
Metaproteomics Approach and Pathway Modulation in Obesity and Diabetes: A Narrative Review

 ,
,  , , , and
, , , and
Abstract
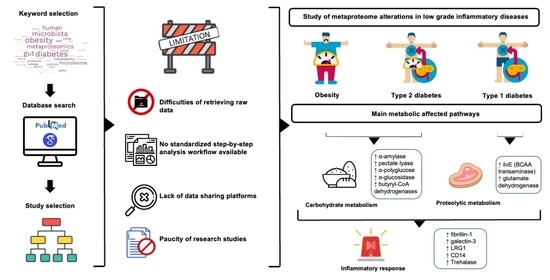
:
1. Introduction
2. Methods
2.1. Searching Strategy
2.2. Inclusion and Exclusion Criteria
2.3. Study Selection
2.4. Data Extraction and Risk of Bias
3. Results
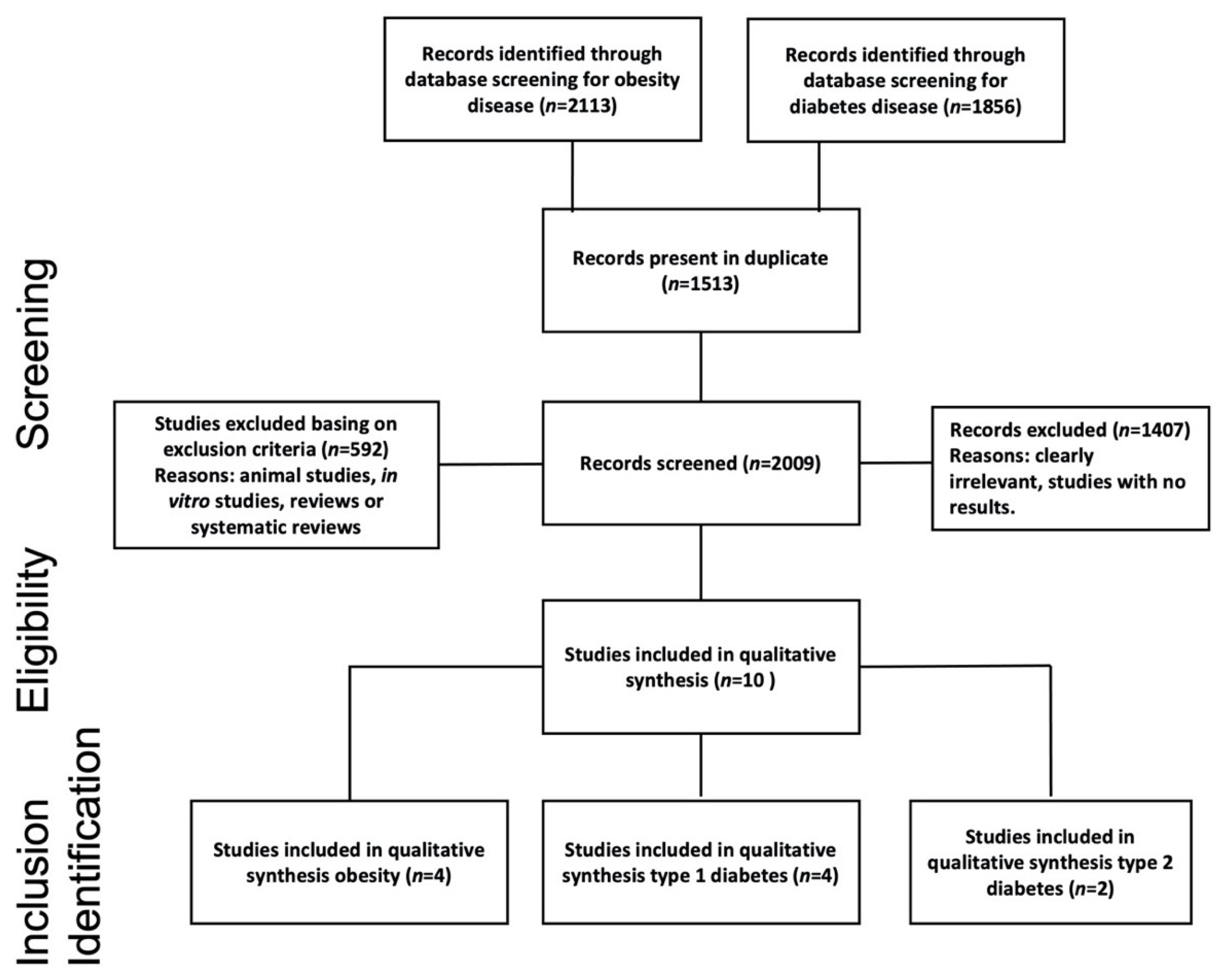
3.1. Literature Search Results and Study Characteristics
3.2. Metaproteome Alteration in Obesity
3.3. Metaproteome Alteration in T1D
3.4. Metaproteome Alteration in T2D
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aron-Wisnewsky, J.; Prifti, E.; Belda, E.; Ichou, F.; Kayser, B.D.; Dao, M.C.; Verger, E.O.; Hedjazi, L.; Bouillot, J.-L.; Chevallier, J.-M.; et al. Major Microbiota Dysbiosis in Severe Obesity: Fate after Bariatric Surgery. Gut 2019, 68, 70–82. [Google Scholar] [CrossRef]
- Abdellatif, A.M.; Sarvetnick, N.E. Current Understanding of the Role of Gut Dysbiosis in Type 1 Diabetes. J. Diabetes 2019, 11, 632–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cénit, M.C.; Matzaraki, V.; Tigchelaar, E.F.; Zhernakova, A. Rapidly Expanding Knowledge on the Role of the Gut Microbiome in Health and Disease. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2014, 1842, 1981–1992. [Google Scholar] [CrossRef] [Green Version]
- Schirmer, M.; Garner, A.; Vlamakis, H.; Xavier, R.J. Microbial Genes and Pathways in Inflammatory Bowel Disease. Nat. Rev. Microbiol. 2019, 17, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Ferrocino, I.; Cagno, R.D.; Angelis, M.D.; Turroni, S.; Vannini, L.; Bancalari, E.; Rantsiou, K.; Cardinali, G.; Neviani, E.; Cocolin, L. Fecal Microbiota in Healthy Subjects Following Omnivore, Vegetarian and Vegan Diets: Culturable Populations and RRNA DGGE Profiling. PLoS ONE 2015, 10, e0128669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ezzeldin, S.; El-Wazir, A.; Enany, S.; Muhammad, A.; Johar, D.; Osama, A.; Ahmed, E.; Shikshaky, H.; Magdeldin, S. Current Understanding of Human Metaproteome Association and Modulation. J. Proteome Res. 2019, 18, 3539–3554. [Google Scholar] [CrossRef]
- Ferreira-Halder, C.V.; Faria, A.V.d.S.; Andrade, S.S. Action and Function of Faecalibacterium Prausnitzii in Health and Disease. Best Pract. Res. Clin. Gastroenterol. 2017, 31, 643–648. [Google Scholar] [CrossRef]
- Round, J.L.; Mazmanian, S.K. The Gut Microbiota Shapes Intestinal Immune Responses during Health and Disease. Nat. Rev. Immunol. 2009, 9, 313–323. [Google Scholar] [CrossRef]
- Erickson, A.R.; Cantarel, B.L.; Lamendella, R.; Darzi, Y.; Mongodin, E.F.; Pan, C.; Shah, M.; Halfvarson, J.; Tysk, C.; Henrissat, B.; et al. Integrated Metagenomics/Metaproteomics Reveals Human Host-Microbiota Signatures of Crohn’s Disease. PLoS ONE 2012, 7, e49138. [Google Scholar] [CrossRef] [Green Version]
- Petriz, B.A.; Franco, O.L. Metaproteomics as a Complementary Approach to Gut Microbiota in Health and Disease. Front. Chem. 2017, 5, 4. [Google Scholar] [CrossRef] [Green Version]
- Winer, D.A.; Luck, H.; Tsai, S.; Winer, S. The Intestinal Immune System in Obesity and Insulin Resistance. Cell Metab. 2016, 23, 413–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snell-Bergeon, J.K.; West, N.A.; Mayer-Davis, E.J.; Liese, A.D.; Marcovina, S.M.; D’Agostino, R.B., Jr.; Hamman, R.F.; Dabelea, D. Inflammatory Markers Are Increased in Youth with Type 1 Diabetes: The SEARCH Case-Control Study. J. Clin. Endocrinol. Metab. 2010, 95, 2868–2876. [Google Scholar] [CrossRef] [PubMed]
- King, G.L. The Role of Inflammatory Cytokines in Diabetes and Its Complications. J. Periodontol. 2008, 79, 1527–1534. [Google Scholar] [CrossRef] [PubMed]
- Balistreri, C.R.; Caruso, C.; Candore, G. The Role of Adipose Tissue and Adipokines in Obesity-Related Inflammatory Diseases. Mediat. Inflamm. 2010, 2010, e802078. [Google Scholar] [CrossRef]
- Monnerie, S.; Comte, B.; Ziegler, D.; Morais, J.A.; Pujos-Guillot, E.; Gaudreau, P. Metabolomic and Lipidomic Signatures of Metabolic Syndrome and Its Physiological Components in Adults: A Systematic Review. Sci. Rep. 2020, 10, 669. [Google Scholar] [CrossRef] [Green Version]
- Asarat, M.; Apostolopoulos, V.; Vasiljevic, T.; Donkor, O. Short-Chain Fatty Acids Regulate Cytokines and Th17/Treg Cells in Human Peripheral Blood Mononuclear Cells in Vitro. Immunol. Investig. 2016, 45, 205–222. [Google Scholar] [CrossRef] [Green Version]
- Triantafilou, M.; Triantafilou, K. Lipopolysaccharide Recognition: CD14, TLRs and the LPS-Activation Cluster. Trends Immunol. 2002, 23, 301–304. [Google Scholar] [CrossRef]
- Zhong, H.; Ren, H.; Lu, Y.; Fang, C.; Hou, G.; Yang, Z.; Chen, B.; Yang, F.; Zhao, Y.; Shi, Z.; et al. Distinct Gut Metagenomics and Metaproteomics Signatures in Prediabetics and Treatment-Naïve Type 2 Diabetics. EBioMedicine 2019, 47, 373–383. [Google Scholar] [CrossRef]
- Xiao, M.; Yang, J.; Feng, Y.; Zhu, Y.; Chai, X.; Wang, Y. Metaproteomic Strategies and Applications for Gut Microbial Research. Appl. Microbiol. Biotechnol. 2017, 101, 3077–3088. [Google Scholar] [CrossRef]
- Pinart, M.; Nimptsch, K.; Forslund, S.K.; Schlicht, K.; Gueimonde, M.; Brigidi, P.; Turroni, S.; Ahrens, W.; Hebestreit, A.; Wolters, M.; et al. Identification and Characterization of Human Observational Studies in Nutritional Epidemiology on Gut Microbiomics for Joint Data Analysis. Nutrients 2021, 13, 3292. [Google Scholar] [CrossRef]
- Pinart, M.; Jeran, S.; Boeing, H.; Stelmach-Mardas, M.; Standl, M.; Schulz, H.; Harris, C.; von Berg, A.; Herberth, G.; Koletzko, S.; et al. Dietary Macronutrient Composition in Relation to Circulating HDL and Non-HDL Cholesterol: A Federated Individual-Level Analysis of Cross-Sectional Data from Adolescents and Adults in 8 European Studies. J. Nutr. 2021, 151, 2317–2329. [Google Scholar] [CrossRef] [PubMed]
- Gaye, A.; Marcon, Y.; Isaeva, J.; LaFlamme, P.; Turner, A.; Jones, E.M.; Minion, J.; Boyd, A.W.; Newby, C.J.; Nuotio, M.-L.; et al. DataSHIELD: Taking the Analysis to the Data, Not the Data to the Analysis. Int. J. Epidemiol. 2014, 43, 1929–1944. [Google Scholar] [CrossRef] [Green Version]
- Marcon, Y.; Bishop, T.; Avraam, D.; Escriba-Montagut, X.; Ryser-Welch, P.; Wheater, S.; Burton, P.; González, J.R. Orchestrating Privacy-Protected Big Data Analyses of Data from Different Resources with R and DataSHIELD. PLoS Comput. Biol. 2021, 17, e1008880. [Google Scholar] [CrossRef]
- Shamseer, L.; Moher, D.; Clarke, M.; Ghersi, D.; Liberati, A.; Petticrew, M.; Shekelle, P.; Stewart, L.A. Preferred Reporting Items for Systematic Review and Meta-Analysis Protocols (PRISMA-P) 2015: Elaboration and Explanation. BMJ 2015, 349, g7647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wells, G.A.; Shea, B.; O’Connell, D.; Peterson, J.; Welch, V.; Losos, M.; Tugwell, P. The Newcastle-Ottawa Scale (NOS) for Assessing the Quality of Nonrandomised Studies in Meta-Analyses 2000. Available online: http://www.ohri.ca/programs/clinical_epidemiology/nosgen.pdf (accessed on 14 October 2021).
- Zhou, W.; Sailani, M.R.; Contrepois, K.; Zhou, Y.; Ahadi, S.; Leopold, S.R.; Zhang, M.J.; Rao, V.; Avina, M.; Mishra, T.; et al. Longitudinal multi-omics of host–microbe dynamics in prediabetes. Nature 2019, 569, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Yu, Y.; Suh, M.-J.; Torralba, M.G.; Stenzel, R.D.; Tovchigrechko, A.; Thovarai, V.; Harkins, D.M.; Rajagopala, S.V.; Osborne, W.; et al. Type 1 Diabetes: Urinary Proteomics and Protein Network Analysis Support Perturbation of Lysosomal Function. Theranostics 2017, 7, 2704–2717. [Google Scholar] [CrossRef]
- Pinto, E.; Anselmo, M.; Calha, M.; Bottrill, A.; Duarte, I.; Andrew, P.W.; Faleiro, M.L. The Intestinal Proteome of Diabetic and Control Children Is Enriched with Different Microbial and Host Proteins. Microbiology 2017, 163, 161–174. [Google Scholar] [CrossRef]
- Ferrer, M.; Ruiz, A.; Lanza, F.; Haange, S.-B.; Oberbach, A.; Till, H.; Bargiela, R.; Campoy, C.; Segura, M.T.; Richter, M.; et al. Microbiota from the Distal Guts of Lean and Obese Adolescents Exhibit Partial Functional Redundancy besides Clear Differences in Community Structure. Environ. Microbiol. 2013, 15, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Kolmeder, C.A.; Ritari, J.; Verdam, F.J.; Muth, T.; Keskitalo, S.; Varjosalo, M.; Fuentes, S.; Greve, J.W.; Buurman, W.A.; Reichl, U.; et al. Colonic Metaproteomic Signatures of Active Bacteria and the Host in Obesity. Proteomics 2015, 15, 3544–3552. [Google Scholar] [CrossRef] [PubMed]
- Heintz-Buschart, A.; May, P.; Laczny, C.C.; Lebrun, L.A.; Bellora, C.; Krishna, A.; Wampach, L.; Schneider, J.G.; Hogan, A.; de Beaufort, C.; et al. Integrated Multi-Omics of the Human Gut Microbiome in a Case Study of Familial Type 1 Diabetes. Nat. Microbiol. 2016, 2, 1–13. [Google Scholar] [CrossRef]
- Sanchez-Carrillo, S.; Ciordia, S.; Rojo, D.; Zubeldia-Varela, E.; Méndez-García, C.; Martínez-Martínez, M.; Barbas, C.; Ruiz-Ruiz, S.; Moya, A.; Garriga, M.; et al. A Body Weight Loss- and Health-Promoting Gut Microbiota Is Established after Bariatric Surgery in Individuals with Severe Obesity. J. Pharm. Biomed. Anal. 2021, 193, 113747. [Google Scholar] [CrossRef] [PubMed]
- Gavin, P.G.; Mullaney, J.A.; Loo, D.; Cao, K.-A.L.; Gottlieb, P.A.; Hill, M.M.; Zipris, D.; Hamilton-Williams, E.E. Intestinal Metaproteomics Reveals Host-Microbiota Interactions in Subjects at Risk for Type 1 Diabetes. Diabetes Care 2018, 41, 2178–2186. [Google Scholar] [CrossRef] [Green Version]
- Hernández, E.; Bargiela, R.; Diez, M.S.; Friedrichs, A.; Pérez-Cobas, A.E.; Gosalbes, M.J.; Knecht, H.; Martínez-Martínez, M.; Seifert, J.; von Bergen, M.; et al. Functional Consequences of Microbial Shifts in the Human Gastrointestinal Tract Linked to Antibiotic Treatment and Obesity. Gut Microbes 2013, 4, 306–315. [Google Scholar] [CrossRef] [Green Version]
- Suh, M.-J.; Tovchigrechko, A.; Thovarai, V.; Rolfe, M.A.; Torralba, M.G.; Wang, J.; Adkins, J.N.; Webb-Robertson, B.-J.M.; Osborne, W.; Cogen, F.R.; et al. Quantitative Differences in the Urinary Proteome of Siblings Discordant for Type 1 Diabetes Include Lysosomal Enzymes. J. Proteome Res. 2015, 14, 3123–3135. [Google Scholar] [CrossRef]
- Xiong, W.; Abraham, P.E.; Li, Z.; Pan, C.; Hettich, R.L. Microbial Metaproteomics for Characterizing the Range of Metabolic Functions and Activities of Human Gut Microbiota. Proteomics 2015, 15, 3424–3438. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.N.; Yao, Y.; Ju, S.Y. Short Chain Fatty Acids and Fecal Microbiota Abundance in Humans with Obesity: A Systematic Review and Meta-Analysis. Nutrients 2019, 11, 2512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, D.J.; Preston, T. Formation of Short Chain Fatty Acids by the Gut Microbiota and Their Impact on Human Metabolism. Gut Microbes 2016, 7, 189–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwiertz, A.; Taras, D.; Schäfer, K.; Beijer, S.; Bos, N.A.; Donus, C.; Hardt, P.D. Microbiota and SCFA in Lean and Overweight Healthy Subjects. Obesity 2010, 18, 190–195. [Google Scholar] [CrossRef]
- Cepeda-Lopez, A.C.; Aeberli, I.; Zimmermann, M.B. Does Obesity Increase Risk for Iron Deficiency? A Review of the Literature and the Potential Mechanisms. Int. J. Vitam. Nutr. Res. 2010, 80, 263–270. [Google Scholar] [CrossRef]
- Femlak, M.; Gluba-Brzózka, A.; Ciałkowska-Rysz, A.; Rysz, J. The Role and Function of HDL in Patients with Diabetes Mellitus and the Related Cardiovascular Risk. Lipids Health Dis. 2017, 16, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell-Thompson, M.; Rodriguez-Calvo, T.; Battaglia, M. Abnormalities of the Exocrine Pancreas in Type 1 Diabetes. Curr. Diab. Rep. 2015, 15, 79. [Google Scholar] [CrossRef] [Green Version]
- Foster, T.P.; Bruggeman, B.; Campbell-Thompson, M.; Atkinson, M.A.; Haller, M.J.; Schatz, D.A. Exocrine Pancreas Dysfunction in Type 1 Diabetes. Endocr. Pract. 2020, 26, 1505–1513. [Google Scholar] [CrossRef] [PubMed]
- Kondrashova, A.; Nurminen, N.; Lehtonen, J.; Hyöty, M.; Toppari, J.; Ilonen, J.; Veijola, R.; Knip, M.; Hyöty, H. Exocrine Pancreas Function Decreases during the Progression of the Beta-Cell Damaging Process in Young Prediabetic Children. Pediatr. Diabetes 2018, 19, 398–402. [Google Scholar] [CrossRef] [Green Version]
- Dozio, N.; Indirli, R.; Giamporcaro, G.M.; Frosio, L.; Mandelli, A.; Laurenzi, A.; Bolla, A.M.; Stabilini, A.; Valle, A.; Locatelli, M.; et al. Impaired Exocrine Pancreatic Function in Different Stages of Type 1 Diabetes. BMJ Open Diabetes Res. Care 2021, 9, e001158. [Google Scholar] [CrossRef] [PubMed]
- Alkanani, A.K.; Hara, N.; Gottlieb, P.A.; Ir, D.; Robertson, C.E.; Wagner, B.D.; Frank, D.N.; Zipris, D. Alterations in Intestinal Microbiota Correlate With Susceptibility to Type 1 Diabetes. Diabetes 2015, 64, 3510–3520. [Google Scholar] [CrossRef] [Green Version]
- Harbison, J.E.; Roth-Schulze, A.J.; Giles, L.C.; Tran, C.D.; Ngui, K.M.; Penno, M.A.; Thomson, R.L.; Wentworth, J.M.; Colman, P.G.; Craig, M.E.; et al. Gut Microbiome Dysbiosis and Increased Intestinal Permeability in Children with Islet Autoimmunity and Type 1 Diabetes: A Prospective Cohort Study. Pediatr. Diabetes 2019, 20, 574–583. [Google Scholar] [CrossRef]
- Hebert, S.L.; Nair, K.S. Protein and Energy Metabolism in Type 1 Diabetes. Clin. Nutr. 2010, 29, 13–17. [Google Scholar] [CrossRef] [Green Version]
- Holeček, M. Branched-Chain Amino Acids and Branched-Chain Keto Acids in Hyperammonemic States: Metabolism and as Supplements. Metabolites 2020, 10, 324. [Google Scholar] [CrossRef]
- Aschner, P.J.; Ruiz, A.J. Metabolic Memory for Vascular Disease in Diabetes. Diabetes Technol. Ther. 2012, 14, S68–S74. [Google Scholar] [CrossRef]
- Timmins-Schiffman, E.; May, D.H.; Mikan, M.; Riffle, M.; Frazar, C.; Harvey, H.R.; Noble, W.S.; Nunn, B.L. Critical Decisions in Metaproteomics: Achieving High Confidence Protein Annotations in a Sea of Unknowns. ISME J. 2017, 11, 309–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werner, J.; Géron, A.; Kerssemakers, J.; Matallana-Surget, S. MPies: A Novel Metaproteomics Tool for the Creation of Relevant Protein Databases and Automatized Protein Annotation. Biol. Direct 2019, 14, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyatt, D.; LoCascio, P.F.; Hauser, L.J.; Uberbacher, E.C. Gene and Translation Initiation Site Prediction in Metagenomic Sequences. Bioinformatics 2012, 28, 2223–2230. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Yang, W.; Chen, G.; Shafiq, M.; Javed, S.; Zaidi, S.S.A.; Shahid, R.; Liu, C.; Bokhari, H. Analysis of Gut Microbiota of Obese Individuals with Type 2 Diabetes and Healthy Individuals. PLoS ONE 2019, 14, e0226372. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
| Authors and Year | Size Sample and Characteristics | Subjects Characteristics (Sex, Age, Country) | Scope of Study | Study Design | Metaproteomics Techniques Used | Other “Omics” Techniques Used | Limitations | NOS Score |
|---|---|---|---|---|---|---|---|---|
| Gavin et al., 2018 | 101 subjects: 33 NO, 17 SP, 29 SN, 22 CO | Denver, Colorado 46 females and 55 males Age: 9–12 | Investigate functional interactions host-microbiota in subjects with T1D risk | Cross-sectional | LC-MS/MS | No information about dietary intake. Wide age range. | 7 | |
| Pinto et al., 2017 | 6 subjects: 3 healthy and 3 T1D children | Portugal 2 males and 4 females Age: T1D children 9.3 ± 1.5 and control children 9.3 ± 0.6 years | Identify differences in the activity of intestinal microbiota between healthy and T1D children | Case-control | SDS-PAGE and LC-MS/MS (using LTQ Orbitrap) | Small number of T1D children. | 6 | |
| Heintz et al., 2016 | 20 subjects from 4 families of at least 2 generations presenting at least 2 cases of T1D | Luxembourg 7 males 13 females Age: 5–62 | Resolution of the taxonomic and functional attributes of gut microbiota and evaluation of the effect of family on gut microbiota composition | Longitudinal study (4 month) | LC (Nano-2D-UPLC-Orbirtap MS system) and MS (TopN-MS/MS method) | Metagenomics and metatranscriptomics | Need for large-scale studies. | 6 |
| Singh et al., 2017 | 223 subjects: 110 T1D children/adolescents and 113 healthy siblings | Washington D.C. 115 males 108 females Age: 13.9–14.5 | Detection of gut microbial differences and evaluation of lysosomal dysfunctions | Case-control | LC-MS/MS | Imperfect glycemic control or subclinical inflammation in T1D patients. No information about eating habits and lifestyle. | 7 | |
| Zhong et al., 2019 | 254 subjects: 77 TN-T2D, 80 Pre-DM, and 97 NGT | Suzhou, China 173 females 81 males Age: 41–86 | Investigate compositional and functional changes of the gut microbiota to characterize different disease stages | Cross-sectional | iTRAQ-coupled- LC-MS/MS | Metagenomics | Limitations of MS-based proteomics. Confounding variables: age, drugs (CCB, hypertension, and dyslipidemia), diet, and health conditions. | 7 |
| Zhou et al., 2019 | 106 subjects: healthy and pre-diabetic adults | Standford, California 55 females and 51 males Age: 25–75 | Understand how healthy individuals and those at risk of T2D, change over time, in response to perturbations and in relation to different molecules and microorganism | Longitudinal study (4 years) | SWATH-MS | Metagenomics, metatranscriptomics, and metabolomics | Limited studies of microbial changes. No information about diet and exercise. Heterogeneous data. | 6 |
| Ferrer et al., 2013 | 2 subjects: 1 lean, 1 obese | Spain 1 female (lean) and 1 male (obese) Age: 15 | Identify and analyze active bacterial members and proteins expressed in lean and obese microbiota | Case-control | 1D-gel electrophoresis and UPLC-LTQ Orbitrap-MS/MS | Metagenetics | No information about dietary intake. | 7 |
| Kolmeder et al., 2015 | 29 subjects: 9 lean, 4 overweight, 16 obese | Spain 21 females 8 males Age: 23.1 ± 2.2 (non-obese); 38.6 ± 2.4 (obese) | Characterization of non-obese and obese fecal metaproteome | Case-control | 1D-gel electrophoresis RP-HPLC online coupled to MS/MS | Regular medication between obese and non-obese group. | 6 | |
| Sanchez-Carrillo et al., 2020 | 40 severely obese adults subjected to BS | Spain Age: 47–60 | Investigation the impact of bariatric surgery | Longitudinal study (3 months) | LC-ESI-MS/MS analysis | Metabolomics | Results biased for using pooling strategy. | 6 |
| Hernandez et al., 2013 | 13 subjects: 2 adults (β-lactam-therapy), 7 obese adolescents, 5 lean adolescents | Germany Obese: 3 females and 4 males Lean: 2 males and 3 females Age: 13–16 | Evaluation of microbial shifts in relation to antibiotic treatment and obesity and measurement of carbohydrate activate enzymes | Cross-sectional | 96-well plates using a BioTek Synergy HT spectrometer in a colorimetric assay | No information about dietary intake. Wide age range. Small number of subjects. | 6 |
| Authors and Year | Disease | Protein Origin | ↑ Proteins | ↓ Proteins | Metabolic Pathway/Functionality |
|---|---|---|---|---|---|
| Gavin et al., 2018 | T1DM | Microbial | 1. Enzymes for mucin degradation 2. Elongation factor 3. Ferredoxin reductase | 4. Transferases (butyrate synthesis) | 3.↑Ferredoxin catabolism 4. ↑ Butyrate anabolism |
| Human | 1. Galectin-3 2. Fibrillin | 3. CELA-3A, 4. CUZD1 5. CLCA1 6. Neutral ceraminidase 7. IGHA1 | 6.↓ Sphingosine (SPH) and sphingosine 1-phosphate (S1P) 3.4.5. ↓ exocrine pancreas functionality 7.↓ IgA | ||
| Pinto et al., 2017 | T1DM | Microbial | 1. ilvE (BCAA transaminase) 2. Glutamate dehydrogenase (AA degradation) 3. Bifunctional GMP synthase 4. Glutamine amido transferase 5. Chaperonin GroEL | 6. Phosphoketolas 7. Glyceraldehyde-3-phosphate dehydrogenase, 8. Transketolase | 1.6.8. ↓ Via penthos phosphate → ↑ BCAA synthesis (Shikmic Acid Pathway) ↓ glycolysis 2.↑ NH4+ (Urea) 7. ↓ Glycolysis →↓ Piruvate ↓ SCFAs |
| Human | MUC2 precursor | CELA-3A | ↑ Intestinal mucin-2 ↓ Exocrine pancreas functionality | ||
| Heintz et al., 2016 | T1DM | Microbial | Thiamine synthesis cofactor | ↓ Thiamine synthesis | |
| Human | ↓ AMY2A, AMY2B, CPA1, and CUDZ1 | ↓ Complex sugar degradation | |||
| Singh et al., 2017 | T1DM | Human urinary proteome | 1. LGR1 2. CD14 3. CPE 4. CTSB 5. CTSD 6. NAGA | 7. Fibronectin-1 8. Pancreatic α-amylase 9. MUC1 10. PTPRN | 1. ↑Inflammatory pathways (TGF-β) 3.↑AA degradation (↑urea production) 8. ↓ Exocrine pancreas functionality and ↓ complex sugar metabolism |
| Zhong et al., 2019 | T2DM | Microbial | 1. PTS 2. ABC transporter 3. FMO3 (TMAO producing enzyme) | 4. Ferredoxin oxidoreductase 5. Bacterial ribosomal proteins | 1.↑Phosphorylation and transport of sugar in microbial cells 2.↑ HDL cholesterol 3.↑TMAO synthesis |
| Zhou et al., 2019 | T2M | Human | 1. IL-1RA 2. CRP 3. A1C | 1.↓IL-1 2.↑immune defense mechanism 3.↑ glycaemia | |
| Ferrer et al., 2013 | Obesity | Microbial | 1. Glycoprotein containing FN3 2. Cobaltochelatases 3. B12-dependent methylmalonyl-CoA mutase 4. PduB 5. 3-hydroxybutyryl-CoA dehydratase 6. Butyryl-CoA dehydrogenases 7. Acetyl-CoA acetyltransferases | 7. Pectate lyase 8. Aldose 1-epimerase 9. SOD 10. Pyridoxal biosynthesis lyases | 1.↑ Fibrin and proteoglycans 2.3. ↑ Vitamin B12 and propionate production 4.↑ Propanediol catabolism 5. Butyrate 10. ↓ Vitamin B6 |
| Kolmeder et al., 2015 | Obesity | Microbial | 1. α-glucosidase 2. Pectate lyase 3. Aminoacyl-histidine dipeptidase 4. Bacteroidetes proteins | 1.2. ↑Starch and pectin metabolism 3. ↑AA metabolism 4. ↑SCFAs | |
| Human | 1. Trehalase (intestinal injury and inflammation marker) 2. Alkaline phosphatase (AP) 3. Serpins (serina protease inhibitors) 4. α-amylase | 1.↑ Threalosie 4.↑Starch digestion | |||
| Sanchez-Carrillo et al., 2020 | Obesity | Microbial (pre-BS) | 1. Enzymes involved in gluconeogenesis (glyceraldehyde 3-phosphate dehydrogenase, pyruvate orthophosphate dikinase, PEP carboxykinase, fructose-bisphosphate aldolase, glutamate dehydrogenase) 2. Enzymes involved in Acetyl-CoA synthesis (Formate C-acetyltransferase, acetyl-CoA synthase, carbon-monoxide dehydrogenase) 3. Ferredoxin oxidoreductase | 4. Ferritin 5. Ferrous ion transport protein 6. Porphobilinogen synthase | 1.↑Pyruvate 2.↑ Acetyl-CoA (WL pathway) 4.5. ↓Iron synthesis |
| Microbial (post-BS) | 1. AdhE 2. OhyA 3. SOD and perodoxins (involved in maintenance of redox balance) | 1. ↑ Acetyl Acteyl-CoA → ethanol 1. Saturated fatty acid | |||
| Hernandez et al., 2013 | Obesity | Microbial | 1. α-polyglucose (refined carbohydrate digestion) 2. Proteins involved in pentose phosphate metabolism (PPP) 3. Proteins involved in TCA cycle | 1.2. ↑ Fructose, mannose, galactose, sucrose, starch, amino sugar, and nucleotide sugar metabolism 3. ↑ Via pentose phosphate |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calabrese, F.M.; Porrelli, A.; Vacca, M.; Comte, B.; Nimptsch, K.; Pinart, M.; Pischon, T.; Pujos-Guillot, E.; De Angelis, M. Metaproteomics Approach and Pathway Modulation in Obesity and Diabetes: A Narrative Review. Nutrients 2022, 14, 47. https://doi.org/10.3390/nu14010047
Calabrese FM, Porrelli A, Vacca M, Comte B, Nimptsch K, Pinart M, Pischon T, Pujos-Guillot E, De Angelis M. Metaproteomics Approach and Pathway Modulation in Obesity and Diabetes: A Narrative Review. Nutrients. 2022; 14(1):47. https://doi.org/10.3390/nu14010047
Chicago/Turabian StyleCalabrese, Francesco Maria, Annalisa Porrelli, Mirco Vacca, Blandine Comte, Katharina Nimptsch, Mariona Pinart, Tobias Pischon, Estelle Pujos-Guillot, and Maria De Angelis. 2022. "Metaproteomics Approach and Pathway Modulation in Obesity and Diabetes: A Narrative Review" Nutrients 14, no. 1: 47. https://doi.org/10.3390/nu14010047