
Endothelial Glycocalyx Preservation—Impact of Nutrition and Lifestyle

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
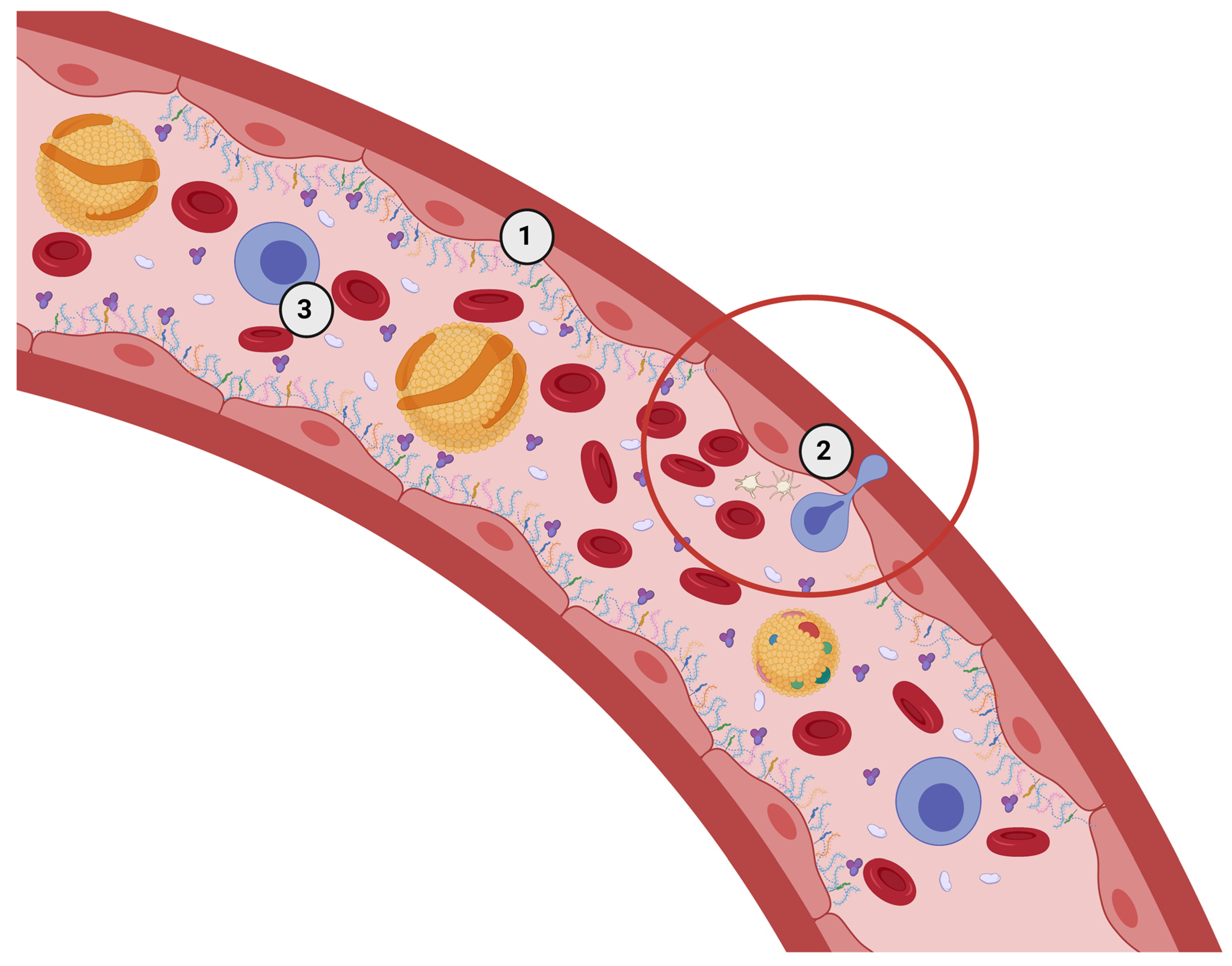
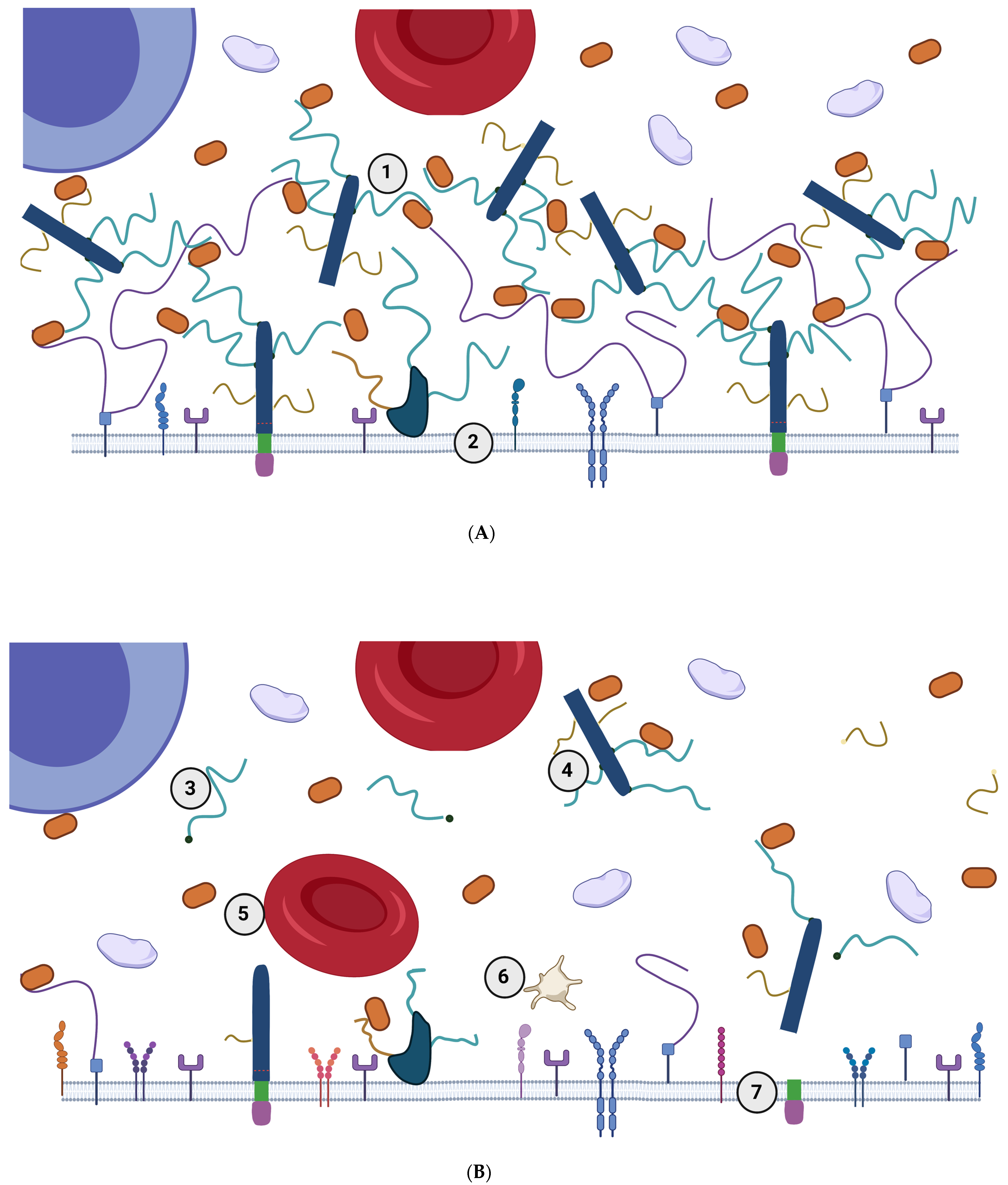
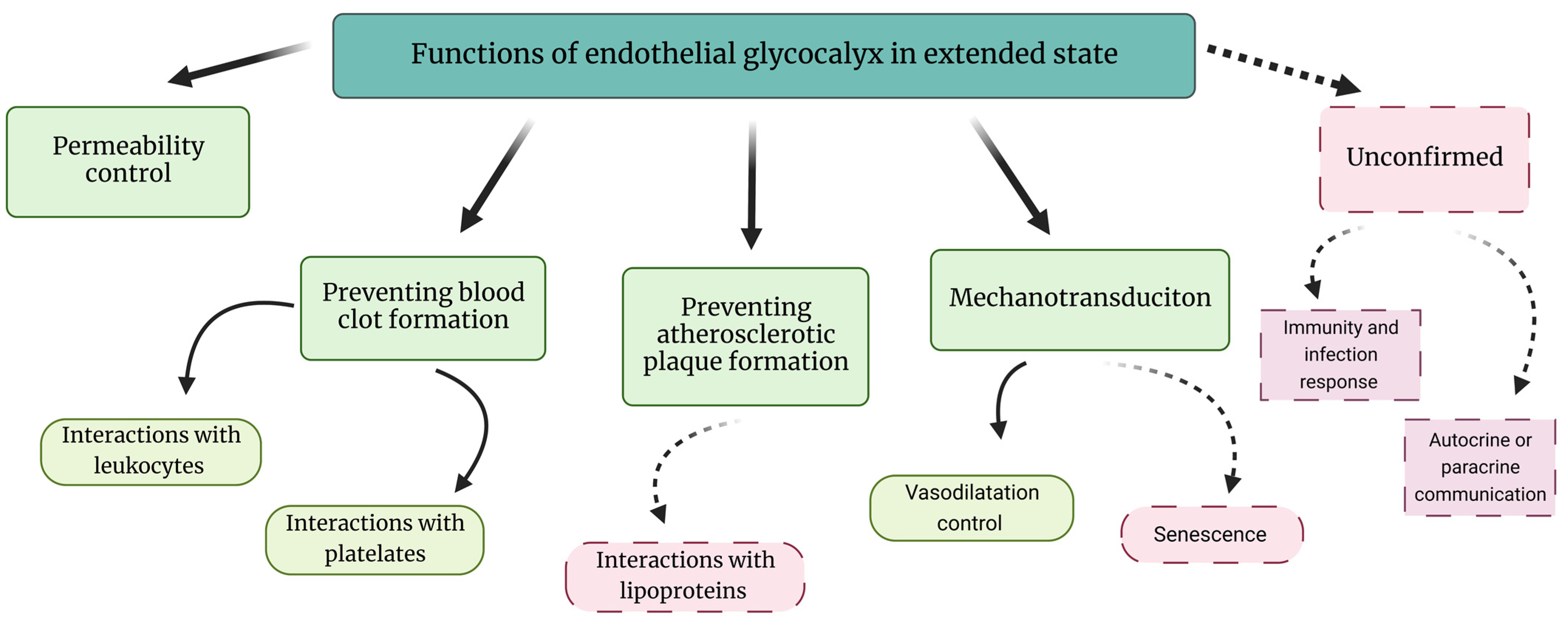
2. The eGC in Healthy Conditions—Extended State of eGC
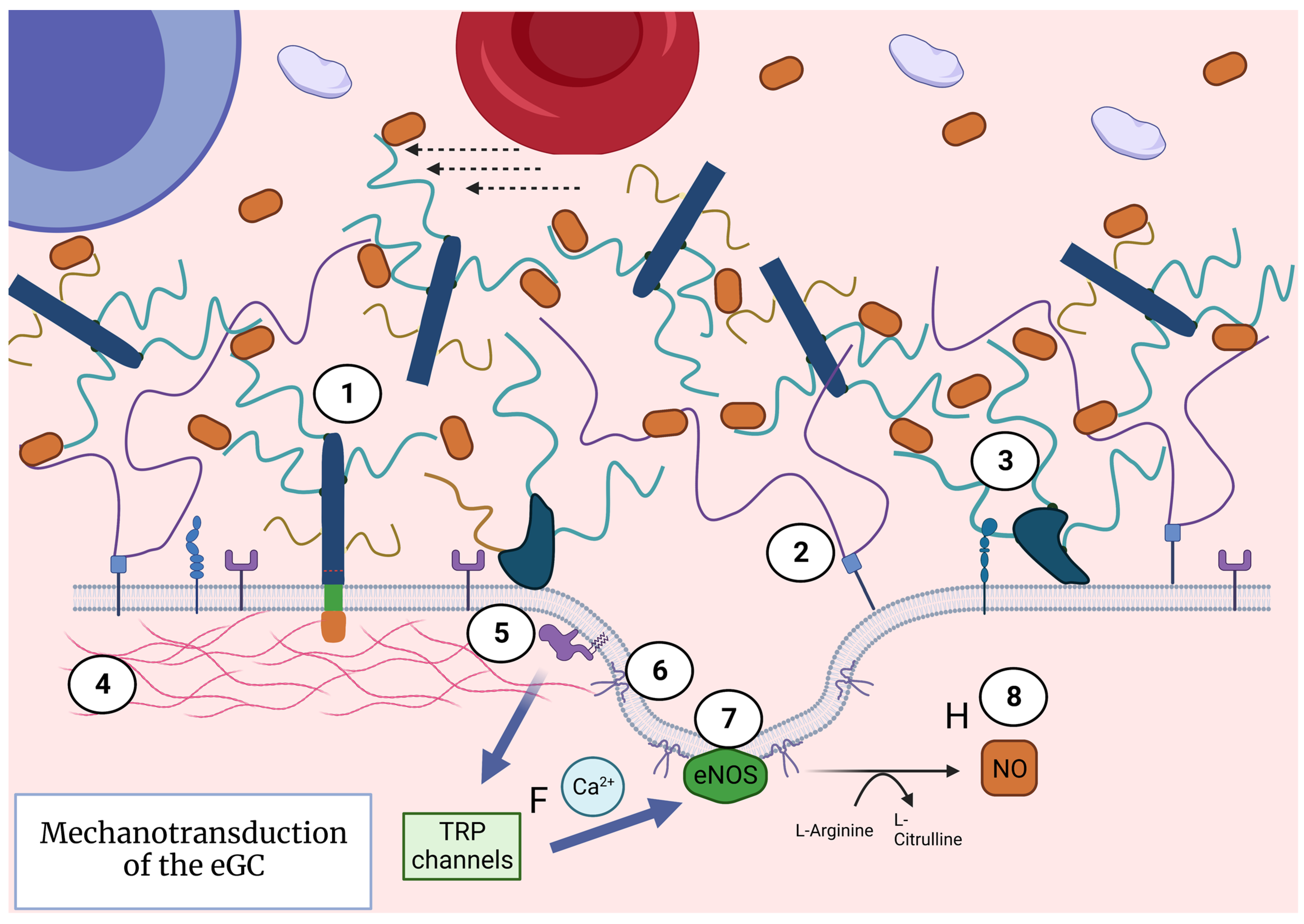
3. Extended eGC Activates Mechanotransduction and Endothelial Nitric Oxide Release
4. The eGC in a State of Low-Grade Inflammation—Diminished eGC
4.1. eGC Response to Inflammation
4.2. Mechanisms of eGC Collapse
5. eGC in Chronic Diseases
5.1. Hypertension
5.2. Atherosclerosis
5.3. Abdominal Obesity
5.4. Hyperglycemia, Type 2 Diabetes, and Metabolic Syndrome
5.5. Chronic Kidney Disease
5.6. Chronic Inflammation
6. Perspectives of Nutritional Therapy for eGC Health
6.1. Preventing Vitamin D Deficiency
6.2. Vitamin D and eGC Connection Hypothesis
6.3. Supplementing with Omega-3 Fatty Acids and Probiotics
6.4. Providing the eGC Building Blocks
6.5. Dietary Sulfur
6.6. Dietary Nitrates
6.7. Lifestyle Changes
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alphonsus, C.S.; Rodseth, R.N. The endothelial glycocalyx: A review of the vascular barrier. Anaesthesia 2014, 69, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Jacob, M.; Rehm, M.; Loetsch, M.; Paul, J.O.; Bruegger, D.; Welsch, U.; Conzen, P.; Becker, B.F. The endothelial glycocalyx prefers albumin for evoking shear stress-induced, nitric oxide-mediated coronary dilatation. J. Vasc. Res. 2007, 44, 435–443. [Google Scholar] [CrossRef] [PubMed]
- DellaValle, B.; Hasseldam, H.; Johansen, F.F.; Iversen, H.K.; Rungby, J.; Hempel, C. Multiple soluble components of the glycocalyx are increased in patient plasma after ischemic stroke. Stroke 2019, 50, 2948–2951. [Google Scholar] [CrossRef]
- Mitra, R.; O’Neil, G.L.; Harding, I.C.; Cheng, M.J.; Mensah, S.A.; Ebong, E.E. Glycocalyx in Atherosclerosis-Relevant Endothelium Function and as a Therapeutic Target. Curr. Atheroscler. Rep. 2017, 1, 63. [Google Scholar] [CrossRef]
- Ikonomidis, I.; Voumvourakis, A.; Makavos, G.; Triantafyllidi, H.; Pavlidis, G.; Katogiannis, K.; Benas, D.; Vlastos, D.; Trivilou, P.; Varoudi, M.; et al. Association of impaired endothelial glycocalyx with arterial stiffness, coronary microcirculatory dysfunction, and abnormal myocardial deformation in untreated hypertensives. J. Clin. Hypertens. 2018, 20, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Nieuwdorp, M.; van Haeften, T.W.; Gouverneur, M.C.L.G.; Mooij, H.L.; van Lieshout, M.H.P.; Levi, M.; Meijers, J.C.M.; Holleman, F.; Hoekstra, J.B.L.; Vink, H.; et al. Loss of endothelial glycocalyx during acute hyperglycemia coincides with endothelial dysfunction and coagulation activation in vivo. Diabetes 2006, 55, 480–486. [Google Scholar] [CrossRef]
- Yilmaz, O.; Afsar, B.; Ortiz, A.; Kanbay, M. The role of endothelial glycocalyx in health and disease. Clin. Kidney J. 2019, 12, 611–619. [Google Scholar] [CrossRef]
- Annecke, T.; Chappell, D.; Chen, C.; Jacob, M.; Welsch, U.; Sommerhoff, C.P.; Rehm, M.; Conzen, P.F.; Becker, B.F. Sevoflurane preserves the endothelial glycocalyx against ischaemia-reperfusion injury. Br. J. Anaesth. 2010, 104, 414–421. [Google Scholar] [CrossRef]
- Iba, T.; Levy, J.H. Derangement of the endothelial glycocalyx in sepsis. J. Thromb. Haemost. 2019, 17, 283–294. [Google Scholar] [CrossRef]
- Astapenko, D.; Benes, J.; Pouska, J.; Lehmann, C.; Islam, S.; Cerny, V. Endothelial glycocalyx in acute care surgery—What anaesthesiologists need to know for clinical practice. BMC Anesthesiol. 2019, 19, 238. [Google Scholar] [CrossRef]
- Maldonado, F.; Morales, D.; Gutiérrez, R.; Barahona, M.; Cerda, O.; Cáceres, M. Effect of sevoflurane and propofol on tourniquet-induced endothelial damage: A pilot randomized controlled trial for knee-ligament surgery. BMC Anesthesiol. 2020, 20, 121. [Google Scholar] [CrossRef] [PubMed]
- Kolářová, H.; Ambrůzová, B.; Švihálková Šindlerová, L.; Klinke, A.; Kubala, L. Modulation of endothelial glycocalyx structure under inflammatory conditions. Mediat. Inflamm. 2014, 2014, 694312. [Google Scholar] [CrossRef]
- Drake-Holland, A.J.; Noble, M.I.M. Update on the Important New Drug Target in Cardiovascular Medicine—The Vascular Glycocalyx. Cardiovasc. Hematol. Disord. Drug Targets 2012, 12, 76–81. [Google Scholar] [CrossRef]
- Groner, W.; Winkelman, J.W.; Harris, A.G.; Ince, C.; Bouma, G.J.; Messmer, K.; Nadeau, R.G. Orthogonal polarization spectral imaging: A new method for study of the microcirculation. Nat. Med. 1999, 5, 1209–1213. [Google Scholar] [PubMed]
- Vlahu, C.A.; Lemkes, B.A.; Struijk, D.G.; Koopman, M.G.; Krediet, R.T.; Vink, H. Damage of the endothelial glycocalyx in dialysis patients. J. Am. Soc. Nephrol. 2012, 23, 1900–1908. [Google Scholar] [CrossRef]
- Chappell, D.; Hofmann-Kiefer, K.; Jacob, M.; Rehm, M.; Briegel, J.; Welsch, U.; Conzen, P.; Becker, B.F. TNF-α induced shedding of the endothelial glycocalyx is prevented by hydrocortisone and antithrombin. Basic Res. Cardiol. 2009, 104, 78–89. [Google Scholar] [CrossRef]
- Oohira, A.; Wight, T.N.; Bornstein, P. Sulfated proteoglycans synthesized by vascular endothelial cells in culture. J. Biol. Chem. 1983, 258, 2014–2021. [Google Scholar] [CrossRef]
- Ushiyama, A.; Kataoka, H.; Iijima, T. Glycocalyx and its involvement in clinical pathophysiologies. J. Intensive Care 2016, 4, 59. [Google Scholar] [CrossRef] [PubMed]
- Dogné, S.; Flamion, B.; Caron, N. Endothelial glycocalyx as a shield against diabetic vascular complications: Involvement of hyaluronan and hyaluronidases. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1427–1439. [Google Scholar] [CrossRef]
- Dogné, S.; Flamion, B. Endothelial Glycocalyx Impairment in Disease: Focus on Hyaluronan Shedding. Am. J. Pathol. Am. Soc. Investig. Pathol. 2020, 190, 768–780. [Google Scholar] [CrossRef]
- Singh, S.S.; Heijmans, R.; Meulen, C.K.E.; Lieverse, A.G.; Gornik, O.; Sijbrands, E.J.G.; Lauc, G.; van Hoek, M. Association of the IgG N-glycome with the course of kidney function in type 2 diabetes. BMJ Open Diabetes Res. Care 2020, 8, e001026. [Google Scholar] [CrossRef]
- Tarbell, J.M.; Pahakis, M.Y. Mechanotransduction and the glycocalyx. J. Intern. Med. 2006, 259, 339–350. [Google Scholar] [CrossRef]
- Mc Clatchey, P.M.; Schafer, M.; Hunter, K.S.; Reusch, J.E. The endothelial glycocalyx promotes homogenous blood flow distribution within the microvasculature. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H168–H176. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.J.; Lillicrap, D. A sticky proposition: The endothelial glycocalyx and von Willebrand factor. J. Thromb. Haemost. 2020, 18, 781–785. [Google Scholar] [CrossRef] [PubMed]
- Oberleithner, H. Vascular endothelium: A vulnerable transit zone for merciless sodium. Nephrol. Dial. Transplant. 2014, 29, 440–446. [Google Scholar] [CrossRef]
- Chappell, D.; Jacob, M.; Paul, O.; Rehm, M.; Welsch, U.; Stoeckelhuber, M.; Conzen, P.; Becker, B.F. The glycocalyx of the human umbilical vein endothelial cell: An impressive structure ex vivo but not in culture. Circ. Res. 2009, 104, 1313–1317. [Google Scholar] [CrossRef] [PubMed]
- Becker, B.F.; Jacob, M.; Leipert, S.; Salmon, A.H.J.; Chappell, D. Degradation of the endothelial glycocalyx in clinical settings: Searching for the sheddases. Br. J. Clin. Pharmacol. 2015, 80, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Pries, A.R.; Secomb, T.W.; Gaehtgens, P. The endothelial surface layer. Pflug. Arch. Eur. J. Physiol. 2000, 440, 653–666. [Google Scholar] [CrossRef]
- Van Haaren, P.M.A.; VanBavel, E.; Vink, H.; Spaan, J.A.E. Charge modification of the endothelial surface layer modulates the permeability barrier of isolated rat mesenteric small arteries. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, 2503–2507. [Google Scholar] [CrossRef]
- Constantinescu, A.A.; Vink, H.; Spaan, J.A.E. Endothelial cell glycocalyx modulates immobilization of leukocytes at the endothelial surface. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1541–1547. [Google Scholar] [CrossRef]
- Dragovich, M.A.; Chester, D.; Fu, B.M.; Wu, C.; Xu, Y.; Goligorsky, M.S.; Zhang, X.F. Mechanotransduction of the endothelial glycocalyx mediates nitric oxide production through activation of TRP channels. Am. J. Physiol. Cell Physiol. 2016, 311, C846–C853. [Google Scholar] [CrossRef] [PubMed]
- Thi, M.M.; Tarbell, J.M.; Weinbaum, S.; Spray, D.C. The role of the glycocalyx in reorganization of the actin cytoskeleton under fluid shear stress: A “bumper-car” model. Proc. Natl. Acad. Sci. USA 2004, 101, 16483–16488. [Google Scholar] [CrossRef] [PubMed]
- Cohen, R.A.; Adachi, T. Nitric-Oxide-Induced Vasodilatation: Regulation by Physiologic S-Glutathiolation and Pathologic Oxidation of the Sarcoplasmic Endoplasmic Reticulum Calcium ATPase. Trends Cardiovasc. Med. 2006, 16, 109–114. [Google Scholar] [CrossRef]
- Yang, X.; Meegan, J.E.; Jannaway, M.; Coleman, D.C.; Yuan, S.Y. A disintegrin and metalloproteinase 15-mediated glycocalyx shedding contributes to vascular leakage during inflammation. Cardiovasc. Res. 2018, 114, 1752–1763. [Google Scholar] [CrossRef]
- Teng, Y.H.F.; Aquino, R.S.; Park, P.W. Molecular functions of syndecan-1 in disease. Matrix Biol. 2012, 31, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Melrose, J. Glycosaminoglycans in Wound Healing. Bone Tissue Regen. Insights 2016, 7, BTRI.S38670. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, C.Z.; Swaroop, M.; Xu, M.; Wang, L.; Lee, J.; Wang, A.Q.; Pradhan, M.; Hagen, N.; Chen, L.; et al. Heparan sulfate assists SARS-CoV-2 in cell entry and can be targeted by approved drugs in vitro. Cell Discov. 2020, 6, 80. [Google Scholar] [CrossRef]
- Parameswaran, N.; Patial, S. Tumor necrosis factor-α signaling in macrophages. Crit. Rev. Eukaryot. Gene Expr. 2010, 20, 87–103. [Google Scholar] [CrossRef]
- Jannaway, M.; Yang, X.; Meegan, J.E.; Coleman, D.C.; Yuan, S.Y. Thrombin-cleaved syndecan-3/-4 ectodomain fragments mediate endothelial barrier dysfunction. PLoS ONE 2019, 14, e0214737. [Google Scholar] [CrossRef]
- Pillinger, N.L.; Kam, P.C.A. Endothelial glycocalyx: Basic science and clinical implications. Anaesth. Intensive Care 2017, 45, 295–307. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Regulation of NF-κB by TNF Family Cytokines. Semin. Immunol. 2014, 26, 253–266. [Google Scholar] [CrossRef]
- Strand, M.E.; Herum, K.M.; Rana, Z.A.; Skrbic, B.; Askevold, E.T.; Dahl, C.P.; Vistnes, M.; Hasic, A.; Kvaløy, H.; Sjaastad, I.; et al. Innate immune signaling induces expression and shedding of the heparan sulfate proteoglycan syndecan-4 in cardiac fibroblasts and myocytes, affecting inflammation in the pressure-overloaded heart. FEBS J. 2013, 280, 2228–2247. [Google Scholar] [CrossRef]
- Lipphardt, M.; Dihazi, H.; Müller, G.A.; Goligorsky, M.S. Fibrogenic secretome of sirtuin 1-deficient endothelial cells: Wnt, notch and glycocalyx rheostat. Front. Physiol. 2018, 9, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sieve, I.; Münster-Kühnel, A.K.; Hilfiker-Kleiner, D. Regulation and function of endothelial glycocalyx layer in vascular diseases. Vasc. Pharmacol. 2018, 100, 26–33. [Google Scholar] [CrossRef]
- Oberleithner, H.; Riethmüller, C.; Schillers, H.; MacGregor, G.A.; de Wardener, H.E.; Hausberg, M. Plasma sodium stiffens vascular endothelium and reduces nitric oxide release. Proc. Natl. Acad. Sci. USA 2007, 104, 16281. [Google Scholar] [CrossRef] [PubMed]
- Oberleithner, H.; Peters, W.; Kusche-Vihrog, K.; Korte, S.; Schillers, H.; Kliche, K.; Oberleithner, K. Salt overload damages the glycocalyx sodium barrier of vascular endothelium. Pflug. Arch. Eur. J. Physiol. 2011, 462, 519–528. [Google Scholar] [CrossRef]
- Rorije, N.M.G.; Rademaker, E.; Schrooten, E.M.; Wouda, R.D.; van der Heide, J.J.H.; van den Born, B.J.H.; Vogt, L. High-salt intake affects sublingual microcirculation and is linked to body weight change in healthy volunteers: A randomized cross-over trial. J. Hypertens. 2019, 37, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, B.M.; Spaan, J.A.E.; Rolf, T.M.; Vink, H. Atherogenic region and diet diminish glycocalyx dimension and increase intima-to-media ratios at murine carotid artery bifurcation. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, 915–920. [Google Scholar] [CrossRef] [PubMed]
- Chiu, J.J.; Usami, S.; Chien, S. Vascular endothelial responses to altered shear stress: Pathologic implications for atherosclerosis. Ann. Med. 2009, 41, 19–28. [Google Scholar] [CrossRef]
- Hahn, R.G.; Patel, V.; Dull, R.O. Human glycocalyx shedding: Systematic review and critical appraisal. Acta Anaesthesiol. Scand. 2021, 65, 590–606. [Google Scholar] [CrossRef]
- Valerio, L.; Peters, R.J.; Zwinderman, A.H.; Pinto-Sietsma, S.J. Sublingual endothelial glycocalyx and atherosclerosis. A cross-sectional study. PLoS ONE 2019, 14, e0213097. [Google Scholar] [CrossRef]
- Giles, J.T.; Wasko, M.C.M.; Chung, C.P.; Szklo, M.; Blumenthal, R.S.; Kao, A.; Bokhari, S.; Zartoshti, A.; Stein, C.M.; Bathon, J.M. Exploring the Lipid Paradox Theory in Rheumatoid Arthritis: Associations of Low Circulating Low-Density Lipoprotein Concentration with Subclinical Coronary Atherosclerosis. Arthritis Rheumatol. 2019, 71, 1426–1436. [Google Scholar] [CrossRef] [PubMed]
- Varvel, S.A.; Dayspring, T.D.; Edmonds, Y.; Thiselton, D.L.; Ghaedi, L.; Voros, S.; McConnell, J.P.; Sasinowski, M.; Dall, T.; Warnick, G.R. Discordance between apolipoprotein B and low-density lipoprotein particle number is associated with insulin resistance in clinical practice. J. Clin. Lipidol. 2015, 9, 247–255. [Google Scholar] [CrossRef]
- Lechner, K.; Mckenzie, A.L.; Kränkel, N.; von Schacky, C.; Worm, N.; Nixdorff, U.; Lechner, B.; Scherr, J.; Weingärtner, O.; Krauss, R.M. High-Risk Atherosclerosis and Metabolic Phenotype: The Roles of Ectopic Adiposity, Atherogenic Dyslipidemia, and Inflammation. Metab. Syndr. Relat. Disord. 2020, 18, 176–185. [Google Scholar] [CrossRef]
- Neeland, I.J.; Ross, R.; Després, J.P.; Matsuzawa, Y.; Yamashita, S.; Shai, I.; Seidell, J.; Magni, P.; Santos, R.D.; Arsenault, B.; et al. Visceral and ectopic fat, atherosclerosis, and cardiometabolic disease: A position statement. Lancet Diabetes Endocrinol. 2019, 7, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Piperi, C.; Adamopoulos, C.; Dalagiorgou, G.; Diamanti-Kandarakis, E.; Papavassiliou, A.G. Crosstalk between advanced glycation and endoplasmic reticulum stress: Emerging therapeutic targeting for metabolic diseases. J. Clin. Endocrinol. Metab. 2012, 97, 2231–2242. [Google Scholar] [CrossRef] [PubMed]
- Jamwal, S.; Sharma, S. Vascular endothelium dysfunction: A conservative target in metabolic disorders. Inflamm. Res. 2018, 67, 391–405. [Google Scholar] [CrossRef]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef]
- Fehsel, K.; Jalowy, A.; Qi, S.; Burkart, V.; Hartmann, B.; Kolb, H. Islet cell DNA is a target of inflammatory attack by nitric oxide. Diabetes 1993, 42, 496–500. [Google Scholar] [CrossRef] [PubMed]
- Udi, S.; Hinden, L.; Ahmad, M.; Drori, A.; Iyer, M.R.; Cinar, R.; Herman-Edelstein, M.; Tam, J. Dual inhibition of cannabinoid CB1 receptor and inducible NOS attenuates obesity-induced chronic kidney disease. Br. J. Pharmacol. 2020, 177, 110–127. [Google Scholar] [CrossRef]
- Łuczak, A.; Madej, M.; Kasprzyk, A.; Doroszko, A. Role of the eNOS Uncoupling and the Nitric Oxide Metabolic Pathway in the Pathogenesis of Autoimmune Rheumatic Diseases. Oxidative Med. Cell. Longev. 2020, 2020, 1417981. [Google Scholar] [CrossRef] [PubMed]
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced glycation end products: Sparking the development of diabetic vascular injury. Circulation 2006, 114, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Gryszczyńska, B.; Budzyń, M.; Begier-Krasińska, B.; Osińska, A.; Boruczkowski, M.; Kaczmarek, M.; Bukowska, A.; Iskra, M.; Kasprzak, M.P. Association between advanced glycation end products, soluble RAGE receptor, and endothelium dysfunction, evaluated by circulating endothelial cells and endothelial progenitor cells in patients with mild and resistant hypertension. Int. J. Mol. Sci. 2019, 20, 3942. [Google Scholar] [CrossRef]
- Mudau, M.; Genis, A.; Lochner, A.; Strijdom, H. Endothelial dysfunction: The early predictor of atherosclerosis. Cardiovasc. J. Afr. 2012, 23, 222–231. [Google Scholar] [CrossRef]
- Cai, W.; He, J.C.; Zhu, L.; Peppa, M.; Lu, C.; Uribarri, J.; Vlassara, H. High levels of dietary advanced glycation end products transform low-density lipoprotein into a potent redox-sensitive mitogen-activated protein kinase stimulant in diabetic patients. Circulation 2004, 110, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Vlassara, H.; Uribarri, J. Advanced glycation end products (AGE) and diabetes: Cause, effect, or both? Curr. Diabetes Rep. 2014, 14, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Hellsten, Y.; Rufener, N.; Nielsen, J.J.; Høier, B.; Krustrup, P.; Bangsbo, J. Passive leg movement enhances interstitial VEGF protein, endothelial cell proliferation, and eNOS mRNA content in human skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.K.; Hevener, A.L.; Barnard, R.J. Metabolic syndrome and insulin resistance: Underlying causes and modification by exercise training. Compr. Physiol. 2013, 3, 1–58. [Google Scholar] [PubMed]
- Pertynska-Marczewska, M.; Merhi, Z. Relationship of advanced glycation end products with cardiovascular disease in menopausal women. Reprod. Sci. 2015, 22, 774–782. [Google Scholar] [CrossRef]
- Nieuwdorp, M.M.; Kroon, H.L.; Atasever, J.; Spaan, B.; Ince, J.A.E.; Holleman, C.; Diamant, F.; Heine, M.; Hoekstra, R.J.; Joost, B.L.; et al. Endothelial Glycocalyx Damage Coincides with Microalbuminuria in Type 1 Diabetes. Diabetes 2005, 55, 1127–1132. [Google Scholar] [CrossRef]
- Primary Causes of End-Stage Renal Disease. Available online: https://www.uspharmacist.com/article/primary-causes-of-endstage-renal-disease (accessed on 30 May 2023).
- Stephen, R.; Jolly, S.E.; Nally, J.V.; Navaneethan, S.D. Albuminuria: When urine predicts kidney and cardiovascular disease. Clevel. Clin. J. Med. 2014, 81, 41–50. [Google Scholar] [CrossRef]
- Van den Hoven, M.J.; Rops, A.L.; Bakker, M.A.; Aten, J.; Rutjes, N.; Roestenberg, P.; Goldschmeding, R.; Zcharia, E.; Vlodavsky, I.; van der Vlag, J.; et al. Increased expression of heparanase in overt diabetic nephropathy. Kidney Int. 2006, 70, 2100–2108. [Google Scholar] [CrossRef]
- Garsen, M.; Rops, A.L.W.M.M.; Dijkman, H.; Willemsen, B.; van Kuppevelt, T.H.; Russel, F.G.; Rabelink, T.J.; Berden, J.H.M.; Reinheckel, T.; van der Vlag, J. Cathepsin L is crucial for the development of early experimental diabetic nephropathy. Kidney Int. 2016, 90, 1012–1022. [Google Scholar] [CrossRef] [PubMed]
- Boels, M.G.S.; Koudijs, A.; Avramut, M.C.; Sol, W.M.P.J.; Wang, G.; van Oeveren-Rietdijk, A.M.; van Zonneveld, A.J.; de Boer, H.C.; van der Vlag, J.; van Kooten, C.; et al. Systemic Monocyte Chemotactic Protein-1 Inhibition Modifies Renal Macrophages and Restores Glomerular Endothelial Glycocalyx and Barrier Function in Diabetic Nephropathy. Am. J. Pathol. 2017, 187, 2430–2440. [Google Scholar] [CrossRef] [PubMed]
- Wassenaar, T.M.; Zimmermann, K. Lipopolysaccharides in Food, Food Supplements, and Probiotics: Should We be Worried? Eur. J. Microbiol. Immunol. 2018, 8, 63. [Google Scholar] [CrossRef] [PubMed]
- Inagawa, R.; Okada, H.; Takemura, G.; Suzuki, K.; Takada, C.; Yano, H.; Ando, Y.; Usui, T.; Hotta, Y.; Miyazaki, N.; et al. Ultrastructural Alteration of Pulmonary Capillary Endothelial Glycocalyx During Endotoxemia. Chest 2018, 154, 317–325. [Google Scholar] [CrossRef]
- Li, H.; Hao, Y.; Yang, L.L.; Wang, X.Y.; Li, X.Y.; Bhandari, S.; Han, J.; Liu, Y.J.; Gong, Y.Q.; Scott, A.; et al. MCTR1 alleviates lipopolysaccharide-induced acute lung injury by protecting lung endothelial glycocalyx. J. Cell. Physiol. 2020, 235, 7283–7294. [Google Scholar] [CrossRef]
- Pan, J.; Li, X.; Wang, X.; Yang, L.; Chen, H.; Su, N.; Wu, C.; Hao, Y.; Jin, S.; Li, H. MCTR1 Intervention Reverses Experimental Lung Fibrosis in Mice. J. Inflamm. Res. 2021, 14, 1873–1881. [Google Scholar] [CrossRef]
- Gardiner, K.R.; Halliday, M.I.; Barclay, G.R.; Milne, L.; Brown, D.; Stephens, S.; Maxwell, R.J.; Rowlands, B.J. Significance of systemic endotoxaemia in inflammatory bowel disease. Gut BMJ 1995, 36, 897. [Google Scholar] [CrossRef]
- Oliveira Magro, D.; Kotze, P.G.; Real Martinez, C.A.; Camargo, M.G.; Guadagnini, D.; Ramos Calixto, A.; Carolina, A.; Vasques, J.; de Lourdes, M.; Ayrizono, S.; et al. Changes in serum levels of lipopolysaccharides and CD26 in patients with Crohn’ s disease. Intest. Res. 2017, 15, 352–357. [Google Scholar] [CrossRef]
- Koivisto, O.; Hanel, A.; Carlberg, C. Key vitamin D target genes with functions in the immune system. Nutrients 2020, 12, 1140. [Google Scholar] [CrossRef]
- Clausen, T.M.; Sandoval, D.R.; Spliid, C.B.; Pihl, J.; Perrett, H.R.; Painter, C.D.; Narayanan, A.; Majowicz, S.A.; Kwong, E.M.; McVicar, R.N.; et al. SARS-CoV-2 Infection Depends on Cellular Heparan Sulfate and ACE2. Cell 2020, 183, 1043–1057.e15. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; McCullough, P.A.; Tecson, K.M. Vitamin D deficiency in association with endothelial dysfunction: Implications for patients with COVID-19. Rev. Cardiovasc. Med. 2020, 21, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Kanikarla-Marie, P.; Jain, S.K. 1,25(OH) 2 D 3 inhibits oxidative stress and monocyte adhesion by mediating the upregulation of GCLC and GSH in endothelial cells treated with acetoacetate (ketosis). J. Steroid Biochem. Mol. Biol. 2016, 159, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Al Mheid, I.; Quyyumi, A.A. Vitamin D and Cardiovascular Disease: Controversy Unresolved. J. Am. Coll. Cardiol. 2017, 70, 89–100. [Google Scholar] [CrossRef]
- Kim, D.H.; Meza, C.A.; Clarke, H.; Kim, J.S.; Hickner, R.C. Vitamin D and endothelial function. Nutrients 2020, 12, 575. [Google Scholar] [CrossRef]
- Stio, M.; Martinesi, M.; Bruni, S.; Treves, C.; Mathieu, C.; Verstuyf, A.; d’Albasio, G.; Bagnoli, S.; Bonanomi, A.G. The Vitamin D analogue TX 527 blocks NF-κB activation in peripheral blood mononuclear cells of patients with Crohn’s disease. J. Steroid Biochem. Mol. Biol. 2007, 103, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Omidian, M.; Mahmoudi, M.; Javanbakht, M.H.; Eshraghian, M.R.; Abshirini, M.; Daneshzad, E.; Hasani, H.; Alvandi, E.; Djalali, M. Effects of vitamin D supplementation on circulatory YKL-40 and MCP-1 biomarkers associated with vascular diabetic complications: A randomized, placebo-controlled, double-blind clinical trial. Diabetes Metab. Syndr. Clin. Res. Rev. 2019, 13, 2873–2877. [Google Scholar] [CrossRef]
- Hutchinson, A.N.; Tingö, L.; Brummer, R.J. The Potential Effects of Probiotics and ω-3 Fatty Acids on Chronic Low-Grade Inflammation. Nutrients 2020, 12, 2402. [Google Scholar] [CrossRef]
- Goodfellow, J.; Bellamy, M.F.; Ramsey, M.W.; Jones, C.J.H.; Lewis, M.J. Dietary supplementation with marine omega-3 fatty acids improve systemic large artery endothelial function in subjects with hypercholesterolemia. J. Am. Coll. Cardiol. 2000, 35, 265–270. [Google Scholar] [CrossRef]
- Miyoshi, T.; Noda, Y.; Ohno, Y.; Sugiyama, H.; Oe, H.; Nakamura, K.; Kohno, K.; Ito, H. Omega-3 fatty acids improve postprandial lipemia and associated endothelial dysfunction in healthy individuals—A randomized cross-over trial. Biomed. Pharmacother. 2014, 68, 1071–1077. [Google Scholar] [CrossRef]
- Huang, F.; del-Río-Navarro, B.E.; Leija-Martinez, J.; Torres-Alcantara, S.; Ruiz-Bedolla, E.; Hernández-Cadena, L.; Barraza-Villarreal, A.; Romero-Nava, R.; Sanchéz-Muñoz, F.; Villafaña, S.; et al. Effect of omega-3 fatty acids supplementation combined with lifestyle intervention on adipokines and biomarkers of endothelial dysfunction in obese adolescents with hypertriglyceridemia. J. Nutr. Biochem. 2019, 64, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Pendyala, S.; Walker, J.M.; Holt, P.R. A High-Fat Diet Is Associated with Endotoxemia That Originates from the Gut. Gastroenterology 2012, 142, 1100–1101.e2. [Google Scholar] [CrossRef] [PubMed]
- André, P.; Laugerette, F.; Féart, C. Metabolic Endotoxemia: A Potential Underlying Mechanism of the Relationship between Dietary Fat Intake and Risk for Cognitive Impairments in Humans? Nutrients 2019, 11, 1887. [Google Scholar] [CrossRef]
- Kazuma, S.; Tokinaga, Y.; Kimizuka, M.; Azumaguchi, R.; Hamada, K.; Yamakage, M. Sevoflurane Promotes Regeneration of the Endothelial Glycocalyx by Upregulating Sialyltransferase. J Surg Res. 2019, 241, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Machin, D.R.; Trott, D.W.; Gogulamudi, V.R.; Islam, M.T.; Bloom, S.I.; Vink, H.; Lesniewski, L.A.; Donato, A.J. Glycocalyx-targeted therapy ameliorates age-related arterial dysfunction. Geroscience 2023, Online ahead of print. [Google Scholar] [CrossRef]
- Ndanuko, R.N.; Tapsell, L.C.; Charlton, K.E.; Neale, E.P.; Batterham, M.J. Dietary Patterns and Blood Pressure in Adults: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Adv. Nutr. 2016, 7, 76. [Google Scholar] [CrossRef]
- Neuhouser, M.L. The Importance of Healthy Dietary Patterns in Chronic Disease Prevention. Nutr. Res. 2019, 70, 3. [Google Scholar] [CrossRef]
- Galbete, C.; Kröger, J.; Jannasch, F.; Iqbal, K.; Schwingshackl, L.; Schwedhelm, C.; Weikert, C.; Boeing, H.; Schulze, M.B. Nordic diet, Mediterranean diet, and the risk of chronic diseases: The EPIC-Potsdam study. BMC Med. 2018, 16, 99. [Google Scholar] [CrossRef]
- Rees, K.; Takeda, A.; Martin, N.; Ellis, L.; Wijesekara, D.; Vepa, A.; Das, A.; Hartley, L.; Stranges, S. Mediterranean-style diet for the primary and secondary prevention of cardiovascular disease. Cochrane Database Syst. Rev. 2019, 3, CD009825. [Google Scholar] [CrossRef]
- Esposito, K.; Maiorino, M.I.; Bellastella, G.; Chiodini, P.; Panagiotakos, D.; Giugliano, D. A journey into a Mediterranean diet and type 2 diabetes: A systematic review with meta-analyses. BMJ Open 2015, 5, e008222. [Google Scholar] [CrossRef]
- Saulle, R.; Lia, L.; de Giusti, M.; la Torre, G. A systematic overview of the scientific literature on the association between Mediterranean Diet and the Stroke prevention. Clin. Ter. 2019, 170, e396–e408. [Google Scholar] [PubMed]
- Nazarian, B.; Fazeli Moghadam, E.; Asbaghi, O.; Zeinali Khosroshahi, M.; Choghakhori, R.; Abbasnezhad, A. Effect of L-arginine supplementation on C-reactive protein and other inflammatory biomarkers: A systematic review and meta-analysis of randomized controlled trials. Complement. Ther. Med. 2019, 47, 102226. [Google Scholar] [CrossRef]
- Bai, Y.; Wang, X.; Zhao, S.; Ma, C.; Cui, J.; Zheng, Y. Sulforaphane Protects against Cardiovascular Disease via Nrf2 Activation. Oxid. Med. Cell. Longev. 2015, 2015, 407580. [Google Scholar] [CrossRef]
- Doleman, J.F.; Grisar, K.; van Liedekerke, L.; Saha, S.; Roe, M.; Tapp, H.S.; Mithen, R.F. The contribution of alliaceous and cruciferous vegetables to dietary sulphur intake. Food Chem. 2017, 234, 38–45. [Google Scholar] [CrossRef]
- Wilson, E.A.; Demmig-Adams, B. Antioxidant, anti-inflammatory, and antimicrobial properties of garlic and onions. Nutr. Food Sci. 2007, 37, 178–183. [Google Scholar] [CrossRef]
- Wlosinska, M.; Nilsson, A.C.; Hlebowicz, J.; Malmsjö, M.; Fakhro, M.; Lindstedt, S. Aged garlic extract preserves cutaneous microcirculation in patients with increased risk for cardiovascular diseases: A double-blinded placebo-controlled study. Int. Wound J. 2019, 16, 1487–1493. [Google Scholar] [CrossRef]
- Axelsson, A.S.; Tubbs, E.; Mecham, B.; Chacko, S.; Nenonen, H.A.; Tang, Y.; Fahey, J.W.; Derry, J.M.J.; Wollheim, C.B.; Wierup, N.; et al. Sulforaphane reduces hepatic glucose production and improves glucose control in patients with type 2 diabetes. Sci. Transl. Med. 2017, 9, 1–13. [Google Scholar] [CrossRef]
- Pereira, A.; Fernandes, R.; Crisóstomo, J.; Seiça, R.M.; Sena, C.M. The Sulforaphane and pyridoxamine supplementation normalize endothelial dysfunction associated with type 2 diabetes. Sci. Rep. 2017, 7, 14375. [Google Scholar] [CrossRef] [PubMed]
- Alyoussef, A.; Taha, M. Antitumor activity of sulforaphane in mice model of skin cancer via blocking sulfatase-2. Exp. Dermatol. 2019, 28, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, S.; Wang, W.; Chen, J.; Zhang, Z.; Zheng, Q.; Liu, Q.; Cai, L. Protection against diabetic cardiomyopathy is achieved using a combination of sulforaphane and zinc in type 1 diabetic OVE26 mice. J. Cell. Mol. Med. 2019, 23, 6319–6330. [Google Scholar] [CrossRef] [PubMed]
- Raubenheimer, K.; Hickey, D.; Leveritt, M.; Fassett, R.; Munoz, J.O.D.Z.; Allen, J.D.; Briskey, D.; Parker, T.J.; Kerr, G.; Peake, J.M.; et al. Acute effects of nitrate-rich beetroot juice on blood pressure, hemostasis and vascular inflammation markers in healthy older adults: A randomized, placebo-controlled crossover study. Nutrients 2017, 9, 1270. [Google Scholar] [CrossRef] [PubMed]
- Zafeiridis, A.; Triantafyllou, A.; Papadopoulos, S.; Koletsos, N.; Touplikioti, P.; Zafeiridis, A.S.; Gkaliagkousi, E.; Dipla, K.; Douma, S. Dietary nitrate improves muscle microvascular reactivity and lowers blood pressure at rest and during isometric exercise in untreated hypertensives. Microcirculation 2019, 26, e12525. [Google Scholar] [CrossRef]
- Kapil, V.; Khambata, R.S.; Robertson, A.; Caulfield, M.J.; Ahluwalia, A. Dietary nitrate provides sustained blood pressure lowering in hypertensive patients: A randomized, phase 2, double-blind, placebo-controlled study. Hypertension 2015, 65, 320–327. [Google Scholar] [CrossRef]
- Oggioni, C.; Jakovljevic, D.G.; Klonizakis, M.; Ashor, A.W.; Ruddock, A.; Ranchordas, M.; Williams, E.; Siervo, M. Dietary nitrate does not modify blood pressure and cardiac output at rest and during exercise in older adults: A randomised cross-over study. Int. J. Food Sci. Nutr. 2018, 69, 74–83. [Google Scholar] [CrossRef]
- Gilchrist, M.; Winyard, P.G.; Aizawa, K.; Anning, C.; Shore, A.; Benjamin, N. Effect of dietary nitrate on blood pressure, endothelial function, and insulin sensitivity in type 2 diabetes. Free Radic. Biol. Med. 2013, 60, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Castro-Barquero, S.; Ruiz-León, A.M.; Sierra-Pérez, M.; Estruch, R.; Casas, R. Dietary strategies for metabolic syndrome: A comprehensive review. Nutrients 2020, 12, 2983. [Google Scholar] [CrossRef]
- Howatson, A.; Wall, C.R.; Turner-Benny, P. The contribution of dietitians to the primary health care workforce. J. Prim. Health Care 2015, 7, 324–332. [Google Scholar] [CrossRef]
- Romero-Gómez, M.; Zelber-Sagi, S.; Trenell, M. Treatment of NAFLD with diet, physical activity and exercise. J. Hepatol. 2017, 67, 829–846. [Google Scholar] [CrossRef]
- Fanti, M.; Mishra, A.; Longo, V.D.; Brandhorst, S. Time-Restricted Eating, Intermittent Fasting, and Fasting-Mimicking Diets in Weight Loss. Curr. Obes. Rep. Curr. Obes. Rep. 2021, 10, 70–80. [Google Scholar] [CrossRef]
- Esmaeilzadeh, F.; van de Borne, P. Does intermittent fasting improve microvascular endothelial function in healthy middle-aged subjects? Biol. Med. 2016, 8, 6. [Google Scholar] [CrossRef]
- Sutton, E.F.; Beyl, R.; Early, K.S.; Cefalu, W.T.; Ravussin, E.; Peterson, C.M. Early Time-Restricted Feeding Improves Insulin Sensitivity, Blood Pressure, and Oxidative Stress Even without Weight Loss in Men with Prediabetes. Cell Metab. Cell Press 2018, 27, 1212–1221.e3. [Google Scholar] [CrossRef] [PubMed]
- Malinowski, B.; Zalewska, K.; Węsierska, A.; Sokołowska, M.M.; Socha, M.; Liczner, G.; Pawlak-Osińska, K.; Wiciński, M. Intermittent fasting in cardiovascular disorders—An overview. Nutrients 2019, 11, 673. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Dalla Man, C.; Morris, C.J.; Cobelli, C.; Scheer, F.A.J.L. Differential effects of the circadian system and circadian misalignment on insulin sensitivity and insulin secretion in humans. Diabetes Obes. Metab. 2018, 20, 2481–2485. [Google Scholar] [CrossRef]
- Poggiogalle, E.; Jamshed, H.; Peterson, C.M. Circadian regulation of glucose, lipid, and energy metabolism in humans. Metab. Clin. Exp. 2018, 84, 11–27. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franceković, P.; Gliemann, L. Endothelial Glycocalyx Preservation—Impact of Nutrition and Lifestyle. Nutrients 2023, 15, 2573. https://doi.org/10.3390/nu15112573
Franceković P, Gliemann L. Endothelial Glycocalyx Preservation—Impact of Nutrition and Lifestyle. Nutrients. 2023; 15(11):2573. https://doi.org/10.3390/nu15112573
Chicago/Turabian StyleFranceković, Paula, and Lasse Gliemann. 2023. "Endothelial Glycocalyx Preservation—Impact of Nutrition and Lifestyle" Nutrients 15, no. 11: 2573. https://doi.org/10.3390/nu15112573