

Inhibitory Effects of Ginsenoside Compound K on Lipopolysaccharide-Stimulated Inflammatory Responses in Macrophages by Regulating Sirtuin 1 and Histone Deacetylase 4

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment
2.2. RNA Extraction and Real-Time Quantitative PCR (qRT-PCR)
2.3. Western Blot Analysis
2.4. Measurements of Cellular ROS Levels
2.5. Small Interfering RNA (siRNA) Transfection
2.6. Energy Metabolism of Cells
2.7. Statistical Analysis
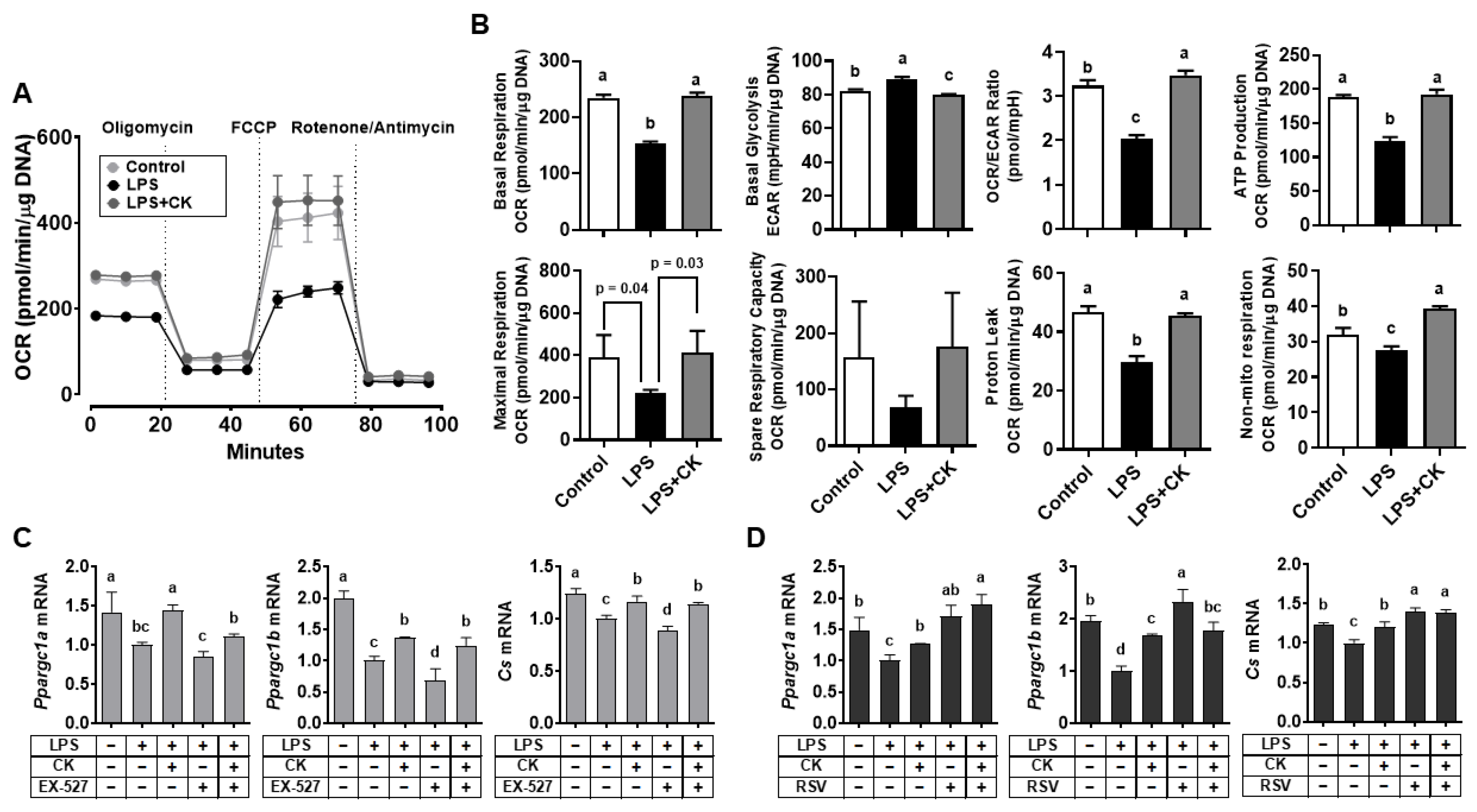
3. Results
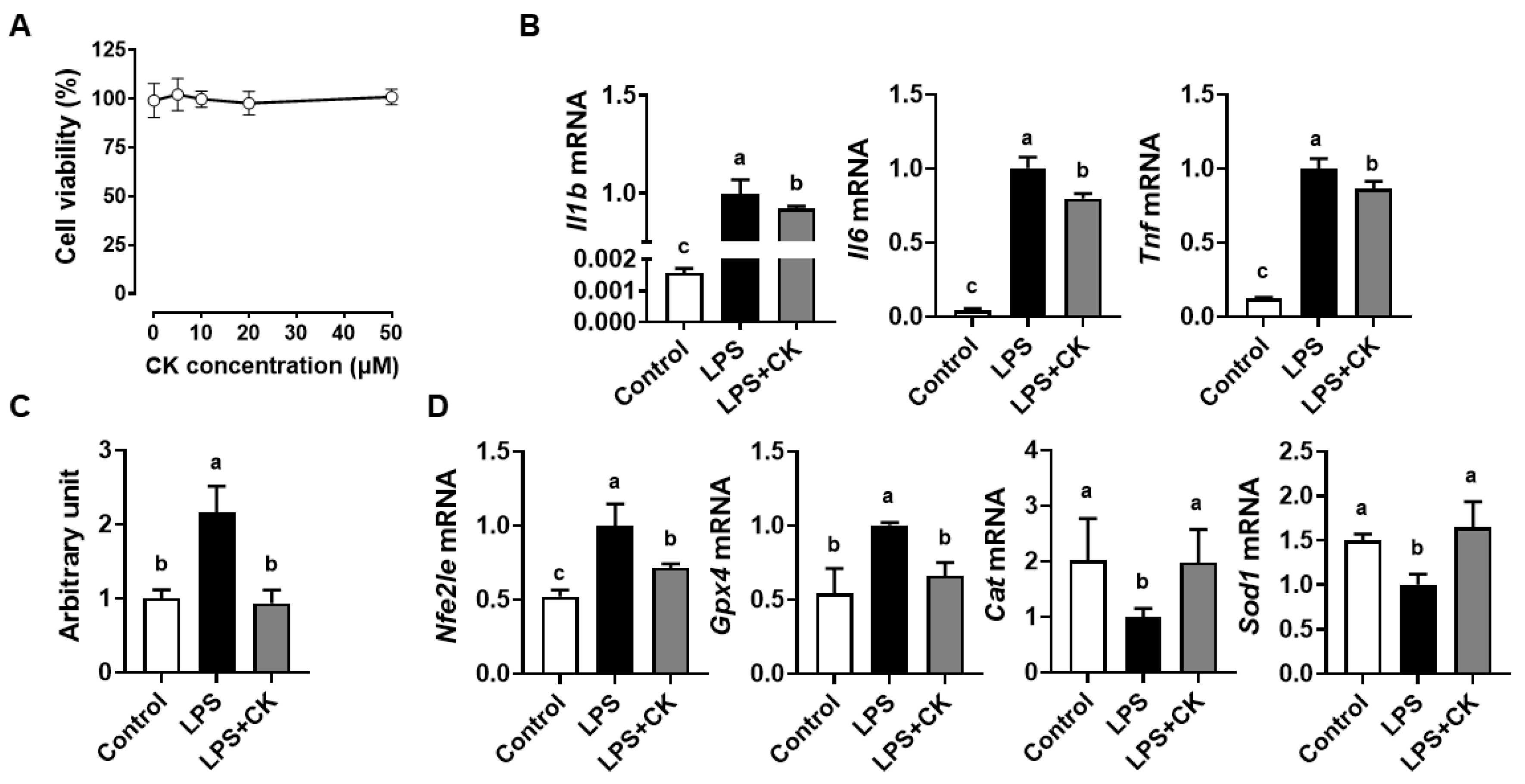
3.1. CK Supressed the LPS-Induced Inflammation and ROS Generation in Macrophages
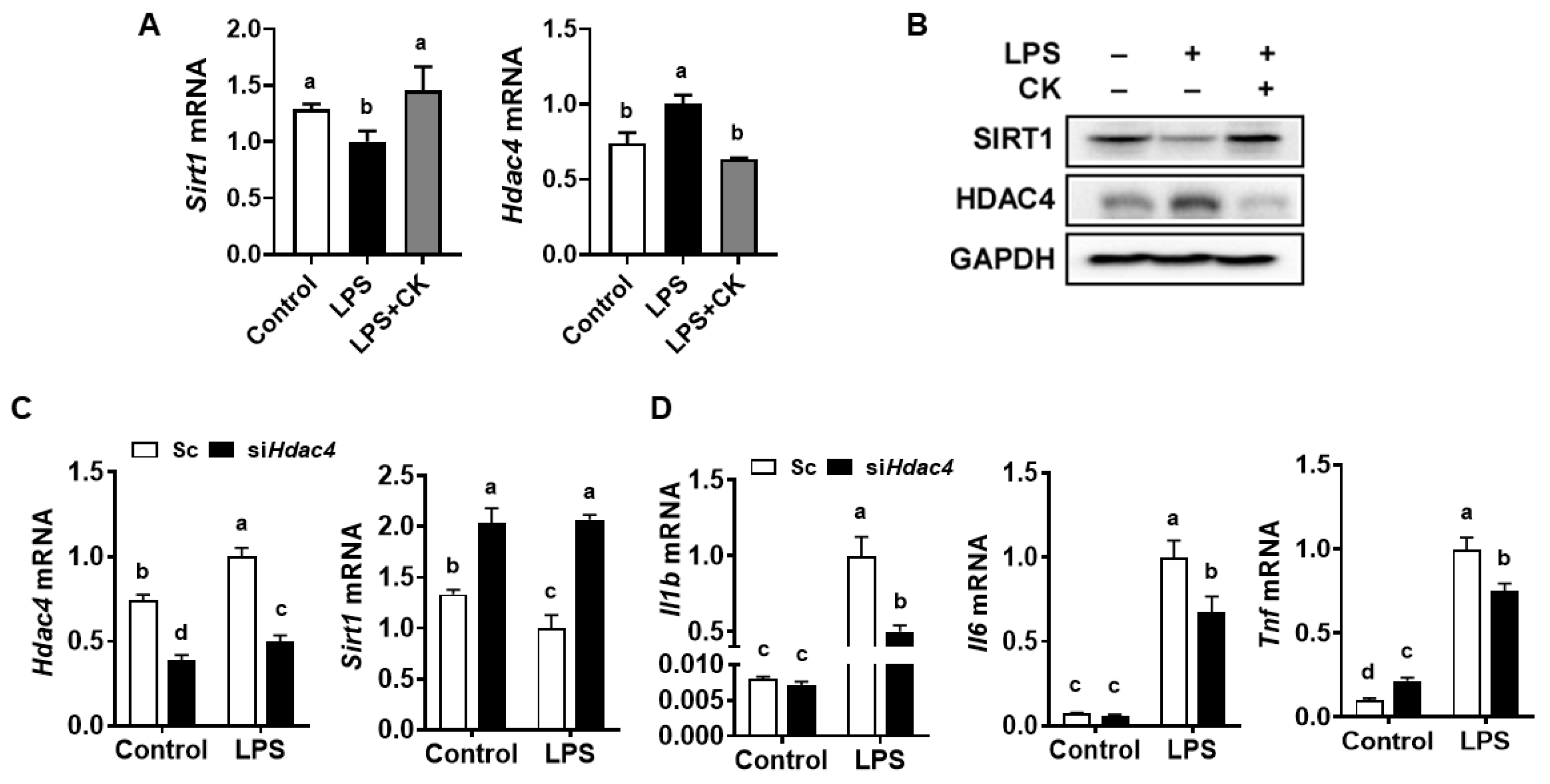
3.2. CK and Hdac4 Knockdown Abolished the LPS-Induced Reduction in Sirt1 and Induction of Pro-Inflammatory Gene Expression in Macrophages
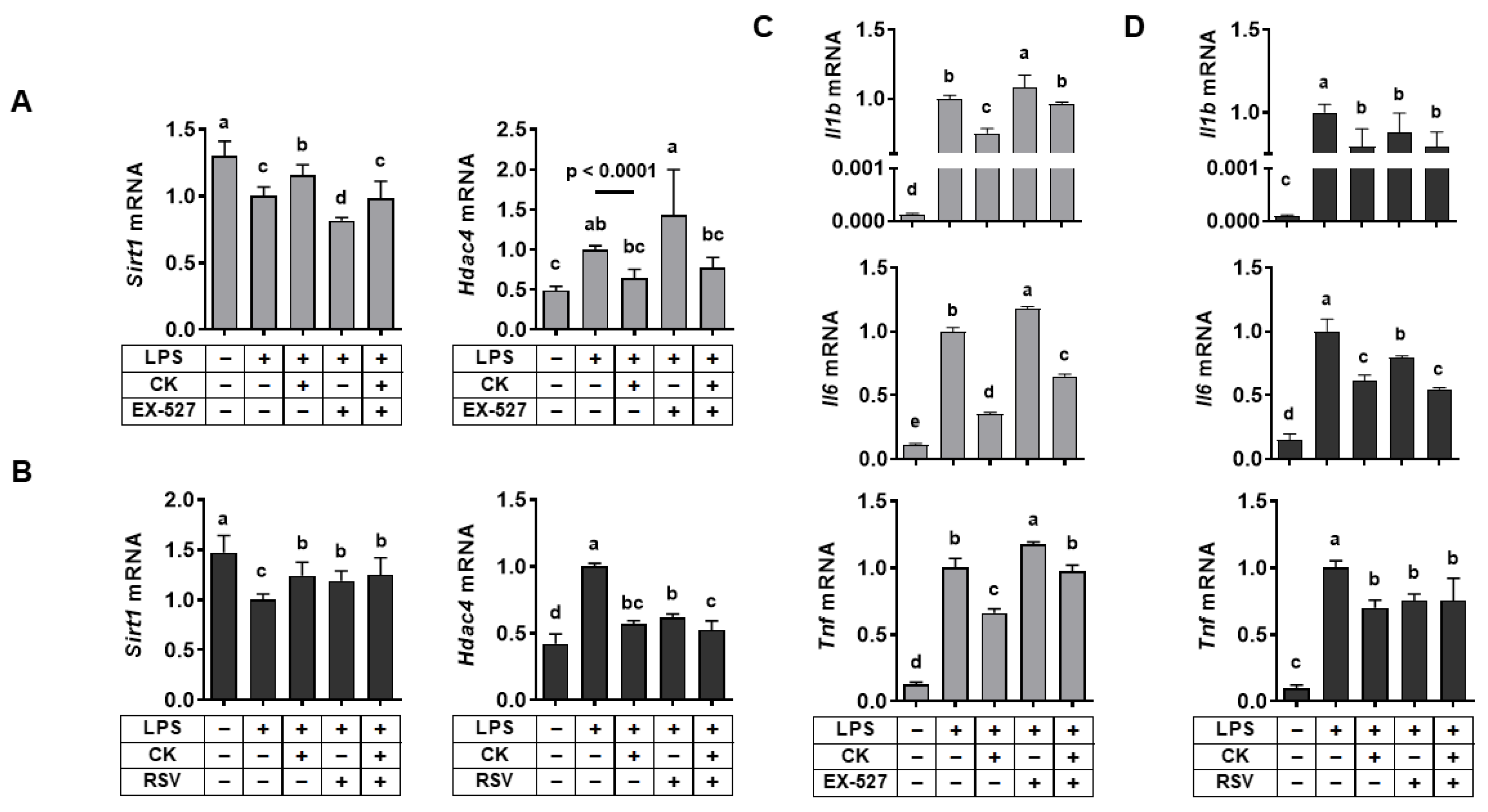
3.3. Regulation of SIRT1 Activity Regulated LPS-Induced Alterations in Hdac4 and Pro-Inflammatory Gene Expression in Macrophages
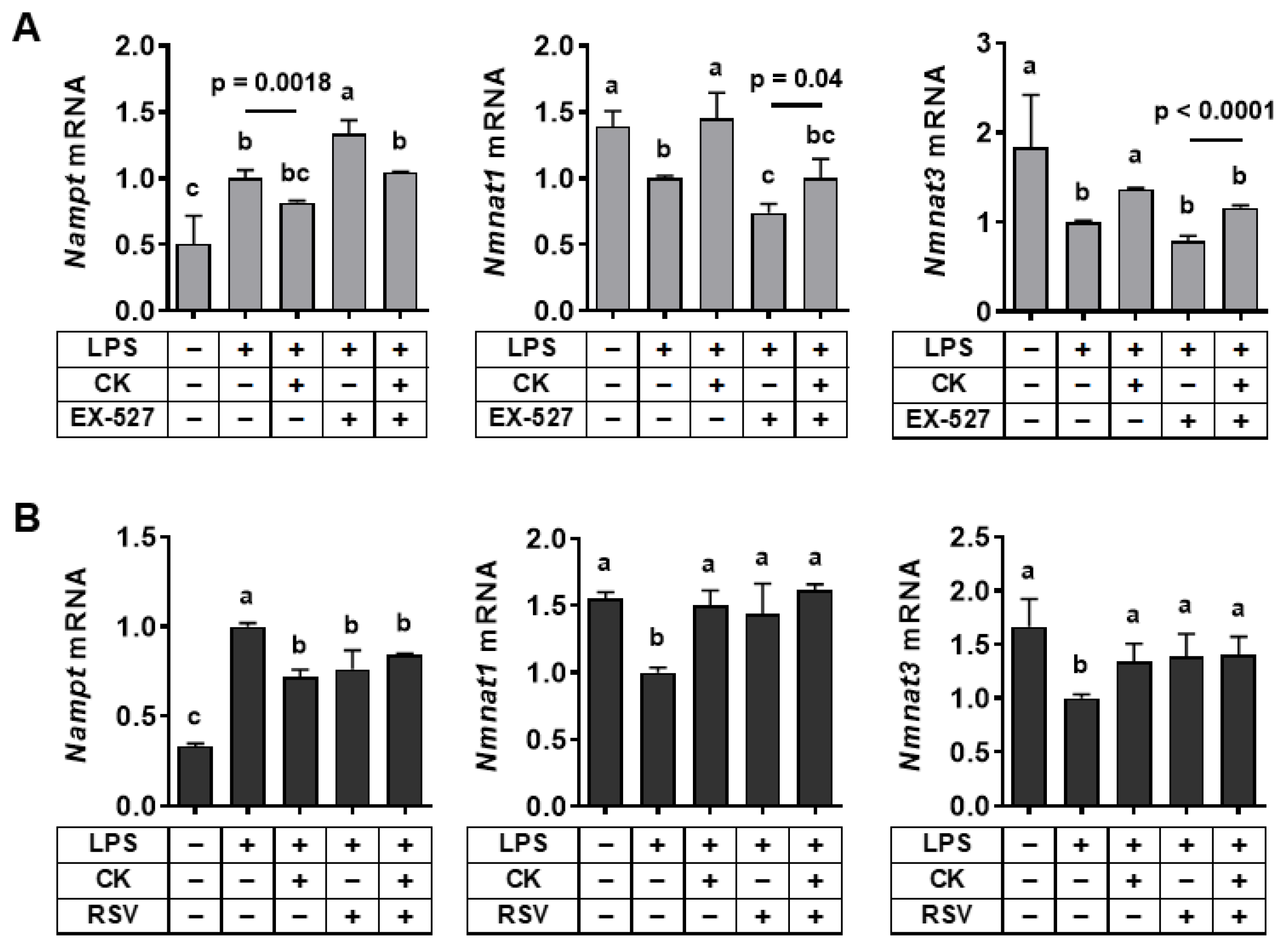
3.4. Regulation of SIRT1 Activity Modulated Gene Expression Associated with the NAD+ Salvage Pathway in LPS-Stimulated Macrophages
3.5. CK Ameliorated the LPS-Induced Glycolytic Capacity of Macrophages through SIRT1 Activation
3.6. CK Regulated the LPS-Altered Mitochondrial Respiration in Macrophages
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shapiro, H.; Lutaty, A.; Ariel, A. Macrophages, meta-inflammation, and immuno-metabolism. Sci. World J. 2011, 11, 2509–2529. [Google Scholar] [CrossRef] [Green Version]
- Sica, A.; Erreni, M.; Allavena, P.; Porta, C. Macrophage polarization in pathology. Cell. Mol. Life Sci. 2015, 72, 4111–4126. [Google Scholar] [CrossRef]
- Brune, B.; Dehne, N.; Grossmann, N.; Jung, M.; Namgaladze, D.; Schmid, T.; von Knethen, A.; Weigert, A. Redox control of inflammation in macrophages. Antioxid. Redox Signal. 2013, 19, 595–637. [Google Scholar] [CrossRef] [Green Version]
- Kominsky, D.J.; Campbell, E.L.; Colgan, S.P. Metabolic shifts in immunity and inflammation. J. Immunol. 2010, 184, 4062–4068. [Google Scholar] [CrossRef] [Green Version]
- Yun, M.; Yi, Y.S. Regulatory roles of ginseng on inflammatory caspases, executioners of inflammasome activation. J. Ginseng Res. 2020, 44, 373–385. [Google Scholar] [CrossRef]
- Kim, J.H. Pharmacological and medical applications of Panax ginseng and ginsenosides: A review for use in cardiovascular diseases. J. Ginseng Res. 2018, 42, 264–269. [Google Scholar] [CrossRef]
- Saw, C.L.; Yang, A.Y.; Cheng, D.C.; Boyanapalli, S.S.; Su, Z.Y.; Khor, T.O.; Gao, S.; Wang, J.; Jiang, Z.H.; Kong, A.N. Pharmacodynamics of ginsenosides: Antioxidant activities, activation of Nrf2, and potential synergistic effects of combinations. Chem. Res. Toxicol. 2012, 25, 1574–1580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Lee, H.J. Ginsenoside Compound K: Insights into Recent Studies on Pharmacokinetics and Health-Promoting Activities. Biomolecules 2020, 10, 1028. [Google Scholar] [CrossRef]
- Baik, I.H.; Kim, K.H.; Lee, K.A. Antioxidant, Anti-Inflammatory and Antithrombotic Effects of Ginsenoside Compound K Enriched Extract Derived from Ginseng Sprouts. Molecules 2021, 26, 4102. [Google Scholar] [CrossRef] [PubMed]
- 1Jiao, H.; Jia, J. Ginsenoside compound K acts via LRP1 to alleviate Amyloid beta(42)-induced neuroinflammation in microglia by suppressing NF-kappaB. Biochem. Biophys. Res. Commun. 2022, 590, 14–19. [Google Scholar] [CrossRef]
- Kang, K.A.; Piao, M.J.; Kim, K.C.; Zheng, J.; Yao, C.W.; Cha, J.W.; Kim, H.S.; Kim, D.H.; Bae, S.C.; Hyun, J.W. Compound K, a metabolite of ginseng saponin, inhibits colorectal cancer cell growth and induces apoptosis through inhibition of histone deacetylase activity. Int. J. Oncol. 2013, 43, 1907–1914. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.J.; Pyun, D.H.; Kim, M.J.; Jeong, J.H.; Abd El-Aty, A.M.; Jung, T.W. Ginsenoside compound K ameliorates palmitate-induced atrophy in C2C12 myotubes via promyogenic effects and AMPK/autophagy-mediated suppression of endoplasmic reticulum stress. J. Ginseng Res. 2022, 46, 444–453. [Google Scholar] [CrossRef]
- Tian, F.; Xu, W.; Chen, L.; Chen, T.; Feng, X.; Chen, J.; Wei, D.; Huang, Q. Ginsenoside compound K increases glucagon-like peptide-1 release and L-cell abundance in db/db mice through TGR5/YAP signaling. Int. Immunopharmacol. 2022, 113, 109405. [Google Scholar] [CrossRef]
- Liu, Z.H.; Li, J.; Xia, J.; Jiang, R.; Zuo, G.W.; Li, X.P.; Chen, Y.; Xiong, W.; Chen, D.L. Ginsenoside 20(s)-Rh2 as potent natural histone deacetylase inhibitors suppressing the growth of human leukemia cells. Chem. Biol. Interact. 2015, 242, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Shan, X.; Fu, Y.S.; Aziz, F.; Wang, X.Q.; Yan, Q.; Liu, J.W. Ginsenoside Rg3 inhibits melanoma cell proliferation through down-regulation of histone deacetylase 3 (HDAC3) and increase of p53 acetylation. PLoS ONE 2014, 9, e115401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, B.; Feng, J.; Yang, N.; Guo, Y.; Chen, C.; Qin, Q. Ginsenoside Rg3 attenuates angiotensin II-induced myocardial hypertrophy through repressing NLRP3 inflammasome and oxidative stress via modulating SIRT1/NF-kappaB pathway. Int. Immunopharmacol. 2021, 98, 107841. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.; Shim, M.K.; Kim, S.; Lee, Y. Red ginseng represses hypoxia-induced cyclooxygenase-2 through sirtuin1 activation. Phytomedicine Int. J. Phytother. Phytopharm. 2015, 22, 597–604. [Google Scholar] [CrossRef]
- Yang, Q.Y.; Lai, X.D.; Ouyang, J.; Yang, J.D. Effects of Ginsenoside Rg3 on fatigue resistance and SIRT1 in aged rats. Toxicology 2018, 409, 144–151. [Google Scholar] [CrossRef]
- Shen, Z.; Ajmo, J.M.; Rogers, C.Q.; Liang, X.; Le, L.; Murr, M.M.; Peng, Y.; You, M. Role of SIRT1 in regulation of LPS- or two ethanol metabolites-induced TNF-alpha production in cultured macrophage cell lines. Am. J. Physiol. Liver Physiol. 2009, 296, G1047–G1053. [Google Scholar] [CrossRef]
- Imai, S.; Armstrong, C.M.; Kaeberlein, M.; Guarente, L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 2000, 403, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P.; et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012, 15, 838–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Sauve, A.A. NAD(+) content and its role in mitochondria. Methods Mol. Biol. 2015, 1241, 39–48. [Google Scholar] [CrossRef]
- Das Gupta, K.; Shakespear, M.R.; Curson, J.E.B.; Murthy, A.M.V.; Iyer, A.; Hodson, M.P.; Ramnath, D.; Tillu, V.A.; von Pein, J.B.; Reid, R.C.; et al. Class IIa Histone Deacetylases Drive Toll-like Receptor-Inducible Glycolysis and Macrophage Inflammatory Responses via Pyruvate Kinase M2. Cell Rep. 2020, 30, 2712–2728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, L.A.; Golenbock, D.; Bowie, A.G. The history of Toll-like receptors—Redefining innate immunity. Nat. Rev. Immunol. 2013, 13, 453–460. [Google Scholar] [CrossRef]
- Shakespear, M.R.; Iyer, A.; Cheng, C.Y.; Das Gupta, K.; Singhal, A.; Fairlie, D.P.; Sweet, M.J. Lysine Deacetylases and Regulated Glycolysis in Macrophages. Trends Immunol. 2018, 39, 473–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.; Park, Y.K.; Lee, J.Y. Inhibition of alcohol-induced inflammation and oxidative stress by astaxanthin is mediated by its opposite actions in the regulation of sirtuin 1 and histone deacetylase 4 in macrophages. Biochimica et biophysica acta. Mol. Cell Biol. Lipids 2021, 1866, 158838. [Google Scholar] [CrossRef]
- Kang, H.; Kim, M.B.; Park, Y.K.; Lee, J.Y. A mouse model of the regression of alcoholic hepatitis: Monitoring the regression of hepatic steatosis, inflammation, oxidative stress, and NAD(+) metabolism upon alcohol withdrawal. J. Nutr. Biochem. 2022, 99, 108852. [Google Scholar] [CrossRef]
- Kang, H.; Lim, J.W.; Kim, H. Inhibitory effect of Korean Red Ginseng extract on DNA damage response and apoptosis in Helicobacter pylori-infected gastric epithelial cells. J. Ginseng Res. 2020, 44, 79–85. [Google Scholar] [CrossRef]
- Yoshizaki, T.; Schenk, S.; Imamura, T.; Babendure, J.L.; Sonoda, N.; Bae, E.J.; Oh, D.Y.; Lu, M.; Milne, J.C.; Westphal, C.; et al. SIRT1 inhibits inflammatory pathways in macrophages and modulates insulin sensitivity. Am. J. Physiol. Metab. 2010, 298, E419–E428. [Google Scholar] [CrossRef] [Green Version]
- Zarzuelo, M.J.; Lopez-Sepulveda, R.; Sanchez, M.; Romero, M.; Gomez-Guzman, M.; Ungvary, Z.; Perez-Vizcaino, F.; Jimenez, R.; Duarte, J. SIRT1 inhibits NADPH oxidase activation and protects endothelial function in the rat aorta: Implications for vascular aging. Biochem. Pharmacol. 2013, 85, 1288–1296. [Google Scholar] [CrossRef]
- Qu, L.; Yu, Y.; Qiu, L.; Yang, D.; Yan, L.; Guo, J.; Jahan, R. Sirtuin 1 regulates matrix metalloproteinase-13 expression induced by Porphyromonas endodontalis lipopolysaccharide via targeting nuclear factor-kappaB in osteoblasts. J. Oral Microbiol. 2017, 9, 1317578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Xu, R.L.; He, P.; Chen, R. MAR1 suppresses inflammatory response in LPS-induced RAW 264.7 macrophages and human primary peripheral blood mononuclear cells via the SIRT1/PGC-1alpha/PPAR-gamma pathway. J. Inflamm. 2021, 18, 8. [Google Scholar] [CrossRef]
- Gertz, M.; Fischer, F.; Nguyen, G.T.T.; Lakshminarasimhan, M.; Schutkowski, M.; Weyand, M.; Steegborn, C. Ex-527 inhibits Sirtuins by exploiting their unique NAD+-dependent deacetylation mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, E2772–E2781. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Tian, R.; Yang, Y.; Jiang, R.; Dai, J.; Tang, L.; Zhang, L. The SIRT1 inhibitor EX-527 suppresses mTOR activation and alleviates acute lung injury in mice with endotoxiemia. Innate Immun. 2017, 23, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.S.; Canto, C. The molecular targets of resveratrol. Biochim. Biophys. Acta 2015, 1852, 1114–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, D.; Wang, M.; Qiu, X.; Liu, D.; Jiang, H.; Yang, N.; Xu, R.M. Structural basis for allosteric, substrate-dependent stimulation of SIRT1 activity by resveratrol. Genes Dev. 2015, 29, 1316–1325. [Google Scholar] [CrossRef] [Green Version]
- Minhas, P.S.; Liu, L.; Moon, P.K.; Joshi, A.U.; Dove, C.; Mhatre, S.; Contrepois, K.; Wang, Q.; Lee, B.A.; Coronado, M.; et al. Macrophage de novo NAD(+) synthesis specifies immune function in aging and inflammation. Nat. Immunol. 2019, 20, 50–63. [Google Scholar] [CrossRef]
- Cameron, A.M.; Castoldi, A.; Sanin, D.E.; Flachsmann, L.J.; Field, C.S.; Puleston, D.J.; Kyle, R.L.; Patterson, A.E.; Hassler, F.; Buescher, J.M.; et al. Inflammatory macrophage dependence on NAD(+) salvage is a consequence of reactive oxygen species-mediated DNA damage. Nat. Immunol. 2019, 20, 420–432. [Google Scholar] [CrossRef]
- Venter, G.; Oerlemans, F.T.; Willemse, M.; Wijers, M.; Fransen, J.A.; Wieringa, B. NAMPT-mediated salvage synthesis of NAD+ controls morphofunctional changes of macrophages. PLoS ONE 2014, 9, e97378. [Google Scholar] [CrossRef] [Green Version]
- Yoshino, J.; Baur, J.A.; Imai, S.I. NAD(+) Intermediates: The Biology and Therapeutic Potential of NMN and NR. Cell Metab. 2018, 27, 513–528. [Google Scholar] [CrossRef] [Green Version]
- Viola, A.; Munari, F.; Sanchez-Rodriguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Freemerman, A.J.; Johnson, A.R.; Sacks, G.N.; Milner, J.J.; Kirk, E.L.; Troester, M.A.; Macintyre, A.N.; Goraksha-Hicks, P.; Rathmell, J.C.; Makowski, L. Metabolic reprogramming of macrophages: Glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J. Biol. Chem. 2014, 289, 7884–7896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, L.R.; Tompkins, S.C.; Taylor, E.B. Regulation of pyruvate metabolism and human disease. Cell. Mol. Life Sci. CMLS 2014, 71, 2577–2604. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Han, J.; Epstein, P.N.; Liu, Y.Q. Regulation of PDK mRNA by high fatty acid and glucose in pancreatic islets. Biochem. Biophys. Res. Commun. 2006, 344, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Wanet, A.; Arnould, T.; Najimi, M.; Renard, P. Connecting Mitochondria, Metabolism, and Stem Cell Fate. Stem Cells Dev. 2015, 24, 1957–1971. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Liu, R.; Liu, Y.; Zhao, M.; Luan, J.; Wang, Y.; Shang, W.; Song, X.; Sun, Y.; Han, F. H55 N variation in citrate synthase leads to decrement in the enzyme activity and transport rate to mitochondria in HEI-OC1 cells. Biochem. Biophys. Res. Commun. 2022, 612, 134–140. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta BBA Mol. Cell Res. 2011, 1813, 1269–1278. [Google Scholar] [CrossRef] [Green Version]
- Larsen, S.; Nielsen, J.; Hansen, C.N.; Nielsen, L.B.; Wibrand, F.; Stride, N.; Schroder, H.D.; Boushel, R.; Helge, J.W.; Dela, F.; et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J. Physiol. 2012, 590, 3349–3360. [Google Scholar] [CrossRef]
- Lou, T.; Huang, Q.; Su, H.; Zhao, D.; Li, X. Targeting Sirtuin 1 signaling pathway by ginsenosides. J. Ethnopharmacol. 2021, 268, 113657. [Google Scholar] [CrossRef]
- Hou, X.; Rooklin, D.; Fang, H.; Zhang, Y. Resveratrol serves as a protein-substrate interaction stabilizer in human SIRT1 activation. Sci. Rep. 2016, 6, 38186. [Google Scholar] [CrossRef] [Green Version]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.; Lee, Y.; Bae, M.; Park, Y.K.; Lee, J.Y. Astaxanthin inhibits alcohol-induced inflammation and oxidative stress in macrophages in a sirtuin 1-dependent manner. J. Nutr. Biochem. 2020, 85, 108477. [Google Scholar] [CrossRef]
- Ma, L.; Zhao, Y.; Wang, R.; Chen, T.; Li, W.; Nan, Y.; Liu, X.; Jin, F. 3,5,4’-Tri-O-acetylresveratrol Attenuates Lipopolysaccharide-Induced Acute Respiratory Distress Syndrome via MAPK/SIRT1 Pathway. Mediat. Inflamm. 2015, 2015, 143074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baird, L.; Yamamoto, M. The Molecular Mechanisms Regulating the KEAP1-NRF2 Pathway. Mol. Cell. Biol. 2020, 40, e00099-20. [Google Scholar] [CrossRef]
- Sng, K.S.; Li, G.; Zhou, L.Y.; Song, Y.J.; Chen, X.Q.; Wang, Y.J.; Yao, M.; Cui, X.J. Ginseng extract and ginsenosides improve neurological function and promote antioxidant effects in rats with spinal cord injury: A meta-analysis and systematic review. J. Ginseng Res. 2022, 46, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.M.; Ji, S.F.; Liu, Y.H.; Xue, X.M.; Xu, J.; Gu, Z.H.; Deng, S.L.; Liu, C.D.; Wang, H.; Chang, Y.M.; et al. Ginsenoside Rd Ameliorates Auditory Cortex Injury Associated With Military Aviation Noise-Induced Hearing Loss by Activating SIRT1/PGC-1alpha Signaling Pathway. Front. Physiol. 2020, 11, 788. [Google Scholar] [CrossRef]
- Canto, C.; Menzies, K.J.; Auwerx, J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef] [Green Version]
- Klimova, N.; Long, A.; Scafidi, S.; Kristian, T. Interplay between NAD(+) and acetylCoA metabolism in ischemia-induced mitochondrial pathophysiology. Biochimica et biophysica acta. Mol. Basis Dis. 2019, 1865, 2060–2067. [Google Scholar] [CrossRef]
- Yu, A.; Zhou, R.; Xia, B.; Dang, W.; Yang, Z.; Chen, X. NAMPT maintains mitochondria content via NRF2-PPARalpha/AMPKalpha pathway to promote cell survival under oxidative stress. Cell. Signal. 2020, 66, 109496. [Google Scholar] [CrossRef]
- Kiss, T.; Nyul-Toth, A.; Balasubramanian, P.; Tarantini, S.; Ahire, C.; Yabluchanskiy, A.; Csipo, T.; Farkas, E.; Wren, J.D.; Garman, L.; et al. Nicotinamide mononucleotide (NMN) supplementation promotes neurovascular rejuvenation in aged mice: Transcriptional footprint of SIRT1 activation, mitochondrial protection, anti-inflammatory, and anti-apoptotic effects. Geroscience 2020, 42, 527–546. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Ma, Q.; Xu, M.; Qiao, Y.; Zhang, Y.; Li, P.; Bi, Y.; Tang, M. Ginsenoside Rb1 Attenuates High Glucose-Induced Oxidative Injury via the NAD-PARP-SIRT Axis in Rat Retinal Capillary Endothelial Cells. Int. J. Mol. Sci. 2019, 20, 4936. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Berrocal, J.G.; Frizzell, K.M.; Gamble, M.J.; DuMond, M.E.; Krishnakumar, R.; Yang, T.; Sauve, A.A.; Kraus, W.L. Enzymes in the NAD+ salvage pathway regulate SIRT1 activity at target gene promoters. J. Biol. Chem. 2009, 284, 20408–20417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Shu, L.; Huang, W.; Song, G.; Ma, H. Resveratrol affects hepatic gluconeogenesis via histone deacetylase 4. Diabetes Metab. Syndr. Obes. Targets Ther. 2019, 12, 401–411. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Hong, W.; Hao, C.; Li, L.; Xu, H.; Li, P.; Xu, Y. Hepatic stellate cell-specific deletion of SIRT1 exacerbates liver fibrosis in mice. Biochimica et biophysica acta. Mol. Basis Dis. 2017, 1863, 3202–3211. [Google Scholar] [CrossRef]
- Fang, M.; Fan, Z.; Tian, W.; Zhao, Y.; Li, P.; Xu, H.; Zhou, B.; Zhang, L.; Wu, X.; Xu, Y. HDAC4 mediates IFN-gamma induced disruption of energy expenditure-related gene expression by repressing SIRT1 transcription in skeletal muscle cells. Biochim. Biophys. Acta 2016, 1859, 294–305. [Google Scholar] [CrossRef]
- Usui, T.; Okada, M.; Mizuno, W.; Oda, M.; Ide, N.; Morita, T.; Hara, Y.; Yamawaki, H. HDAC4 mediates development of hypertension via vascular inflammation in spontaneous hypertensive rats. American journal of physiology. Heart Circ. Physiol. 2012, 302, H1894–H1904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.; Lee, Y.; Kim, M.B.; Hu, S.; Jang, H.; Park, Y.K.; Lee, J.Y. The loss of histone deacetylase 4 in macrophages exacerbates hepatic and adipose tissue inflammation in male but not in female mice with diet-induced non-alcoholic steatohepatitis. J. Pathol. 2021, 255, 319–329. [Google Scholar] [CrossRef]
- Li, K.; Lv, G.; Pan, L. Sirt1 alleviates LPS induced inflammation of periodontal ligament fibroblasts via downregulation of TLR4. Int. J. Biol. Macromol. 2018, 119, 249–254. [Google Scholar] [CrossRef]
- Okazaki, M.; Iwasaki, Y.; Nishiyama, M.; Taguchi, T.; Tsugita, M.; Nakayama, S.; Kambayashi, M.; Hashimoto, K.; Terada, Y. PPARbeta/delta regulates the human SIRT1 gene transcription via Sp1. Endocr. J. 2010, 57, 403–413. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Pore, N.; Kim, M.; Voong, K.R.; Dowling, M.; Maity, A.; Kao, G.D. Regulation of histone deacetylase 4 expression by the SP family of transcription factors. Mol. Biol. Cell 2006, 17, 585–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Bossche, J.; Baardman, J.; de Winther, M.P. Metabolic Characterization of Polarized M1 and M2 Bone Marrow-derived Macrophages Using Real-time Extracellular Flux Analysis. J. Vis. Exp. JoVE 2015, 105, e53424. [Google Scholar] [CrossRef] [Green Version]
- Mouton, A.J.; Li, X.; Hall, M.E.; Hall, J.E. Obesity, Hypertension, and Cardiac Dysfunction: Novel Roles of Immunometabolism in Macrophage Activation and Inflammation. Circ. Res. 2020, 126, 789–806. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.W.; Bae, M.K.; Ahn, M.Y.; Kim, S.H.; Sohn, T.K.; Bae, M.H.; Yoo, M.A.; Song, E.J.; Lee, K.J.; Kim, K.W. Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell 2002, 111, 709–720. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.H.; Lee, Y.M.; Chun, Y.S.; Chen, J.; Kim, J.E.; Park, J.W. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol. Cell 2010, 38, 864–878. [Google Scholar] [CrossRef]
- Seo, H.W.; Kim, E.J.; Na, H.; Lee, M.O. Transcriptional activation of hypoxia-inducible factor-1alpha by HDAC4 and HDAC5 involves differential recruitment of p300 and FIH-1. FEBS Lett. 2009, 583, 55–60. [Google Scholar] [CrossRef] [Green Version]
- Eguchi, K.; Nakayama, K. Prolonged hypoxia decreases nuclear pyruvate dehydrogenase complex and regulates the gene expression. Biochem. Biophys. Res. Commun. 2019, 520, 128–135. [Google Scholar] [CrossRef]
- Lee, D.C.; Sohn, H.A.; Park, Z.Y.; Oh, S.; Kang, Y.K.; Lee, K.M.; Kang, M.; Jang, Y.J.; Yang, S.J.; Hong, Y.K.; et al. A lactate-induced response to hypoxia. Cell 2015, 161, 595–609. [Google Scholar] [CrossRef] [Green Version]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Galvan-Pena, S.; O’Neill, L.A. Metabolic reprograming in macrophage polarization. Front. Immunol. 2014, 5, 420. [Google Scholar] [CrossRef] [Green Version]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Locher, L.; Huber, K.; Rehage, J.; Tienken, R.; Meyer, U.; Danicke, S.; Webb, L.; Sauerwein, H.; Mielenz, M. Longitudinal changes in adipose tissue of dairy cows from late pregnancy to lactation. Part 2: The SIRT-PPARGC1A axis and its relationship with the adiponectin system. J. Dairy Sci. 2016, 99, 1560–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.; Park, Y.K.; Lee, J.Y. Nicotinamide riboside, an NAD(+) precursor, attenuates inflammation and oxidative stress by activating sirtuin 1 in alcohol-stimulated macrophages. Lab. Investig. A J. Tech. Methods Pathol. 2021, 101, 1225–1237. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, H.; Kim, S.; Lee, J.-Y.; Kim, B. Inhibitory Effects of Ginsenoside Compound K on Lipopolysaccharide-Stimulated Inflammatory Responses in Macrophages by Regulating Sirtuin 1 and Histone Deacetylase 4. Nutrients 2023, 15, 1626. https://doi.org/10.3390/nu15071626
Kang H, Kim S, Lee J-Y, Kim B. Inhibitory Effects of Ginsenoside Compound K on Lipopolysaccharide-Stimulated Inflammatory Responses in Macrophages by Regulating Sirtuin 1 and Histone Deacetylase 4. Nutrients. 2023; 15(7):1626. https://doi.org/10.3390/nu15071626
Chicago/Turabian StyleKang, Hyunju, Shin Kim, Jin-Young Lee, and Bohkyung Kim. 2023. "Inhibitory Effects of Ginsenoside Compound K on Lipopolysaccharide-Stimulated Inflammatory Responses in Macrophages by Regulating Sirtuin 1 and Histone Deacetylase 4" Nutrients 15, no. 7: 1626. https://doi.org/10.3390/nu15071626