
New-Generation Ektacytometry Study of Red Blood Cells in Different Hemoglobinopathies and Thalassemia

Abstract
:1. Introduction
2. Patients
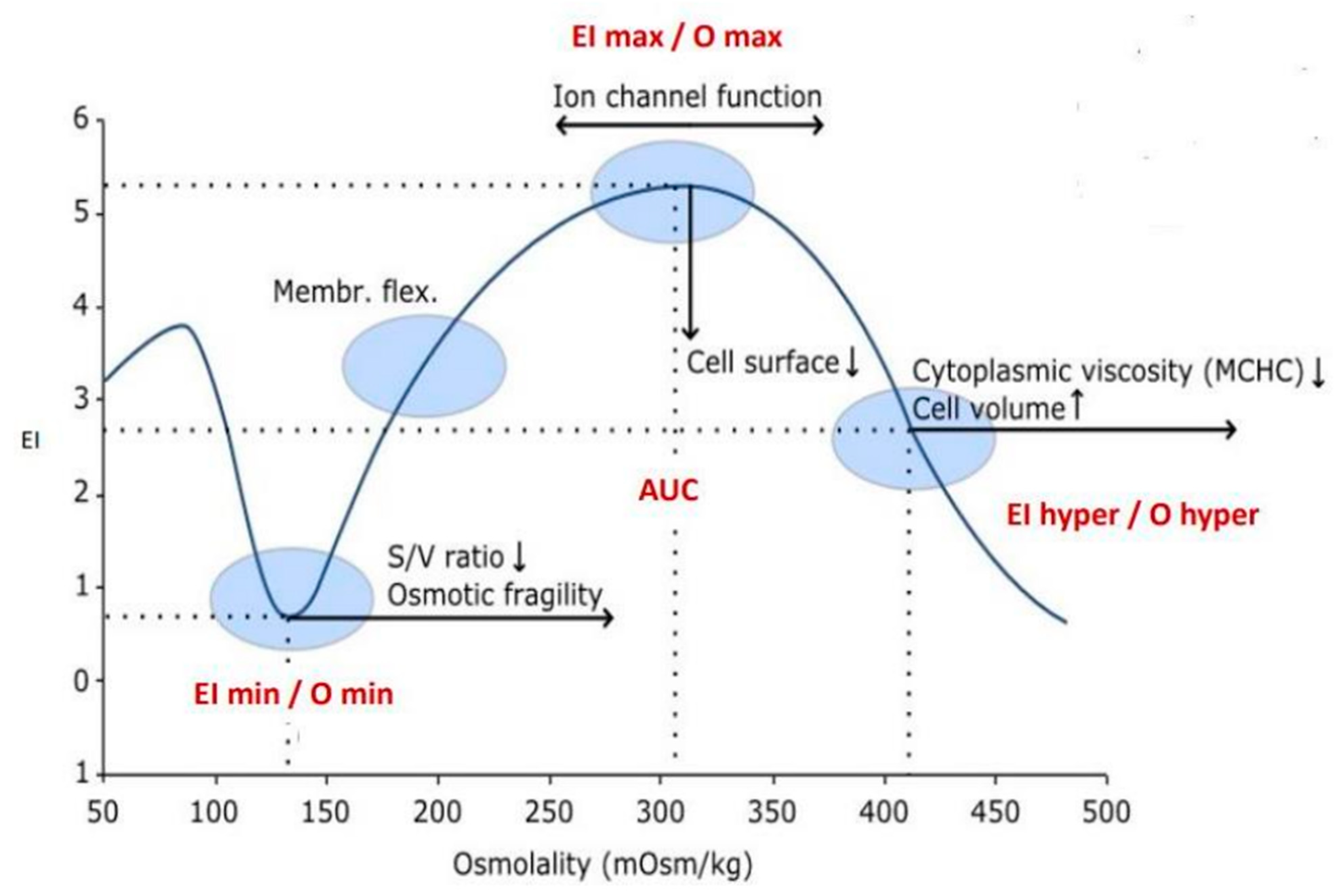
3. Methods
4. Results
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vives-Corrons, J. The Rare Anaemias. In Rare Diseases [Internet]; Wu, Z.H., Ed.; IntechOpen: London, UK, 2019; Available online: https://www.intechopen.com/chapters/69673 (accessed on 13 February 2022). [CrossRef] [Green Version]
- Mohandas, N.J. Red cell membrane disorders. Int. J. Lab. Hematol. 2017, 39 (Suppl. 1), 47. [Google Scholar]
- Kohne, E. Hemoglobinopathies: Clinical manifestations, diagnosis, and treatment. Dtsch. Ärzteblatt Int. 2011, 108, 532–540. [Google Scholar]
- Brancaleoni, V.; Di Pierro, E.; Motta ICappellini, M.D. Laboratory diagnosis of thalassemia. Int. J. Lab. Hematol. 2016, 38, 32–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polin, R.A.; Steven, H.; Abman, D.; David Rowitch, F. Fetal and Neonatal Physiology, 5th ed.; Elsevier: Amsterdam, The Netherlands, 2017; Volume 2. [Google Scholar]
- Andolfo, I.; Russo, R.; Gambale, A.; Iolascon, A. New insights on hereditary erythrocyte membrane defects. Haematologica 2016, 101, 1284–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolton-Maggs, P.H.; Langer, J.C.; Iolascon, A. General Haematology Task Force of the British Committee for Standards in H. Guidelines for the diagnosis and management of hereditary spherocytosis—2011 Update. Br. J. Haematol. 2011, 2012, 37–49. [Google Scholar]
- Bianchi, P.; Vercellati, C.; Fermo, E. How will next generation sequencing (NGS) improve the diagnosis of congenital hemolytic anemia? Ann. Transl. Med. 2020, 8, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Llaudet-Planas, E.; Vives-Corrons, J.L.; Rizzuto, V.; Gómez-Ramírez, P.; Sevilla Navarro, J.; Coll Sibina, M.T.; García-Bernal, M.; Ruiz Llobet, D.; Badell, I.; Velasco-Puyó, P.; et al. Osmotic gradient ektacytometry: A valuable screening test for hereditary spherocytosis and other red blood cell membrane disorders. Int. J. Lab. Hematol. 2018, 40, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Zaninoni, A.; Fermo, E.; Vercellati, C.; Consonni, D.; Marcello, A.P.; Zanella, A.; Cortelezzi, A.; Barcellini, W.; Bianchi, P. Use of laser assisted optical rotational cell analyzer (LoRRca MaxSis) in the diagnosis of RBC membrane disorders, enzyme defects, and congenital dyserythropoietic anemias: A monocentric study on 202 patients. Front. Physiol. 2018, 9, 451. [Google Scholar] [CrossRef] [PubMed]
- Vives-Corrons, J.L.; Krishnevskaya, E.; Rodriguez, I.H.; Ancochea, A. Characterization of hereditary red blood cell membranopathies using combined targeted next-generation sequencing and osmotic gradient ektacytometry. Int. J. Hematol. 2021, 113, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Vives Corrons, L.; Bascompte, A. Technical Manual for Hematology Diagnosis, 4th ed.; Elsevier-Masson: Amsterdam, The Netherlands, 2014. (In Spanish) [Google Scholar]
- Huisjes, R.; Bogdanova, A.; van Solinge, W.W.; Schiffelers, R.M.; Kaestner, L.; van Wijk, R. Squeezing for Life—Properties of Red Blood Cell Deformability. Front. Physiol. 2018, 9, 656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berga, L.; Feliu, E.; Vives Corrons, J.L. Deformabilidad Eritrocitaria y Anemias Hemolíticas. Rev. Obras Publicas 1989, 3285, 825–838. [Google Scholar]
- Ilesanmi, O.O. Pathological basis of symptoms and crises in sickle cell disorder: Implications for counseling and psychotherapy. Hematol. Rep. 2010, 2, e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rees, D.C.; Williams, T.N.; Gladwin, M.T. Sickle-cell disease. Lancet 2010, 376, 2018–2031. [Google Scholar] [CrossRef] [PubMed]
- Krishnevskaya, E.; Payán-Pernía, S.; Hernández-Rodríguez, I.; Remacha Sevilla, Á.F.; Ancochea Serra, Á.; Morales-Indiano, C.; Serra Ferrer, M.; Vives-Corrons, J.L. Distinguishing iron deficiency from beta-thalassemia trait by new generation ektacytometry. Int. J. Lab. Hematol. 2021, 43, e58–e60. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Mora, L.; Cabello-Fusarés, M.; Ferré-Torres, J.; Riera-Llobet, C.; Krishnevskaya, E.; Trejo-Soto, C.; Payán-Pernía, S.; Hernández-Rodríguez, I.; Morales-Indiano, C.; Alarcón, T.; et al. Blood Rheological Characterization of β-Thalassemia Trait and Iron Deficiency Anemia Using Front Microrheometry. Front. Physiol. 2021, 12, 761411. [Google Scholar] [CrossRef] [PubMed]
- Bunn, H.F. Pathogenesis and treatment of sickle cell disease. N. Engl. J. Med. 1997, 337, 762–769. [Google Scholar] [CrossRef] [PubMed]
- Barabino, G.A.; Platt, M.O.; Kaul, D.K. Sickle cell biomechanics. Annu. Rev. Biomed. Eng. 2010, 12, 345–367. [Google Scholar] [CrossRef] [PubMed]
- Lazarova, E.; Gulbis, B.; Oirschot, B.V.; van Wijk, R. Next-generation osmotic gradient ektacytometry for the diagnosis of hereditary spherocytosis: Interlaboratory method validation and experience. Clin. Chem. Lab. Med. 2017, 55, 394–402. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| n = 96 | RBCs (×1012/L) | Hb (g/L) | Reticulocytes (×109/L) | MCV (fL) | MCH (pg) | MCHC (g/L) | RDW (%) | |
|---|---|---|---|---|---|---|---|---|
| HbD | 4 | 5.32 ± 0.5 | 141.8 ± 5.7 | Normal | 79.95 ± 5. | 26.80 ± 1.9 | 335.3 ± 6.0 | 14.25 ± 1.5 |
| HbC | 7 | 5.01 ± 0.76 | 134.6 ± 19.4 | Normal | 78.94 ± 8. | 27.15 ± 3.5 | 340.4 ± 12.0 | 15.59 ± 5.1 |
| HbC+ Hb O-Arab * | 1 | 6.02 | 156 | 205 | 76.3 | 25.9 | 339 | 17.5 |
| HbE | 5 | 5.37 ± 0.48 | 138.7 ± 9.3 | Normal | 80.47 ± 4 | 25.9 ± 1.0 | 322.3 ± 8.0 | 15.20 ± 0.8 |
| HbS | 24 | 4.71 ± 0.92 | 131.1 ± 23.4 | Normal | 83.93 ± 5 | 28.02 ± 2.1 | 333.5 ± 9.2 | 14.65 ± 1.9 |
| HbSS * | 7 | 3.09 ± 0.92 | 88.0 ± 5.2 | 135.2 ± 9.2 | 87.57 ± 14 | 29.92 ± 5.5 | 341.0 ± 14.5 | 20.67 ± 1.9 |
| HbSC | 2 | 4.62 ± 0.2 | 139 ± 14 | Normal | 87.3 ± 6 | 30.05 ± 1.0 | 345± 9.0 | 15.60 ± 0.7 |
| HbS+ Hb O-Arab * | 1 | 2.71 | 76 | 231.2 | 85.2 | 28 | 329 | 20.4 |
| β-thal * | 41 | 5.30 ± 1.01 | 106.5 ± 21.3 | 115.3 ± 7.8 | 63.38 ± 4 | 20.11 ± 1.3 | 317.4 ± 5.3 | 17.33 ± 1.5 |
| δβ-thal | 3 | 5.21 ± 0.49 | 148.8 ± 2.5 | Normal | 80.05 ± 51 | 27.10 ± 1.9 | 329.1 ± 6.1 | 14.55 ± 09 |
| α-thal * | 1 | 4.66 | 103 | 113.6 | 69 | 22.1 | 323 | 15.3 |
| Controls | 50 | 4.50–5.1 | 135–160 | 40–85 | 80–95 | 27–32 | 300–350 | 10–15 |
| n = 96 | Omín (mOsm/kg) | EImax | Ohyper (mOsm/kg) | AUC | |
|---|---|---|---|---|---|
| HbD | 4 | 138 (128–148) | 0.615 (0.607–0.622) | ** 443 (428–458) | 169 (156–182) |
| HbC | 7 | ** 128 (110–146) | ** 0.597 (0.577–0,616) | ** 415 (403–136) | ** 152 (136–168) |
| HbC+ Hb O-Arab | 1 | 133 | 0.53 | 345 | 96.9 |
| HbE | 5 | * 136.5 (126–146) | 0.609 (0.597–0.619) | 463 (440–487) | * 173 (166–181) |
| HbS | 24 | ** 132.7 (110–155) | 0.614 (0.596–0,63) | ** 436.7 (406–467) | 162 (144–179) |
| HbSS | 7 | ** 108 (78–139) | ** 0.572 (0.513–0.631) | ** 385 (290–479) | ** 146 (114–176) |
| HbSC | 2 | ** 102 (98–106) | 0.555 (0.496–0.605) | ** 364 (352–356) | * 116 (107–124) |
| HbS+ Hb O-Arab | 1 | 100 | 0.545 | 403 | 144.6 |
| β-thal | 41 | ** 119 (98–139) | ** 0.597 (0.567–0.623) | ** 411 (411–482) | ** 171 (155–185) |
| δβ-thal | 3 | 124 (100–148) | 0.588 (0.574–0.603) | ** 452 (405–498.5) | 168 |
| α-thal (α-/α-) | 1 | 120 | 0.61 | 474 | 189.8 |
| Controls | 50 | 145.5 (130–161) | 0.614 (0.6–0.628) | 465.5 (450–481) | 166.8 (160–175) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krishnevskaya, E.; Molero, M.; Ancochea, Á.; Hernández, I.; Vives-Corrons, J.-L. New-Generation Ektacytometry Study of Red Blood Cells in Different Hemoglobinopathies and Thalassemia. Thalass. Rep. 2023, 13, 70-76. https://doi.org/10.3390/thalassrep13010007
Krishnevskaya E, Molero M, Ancochea Á, Hernández I, Vives-Corrons J-L. New-Generation Ektacytometry Study of Red Blood Cells in Different Hemoglobinopathies and Thalassemia. Thalassemia Reports. 2023; 13(1):70-76. https://doi.org/10.3390/thalassrep13010007
Chicago/Turabian StyleKrishnevskaya, Elena, Marta Molero, Águeda Ancochea, Ines Hernández, and Joan-Lluis Vives-Corrons. 2023. "New-Generation Ektacytometry Study of Red Blood Cells in Different Hemoglobinopathies and Thalassemia" Thalassemia Reports 13, no. 1: 70-76. https://doi.org/10.3390/thalassrep13010007