
Inhibition Kinetics and Theoretical Studies on Zanthoxylum chalybeum Engl. Dual Inhibitors of α-Glucosidase and α-Amylase
 ,
,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolation of Study Compounds and Their Kinetic Analyses
2.2. Statistical Analysis
2.3. In Silico Method
2.3.1. Ligands Preparation
2.3.2. Drug-Likeness Predictions and Structural Skeleton Similarity Analysis
2.3.3. ADME/Tox Prediction
2.3.4. 3D Protein Structures Preparation
2.3.5. Docking Simulation
3. Results and Discussion
3.1. Kinetic Analyses
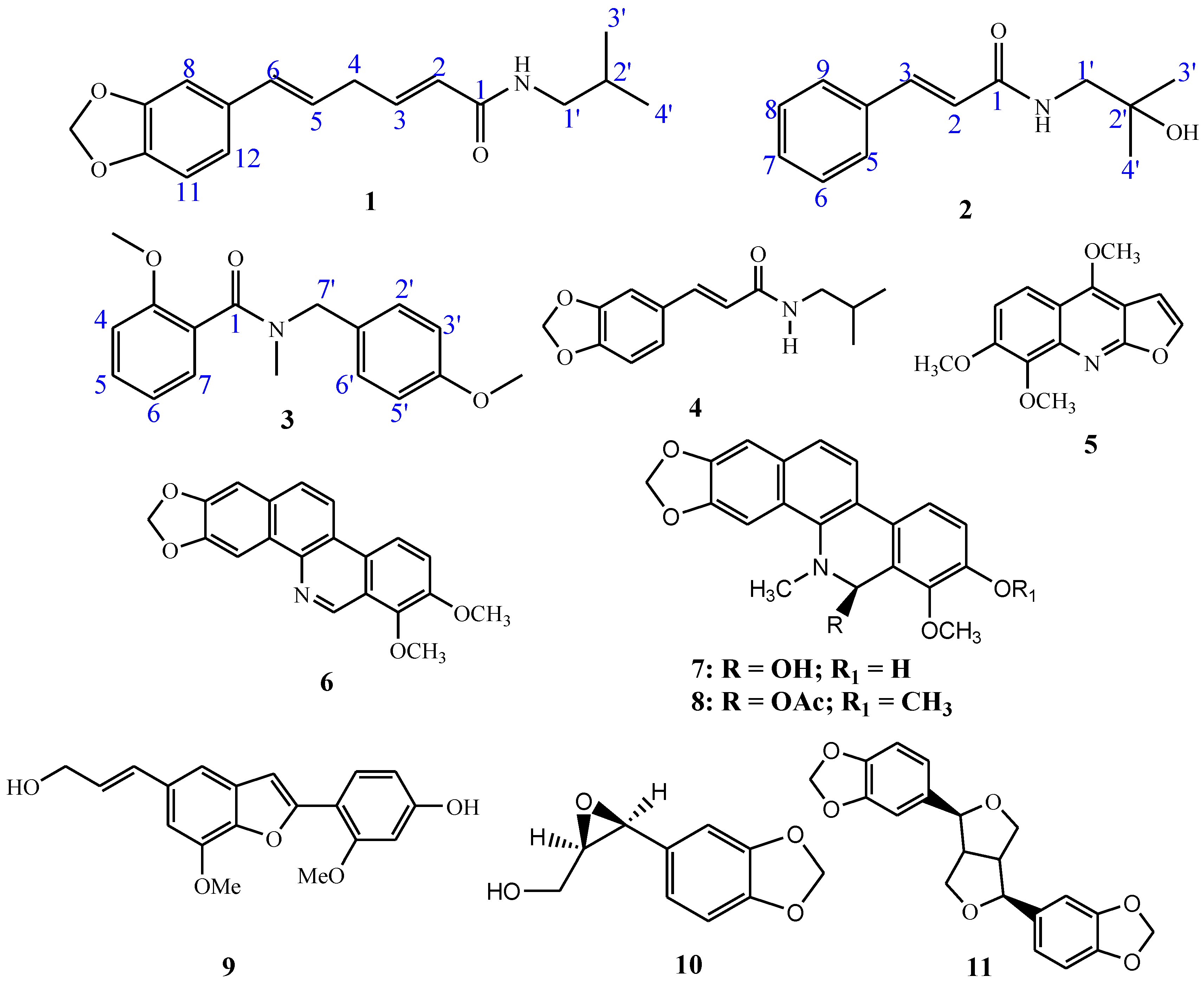
3.2. Drug-Likeness Predictions and Structural Skeleton Similarity Analysis
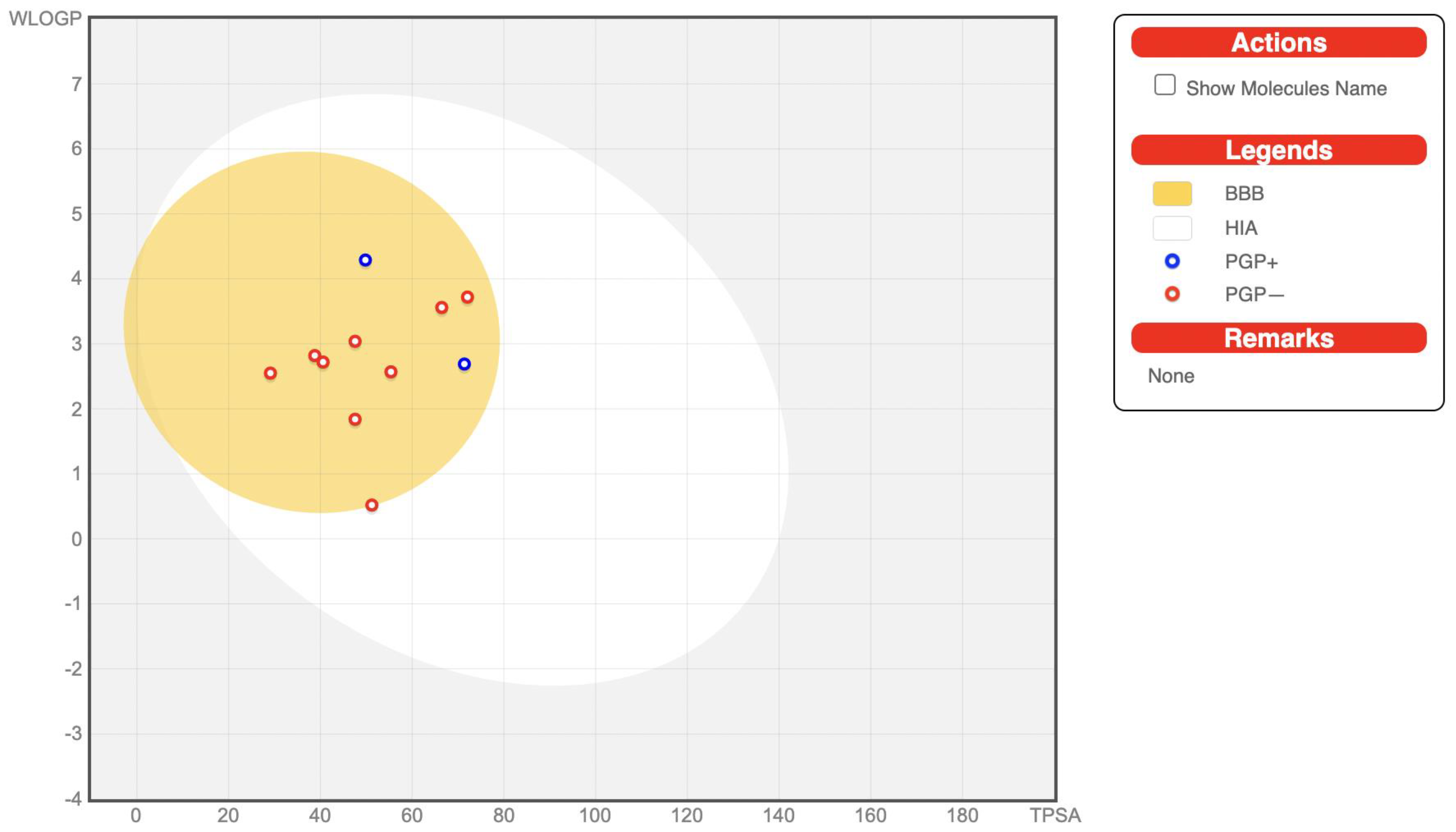
3.3. ADME/Tox Prediction
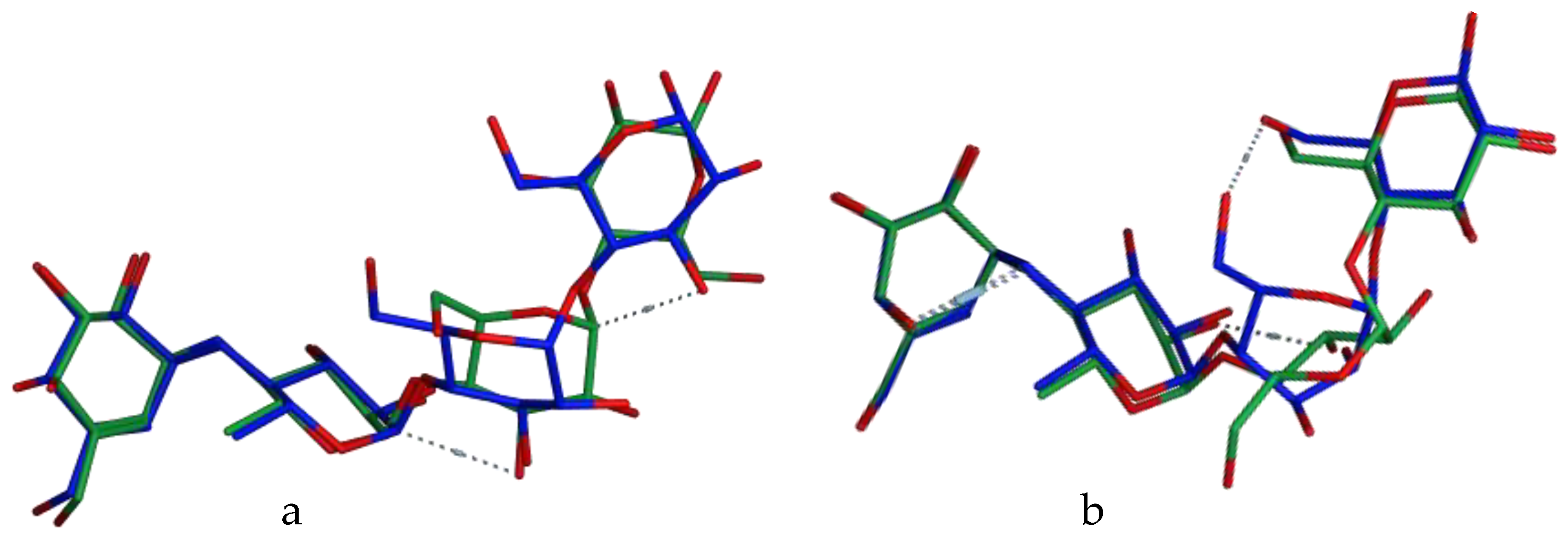
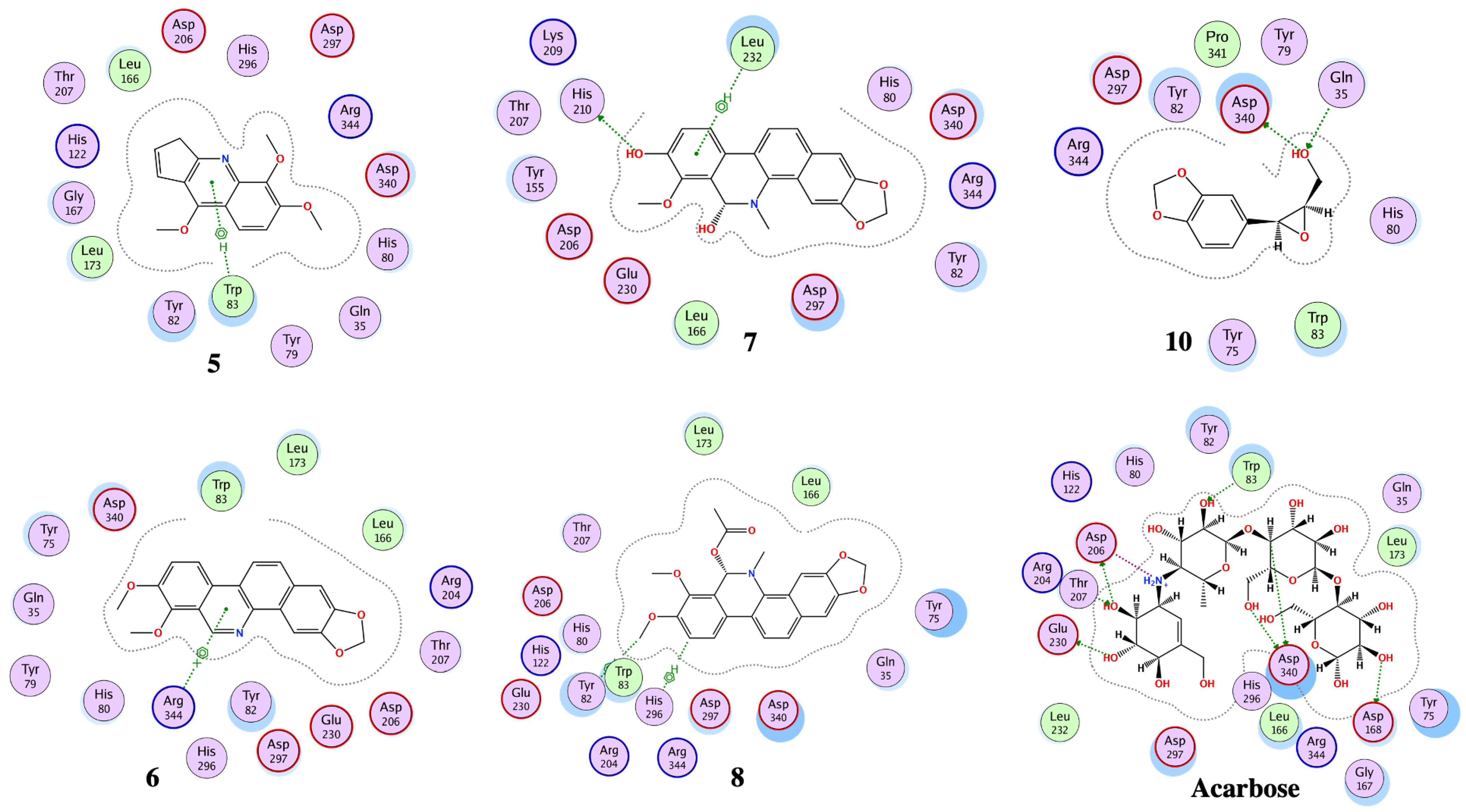
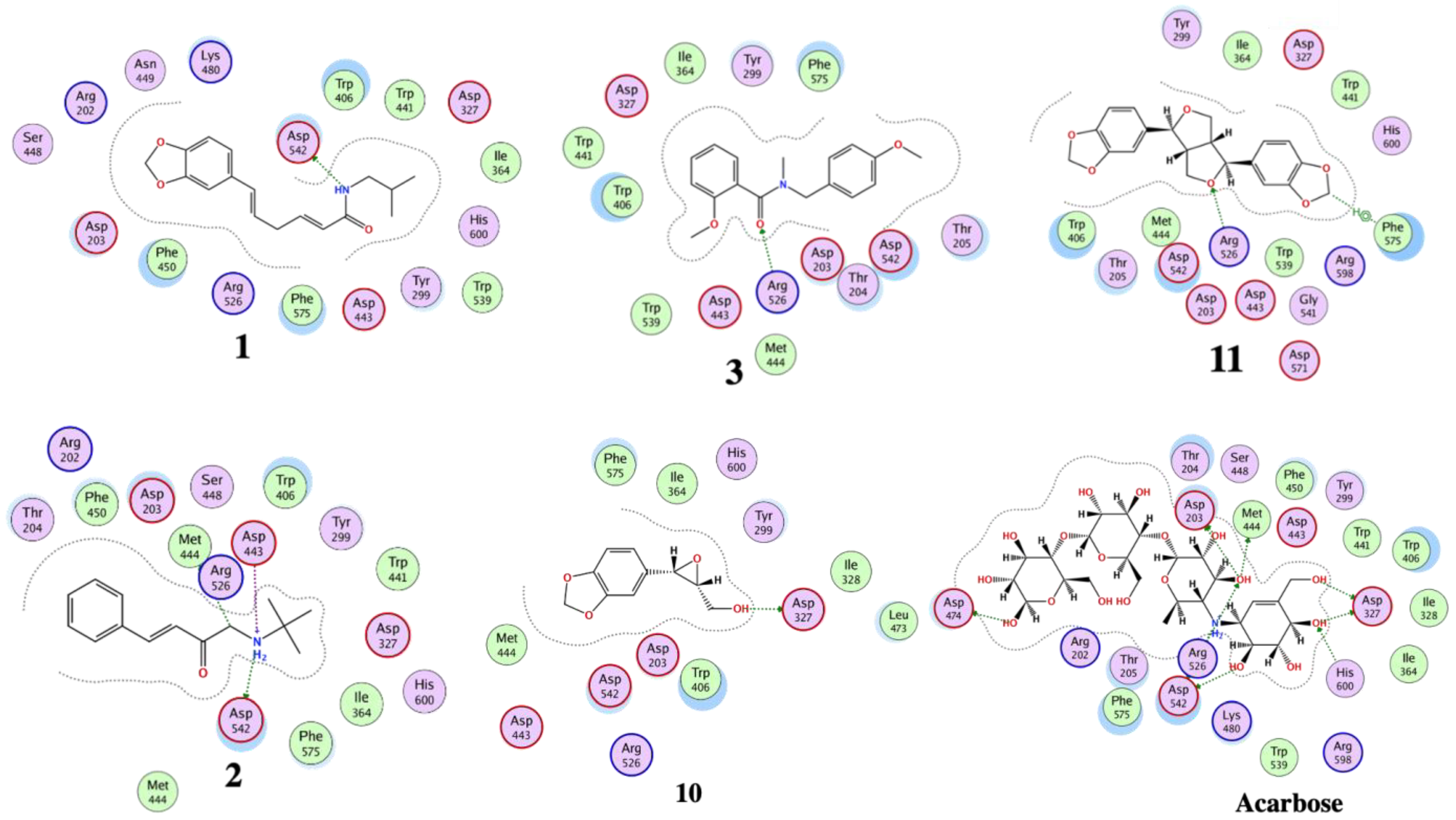
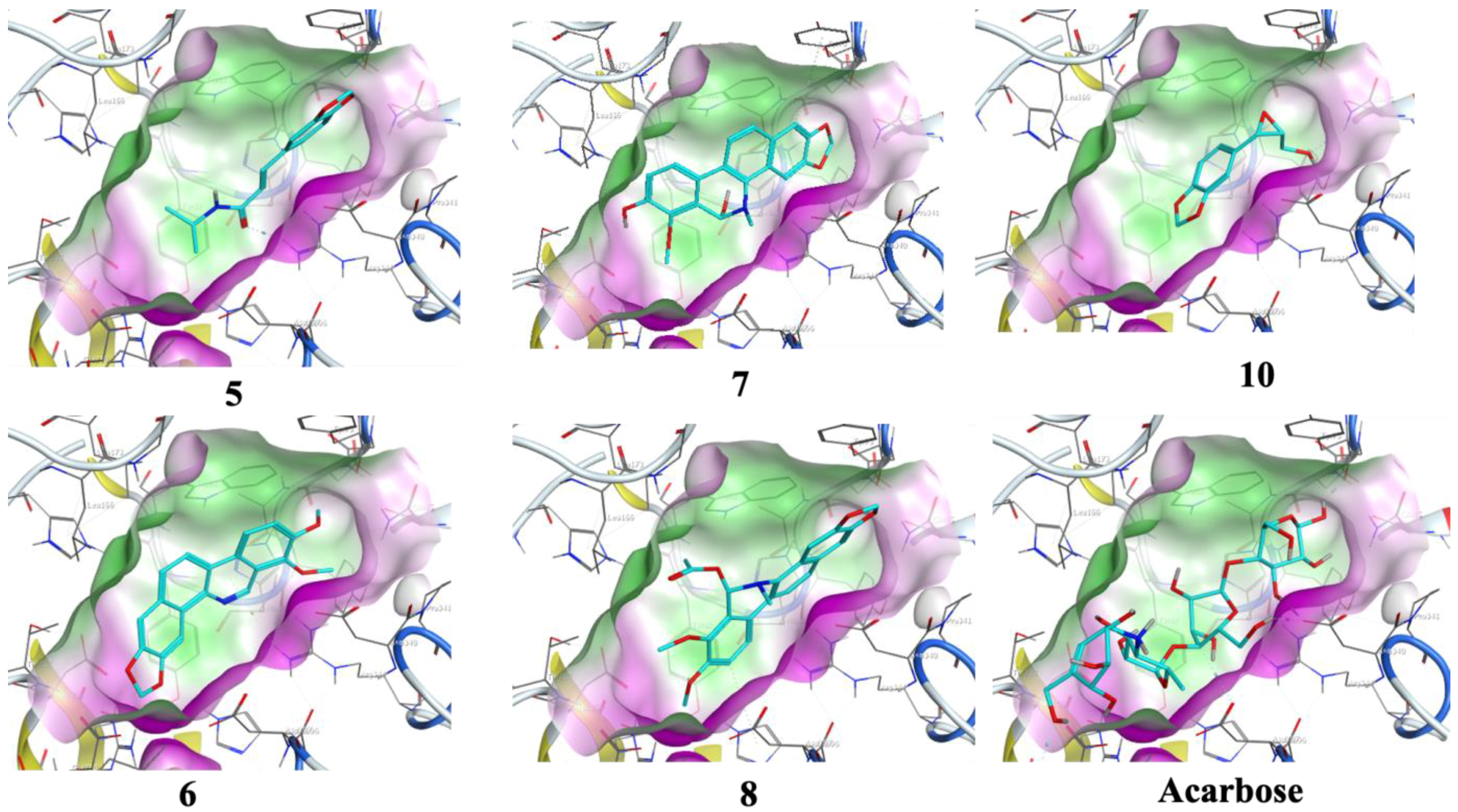
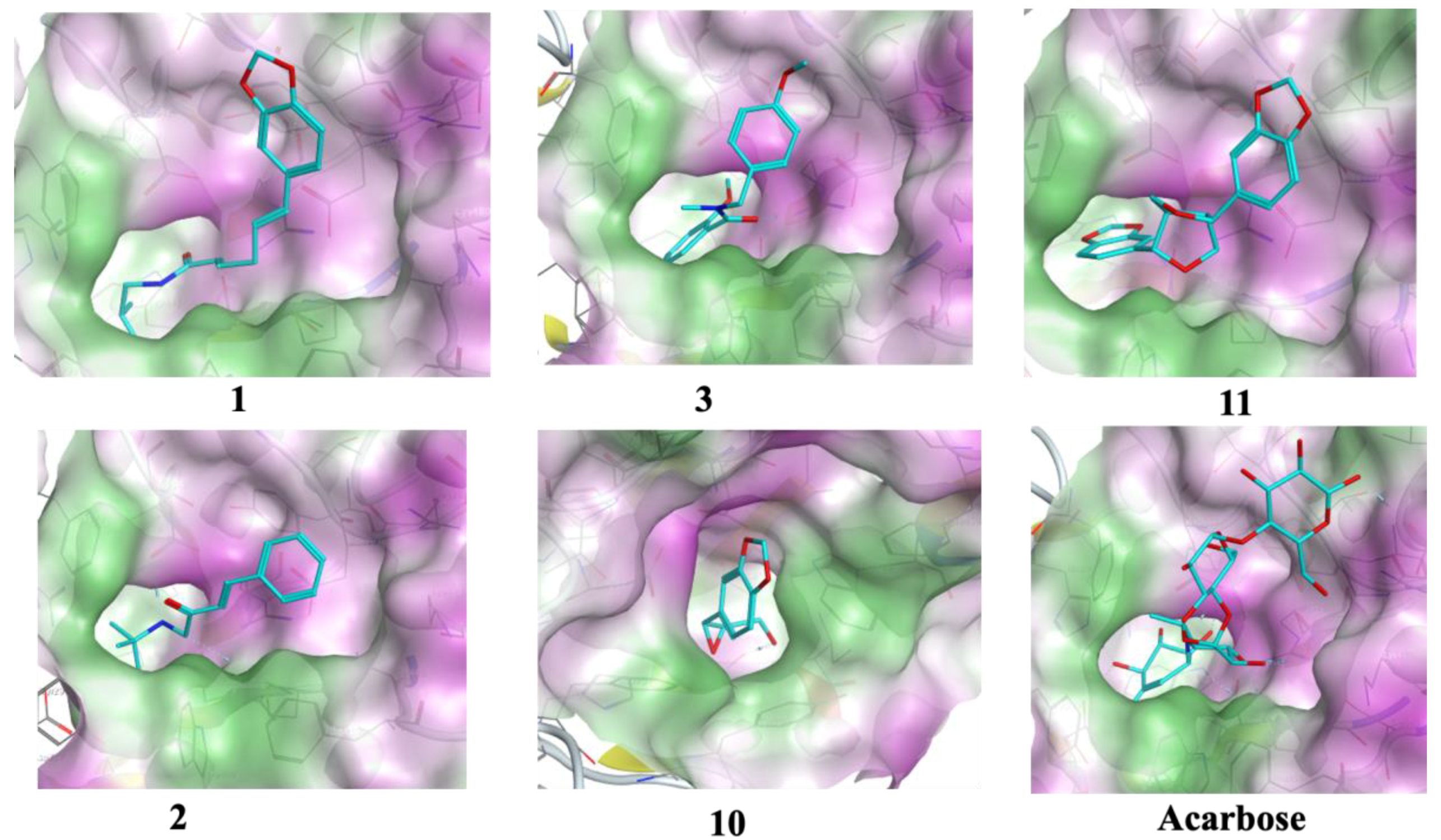
3.4. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marchetti, P.; Lupi, R.; del Guerra, S.; Bugliani, M.; D’Aleo, V.; Occhipinti, M.; Boggi, U.; Marselli, L.; Masini, M. Goals of Treatment for Type 2 Diabetes: Beta-Cell Preservation for Glycemic Control. Diabetes Care 2009, 32 (Suppl. 2), S178–S183. [Google Scholar] [CrossRef] [Green Version]
- Wild, S.; Roglic, G.; Green, A.; Sicree, R.; King, H. Global Prevalence of Diabetes: Estimates for the Year 2000 and Projections for 2030. Diabetes Care 2004, 27, 1047–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, S.F.; Mwangi, M.; Mutua, M.K.; Kibachio, J.; Hussein, A.; Ndegwa, Z.; Owondo, S.; Asiki, G.; Kyobutungi, C. Prevalence and Factors Associated with Pre-Diabetes and Diabetes Mellitus in Kenya: Results from a National Survey. BMC Public Health 2018, 18, 1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vecchio, I.; Tornali, C.; Bragazzi, N.L.; Martini, M. The Discovery of Insulin: An Important Milestone in the History of Medicine. Front. Endocrinol. 2018, 9, 613. [Google Scholar] [CrossRef] [Green Version]
- Chaudhury, A.; Duvoor, C.; Dendi, V.S.R.; Kraleti, S.; Chada, A.; Ravilla, R.; Marco, A.; Shekhawat, N.S.; Montales, M.T.; Kuriakose, K.; et al. Clinical Review of Antidiabetic Drugs: Implications for Type 2 Diabetes Mellitus Management. Front. Endocrinol. 2017, 8, 6. [Google Scholar] [CrossRef] [Green Version]
- Derosa, G.; Maffioli, P. α-Glucosidase Inhibitors and Their Use in Clinical Practice. Arch. Med. Sci. 2012, 8, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Inzucchi, S.E.; Lipska, K.J.; Mayo, H.; Bailey, C.J.; McGuire, D.K. Metformin in Patients with Type 2 Diabetes and Kidney Disease: A Systematic Review. JAMA 2014, 312, 2668–2675. [Google Scholar] [CrossRef] [Green Version]
- Swilam, N.; Nawwar, M.A.M.; Radwan, R.A.; Mostafa, E.S. Antidiabetic Activity and In Silico Molecular Docking of Polyphenols from Ammannia baccifera L. subsp. Aegyptiaca (Willd.) Koehne Waste: Structure Elucidation of Undescribed Acylated Flavonol Diglucoside. Plants 2022, 11, 452. [Google Scholar] [CrossRef]
- Olila, D.; Opuda-Asibo, J. Antibacterial and Antifungal Activities of Extracts of Zanthoxylum Chalybeum and Warburgia Ugandensis, Ugandan Medicinal Plants. Afr. Health Sci. 2001, 1, 66. [Google Scholar]
- Agwaya, M.S.; Vuzi, P.C.; Nandutu, A.M. Hypoglycemic Activity of Aqueous Root Bark Extract Zanthoxylum Chalybeum in Alloxan-Induced Diabetic Rats. J. Diabetes Res. 2016, 2016, 8727590. [Google Scholar] [CrossRef] [Green Version]
- Agwaya, M.; Nandutu, A.; Vuzi, P. Protective Effects of Zanthoxylum Chalybeum in Diabetes-Induced Myocardial Dysfunction in Rats. European J. Med. Plants 2016, 12, 1–10. [Google Scholar] [CrossRef]
- Ochieng, C.O.; Nyongesa, D.W.; Yamo, K.O.; Onyango, J.O.; Langat, M.K.; Manguro, L.A.O. α-Amylase and α-Glucosidase Inhibitors from Zanthoxylum Chalybeum Engl. Root Bark. Fitoterapia 2020, 146, 104719. [Google Scholar] [CrossRef]
- Bharathi, A.; Roopan, S.M.; Vasavi, C.S.; Munusami, P.; Gayathri, G.A.; Gayathri, M. Antidiabetic Studies of Dihydropyrimido [4,5-a]Acridin-2-Amines. BioMed Res. Int. 2014, 2014, 971569. [Google Scholar] [CrossRef] [Green Version]
- Bharatam, P.; Patel, D.; Adane, L.; Mittal, A.; Sundriyal, S. Modeling and Informatics in Designing Anti-Diabetic Agents. Curr. Pharm. Des. 2007, 13, 3518–3530. [Google Scholar] [CrossRef]
- Segel, I.H. Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems; John Wiley and Sons: Hoboken, NJ, USA, 1993; ISBN 978-0-471-30309-1. [Google Scholar]
- Halgren, T.A. Merck Molecular Force Field. I. Basis, Form, Scope, Parameterization, and Performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/#query=acarbose (accessed on 12 August 2022).
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug. Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- www.Openmolecules.Org. Available online: https://openmolecules.org/propertyexplorer/druglikeness.html (accessed on 12 August 2022).
- PreADMET|Prediction of ADME/Tox—Just Another BMDRC Sites Site. Available online: https://preadmet.webservice.bmdrc.org/ (accessed on 12 May 2022).
- SwissADME. Available online: http://www.swissadme.ch/ (accessed on 12 May 2022).
- ProTox-II—Prediction of TOXicity of Chemicals. Available online: https://tox-new.charite.de/protox_II/index.php?site=compound_search_similarity (accessed on 12 May 2022).
- Sim, L.; Quezada-Calvillo, R.; Sterchi, E.E.; Nichols, B.L.; Rose, D.R. Human Intestinal Maltase-Glucoamylase: Crystal Structure of the N-Terminal Catalytic Subunit and Basis of Inhibition and Substrate Specificity. J. Mol. Biol. 2008, 375, 782–792. [Google Scholar] [CrossRef] [PubMed]
- Brzozowski, A.M.; Davies, G.J. Structure of the Aspergillus Oryzae α-Amylase Complexed with the Inhibitor Acarbose at 2.0 Å Resolution. Biochemistry 1997, 36, 10837–10845. [Google Scholar] [CrossRef] [PubMed]
- RCSB PDB: Homepage. Available online: https://www.rcsb.org/ (accessed on 12 August 2022).
- Cruz, J.V.; Giuliatti, S.; Alves, L.B.; Silva, R.C.; Ferreira, E.F.B.; Kimani, N.M.; Silva, C.H.T.P.; de Souza, J.S.N.; Espejo-Román, J.M.; Santos, C.B.R. Identification of Novel Potential Cyclooxygenase-2 Inhibitors Using Ligand- and Structure-Based Virtual Screening Approaches. J. Biomol. Struct. Dyn. 2022, 40, 5386–5408. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.C.; Freitas, H.F.; Campos, J.M.; Kimani, N.M.; Silva, C.H.T.P.; Borges, R.S.; Pita, S.S.R.; Santos, C.B.R. Natural Products-Based Drug Design against SARS-CoV-2 Mpro 3clpro. Int. J. Mol. Sci. 2021, 22, 11739. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.B.; Ferreira, E.F.B.; Espejo-Román, J.M.; Costa, G.V.; Cruz, J.V.; Kimani, N.M.; Costa, J.S.; Bittencourt, J.A.H.M.; Cruz, J.N.; Campos, J.M.; et al. Galantamine Based Novel Acetylcholinesterase Enzyme Inhibitors: A Molecular Modeling Design Approach. Molecules 2023, 28, 1035. [Google Scholar] [CrossRef]
- Rossino, G.; Rui, M.; Pozzetti, L.; Schepmann, D.; Wünsch, B.; Zampieri, D.; Pellavio, G.; Laforenza, U.; Rinaldi, S.; Colombo, G.; et al. Setup and Validation of a Reliable Docking Protocol for the Development of Neuroprotective Agents by Targeting the Sigma-1 Receptor (S1R). Int. J. Mol. Sci. 2020, 21, 7708. [Google Scholar] [CrossRef] [PubMed]
- Al-Khodairy, F.M.; Khan, M.K.A.; Kunhi, M.; Pulicat, M.S.; Akhtar, S.; Arif, J.M. In Silico Prediction of Mechanism of Erysolin-Induced Apoptosis in Human Breast Cancer Cell Lines. Am. J. Bioinform. Res. 2013, 3, 62–71. [Google Scholar] [CrossRef]
- Hevener, K.E.; Zhao, W.; Ball, D.M.; Babaoglu, K.; Qi, J.; White, S.W.; Lee, R.E. Validation of Molecular Docking Programs for Virtual Screening against Dihydropteroate Synthase. J. Chem. Inf. Model 2009, 49, 444–460. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Lead- and Drug-like Compounds: The Rule-of-Five Revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Protti, Í.F.; Rodrigues, D.R.; Fonseca, S.K.; Alves, R.J.; de Oliveira, R.B.; Maltarollo, V.G. Do Drug-Likeness Rules Apply to Oral Prodrugs? ChemMedChem 2021, 16, 1446–1456. [Google Scholar] [CrossRef]
- Ertl, P.; Rohde, B.; Selzer, P. Fast Calculation of Molecular Polar Surface Area as a Sum of Fragment-Based Contributions and Its Application to the Prediction of Drug Transport Properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef]
- Kalita, J.; Chetia, D.; Rudrapal, M. Molecular Docking, Drug-Likeness Studies and ADMET Prediction of Quinoline Imines for Antimalarial Activity. J. Med. Chem. Drug. Des. 2019, 2, 208–218. [Google Scholar] [CrossRef]
- Hansch, C.; Fujita, T. P-σ-π Analysis. A Method for the Correlation of Biological Activity and Chemical Structure. J. Am. Chem. Soc. 1964, 86, 1616–1626. [Google Scholar] [CrossRef]
- Box, K.; Comer, J. Using Measured PKa, LogP and Solubility to Investigate Supersaturation and Predict BCS Class. Curr. Drug Metab. 2008, 9, 869–878. [Google Scholar] [CrossRef]
- Zhao, Y.H.; Le, J.; Abraham, M.H.; Hersey, A.; Eddershaw, P.J.; Luscombe, C.N.; Boutina, D.; Beck, G.; Sherborne, B.; Cooper, I.; et al. Evaluation of Human Intestinal Absorption Data and Subsequent Derivation of a Quantitative Structure–Activity Relationship (QSAR) with the Abraham Descriptors. J. Pharm. Sci. 2001, 90, 749–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.L.; Chen, C.; Yang, J. Predictive Model of Blood-Brain Barrier Penetration of Organic Compounds. Acta Pharmacol. Sin. 2005, 26, 500–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, D.; Schmidt, S.; Derendorf, H. Importance of Relating Efficacy Measures to Unbound Drug Concentrations for Anti-Infective Agents. Clin. Microbiol. Rev. 2013, 26, 274–288. [Google Scholar] [CrossRef] [Green Version]
- Wairata, J.; Sukandar, E.R.; Fadlan, A.; Purnomo, A.S.; Taher, M.; Ersam, T. Evaluation of the Antioxidant, Antidiabetic, and Antiplasmodial Activities of Xanthones Isolated from Garcinia forbesii and Their In Silico Studies. Biomedicines 2021, 9, 1380. [Google Scholar] [CrossRef]
- Ogu, C.C.; Maxa, J.L. Drug Interactions Due to Cytochrome P450. Bayl. Univ. Med. Cent. Proc. 2017, 13, 421–423. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A Webserver for the Prediction of Toxicity of Chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zafar, F.; Gupta, A.; Thangavel, K.; Khatana, K.; Sani, A.A.; Ghosal, A.; Tandon, P.; Nishat, N. Physicochemical and Pharmacokinetic Analysis of Anacardic Acid Derivatives. ACS Omega 2020, 5, 6021–6030. [Google Scholar] [CrossRef]
- Rehman, N.U.; Rafiq, K.; Khan, A.; Halim, S.A.; Ali, L.; Al-Saady, N.; Al-Balushi, A.H.; Al-Busaidi, H.K.; Al-Harrasi, A. α-Glucosidase Inhibition and Molecular Docking Studies of Natural Brominated Metabolites from Marine Macro Brown Alga Dictyopteris Hoytii. Mar. Drugs 2019, 17, 666. [Google Scholar] [CrossRef] [Green Version]
- Aminu, K.S.; Uzairu, A.; Umar, A.B.; Ibrahim, M.T. Salicylic Acid Derivatives as Potential α-Glucosidase Inhibitors: Drug Design, Molecular Docking and Pharmacokinetic Studies. Bull. Natl. Res. Cent. 2022, 46, 162. [Google Scholar] [CrossRef]
- Kulkarni, S.; Gupta, P.; Pallavi, A.; Zeng, W. Investigation of Enzymes Binding to “Voglibose- an Antidiabetic Drug” and the Choice of Enzyme to Be Used for Biosensing. J. Pharm. Res. Int. 2016, 14, 30369. [Google Scholar] [CrossRef]
- Mohapatra, S.; Prasad, A.; Haque, F.; Ray, S.; De, B.; Ray, S.S. In Silico Investigation of Black Tea Components on α-Amylase, α-Glucosidase and Lipase. J. Appl. Pharm. Sci. 2015, 5, 042–047. [Google Scholar] [CrossRef] [Green Version]
- Lakehal, S.; Ferkous, F.; Kraim, K.; Attoui Yahia, O.; Saihi, Y. Molecular Docking Study on Xanthone Derivatives toward Alpha-Glucosidase. Res. J. Pharm. Biol. Chem. Sci. 2016, 7, 1739–1750. [Google Scholar]
- Saddique, F.A.; Ahmad, M.; Ashfaq, U.A.; Mansha, A.; Gul Khan, S. Alpha-Glucosidase Inhibition and Molecular Docking Studies of 4-Hydroxy-N’-[Benzylidene/1-Phenylethylidene]-2H-1,2-Benzothiazine-3-Carbohydrazide 1,1-Dioxides. Chiang Mai J. Sci. 2021, 48, 460–469. [Google Scholar]
- Saddique, F.A.; Ahmad, M.; Ashfaq, U.A.; Muddassar, M.; Sultan, S.; Zaki, M.E.A. Identification of Cyclic Sulfonamides with an N-Arylacetamide Group as α-Glucosidase and α-Amylase Inhibitors: Biological Evaluation and Molecular Modeling. Pharmaceuticals 2022, 15, 106. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | α-Amylase | α-Glucosidase | ||||
|---|---|---|---|---|---|---|
| Inhibition Mode | Ki (mM) | R2 | Inhibition Mode | Ki (mM) | R2 | |
| 1 | Non-competitive | 13.36 ± 2.43 ** | 0.9345 | Non-competitive | 20.95 ± 2.14 | 0.9824 |
| 2 | Non-competitive | 11.05 ± 0.58 ** | 0.9424 | Uncompetitive | 44.58 ± 1.65 | 0.9207 |
| 3 | Non-competitive | 14.83 ± 0.50 ** | 0.9381 | Non-competitive | 17.56 ± 0.24 | 0.9824 |
| 4 | Non-competitive | 26.69 ± 2.13 ** | 0.9899 | Non-competitive | 34.73 ± 0.79 ** | 0.9198 |
| 5 | Mixed | 2.74 ± 0.06 | 0.9572 | Mixed | 7.64 ± 0.02 ** | 0.9544 |
| 6 | Mixed | 7.57 ± 0.59 | 0.9527 | Mixed | 7.68 ± 0.04 ** | 0.9578 |
| 7 | Mixed | 3.34 ± 0.03 | 0.9978 | Mixed | 4.73 ± 0.10 ** | 0.9966 |
| 8 | Mixed | 3.10 ± 0.20 | 0.9753 | Mixed | 9.17 ± 0.10 ** | 0.9913 |
| 9 | Uncompetitive | 26.28 ± 1.47 ** | 0.9619 | Uncompetitive | 20.62 ± 1.94 | 0.9929 |
| 10 | Competitive | 5.54 ± 1.02 | 0.9850 | Competitive | 17.21 ± 0.16 | 0.8692 |
| 11 | Non-competitive | 12.53 ± 1.957 ** | 0.9552 | non-competitive | 24.33 ± 1.93 | 0.8748 |
| Acarbose | Competitive | 6.14 ± 0.01 | 0.9606 | Competitive | 22.40 ± 1.23 | 0.8387 |
| Molecule | MW a | LogP b | HBA c | HBD d | RB e | Electronegative Atoms | Rings Closures | Carbo-rings | Hetero-rings | Aromatic Atoms | TPSA f (Å) | Mutagenic | Tumorigenic | Reproductive Effect | Irritant |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 287.358 | 3.7 | 4 | 1 | 6 | 2 | 1 | 1 | 0 | 6 | 47.6 | none | none | high | none |
| 2 | 218.319 | 1.1 | 2 | 1 | 5 | 4 | 2 | 1 | 1 | 6 | 33.7 | none | none | none | none |
| 3 | 285.342 | 2.6 | 4 | 0 | 5 | 4 | 2 | 1 | 1 | 12 | 38.8 | none | none | none | none |
| 4 | 233.266 | 2.3 | 4 | 1 | 3 | 4 | 2 | 1 | 2 | 6 | 47.6 | none | none | high | none |
| 5 | 257.288 | 2.5 | 4 | 0 | 3 | 4 | 3 | 1 | 3 | 10 | 40.6 | none | high | none | none |
| 6 | 333.342 | 4.3 | 5 | 0 | 2 | 4 | 3 | 2 | 0 | 18 | 49.8 | low | none | none | none |
| 7 | 351.357 | 3.3 | 6 | 2 | 1 | 5 | 5 | 2 | 1 | 16 | 71.4 | high | high | none | none |
| 8 | 407.421 | 4.1 | 7 | 0 | 4 | 5 | 3 | 2 | 1 | 16 | 66.5 | high | high | none | none |
| 9 | 326.347 | 3.5 | 5 | 2 | 5 | 6 | 5 | 2 | 4 | 15 | 72.1 | none | none | high | high |
| 10 | 194.185 | 0.9 | 4 | 1 | 2 | 6 | 6 | 3 | 2 | 6 | 51.2 | low | none | none | none |
| 11 | 354.357 | 3.2 | 6 | 0 | 2 | 7 | 5 | 3 | 2 | 12 | 55.4 | none | none | none | none |
| 12 | 646.613 | −8.4 | 19 | 14 | 9 | 19 | 4 | 3 | 2 | 0 | 325.8 | none | none | none | none |
| Property\Compound | BBB | HIA (%) | Plasma Protein Binding (%) | CYP3A4 Inhibitor | CYP2C9 Inhibitor | CYP2D6 Inhibitor | CYP2C19 Inhibitor | Skin Permeability (cm/h) | Caco2 | Hepatotoxicity | Carcinogenicity | Immunotoxicity | Cytotoxicity | Toxicity Class |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1.13 | 95.34 | 87.74 | non | non | non | non | −2.86 | 44.84 | Inactive | Inactive | Active | Inactive | 4 |
| 2 | 0.79 | 96.22 | 40.15 | non | non | yes | non | −1.03 | 54.93 | Inactive | Inactive | Inactive | Inactive | 5 |
| 3 | 0.17 | 98.11 | 87.07 | non | non | non | non | −2.92 | 54.35 | Inactive | Inactive | Inactive | Inactive | 4 |
| 4 | 0.14 | 94.94 | 53.54 | non | non | non | non | −3.43 | 38.10 | Inactive | Inactive | Active | Inactive | 4 |
| 5 | 2.68 | 97.93 | 90.19 | yes | yes | non | yes | −3.74 | 56.83 | Active | Inactive | Active | Inactive | 4 |
| 6 | 0.05 | 97.64 | 90.55 | yes | yes | non | yes | −3.98 | 52.67 | Inactive | Active | Active | Inactive | 4 |
| 7 | 0.20 | 94.62 | 87.76 | yes | yes | non | non | −4.02 | 21.94 | Inactive | Inactive | Active | Active | 4 |
| 8 | 0.02 | 97.55 | 89.45 | yes | yes | non | non | −3.88 | 48.50 | Inactive | Active | Active | Active | 4 |
| 9 | 0.28 | 94.23 | 88.29 | yes | yes | non | yes | −2.92 | 30.85 | Inactive | Inactive | Active | Inactive | 5 |
| 10 | 0.58 | 93.66 | 45.96 | yes | yes | non | yes | −3.97 | 24.39 | Inactive | Active | Inactive | Inactive | 3 |
| 11 | 0.05 | 97.12 | 83.12 | yes | yes | non | yes | −4.42 | 57.03 | Inactive | Active | Active | Inactive | 3 |
| Acarbose | 0.03 | 0.00 | 31.61 | yes | non | yes | non | −5.19 | 0.81 | Active | Inactive | Active | Inactive | 6 |
| α-Amylase | α-Glucosidase | |||||||
|---|---|---|---|---|---|---|---|---|
| Compound No. | Docking Score (kcal/mol) | RMSD Value (Å) | Binding Residues | Interaction | Docking Score (kcal/mol) | RMSD Value (Å) | Binding Residues | Interaction |
| 1 | −11.2 | 1.5 | ASP 297 ARG 344 | H-donor H-acceptor | −9.3 | 2.0 | ASP 542 | H-donor |
| 2 | -13.8 | 0.8 | ASP 340 ARG 344 ASP 340 | H-donor H-acceptor Ionic | -12.6 | 1.8 | MET 444 ASP 542 ASP 443 ASP 542 | H-donor H-donor Ionic ionic |
| 3 | −10.1 | 0.8 | HIS 296 TYR 82 | H-π π-π | −10.6 | 0.9 | ARG 526 | H-acceptor |
| 4 | −9.4 | 0.8 | ASP 340 | H-donor | −9.0 | 1.2 | MET 444 ASP 542 PHE 575 | H-donor H-donor H-π |
| 5 | −10.0 | 1.9 | TRP 83 | π-H | −9.5 | 0.8 | ASP 542 TRP 406 | H-donor π-H |
| 6 | −11.9 | 1.9 | ARG | π-cation | −9.8 | 0.9 | - | - |
| 7 | −10.9 | 0.8 | HIS 210 LEU 232 | H-donor π-H | −13.8 | 0.8 | ASP 443 ASP 542 | H-donor H-donor |
| 8 | −10.8 | 1.3 | HIS 296 TYR 82 | H-π H-π | −8.0 | 2.0 | THR 204 | π-H |
| 9 | −11.4 | 1.0 | ASP 340 GLN 35 TYR 79 HIS 296 | H-donor H-acceptor H-acceptor H-π | −10.3 | 2.8 | ASP 327 | H-donor |
| 10 | −10.1 | 1.1 | ASP 340 GLN 35 | H-donor H-acceptor | −10.0 | 0.6 | ASP 327 | H-donor |
| 11 | −12.3 | 1.2 | ARG 204 TRP 83 | H-acceptor H-acceptor | −11.0 | 1.5 | ARG 526 PHE 575 | H-acceptor H-π |
| Acarbose | −17.6 | 1.7 | ASP 206 GLU 230 ASP 340 ASP 168 ARG 204 TRP 83 ASP 206 | H-donor H-donor H-donor H-donor H-acceptor H-acceptor ionic | −20.5 | 1.6 | ASP 542 ASP 327 ASP 203 MET 444 ASP 474 HIS 600 ARG 526 ASP 542 | H-donor H-donor H-donor H-donor H-donor H-acceptor H-acceptor ionic |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kimani, N.M.; Ochieng, C.O.; Ogutu, M.D.; Yamo, K.O.; Onyango, J.O.; Santos, C.B.R. Inhibition Kinetics and Theoretical Studies on Zanthoxylum chalybeum Engl. Dual Inhibitors of α-Glucosidase and α-Amylase. J. Xenobiot. 2023, 13, 102-120. https://doi.org/10.3390/jox13010009
Kimani NM, Ochieng CO, Ogutu MD, Yamo KO, Onyango JO, Santos CBR. Inhibition Kinetics and Theoretical Studies on Zanthoxylum chalybeum Engl. Dual Inhibitors of α-Glucosidase and α-Amylase. Journal of Xenobiotics. 2023; 13(1):102-120. https://doi.org/10.3390/jox13010009
Chicago/Turabian StyleKimani, Njogu M., Charles O. Ochieng, Mike Don Ogutu, Kevin Otieno Yamo, Joab Otieno Onyango, and Cleydson B. R. Santos. 2023. "Inhibition Kinetics and Theoretical Studies on Zanthoxylum chalybeum Engl. Dual Inhibitors of α-Glucosidase and α-Amylase" Journal of Xenobiotics 13, no. 1: 102-120. https://doi.org/10.3390/jox13010009