
Ellagic Acid and Its Metabolites as Potent and Selective Allosteric Inhibitors of Liver Pyruvate Kinase

 , , , , and add
Show full author list
, , , , and add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
- 3,4,8,9,10-Pentahydroxy-6H-benzo[c]chromen-6-one (4)

- 2-Bromo-4,5-Dimethoxybenzoic Acid (b)
- tert-Butyl 2-bromo-4,5-dimethoxybenzoate (c)
- (E)-tert-Butyl 2-(2-ethoxyvinyl)-4,5-dimethoxybenzoate (d)
- 6,7-Dimethoxy-1H-isochromen-1-one (e)
- 6,7-dihydroxy-1H-isochromen-1-one (6)
- [1,1’-Biphenyl]-2,2’,3,3’,4,4’-hexaol (8)

- 3,3’,4,4’-tetramethoxy-1,1’-biphenyl (h)
- [1,1’-biphenyl]-3,3’,4,4’-tetraol (9)

- 3-Hydroxy-8,9-dimethoxy-6H-benzo[c]chromen-6-one (i)
- 3,8,9-trihydroxy-6H-benzo[c]chromen-6-one (10)
- 2-Methoxyphenyl 2-bromo-4,5-dimethoxybenzoate (j)
- 4,8,9-Trimethoxy-6H-benzo[c]chromen-6-one (k)
- 4,8,9-Trihydroxy-6H-benzo[c]chromen-6-one (13)

- 2,3-Dimethoxyphenyl 2-bromo-4-methoxybenzoate (o)
- 3,4,9-Trimethoxy-6H-benzo[c]chromen-6-one (q)
- 3,4,9-Trihydroxy-6H-benzo[c]chromen-6-one (14)
- 2,3-Dimethoxyphenyl 2-bromo-5-methoxybenzoate (p)
- 3,4,8-Trimethoxy-6H-benzo[c]chromen-6-one (r)
- 3,4,8-Trihydroxy-6H-benzo[c]chromen-6-one (15)
2.2. Kinase Assay
2.3. Molecular Modelling
2.3.1. Homology Modelling
2.3.2. Induced Fit Docking
2.3.3. Prime-MMGBSA Energy Calculations
3. Results
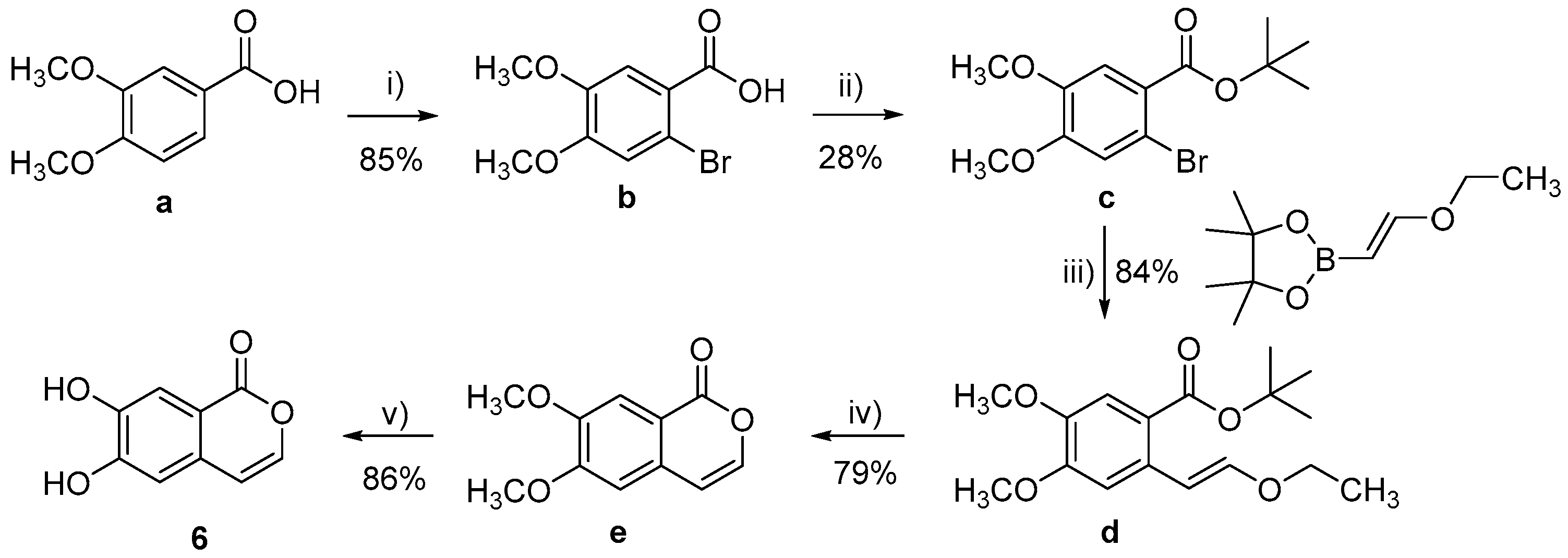
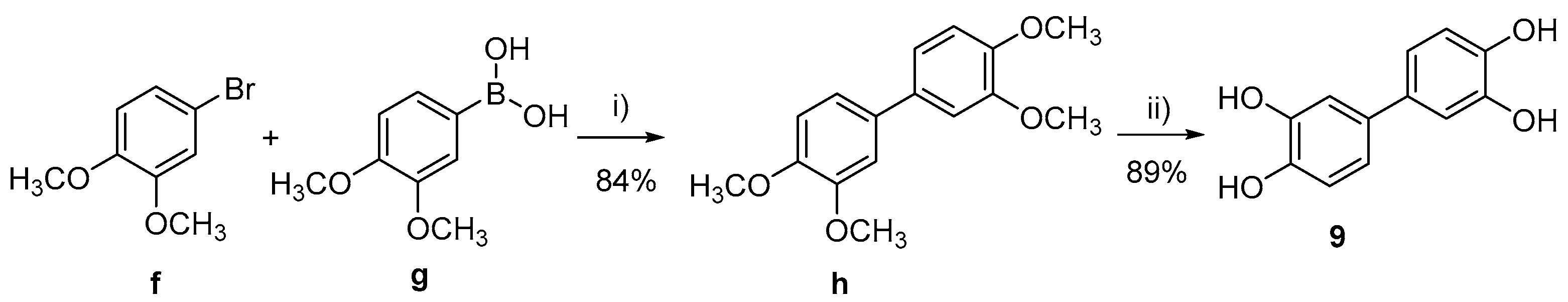
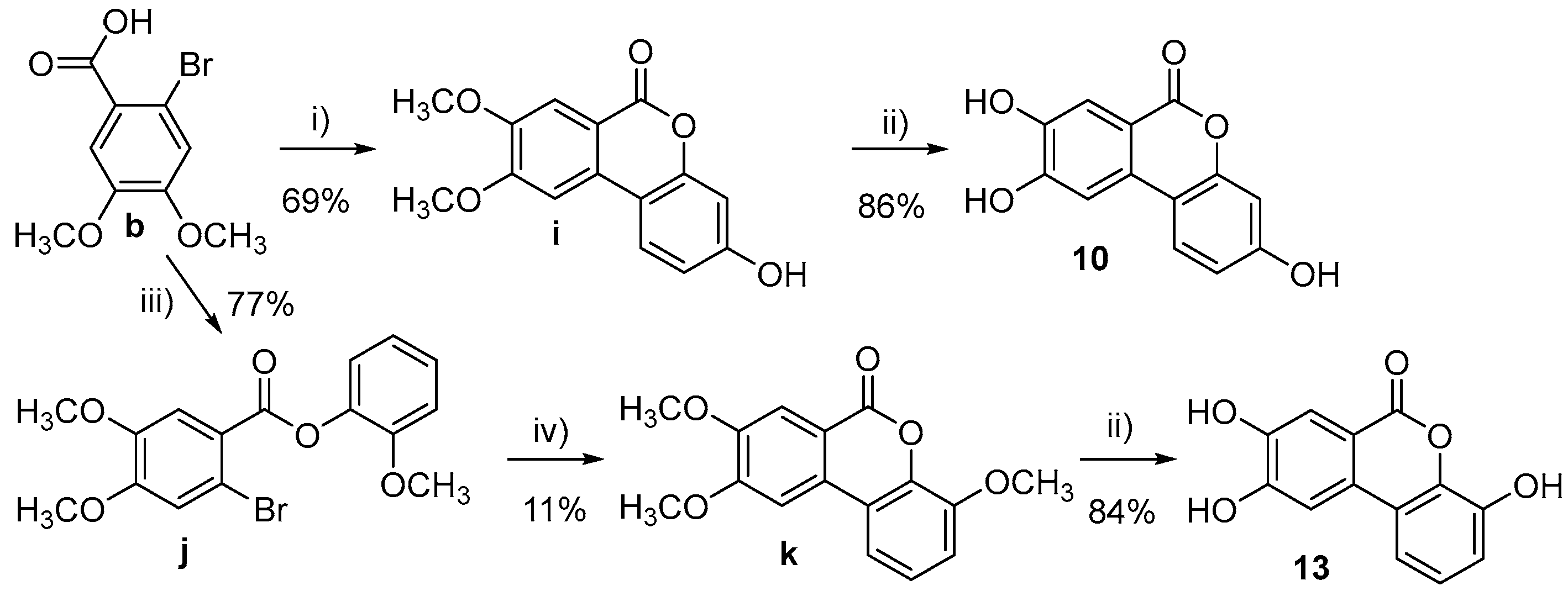
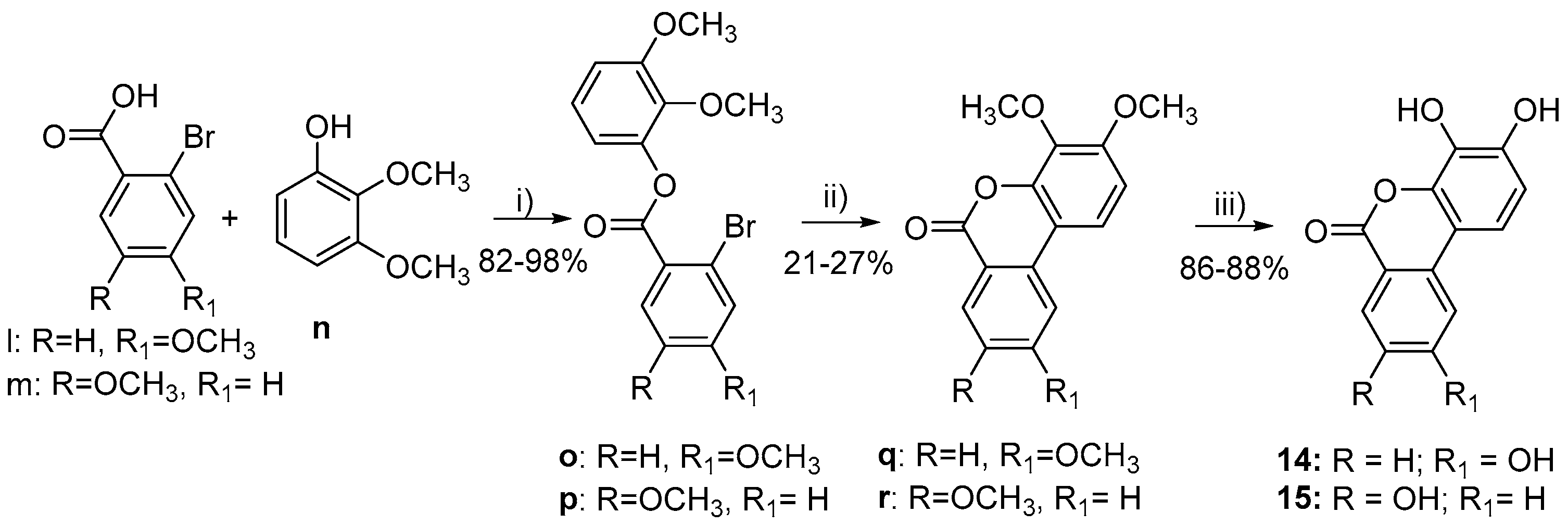
3.1. Chemistry
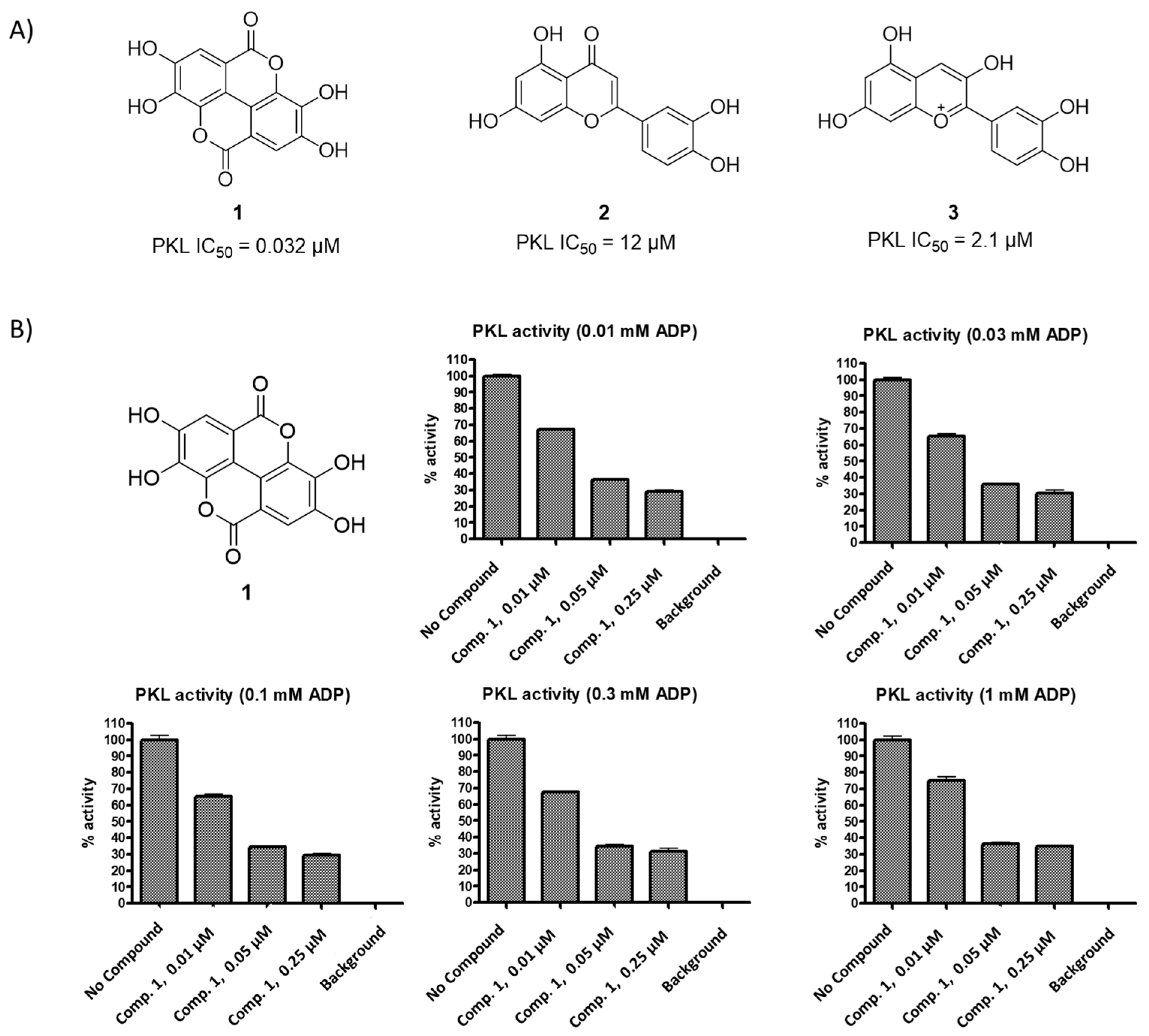
3.2. Initial Screening Results
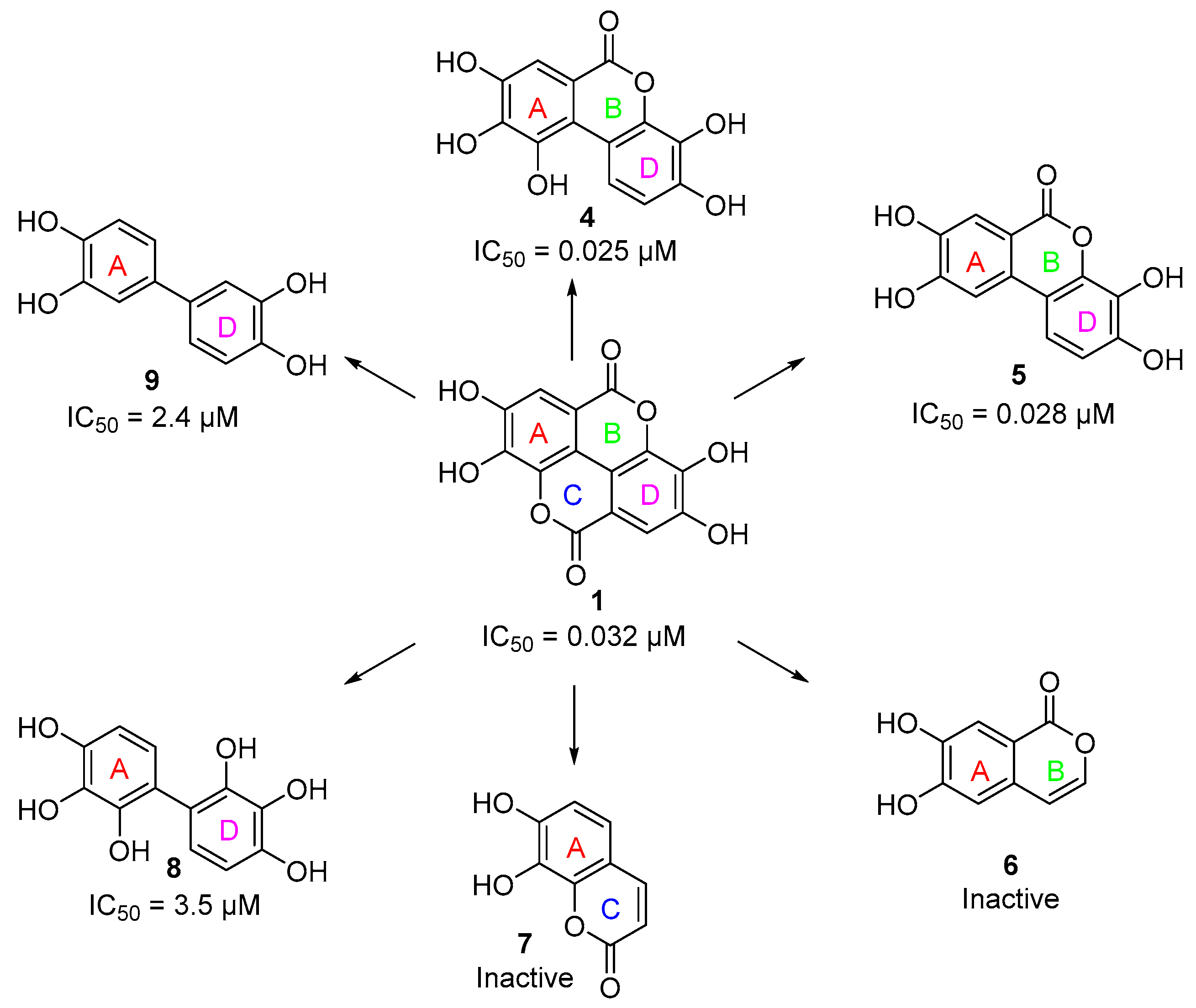
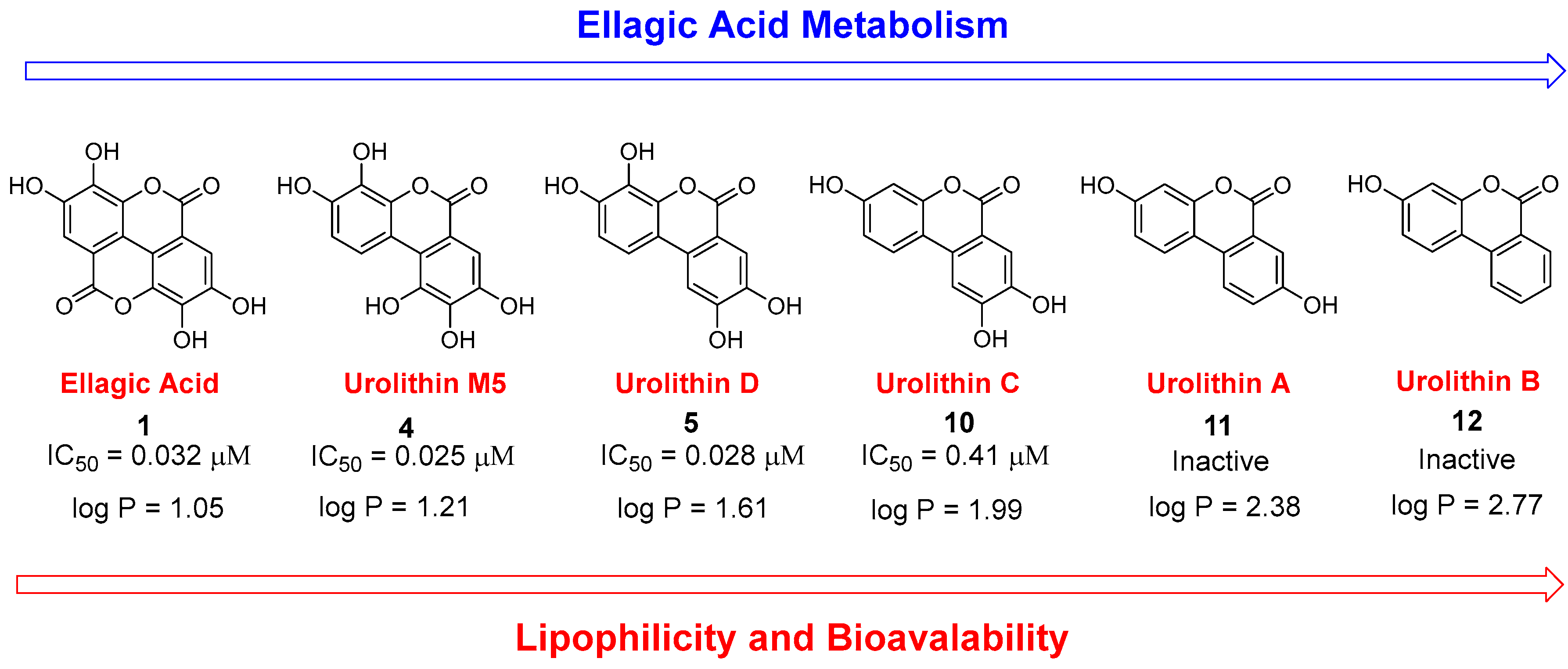
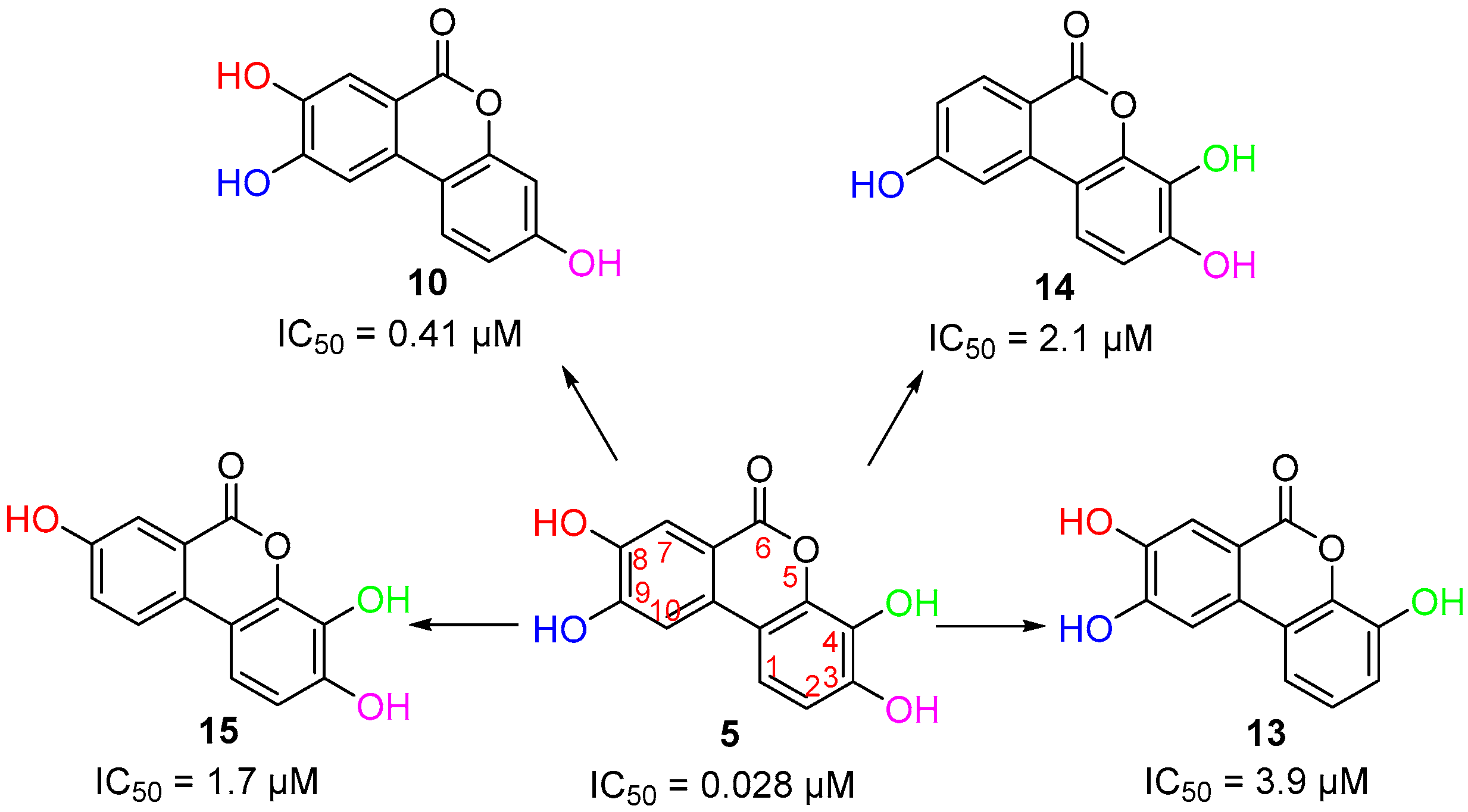
3.3. Deconstruction of EA and Its Metabolites
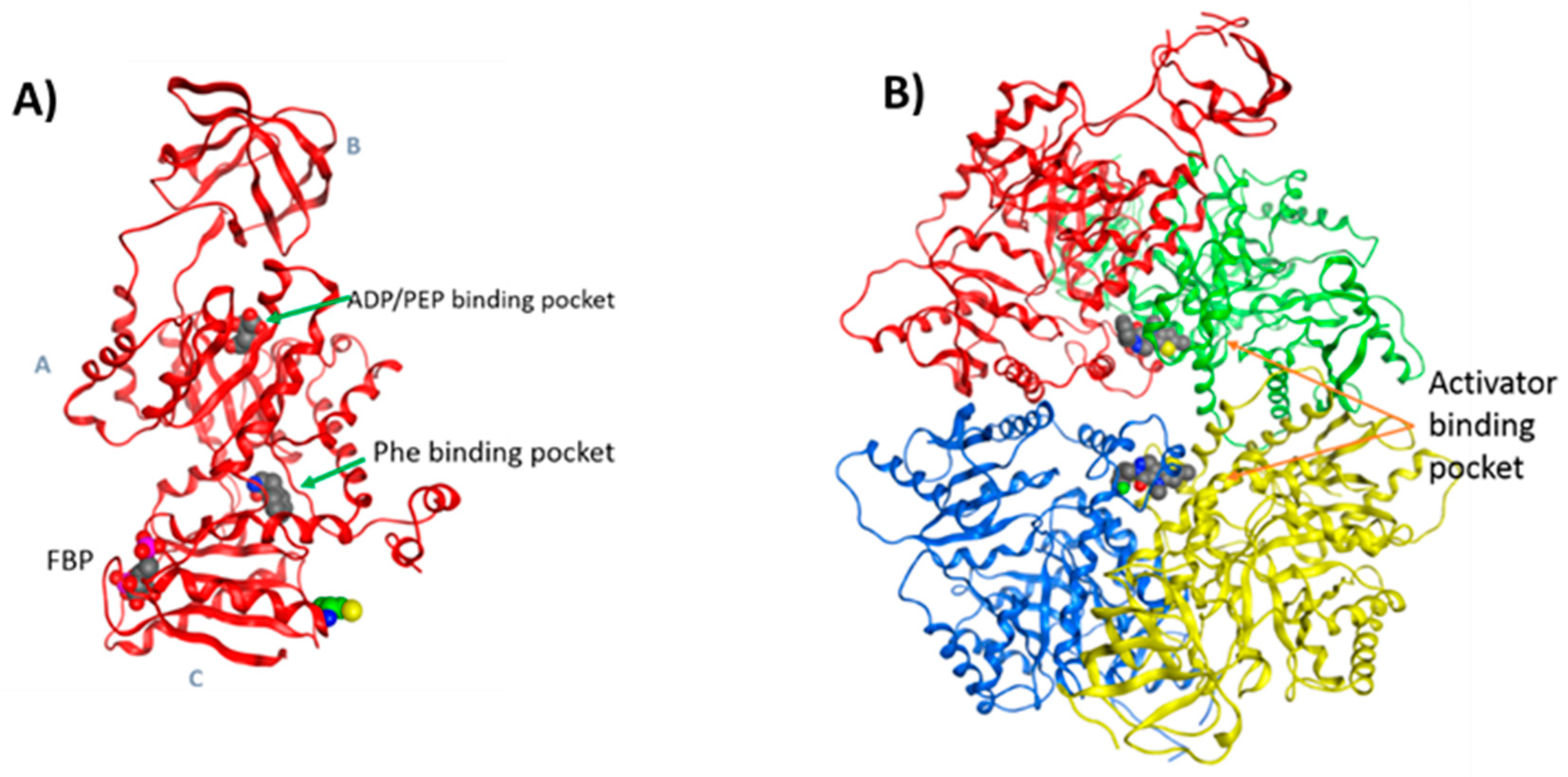
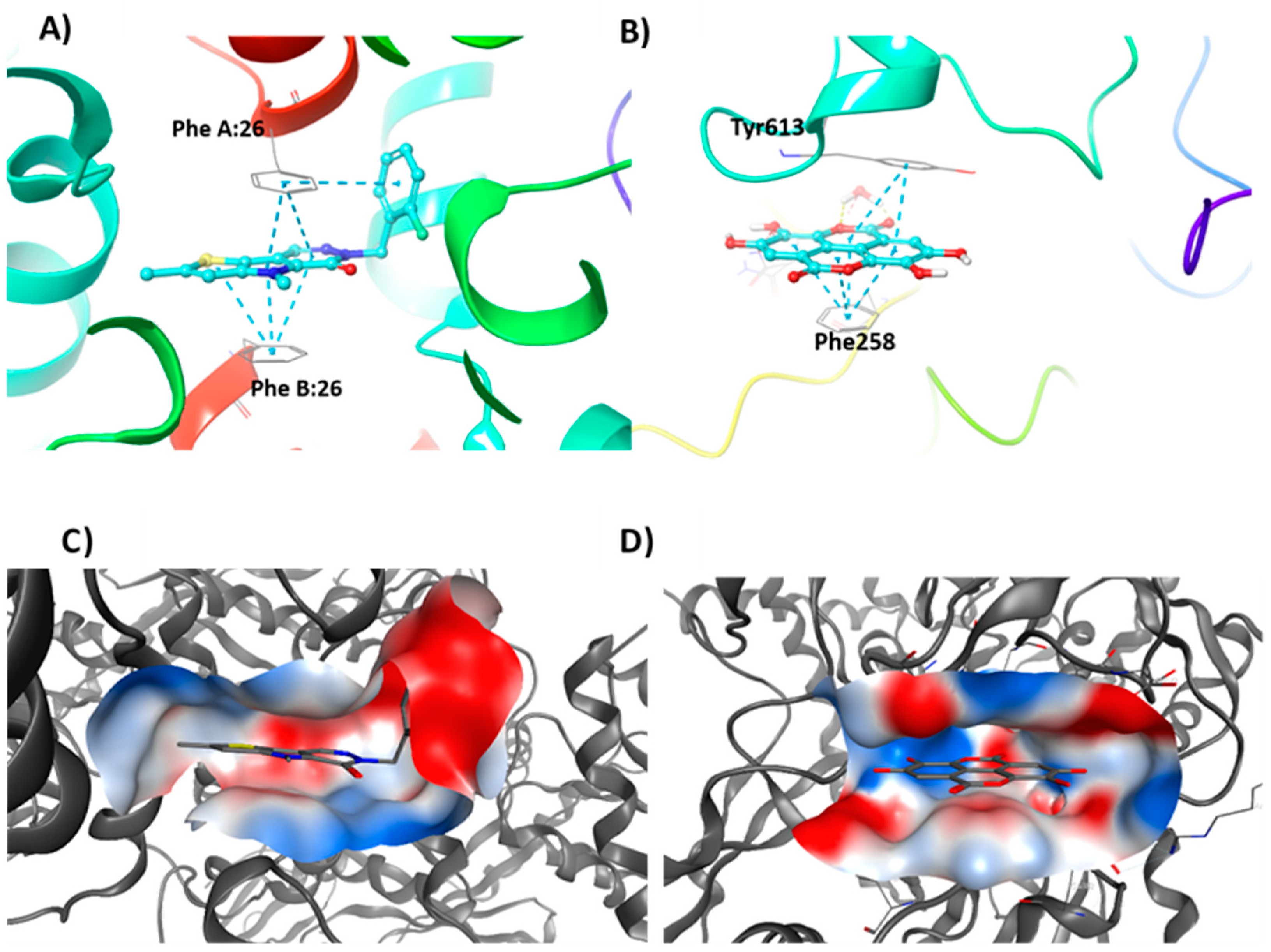
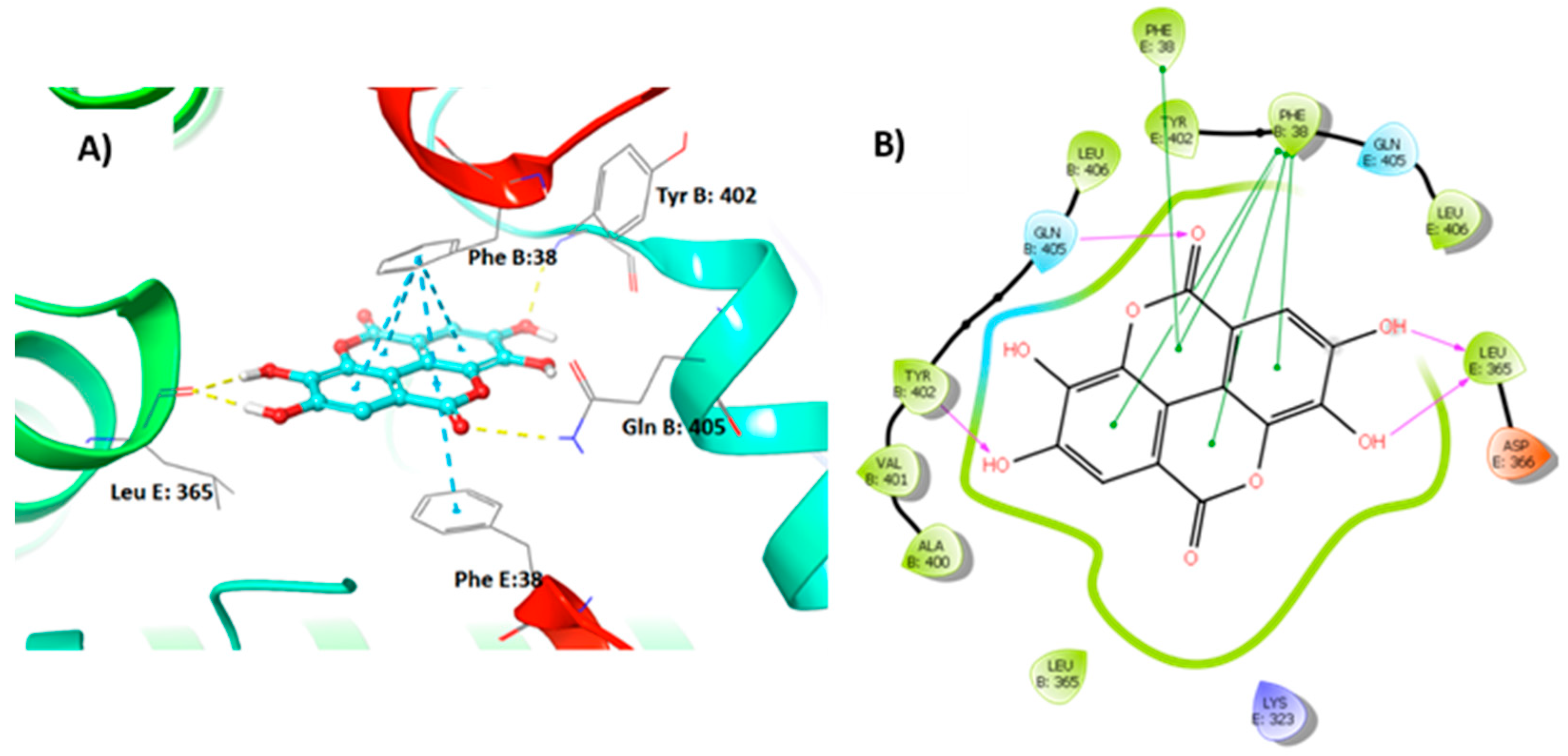
3.4. Molecular Modelling Studies
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Benedict, M.; Zhang, X. Non-alcoholic fatty liver disease: An expanded review. World J. Hepatol. 2017, 9, 715–732. [Google Scholar] [CrossRef]
- Albhaisi, S.; Sanyal, A. Recent advances in understanding and managing non-alcoholic fatty liver disease. F1000Research 2018, 7, 720. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Human Fatty Liver Disease: Old Questions and New Insights. Science 2011, 332, 1519–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The Diagnosis and Management of Non-alcoholic Fatty Liver Disease: Practice Guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology 2012, 142, 1592–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Lee, S.; Zhang, C.; Liu, Z.; Klevstig, M.; Mukhopadhyay, B.; Bergentall, M.; Cinar, R.; Stahlman, M.; Sikanic, N.; Park, J.K.; et al. Network analyses identify liver-specific targets for treating liver diseases. Mol. Syst. Biol. 2017, 13, 938. [Google Scholar] [CrossRef] [PubMed]
- Chella Krishnan, K.; Kurt, Z.; Barrere-Cain, R.; Sabir, S.; Das, A.; Floyd, R.; Vergnes, L.; Zhao, Y.; Che, N.; Charugundla, S.; et al. Integration of Multi-omics Data from Mouse Diversity Panel Highlights Mitochondrial Dysfunction in Non-alcoholic Fatty Liver Disease. Cell Syst. 2018, 6, 103–115.E7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mardinoglu, A.; Uhlen, M.; Borén, J. Broad Views of Non-alcoholic Fatty Liver Disease. Cell Syst. 2018, 6, 7–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muirhead, H. Isoenzymes of pyruvate kinase. Biochem. Soc. Trans. 1990, 18, 193–196. [Google Scholar] [CrossRef]
- Cardenas, J.M.; Dyson, R.D. Mammalian pyruvate kinase hybrid isozymes: Tissue distribution and physiological significance. J. Exp. Zool. 1978, 204, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Imamura, K.; Tanaka, T. Multimolecular Forms of Pyruvate Kinase from Rat and Other Mammalian Tissues I. Electrophoretic Studies. J. Biochem. 1972, 71, 1043–1051. [Google Scholar] [CrossRef] [PubMed]
- Strandholm, J.J.; Dyson, R.D.; Cardenas, J.M. Bovine pyruvate kinase isozymes and hybrid isozymes: Electrophoretic studies and tissue distribution. Arch. Biochem. Biophys. 1976, 173, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Noguchi, T. Nutrient and hormonal regulation of pyruvate kinase gene expression. Biochem. J. 1999, 337, 1–11. [Google Scholar] [CrossRef]
- Aslan, E.; Guler, C.; Adem, S. In vitro effects of some flavonoids and phenolic acids on human pyruvate kinase isoenzyme M2. J. Enzyme Inhib. Med. Chem. 2016, 31, 314–317. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Ding, G.-B.; Liu, W.; Fu, R.; Sajid, A.; Li, Z. Tannic acid directly targets pyruvate kinase isoenzyme M2 to attenuate colon cancer cell proliferation. Food Funct. 2018, 9, 5547–5559. [Google Scholar] [CrossRef]
- Adem, S.; Aslan, A.; Ahmed, I.; Krohn, K.; Guler, C.; Comaklı, V.; Demirdag, R.; Kuzu, M. Inhibitory and Activating Effects of Some Flavonoid Derivatives on Human Pyruvate Kinase Isoenzyme M2. Arch. Pharm. 2015, 349, 132–136. [Google Scholar] [CrossRef]
- Rodriguez-Ramiro, I.; Vauzour, D.; Minihane, A.M. Polyphenols and non-alcoholic fatty liver disease: Impact and mechanisms. Proc. Nutr. Soc. 2016, 75, 47–60. [Google Scholar] [CrossRef] [Green Version]
- Ludovico, A.; Natasa, M.; Francesco, L.; Luigi, B.; Antonino De, L. Polyphenols treatment in patients with nonalcoholic fatty liver disease. J. Transl. Int. Med. 2017, 5, 144–147. [Google Scholar]
- Li, S.; Tan, H.Y.; Wang, N.; Cheung, F.; Hong, M.; Feng, Y. The Potential and Action Mechanism of Polyphenols in the Treatment of Liver Diseases. Oxid. Med. Cell. Longev. 2018, 2018, 25. [Google Scholar] [CrossRef] [Green Version]
- Van De Wier, B.; Koek, G.H.; Bast, A.; Haenen, G.R.M.M. The potential of flavonoids in the treatment of non-alcoholic fatty liver disease. Crit. Rev. Food Sci. Nutr. 2017, 57, 834–855. [Google Scholar] [CrossRef] [PubMed]
- Sharifi-Rad, J.; Quispe, C.; Castillo, C.M.S.; Caroca, R.; Lazo-Vélez, M.A.; Antonyak, H.; Polishchuk, A.; Lysiuk, R.; Oliinyk, P.; De Masi, L.; et al. Ellagic Acid: A Review on Its Natural Sources, Chemical Stability, and Therapeutic Potential. Oxid. Med. Cell. Longev. 2022, 2022, 3848084. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Bonvini, P.; Zorzi, E.; Poletto, G.; Pagano, M.A.; Sarno, S.; Donella-Deana, A.; Zagotto, G.; Rosolen, A.; Pinna, L.A.; et al. Identification of ellagic acid as potent inhibitor of protein kinase CK2: A successful example of a virtual screening application. J. Med. Chem. 2006, 49, 2363–2366. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Gianoncelli, A.; Bonvini, P.; Zorzi, E.; Pasquale, R.; Rosolen, A.; Pinna, L.A.; Meggio, F.; Zagotto, G.; Moro, S. Urolithin as a Converging Scaffold Linking Ellagic acid and Coumarin Analogues: Design of Potent Protein Kinase CK2 Inhibitors. ChemMedChem 2011, 6, 2273–2286. [Google Scholar] [CrossRef]
- Dasaradhan, C.; Kumar, Y.S.; Prabakaran, K.; Khan, F.-R.N.; Jeong, E.D.; Chung, E.H. Efficient and convenient copper-free Pd(OAc)2/Ruphos-catalyzed Sonogashira coupling in the preparation of corfin analogues. Tetrahedron Lett. 2015, 56, 784–788. [Google Scholar] [CrossRef]
- Greenberg, J.A.; Sammakia, T. The Conversion of tert-Butyl Esters to Acid Chlorides Using Thionyl Chloride. J. Org. Chem. 2017, 82, 3245–3251. [Google Scholar] [CrossRef]
- Toure, M.; Jaime-Figueroa, S.; Burslem, G.M.; Crews, C.M. Expeditious Synthesis of Isoquinolones and Isocoumarins with a Vinyl Borane as an Acetylene Equivalent. Eur. J. Org. Chem. 2016, 2016, 4171–4175. [Google Scholar] [CrossRef] [Green Version]
- Battisti, U.M.; Jozwiak, K.; Cannazza, G.; Puia, G.; Stocca, G.; Braghiroli, D.; Parenti, C.; Brasili, L.; Carrozzo, M.M.; Citti, C.; et al. 5-Arylbenzothiadiazine Type Compounds as Positive Allosteric Modulators of AMPA/Kainate Receptors. ACS Med. Chem. Lett. 2012, 3, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.-F.; Li, X.; Wang, X.; Zou, M.; Tang, C.; Liang, Y.; Song, S.; Jiao, N. Conversion of Simple Cyclohexanones into Catechols. J. Am. Chem. Soc. 2016, 138, 12271–12277. [Google Scholar] [CrossRef]
- Goins, C.M.; Dajnowicz, S.; Thanna, S.; Sucheck, S.J.; Parks, J.M.; Ronning, D.R. Exploring Covalent Allosteric Inhibition of Antigen 85C from Mycobacterium tuberculosis by Ebselen Derivatives. ACS Infect. Dis. 2017, 3, 378–387. [Google Scholar] [CrossRef]
- Weidner-Wells, M.A.; Altom, J.; Fernandez, J.; Fraga-Spano, S.A.; Hilliard, J.; Ohemeng, K.; Barrett, J.F. DNA gyrase inhibitory activity of ellagic acid derivatives. Bioorg. Med. Chem. Lett. 1998, 8, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; McNae, I.W.; Chen, Y.; Blackburn, E.A.; Wear, M.A.; Michels, P.A.M.; Fothergill-Gilmore, L.A.; Hupp, T.; Walkinshaw, M.D. An allostatic mechanism for M2 pyruvate kinase as an amino-acid sensor. Biochem. J. 2018, 475, 1821–1837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holyoak, T.; Zhang, B.; Deng, J.; Tang, Q.; Prasannan, C.B.; Fenton, A.W. Energetic coupling between an oxidizable cysteine and the phosphorylatable N-terminus of human liver pyruvate kinase. Biochem. 2013, 52, 466–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, H.P.; O’Reilly, F.J.; Wear, M.A.; O’Neill, J.R.; Fothergill-Gilmore, L.A.; Hupp, T.; Walkinshaw, M.D. M2 pyruvate kinase provides a mechanism for nutrient sensing and regulation of cell proliferation. Proc. Natl. Acad. Sci. USA 2013, 110, 5881–5886. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the Role of the Crystal Environment in Determining Protein Side-chain Conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef]
- García-Niño, W.R.; Zazueta, C. Ellagic acid: Pharmacological activities and molecular mechanisms involved in liver protection. Pharmacol. Res. 2015, 97, 84–103. [Google Scholar] [CrossRef]
- Cozza, G. The Development of CK2 Inhibitors: From Traditional Pharmacology to in Silico Rational Drug Design. Pharmaceuticals 2017, 10, 26. [Google Scholar] [CrossRef] [Green Version]
- Tang, Q.; Villar, M.T.; Artigues, A.; Thyfault, J.P.; Apte, U.; Zhu, H.; Peterson, K.R.; Fenton, A.W. Mutational mimics of allosteric effectors: A genome editing design to validate allosteric drug targets. Sci. Rep. 2019, 9, 9031. [Google Scholar] [CrossRef] [Green Version]
- Anastasiou, D.; Yu, Y.; Israelsen, W.J.; Jiang, J.-K.; Boxer, M.B.; Hong, B.S.; Tempel, W.; Dimov, S.; Shen, M.; Jha, A.; et al. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat. Chem. Biol. 2012, 8, 839. [Google Scholar] [CrossRef] [Green Version]
- Kyriakis, E.; Stravodimos, G.A.; Kantsadi, A.L.; Chatzileontiadou, D.S.M.; Skamnaki, V.T.; Leonidas, D.D. Natural flavonoids as antidiabetic agents. The binding of gallic and ellagic acids to glycogen phosphorylase b. FEBS Lett. 2015, 589, 1787–1794. [Google Scholar] [CrossRef]
- Sekiguchi, Y.; Nakaniwa, T.; Kinoshita, T.; Nakanishi, I.; Kitaura, K.; Hirasawa, A.; Tsujimoto, G.; Tada, T. Structural insight into human CK2α in complex with the potent inhibitor ellagic acid. Bioorg. Med. Chem. Lett. 2009, 19, 2920–2923. [Google Scholar] [CrossRef] [PubMed]
- Evtyugin, D.D.; Magina, S.; Evtuguin, D.V. Recent advances in the production and applications of ellagic acid and its derivatives. A review. Molecules 2020, 25, 2745. [Google Scholar] [CrossRef]
- Seo, C.S.; Jeong, S.J.; Yoo, S.R.; Lee, N.R.; Shin, H.K. Quantitative analysis and in vitro anti-inflammatory effects of gallic acid, ellagic acid, and quercetin from radix sanguisorbae. Pharmacogn Mag. 2016, 12, 104–108. [Google Scholar] [CrossRef] [Green Version]
- Polce, S.A.; Burke, C.; França, L.M.; Kramer, B.; de Andrade Paes, A.M.; Carrillo-Sepulveda, M.A. Ellagic acid alleviates hepatic oxidative stress and insulin resistance in diabetic female rats. Nutrients 2018, 10, 531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Yan, Y.; Jiang, Y.; Meng, X. Ellagic Acid and Its Anti-Aging Effects on Central Nervous System. Int. J. Mol. Sci. 2022, 23, 10937. [Google Scholar] [CrossRef] [PubMed]
- Okla, M.; Kang, I.; Kim, D.M.; Gourineni, V.; Shay, N.; Gu, L.; Chung, S. Ellagic acid modulates lipid accumulation in primary human adipocytes and human hepatoma Huh7 cells via discrete mechanisms. J. Nutr. Biochem. 2015, 26, 82–90. [Google Scholar] [CrossRef]
- Zhang, C.; Hu, J.; Sheng, L.; Yuan, M.; Wu, Y.; Chen, L.; Wang, G.; Qiu, Z. Ellagic acid ameliorates AKT-driven hepatic steatosis in mice by suppressing de novo lipogenesis via the AKT/SREBP-1/FASN pathway. Food Funct. 2019, 10, 3410–3420. [Google Scholar] [CrossRef] [PubMed]
- Poulose, N.; Vishnu Prasad, C.N.; Nidhina Haridas, P.A.; Anilkumar, G. Ellagic Acid Stimulates Glucose Transport in Adipocytes and Muscles through AMPK Mediated Pathway. J. Diabetes Metab. 2011, 2, 149. [Google Scholar] [CrossRef]
- Kang, I.; Kim, Y.; Tomás-Barberán, F.A.; Espín, J.C.; Chung, S. Urolithin A, C, and D, but not iso-urolithin A and urolithin B, attenuate triglyceride accumulation in human cultures of adipocytes and hepatocytes. Mol. Nutr. Food Res. 2016, 60, 1129–1138. [Google Scholar] [CrossRef] [Green Version]
- ALTamimi, J.Z.; Alshammari, G.M.; AlFaris, N.A.; Alagal, R.I.; Aljabryn, D.H.; Albekairi, N.A.; Alkhateeb, M.A.; Yahya, M.A. Ellagic acid protects against non-alcoholic fatty liver disease in streptozotocin-diabetic rats by activating AMPK. Pharm. Biol. 2022, 60, 25–37. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | PKL IC50 | PKR IC50 | PKM1 IC50 | PKM2 IC50 |
|---|---|---|---|---|
| 1 | 0.032 µM | 4.7 µM | 81 µM | 0.07 µM a |
| 2 | 12 µM | >100 µM | N.D. | N.D. |
| 3 | 2.1 µM | 7.4 µM | N.D. | N.D. |
| 4 | 0.025 µM | 0.49 µM | 8 µM | >100 µM |
| 5 | 0.028 µM | 1.27 µM | 36 µM | 0.03 µM a |
| 6 | Inactive b | Inactive b | N.D. | N.D. |
| 7 | Inactive b | Inactive b | N.D. | N.D. |
| 8 | 3.5 µM | 12 µM | N.D. | N.D. |
| 9 | 2.4 µM | 5.1 µM | N.D. | N.D. |
| 10 | 0.41 µM | 2.6 µM | N.D. | N.D. |
| 11 | Inactive b | Inactive b | N.D. | N.D. |
| 12 | Inactive b | Inactive b | N.D. | N.D. |
| 13 | 3.9 µM | 43 µM | N.D. | N.D. |
| 14 | 2.1 µM | 68 µM | N.D. | N.D. |
| 15 | 1.7 µM | >100 µM | N.D. | N.D. |
| FBP * | 0.02 µM a | 0.01 µM a | Inactive | 0.05 µM a |
| Binding Site | ∆Gbind (kcal/mol) a |
|---|---|
| FBP | −66.56 |
| PEP | −39.51 |
| Phe | −72.11 |
| Inactive tetramer interface | −75.36 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Battisti, U.M.; Gao, C.; Akladios, F.; Kim, W.; Yang, H.; Bayram, C.; Bolat, I.; Kiliclioglu, M.; Yuksel, N.; Tozlu, O.O.; et al. Ellagic Acid and Its Metabolites as Potent and Selective Allosteric Inhibitors of Liver Pyruvate Kinase. Nutrients 2023, 15, 577. https://doi.org/10.3390/nu15030577
Battisti UM, Gao C, Akladios F, Kim W, Yang H, Bayram C, Bolat I, Kiliclioglu M, Yuksel N, Tozlu OO, et al. Ellagic Acid and Its Metabolites as Potent and Selective Allosteric Inhibitors of Liver Pyruvate Kinase. Nutrients. 2023; 15(3):577. https://doi.org/10.3390/nu15030577
Chicago/Turabian StyleBattisti, Umberto Maria, Chunixa Gao, Fady Akladios, Woonghee Kim, Hong Yang, Cemil Bayram, Ismail Bolat, Metin Kiliclioglu, Nursena Yuksel, Ozlem Ozdemir Tozlu, and et al. 2023. "Ellagic Acid and Its Metabolites as Potent and Selective Allosteric Inhibitors of Liver Pyruvate Kinase" Nutrients 15, no. 3: 577. https://doi.org/10.3390/nu15030577