STZ-Diabetic Rats by Stimulating AMPK
3.3. AKBA Attenuated Weight Loss, Hyperglycemia, Hyperlipidemia, and Lipid Accumulation in the Heart and Liver of STZ Rats by Activating AMPK
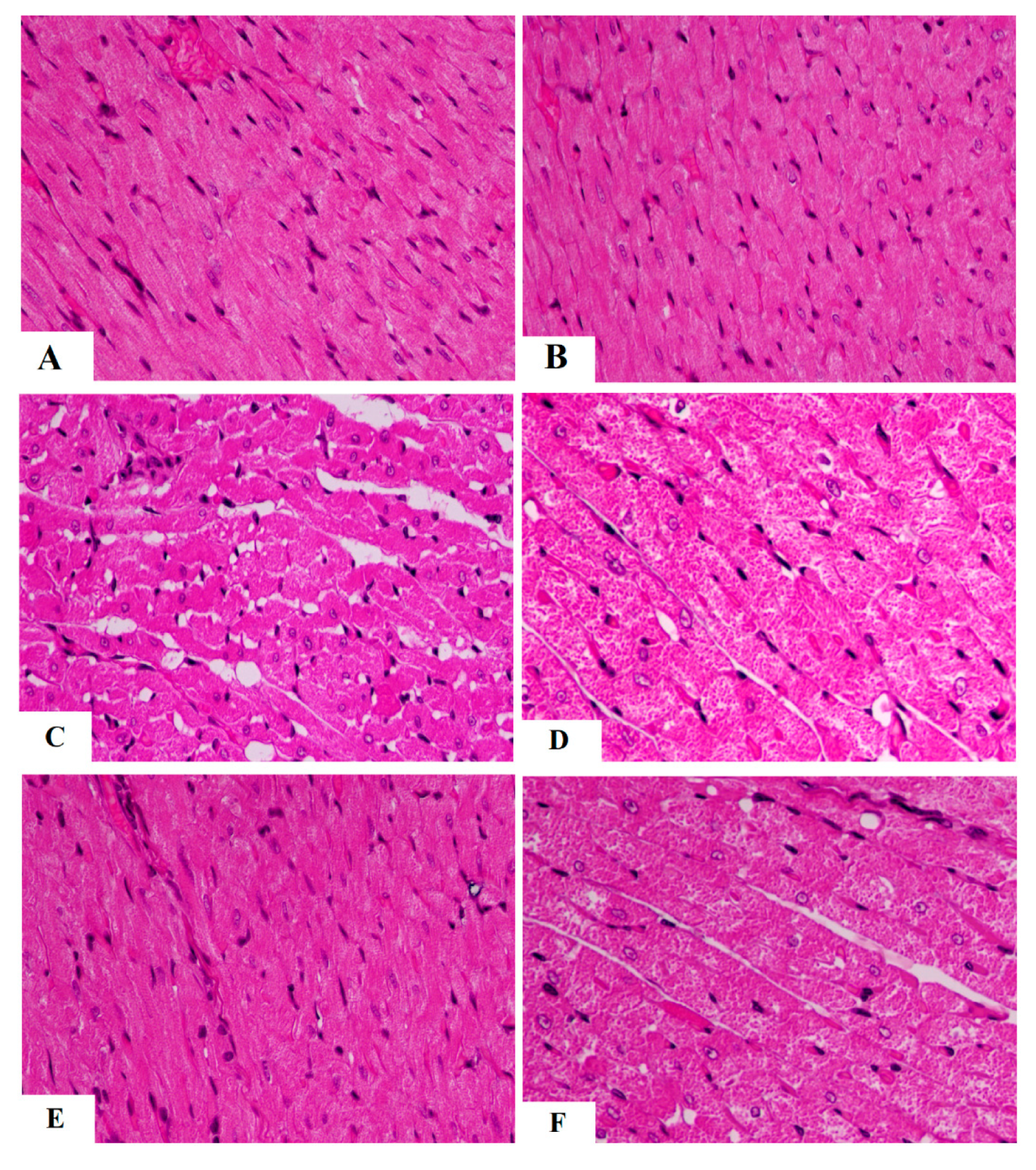
3.4. The Protective Effect of AKBA on Cardiac Structure and Function Required the Activation of AMPK
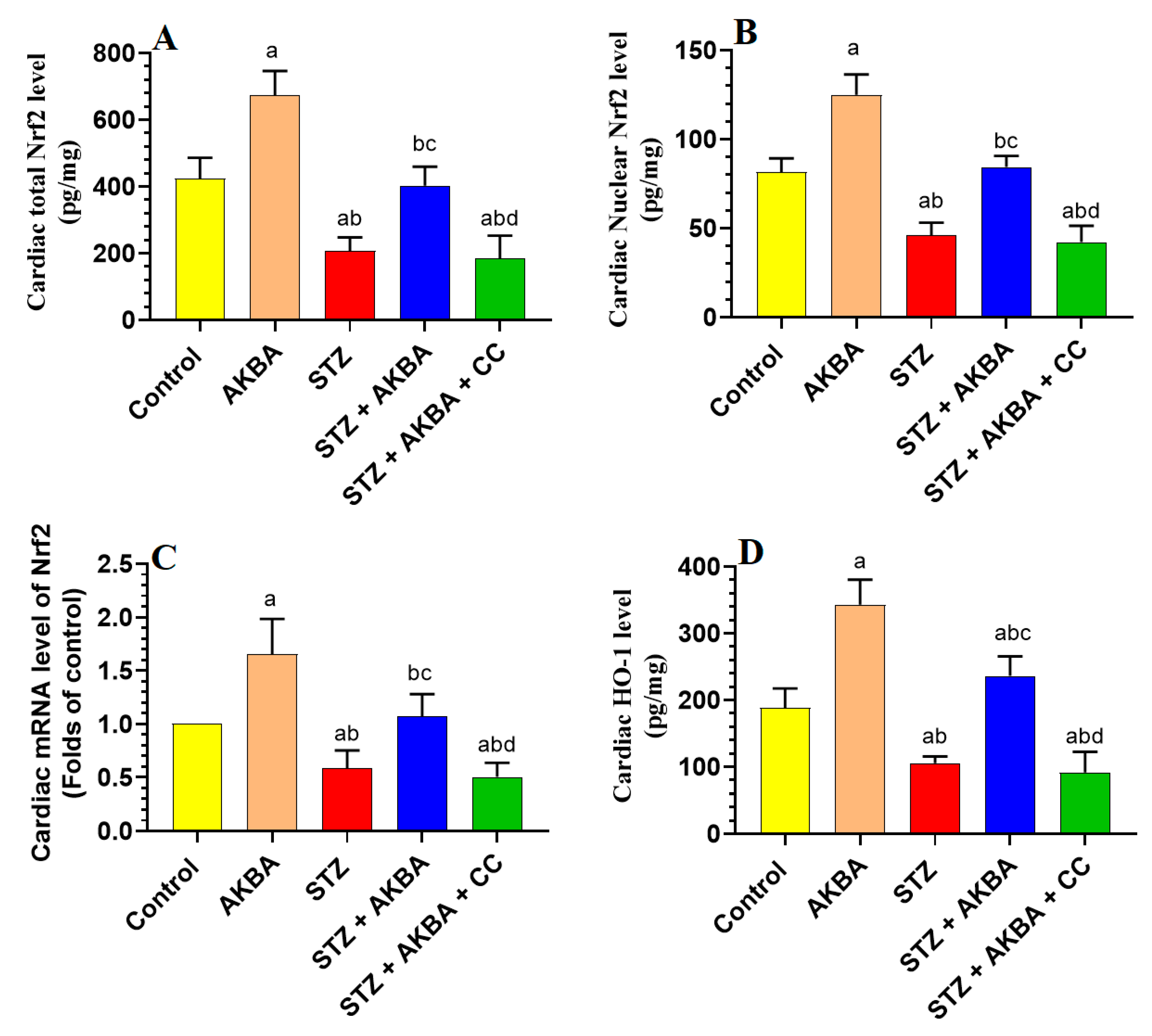
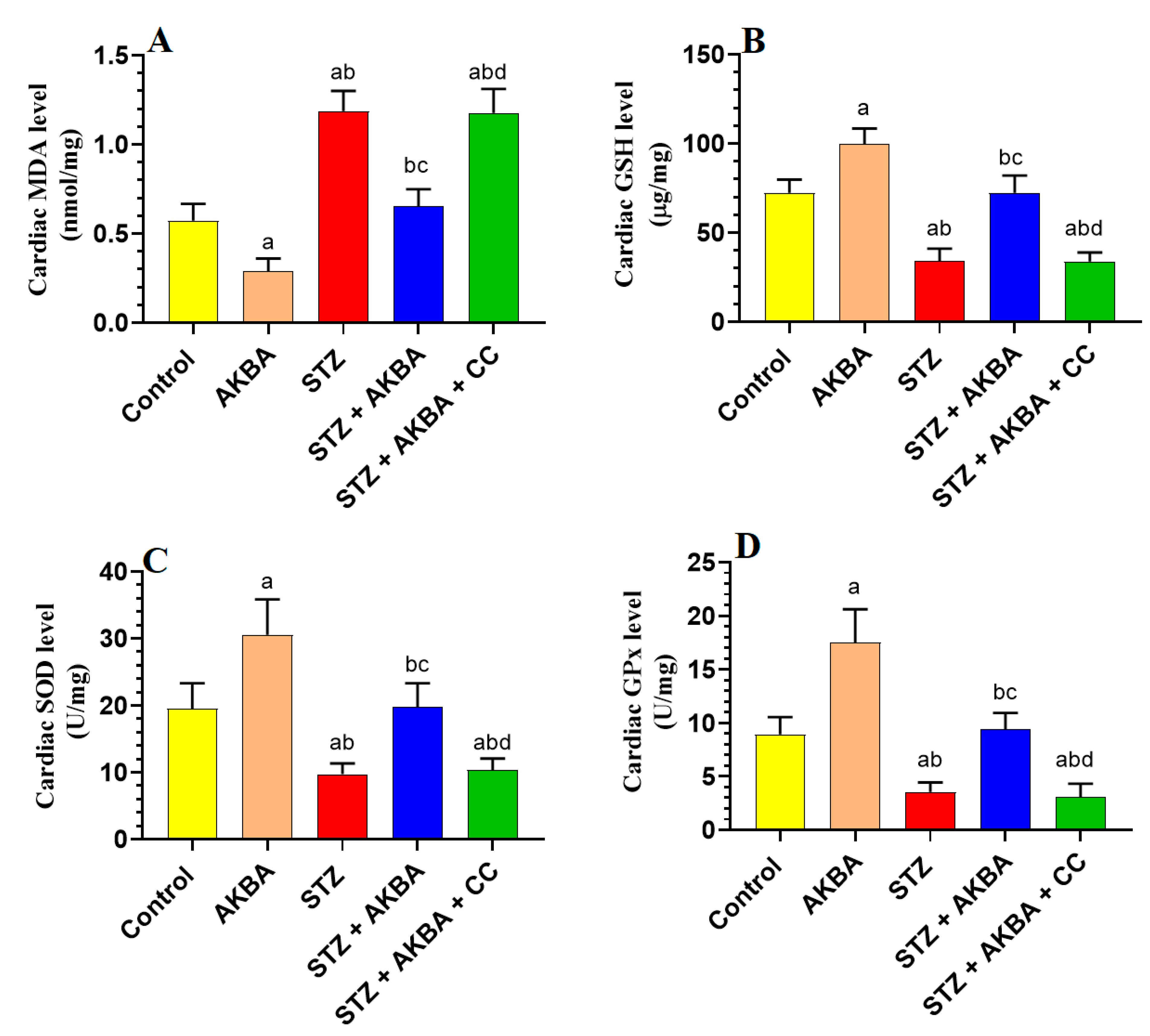
3.5. AKBA Stimulated the Nrf2/Antioxidant Axis in the LV of the Control and STZ-Diabetic Rats in an AMPK-Dependent Manner
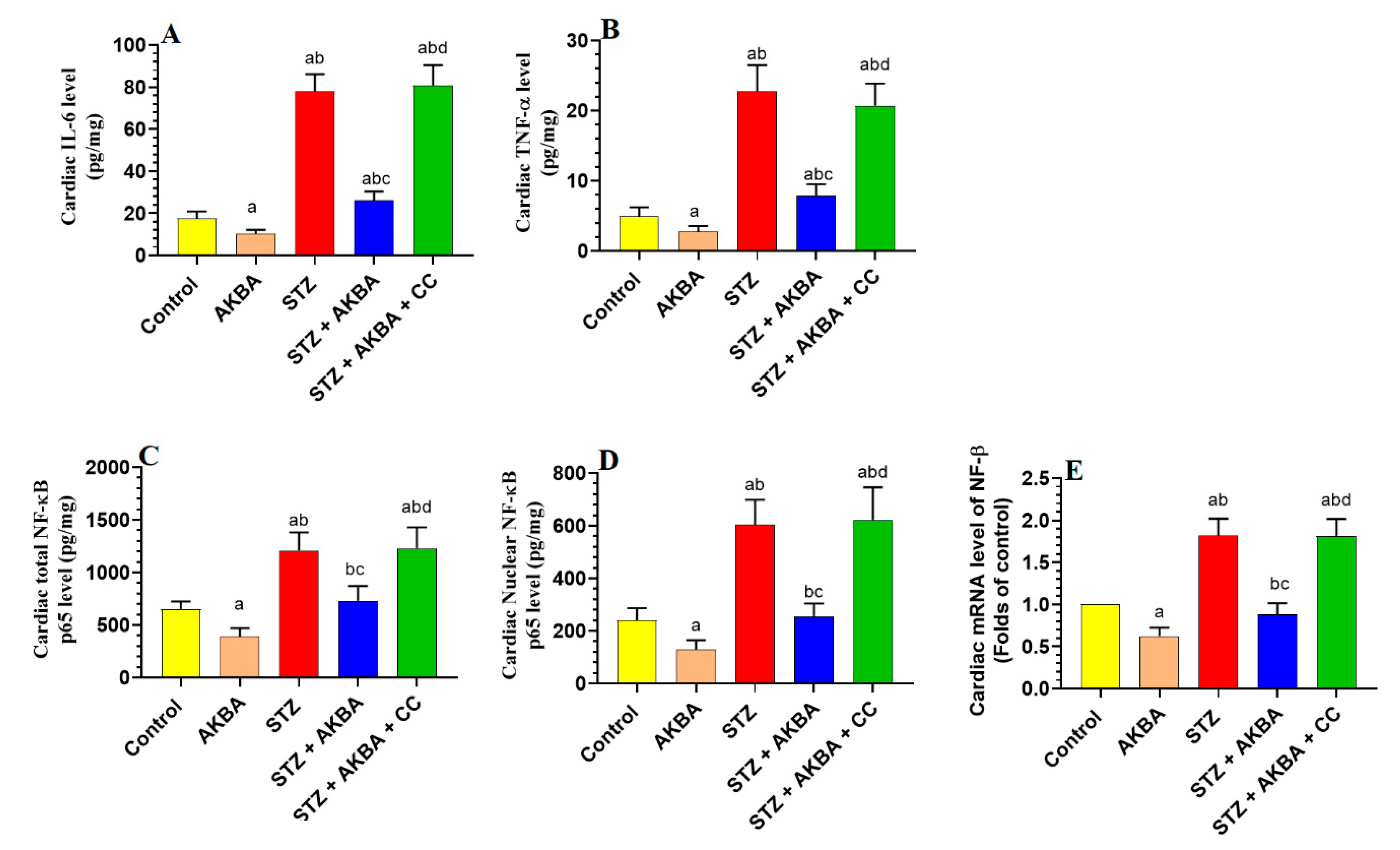
3.6. AKBA Suppressed NF-κB p65 and Cytokines Levels in the LVs of the Control and STZ-Diabetic Rats in an AMPK-Dependent Manner
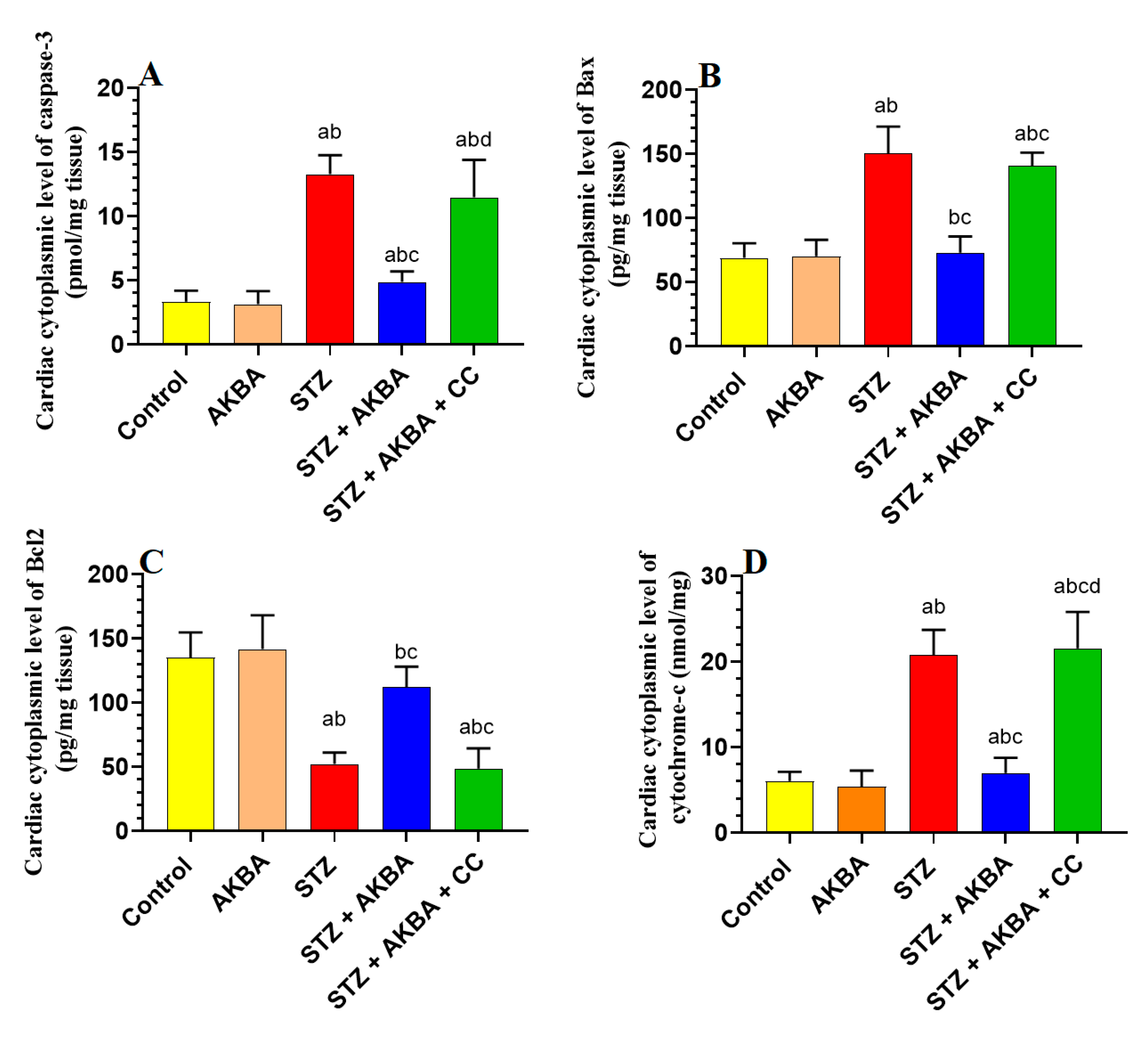
3.7. AKBA Inhibited Intrinsic Cell Death in the LV of STZ-Diabetic Rats in an AMPK-Dependent Manner
4. Discussion
Conclusions, Study Limitations, and Future Studies
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ritterhoff, J.; Tian, R. Metabolism in cardiomyopathy: Every substrate matters. Cardiovasc. Res. 2017, 113, 411–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, T.; Sano, M. Deranged Myocardial Fatty Acid Metabolism in Heart Failure. Int. J. Mol. Sci. 2022, 23, 996. [Google Scholar] [CrossRef] [PubMed]
- Abel, E.D. Insulin signaling in the heart. Am. J. Physiol.-Endocrinol. Metab. 2021, 321, E130–E145. [Google Scholar] [CrossRef]
- An, D.; Rodrigues, B. Role of changes in cardiac metabolism in development of diabetic cardiomyopathy. Am. J. Physiol.-Heart Circ. Physiol. 2006, 291, H1489–H1506. [Google Scholar] [CrossRef] [Green Version]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Liu, S.; Wang, X.; Chen, Y.; Pang, P.; Yang, Q.; Lin, J.; Deng, S.; Wu, S.; Fan, G. Diabetic cardiomyopathy: Clinical phenotype and practice. Front. Endocrinol. 2022, 13, 1032268. [Google Scholar] [CrossRef]
- Tan, Y.; Zhang, Z.; Zheng, C.; Wintergerst, K.A.; Keller, B.B.; Cai, L. Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: Preclinical and clinical evidence. Nat. Rev. Cardiol. 2020, 17, 585–607. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, S.; Cai, L. Diabetic cardiomyopathy and its mechanisms: Role of oxidative stress and damage. J. Diabetes Investig. 2014, 5, 623–634. [Google Scholar] [CrossRef]
- Kaludercic, N.; Di Lisa, F. Mitochondrial ROS formation in the pathogenesis of diabetic cardiomyopathy. Front. Cardiovasc. Med. 2020, 7, 12. [Google Scholar] [CrossRef] [Green Version]
- Karwi, Q.G.; Wagg, C.S.; Altamimi, T.R.; Uddin, G.M.; Ho, K.L.; Darwesh, A.M.; Seubert, J.M.; Lopaschuk, G.D. Insulin directly stimulates mitochondrial glucose oxidation in the heart. Cardiovasc. Diabetol. 2020, 19, 207. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.-L.; Fu, Y.; Wu, C.-W.; Zhang, Y.; Ren, H.; Zhou, S. Signaling pathways related to oxidative stress in diabetic cardiomyopathy. Front. Endocrinol. 2022, 13, 907757. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, M.Y.; Choi, H.K.; Hwang, J.T. AMPK Activity: A Primary Target for Diabetes Prevention with Therapeutic Phytochemicals. Nutrients. 2021, 13, 4050. [Google Scholar] [CrossRef]
- Jeon, S.M. Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 2016, 48, e245. [Google Scholar] [CrossRef] [Green Version]
- Trefts, E.; Shaw, R.J. AMPK: Restoring metabolic homeostasis over space and time. Mol. Cell 2021, 81, 3677–3690. [Google Scholar] [CrossRef]
- Bairwa, S.C.; Parajuli, N.; Dyck, J.R. The role of AMPK in cardiomyocyte health and survival. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2016, 1862, 2199–2210. [Google Scholar] [CrossRef]
- Wu, S.; Zou, M.-H. AMPK, mitochondrial function, and cardiovascular disease. Int. J. Mol. Sci. 2020, 21, 4987. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Qin, Y.; Wang, Y.; Meng, S.; Xian, H.; Che, H.; Lv, J.; Li, Y.; Yu, Y.; Bai, Y. Metformin inhibits the NLRP3 inflammasome via AMPK/mTOR-dependent effects in diabetic cardiomyopathy. Int. J. Biol. Sci. 2019, 15, 1010–1019. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Dong, S.; Xu, Q.; Shi, B.; Li, L.; Zhang, W.; Zhu, J.; Cheng, Y.; Zhang, G.; Zhong, M. Sleeve Gastrectomy-Induced AMPK Activation Attenuates Diabetic Cardiomyopathy by Maintaining Mitochondrial Homeostasis via NR4A1 Suppression in Rats. Front. Physiol. 2022, 13, 837798. [Google Scholar] [CrossRef]
- Sung, M.M.; Zordoky, B.N.; Bujak, A.L.; Lally, J.S.; Fung, D.; Young, M.E.; Horman, S.; Miller, E.J.; Light, P.E.; Kemp, B.E. AMPK deficiency in cardiac muscle results in dilated cardiomyopathy in the absence of changes in energy metabolism. Cardiovasc. Res. 2015, 107, 235–245. [Google Scholar] [CrossRef] [Green Version]
- Tokarska-Schlattner, M.; Kay, L.; Perret, P.; Isola, R.; Attia, S.; Lamarche, F.; Tellier, C.; Cottet-Rousselle, C.; Uneisi, A.; Hininger-Favier, I. Role of Cardiac AMP-Activated Protein Kinase in a Non-pathological Setting: Evidence From Cardiomyocyte-Specific, Inducible AMP-Activated Protein Kinase α1α2-Knockout Mice. Front. Cell Dev. Biol. 2021, 9, 2903. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; He, C.; Zou, M.-H. AMP-activated protein kinase modulates cardiac autophagy in diabetic cardiomyopathy. Autophagy 2011, 7, 1254–1255. [Google Scholar] [CrossRef] [Green Version]
- Kumar, J.; Gupta, D.S.; Gupta, S.; Dubey, S.; Gupta, P.; Kumar, S. Quantitative trait loci from identification to exploitation for crop improvement. Plant Cell Rep. 2017, 36, 1187–1213. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Feng, A.; Lin, S.; Yu, L.; Lin, X.; Yan, X.; Lu, X.; Zhang, C. Fibroblast growth factor-21 prevents diabetic cardiomyopathy via AMPK-mediated antioxidation and lipid-lowering effects in the heart. Cell Death Dis. 2018, 9, 227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, W.; Bai, T.; Zeng, J.; Niu, Z.; Fan, D.; Xu, X.; Luo, M.; Wang, P.; Zou, Q.; Dai, X. Combined administration of metformin and atorvastatin attenuates diabetic cardiomyopathy by inhibiting inflammation, apoptosis, and oxidative stress in type 2 diabetic mice. Front. Cell Dev. Biol. 2021, 9, 634900. [Google Scholar] [CrossRef]
- Jubaidi, F.F.; Zainalabidin, S.; Taib, I.S.; Hamid, Z.A.; Budin, S.B. The potential role of flavonoids in ameliorating diabetic cardiomyopathy via alleviation of cardiac oxidative stress, inflammation and apoptosis. Int. J. Mol. Sci. 2021, 22, 5094. [Google Scholar] [CrossRef] [PubMed]
- Moussaieff, A.; Mechoulam, R. Boswellia resin: From religious ceremonies to medical uses; a review of in-vitro, in-vivo and clinical trials. J. Pharm. Pharmacol. 2009, 61, 1281–1293. [Google Scholar] [CrossRef]
- Roy, N.K.; Parama, D.; Banik, K.; Bordoloi, D.; Devi, A.K.; Thakur, K.K.; Padmavathi, G.; Shakibaei, M.; Fan, L.; Sethi, G. An update on pharmacological potential of boswellic acids against chronic diseases. Int. J. Mol. Sci. 2019, 20, 4101. [Google Scholar] [CrossRef] [Green Version]
- Shang, P.; Liu, W.; Liu, T.; Zhang, Y.; Mu, F.; Zhu, Z.; Liang, L.; Zhai, X.; Ding, Y.; Li, Y. Acetyl-11-keto-β-boswellic acid attenuates prooxidant and profibrotic mechanisms involving transforming growth factor-β1, and improves vascular remodeling in spontaneously hypertensive rats. Sci. Rep. 2016, 6, 39809. [Google Scholar] [CrossRef] [Green Version]
- Lv, M.; Shao, S.; Zhang, Q.; Zhuang, X.; Qiao, T. Acetyl-11-Keto-β-Boswellic acid exerts the anti-cancer effects via cell cycle arrest, apoptosis induction and autophagy suppression in non-small cell lung cancer cells. OncoTargets Ther. 2020, 13, 733. [Google Scholar] [CrossRef] [Green Version]
- Marefati, N.; Beheshti, F.; Memarpour, S.; Bayat, R.; Shafei, M.N.; Sadeghnia, H.R.; Ghazavi, H.; Hosseini, M. The effects of acetyl-11-keto-β-boswellic acid on brain cytokines and memory impairment induced by lipopolysaccharide in rats. Cytokine 2020, 131, 155107. [Google Scholar] [CrossRef]
- Gong, Y.; Jiang, X.; Yang, S.; Huang, Y.; Hong, J.; Ma, Y.; Fang, X.; Fang, Y.; Wu, J. The Biological Activity of 3-O-Acetyl-11-keto-β-Boswellic Acid in Nervous System Diseases. Neuromol. Med. 2022, 24, 374–384. [Google Scholar] [CrossRef]
- Taherzadeh, D.; Baradaran Rahimi, V.; Amiri, H.; Ehtiati, S.; Yahyazadeh, R.; Hashemy, S.I.; Askari, V.R. Acetyl-11-Keto-β-Boswellic acid (AKBA) prevents lipopolysaccharide-induced inflammation and cytotoxicity on H9C2 cells. Evid. Based Complement. Alternat. Med. 2022, 2022, 2620710. [Google Scholar] [CrossRef] [PubMed]
- Ammon, H. Boswellic extracts and 11-keto-ß-boswellic acids prevent type 1 and type 2 diabetes mellitus by suppressing the expression of proinflammatory cytokines. Phytomedicine 2019, 63, 153002. [Google Scholar] [CrossRef] [PubMed]
- Gomaa, A.A.; Farghaly, H.A.; Abdel-Wadood, Y.A.; Gomaa, G.A. Potential therapeutic effects of boswellic acids/Boswellia serrata extract in the prevention and therapy of type 2 diabetes and Alzheimer’s disease. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2021, 394, 2167–2185. [Google Scholar] [CrossRef] [PubMed]
- Kherouf, A.; Aouacheri, O.; Tichati, L.; Tebboub, I.; Kherouf, M.; Saka, S. Potential antioxidant properties and anti-diabetic and hepatic/pancreatic protective effects of dietary Boswellia serrata gum resin powder against oxidative damage in streptozotocin-induced diabetic rats. Comp. Clin. Path. 2021, 30, 891–904. [Google Scholar] [CrossRef]
- Khan, A.; Khan, I.; Halim, S.A.; Rehman, N.U.; Karim, N.; Ahmad, W.; Khan, M.; Csuk, R.; Al-Harrasi, A. Anti-diabetic potential of β-boswellic acid and 11-keto-β-boswellic acid: Mechanistic insights from computational and biochemical approaches. Biomed. Pharmacother. 2022, 147, 112669. [Google Scholar] [CrossRef]
- Umemneku-Chikere, C.M.; Ayodele, O.; Soares, M.; Khan, S.; Abrams, K.; Owen, R.; Bujkiewicz, S. Comparative review of pharmacological therapies in individuals with HER2-positive advanced breast cancer with focus on hormone receptor subgroups. Front. Oncol. 2022, 12, 4191. [Google Scholar] [CrossRef]
- Shadfar, S.; Khanal, S.; Bohara, G.; Kim, G.; Sadigh-Eteghad, S.; Ghavami, S.; Choi, H.; Choi, D.-Y. Methanolic extract of Boswellia serrata gum protects the nigral dopaminergic neurons from rotenone-induced neurotoxicity. Mol. Neurobiol. 2022, 59, 5874–5890. [Google Scholar] [CrossRef]
- Ali, T.M.; Abo-Salem, O.M.; El Esawy, B.H.; El Askary, A. The Potential Protective Effects of Diosmin on Streptozotocin-Induced Diabetic Cardiomyopathy in Rats. Am. J. Med. Sci. 2020, 359, 32–41. [Google Scholar] [CrossRef]
- Chen, X.; Yun, C.; Zheng, H.; Chen, X.; Han, Q.; Pan, H.; Wang, Y.; Zhong, J. The protective effects of S14G-humanin (HNG) against streptozotocin (STZ)-induced cardiac dysfunction. Bioengineered. 2021, 12, 5491–5503. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Peng, J.; Feng, H.; Yang, Y.; Gao, J.; Liu, C.; Xu, J.; Zhao, Y.; Pan, S.; Wang, Y.; et al. KLF9 Aggravates Streptozotocin-Induced Diabetic Cardiomyopathy by Inhibiting PPARγ/NRF2 Signalling. Cells 2022, 11, 3393. [Google Scholar] [CrossRef] [PubMed]
- National Research Council (US) Institute for Laboratory Animal Research. Guide for the Care and Use of Laboratory Animals; National Academies Press (US): Washington, DC, USA, 1996; ISBN -10 0-309-05377-3. [Google Scholar]
- Altamimi, J.; Alfaris, N.; Alshammari, G.; Alagal, R.; Aljabryn, D.; Aldera, H.; Alkhateeb, M.; Yahya, M. Ellagic acid protects against diabetic cardiomyopathy in rats by stimulating cardiac silent information regulator 1 signaling. J. Physiol. Pharmacol. 2020, 71, 891–904. [Google Scholar]
- Albasher, G.; Alkahtani, S.; Al-Harbi, L.N. Urolithin A prevents streptozotocin-induced diabetic cardiomyopathy in rats by activating SIRT1. Saudi J. Biol. Sci. 2022, 29, 1210–1220. [Google Scholar] [CrossRef]
- Almohawes, Z.N.; El-Kott, A.; Morsy, K.; Shati, A.A.; El-Kenawy, A.E.; Khalifa, H.S.; Elsaid, F.G.; Abd-Lateif, A.-E.-K.M.; Abu-Zaiton, A.; Ebealy, E.R. Salidroside inhibits insulin resistance and hepatic steatosis by downregulating miR-21 and subsequent activation of AMPK and upregulation of PPARα in the liver and muscles of high fat diet-fed rats. Arch. Physiol. Biochem. 2022, 1–18. [Google Scholar] [CrossRef]
- Yahya, M.A.; Alshammari, G.M.; Osman, M.A.; Al-Harbi, L.N.; Yagoub, A.E.A.; AlSedairy, S.A. Liquorice root extract and isoliquiritigenin attenuate high-fat diet-induced hepatic steatosis and damage in rats by regulating AMPK. Arch. Physiol. Biochem. 2022, 1–16. [Google Scholar] [CrossRef]
- Eid, R.A.; Alkhateeb, M.A.; Al-Shraim, M.; Eleawa, S.M.; Shatoor, A.S.; El-Kott, A.F.; Zaki, M.S.A.; Shatoor, K.A.; Bin-Jaliah, I.; Al-Hashem, F.H. Ghrelin prevents cardiac cell apoptosis during cardiac remodelling post experimentally induced myocardial infarction in rats via activation of Raf-MEK1/2-ERK1/2 signalling. Arch. Physiol. Biochem. 2019, 125, 93–103. [Google Scholar] [CrossRef]
- Eid, R.A.; Khalil, M.A.; Alkhateeb, M.A.; Eleawa, S.M.; Zaki, M.S.A.; El-Kott, A.F.; Al-Shraim, M.; El-Sayed, F.; Eldeen, M.A.; Bin-Meferij, M.M. Exendin-4 attenuates remodeling in the remote myocardium of rats after an acute myocardial infarction by activating β-arrestin-2, protein phosphatase 2A, and glycogen synthase kinase-3 and inhibiting β-catenin. Cardiovasc. Drugs Ther. 2021, 35, 1095–1110. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipids from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]
- Dedkova, E.N.; Blatter, L.A. Measuring mitochondrial function in intact cardiac myocytes. J. Mol. Cell Cardiol. 2012, 52, 48–61. [Google Scholar] [CrossRef] [Green Version]
- Carnevali, L.; Mastorci, F.; Graiani, G.; Razzoli, M.; Trombini, M.; Pico-Alfonso, M.-A.; Arban, R.; Grippo, A.J.; Quaini, F.; Sgoifo, A. Social defeat and isolation induce clear signs of a depression-like state, but modest cardiac alterations in wild-type rats. Physiol. Behav. 2012, 106, 142–150. [Google Scholar] [CrossRef]
- Jia, G.; Whaley-Connell, A.; Sowers, J.R. Diabetic cardiomyopathy: A hyperglycaemia- and insulin-resistance-induced heart disease. Diabetologia 2018, 61, 21–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, U.; Chakraborty, M.; Chutia, D.; Bhuyan, N.R. Cellular and molecular mechanisms, genetic predisposition and treatment of diabetes-induced cardiomyopathy. Curr. Res. Pharmacol. Drug Discov. 2022, 3, 100126. [Google Scholar] [CrossRef] [PubMed]
- Adeghate, E.; Singh, J. Structural changes in the myocardium during diabetes-induced cardiomyopathy. Heart Fail Rev. 2014, 19, 15–23. [Google Scholar] [CrossRef]
- Nakamura, K.; Miyoshi, T.; Yoshida, M.; Akagi, S.; Saito, Y.; Ejiri, K.; Matsuo, N.; Ichikawa, K.; Iwasaki, K.; Naito, T.; et al. Pathophysiology and Treatment of Diabetic Cardiomyopathy and Heart Failure in Patients with Diabetes Mellitus. Int. J. Mol. Sci. 2022, 23, 3587. [Google Scholar] [CrossRef]
- Makino, A.; Dai, A.; Han, Y.; Youssef, K.D.; Wang, W.; Donthamsetty, R.; Scott, B.T.; Wang, H.; Dillmann, W.H. O-GlcNAcase overexpression reverses coronary endothelial cell dysfunction in type 1 diabetic mice. Am. J. Physiol. Cell Physiol. 2015, 309, C593-9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, J.; Pan, J.; Lin, H.; Gu, J. Diabetic cardiomyopathy: A brief summary on lipid toxicity. ESC Heart Fail. 2022. [Google Scholar] [CrossRef]
- Levelt, E.; Gulsin, G.; Neubauer, S.; McCann, G.P. Mechanisms in Endocrinology: Diabetic cardiomyopathy: Pathophysiology and potential metabolic interventions state of the art review. Eur. J. Endocrinol. 2018, 178, R127–R139. [Google Scholar] [CrossRef] [Green Version]
- Hatting, M.; Tavares, C.D.J.; Sharabi, K.; Rines, A.K.; Puigserver, P. Insulin regulation of gluconeogenesis. Ann. N. Y. Acad. Sci. 2018, 1411, 21–35. [Google Scholar] [CrossRef]
- Zheng, T.; Yang, X.; Li, W.; Wang, Q.; Chen, L.; Wu, D.; Bian, F.; Xing, S.; Jin, S. Salidroside Attenuates High-Fat Diet-Induced Nonalcoholic Fatty Liver Disease via AMPK-Dependent TXNIP/NLRP3 Pathway. Oxidative Med. Cell. Longev. 2018, 2018, 8597897. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Jian, T.; Li, J.; Lv, H.; Tong, B.; Li, J.; Meng, X.; Ren, B.; Chen, J. Chicoric Acid Ameliorates Nonalcoholic Fatty Liver Disease via the AMPK/Nrf2/NFκB Signaling Pathway and Restores Gut Microbiota in High-Fat-Diet-Fed Mice. Oxidative Med. Cell. Longev. 2020, 2020, 9734560. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.K.; Marcinko, K.; Desjardins, E.M.; Lally, J.S.; Ford, R.J.; Steinberg, G.R. Treatment of nonalcoholic fatty liver disease: Role of AMPK. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E730–E740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, F.L.; Huang, C.F.; Chen, Y.W.; Yen, Y.P.; Wu, C.T.; Uang, B.J.; Yang, R.S.; Liu, S.H. Antidiabetic effects of pterosin A, a small-molecular-weight natural product, on diabetic mouse models. Diabetes 2013, 62, 628–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, H.; Wu, H. Maslinic acid activates renal AMPK/SIRT1 signaling pathway and protects against diabetic nephropathy in mice. BMC Endocr. Disord. 2022, 22, 25. [Google Scholar] [CrossRef] [PubMed]
- Cool, B.; Zinker, B.; Chiou, W.; Kifle, L.; Cao, N.; Perham, M.; Dickinson, R.; Adler, A.; Gagne, G.; Iyengar, R.; et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006, 3, 403–416. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, H.M. AMPK and Exercise: Glucose Uptake and Insulin Sensitivity. Diabetes Metab. J. 2013, 37, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Habegger, K.M.; Hoffman, N.J.; Ridenour, C.M.; Brozinick, J.T.; Elmendorf, J.S. AMPK enhances insulin-stimulated GLUT4 regulation via lowering membrane cholesterol. Endocrinology 2012, 153, 2130–2141. [Google Scholar] [CrossRef]
- Lochhead, P.A.; Salt, I.P.; Walker, K.S.; Hardie, D.G.; Sutherland, C. 5-aminoimidazole-4-carboxamide riboside mimics the effects of insulin on the expression of the 2 key gluconeogenic genes PEPCK and glucose-6-phosphatase. Diabetes 2000, 49, 896–903. [Google Scholar] [CrossRef] [Green Version]
- Johanns, M.; Hue, L.; Rider, M.H. AMPK inhibits liver gluconeogenesis: Fact or fiction? Biochem. J. 2023, 480, 105–125. [Google Scholar] [CrossRef]
- Sansome, D.J.; Xie, C.; Veedfald, S.; Horowitz, M.; Rayner, C.K.; Wu, T. Mechanism of glucose-lowering by metformin in type 2 diabetes: Role of bile acids. Diabetes Obes. Metab. 2020, 22, 141–148. [Google Scholar] [CrossRef]
- Hasanvand, A.; Amini-Khoei, H.; Jahanabadi, S.; Hadian, M.; Abdollahi, A.; Tavangar, S.M.; Mehr, S.J.; Dehpour, A.R. Metformin attenuates streptozotocin-induced diabetic nephropathy in rats through activation of AMPK signaling pathway. J. Nephropathol. 2018, 7, 37–42. [Google Scholar] [CrossRef] [Green Version]
- Li, L.X.; Skorpen, F.; Egeberg, K.; Jorgensen, I.H.; Grill, V. Induction of uncoupling protein 2 mRNA in beta-cells is stimulated by oxidation of fatty acids but not by nutrient oversupply. Endocrinology 2002, 143, 1371–1377. [Google Scholar] [CrossRef] [PubMed]
- Nyblom, H.K.; Sargsyan, E.; Bergsten, P. AMP-activated protein kinase agonist dose dependently improves function and reduces apoptosis in glucotoxic beta-cells without changing triglyceride levels. J. Mol. Endocrinol. 2008, 41, 187–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, M.; Liu, Y.; Zhou, F.; Zhang, Y.; Zhu, Q.; Zhang, L.; Zhang, Q.; Wang, S.; Zhu, K.; Wang, X.; et al. Berberine inhibits glucose oxidation and insulin secretion in rat islets. Endocr. J. 2018, 65, 469–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villanueva-Paz, M.; Cotán, D.; Garrido-Maraver, J.; Oropesa-Ávila, M.; Mata, M.d.l.; Delgado-Pavón, A.; Lavera, I.d.; Alcocer-Gómez, E.; Álvarez-Córdoba, M.; Sánchez-Alcázar, J.A. AMPK regulation of cell growth, apoptosis, autophagy, and bioenergetics. AMP-Act. Protein Kinase 2016, 107, 45–71. [Google Scholar]
- Brahma, M.K.; Pepin, M.E.; Wende, A.R. My sweetheart is broken: Role of glucose in diabetic cardiomyopathy. Diabetes Metab. J. 2017, 41, 1–9. [Google Scholar] [CrossRef]
- Lee, J.O.; Lee, S.K.; Kim, J.H.; Kim, N.; You, G.Y.; Moon, J.W.; Kim, S.J.; Park, S.H.; Kim, H.S. Metformin regulates glucose transporter 4 (GLUT4) translocation through AMP-activated protein kinase (AMPK)-mediated Cbl/CAP signaling in 3T3-L1 preadipocyte cells. J. Biol. Chem. 2012, 287, 44121–44129. [Google Scholar] [CrossRef] [Green Version]
- Marsin, A.-S.; Bertrand, L.; Rider, M.H.; Deprez, J.; Beauloye, C.; Vincent, M.F.; Van den Berghe, G.; Carling, D.; Hue, L. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr. Biol. 2000, 10, 1247–1255. [Google Scholar] [CrossRef] [Green Version]
- Kewalramani, G.; Rodrigues, B. AMP-activated protein kinase in the heart: Role in cardiac glucose and fatty acid metabolism. Clin. Lipidol. 2009, 4, 643–661. [Google Scholar] [CrossRef]
- Kishton, R.J.; Barnes, C.E.; Nichols, A.G.; Cohen, S.; Gerriets, V.A.; Siska, P.J.; Macintyre, A.N.; Goraksha-Hicks, P.; de Cubas, A.A.; Liu, T. AMPK is essential to balance glycolysis and mitochondrial metabolism to control T-ALL cell stress and survival. Cell Metab. 2016, 23, 649–662. [Google Scholar] [CrossRef] [Green Version]
- Nellaiappan, K.; Yerra, V.G.; Kumar, A. Role of AMPK in diabetic cardiovascular complications: An overview. Cardiovasc. Haematol. Disord.-Drug Targets 2019, 19, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Volpe, C.M.O.; Villar-Delfino, P.H.; Dos Anjos, P.M.F.; Nogueira-Machado, J.A. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018, 9, 119. [Google Scholar] [CrossRef] [Green Version]
- De Geest, B.; Mishra, M. Role of oxidative stress in diabetic cardiomyopathy. Antioxidants 2022, 11, 784. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, Z.; Cai, L. Diabetic cardiomyopathy and its prevention by nrf2: Current status. Diabetes Metab. J. 2014, 38, 337–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.-M.; Desai, L.P. Reciprocal regulation of TGF-β and reactive oxygen species: A perverse cycle for fibrosis. Redox Biol. 2015, 6, 565–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, O.; Picatoste, B.; Ares-Carrasco, S.; Ramírez, E.; Egido, J.; Tuñón, J. Potential role of nuclear factor κB in diabetic cardiomyopathy. Mediators Inflamm. 2011, 2011, 652097. [Google Scholar] [CrossRef] [Green Version]
- Raish, M.; Ahmad, A.; Bin Jardan, Y.A.; Shahid, M.; Alkharfy, K.M.; Ahad, A.; Ansari, M.A.; Abdelrahman, I.A.; Al-Jenoobi, F.I. Sinapic acid ameliorates cardiac dysfunction and cardiomyopathy by modulating NF-κB and Nrf2/HO-1 signaling pathways in streptozocin induced diabetic rats. Biomed. Pharmacother. 2022, 145, 112412. [Google Scholar] [CrossRef]
- Barakat, B.M.; Ahmed, H.I.; Bahr, H.I.; Elbahaie, A.M. Protective effect of boswellic acids against doxorubicin-induced hepatotoxicity: Impact on Nrf2/HO-1 defense pathway. Oxidative Med. Cell. Longev. 2018, 2018, 8296451. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Zhou, B.; Xu, W.; Xue, F.; Nisar, M.F.; Bian, C.; Huang, X.; Yang, L.; Zhang, Y.; Bartsch, J.W. Nrf2-and Bach1 may play a role in the modulation of ultraviolet A-induced oxidative stress by acetyl-11-keto-β-boswellic acid in skin keratinocytes. Ski. Pharmacol. Physiol. 2017, 30, 13–23. [Google Scholar] [CrossRef]
- Wei, C.; Fan, J.; Sun, X.; Yao, J.; Guo, Y.; Zhou, B.; Shang, Y. Acetyl-11-keto-β-boswellic acid ameliorates cognitive deficits and reduces amyloid-β levels in APPswe/PS1dE9 mice through antioxidant and anti-inflammatory pathways. Free Radic. Biol. Med. 2020, 150, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Lin, X.; Li, H.; Zhou, X.; Fan, F.; Yang, J.; Luo, Y.; Liu, X. Acetyl-11-Keto-Beta Boswellic Acid (AKBA) Protects Lens Epithelial Cells Against H2O2-Induced Oxidative Injury and Attenuates Cataract Progression by Activating Keap1/Nrf2/HO-1 Signaling. Front. Pharmacol. 2022, 13, 927871. [Google Scholar] [CrossRef]
- Syrovets, T.; Büchele, B.; Krauss, C.; Laumonnier, Y.; Simmet, T. Acetyl-boswellic acids inhibit lipopolysaccharide-mediated TNF-α induction in monocytes by direct interaction with IκB kinases. J. Immunol. 2005, 174, 498–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takada, Y.; Ichikawa, H.; Badmaev, V.; Aggarwal, B.B. Acetyl-11-keto-β-boswellic acid potentiates apoptosis, inhibits invasion, and abolishes osteoclastogenesis by suppressing NF-κB and NF-κB-regulated gene expression. J. Immunol. 2006, 176, 3127–3140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuaz-Pérolin, C.; Billiet, L.; Baugé, E.; Copin, C.; Scott-Algara, D.; Genze, F.; Büchele, B.; Syrovets, T.; Simmet, T.; Rouis, M. Antiinflammatory and antiatherogenic effects of the NF-κB inhibitor acetyl-11-keto-β-boswellic acid in LPS-challenged ApoE−/− mice. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 272–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Syrovets, T.; Kess, D.; Büchele, B.; Hainzl, H.; Lunov, O.; Weiss, J.M.; Scharffetter-Kochanek, K.; Simmet, T. Targeting NF-κB with a natural triterpenoid alleviates skin inflammation in a mouse model of psoriasis. J. Immunol. 2009, 183, 4755–4763. [Google Scholar] [CrossRef] [Green Version]
- Mo, C.; Wang, L.; Zhang, J.; Numazawa, S.; Tang, H.; Tang, X.; Han, X.; Li, J.; Yang, M.; Wang, Z. The crosstalk between Nrf2 and AMPK signal pathways is important for the anti-inflammatory effect of berberine in LPS-stimulated macrophages and endotoxin-shocked mice. Antioxid. Redox Signal. 2014, 20, 574–588. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Zhao, T.; Xiao, H. The implication of oxidative stress and AMPK-Nrf2 antioxidative signaling in pneumonia pathogenesis. Front. Endocrinol. 2020, 11, 400. [Google Scholar] [CrossRef]
- Zimmermann, K.; Baldinger, J.; Mayerhofer, B.; Atanasov, A.G.; Dirsch, V.M.; Heiss, E.H. Activated AMPK boosts the Nrf2/HO-1 signaling axis—A role for the unfolded protein response. Free Radic. Biol. Med. 2015, 88, 417–426. [Google Scholar] [CrossRef] [Green Version]
- Joo, M.S.; Kim, W.D.; Lee, K.Y.; Kim, J.H.; Koo, J.H.; Kim, S.G. AMPK facilitates nuclear accumulation of Nrf2 by phosphorylating at serine 550. Mol. Cell. Biol. 2016, 36, 1931–1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salminen, A.; Hyttinen, J.M.; Kaarniranta, K. AMP-activated protein kinase inhibits NF-κB signaling and inflammation: Impact on healthspan and lifespan. J. Mol. Med. 2011, 89, 667–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | ABKA | STZ | STZ + ABKA | STZ + ABKA + CC | ||
|---|---|---|---|---|---|---|
| Final body weight | 411.5 ± 36.7 | 421.5 ± 31.2 | 301 ± 25.7 ab | 398 ± 31.3 abc | 318 ± 29.2 abd | |
| Plasma | Glucose (mg/dl) | 106.3 ± 9.3 | 102.1 ± 8.2 | 341 ± 33.2 ab | 143 ± 12.5 abc | 328 ± 27.8 abd |
| Insulin (ng/mL) | 4.7 ± 0.7 | 4.4 ± 0.5 | 2.1 ± 0.3 ab | 3.25 ± 0.4 abc | 2.18 ± 0.3 abd | |
| Serum | TGs (mg/dl) | 66.4 ± 4.3 | 52.5 ± 45 a | 137 ± 11.2 ab | 79.4 ± 6.8 abc | 129 ± 11.4 abd |
| CHOL (mg/dl) | 78.4 ± 7.9 | 59.7 ± 6.6 a | 168 ± 15.6 ab | 86.8 ± 6.1 abc | 183 ± 4.5 abd | |
| LDL-c (mg/dl) | 44.1 ± 5.8 | 31.2 ± 3.7 a | 97.6 ± 8.4 ab | 51.8 ± 5.9 abc | 104 ± 9.3 abd | |
| FFAs (μmol/mg) | 398 ± 43.2 | 376.1± 29.8 | 893.2 ± 77.3 ab | 484.2 ± 36.8 abc | 991 ± 86.5 abcd | |
| Liver | TGs (mg/g) | 5.8 ± 0.7 | 4.1 ± 0.5 a | 11.3 ± 1.6 ab | 7.1 ± 0.9 abc | 12.4 ± 2.7 abd |
| CHOL (mg/dl) | 4.9 ± 0.39 | 3.1 ± 0.4 a | 8.8 ± 0.6 ab | 5.1 ± 0.4 abc | 9.3 ± 0.9 abd | |
| FFAs (μmol/mg) | 97.5 ± 8.4 | 76.2 ± 6.4 a | 388.1 ± 29.6 ab | 129 ± 11.3 abc | 369.2 ± 26.8 abd | |
| Heart | TGs (μg/g) | 50.4 ± 5.1 | 36.7 ± 3.5 a | 111.4 ± 9.7 ab | 61.3 ± 5.7 abc | 119.4 ± 10.1 abd |
| FFAs (nmol/mg) | 123.2 ± 11.3 | 88.2 ± 7.3 a | 365.3 ± 22.5 ab | 162.2 ± 13.8 abc | 389.9 ± 33.5 abd |
| Control | ABKA | STZ | STZ + ABKA | STZ + ABKA + CC | ||
|---|---|---|---|---|---|---|
| Heart weight (g) | 1.2 ± 0.2 | 1.15 ± 0.18 | 1.46 ± 0.31 ab | 1.14 ± 0.27 abc | 1.52 ± 0.5 abd | |
| Serum | Troponin-I (pg/mL) | 76.3 ± 6.5 | 71.7 ± 7.5 | 583 ± 42.5 ab | 112.5 ± 11.9 abc | 612 ± 58.3 abd |
| CK-MB (pg/mL) | 212.1 ± 18.7 | 209.5 ± 20.4 | 633.4 ± 54.9 ab | 298.6 ± 25.4 abc | 618 ± 52.2 abd | |
| LV function | dp/dtmax (mmHg) | 5994 ± 439 | 5778 ± 593 | 2934 ± 209 ab | 4938 ± 398 abc | 2609 ± 211 abd |
| dp/dtmin (mmHg) | 4983 ± 375 | 4819 ± 478 | 2022 ± 199 ab | 3772 ± 289 abc | 2231 ± 245 abd | |
| LVSP (mmHg) | 122.4 ± 11.3 | 125.1± 12.7 | 75.3 ± 6.9 ab | 108.5 ± 9.9 abc | 69.6 ± 5.8 abd | |
| LVEDP (mmHg) | 2.9 ± 0.3 | 3.1 ± 0.3 | 11.5 ± 1.6 ab | 3.7 ± 0.4 abc | 13.1 ± 2.7 abd |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
AlTamimi, J.Z.; AlFaris, N.A.; Alshammari, G.M.; Alagal, R.I.; Aljabryn, D.H.; Yahya, M.A. The Protective Effect of 11-Keto-β-Boswellic Acid against Diabetic Cardiomyopathy in Rats Entails Activation of AMPK. Nutrients 2023, 15, 1660. https://doi.org/10.3390/nu15071660
AlTamimi JZ, AlFaris NA, Alshammari GM, Alagal RI, Aljabryn DH, Yahya MA. The Protective Effect of 11-Keto-β-Boswellic Acid against Diabetic Cardiomyopathy in Rats Entails Activation of AMPK. Nutrients. 2023; 15(7):1660. https://doi.org/10.3390/nu15071660
Chicago/Turabian StyleAlTamimi, Jozaa Z., Nora A. AlFaris, Ghedeir M. Alshammari, Reham I. Alagal, Dalal H. Aljabryn, and Mohammed Abdo Yahya. 2023. "The Protective Effect of 11-Keto-β-Boswellic Acid against Diabetic Cardiomyopathy in Rats Entails Activation of AMPK" Nutrients 15, no. 7: 1660. https://doi.org/10.3390/nu15071660