
Alpha-Thalassemia: Diversity of Clinical Phenotypes and Update on the Treatment

Abstract
:1. Introduction
2. Phenotypic Diversity of HbH Disease
2.1. Effects of α-Globin Genotypes on Severity of HbH
2.2. Effects of Co-Inheritance of β-Thalassemia
2.3. Other Genetic Modifiers of HbH
3. Diagnosis of HbH Disease
3.1. Hb Levels and Red Cell Indices
3.2. Inclusion Bodies
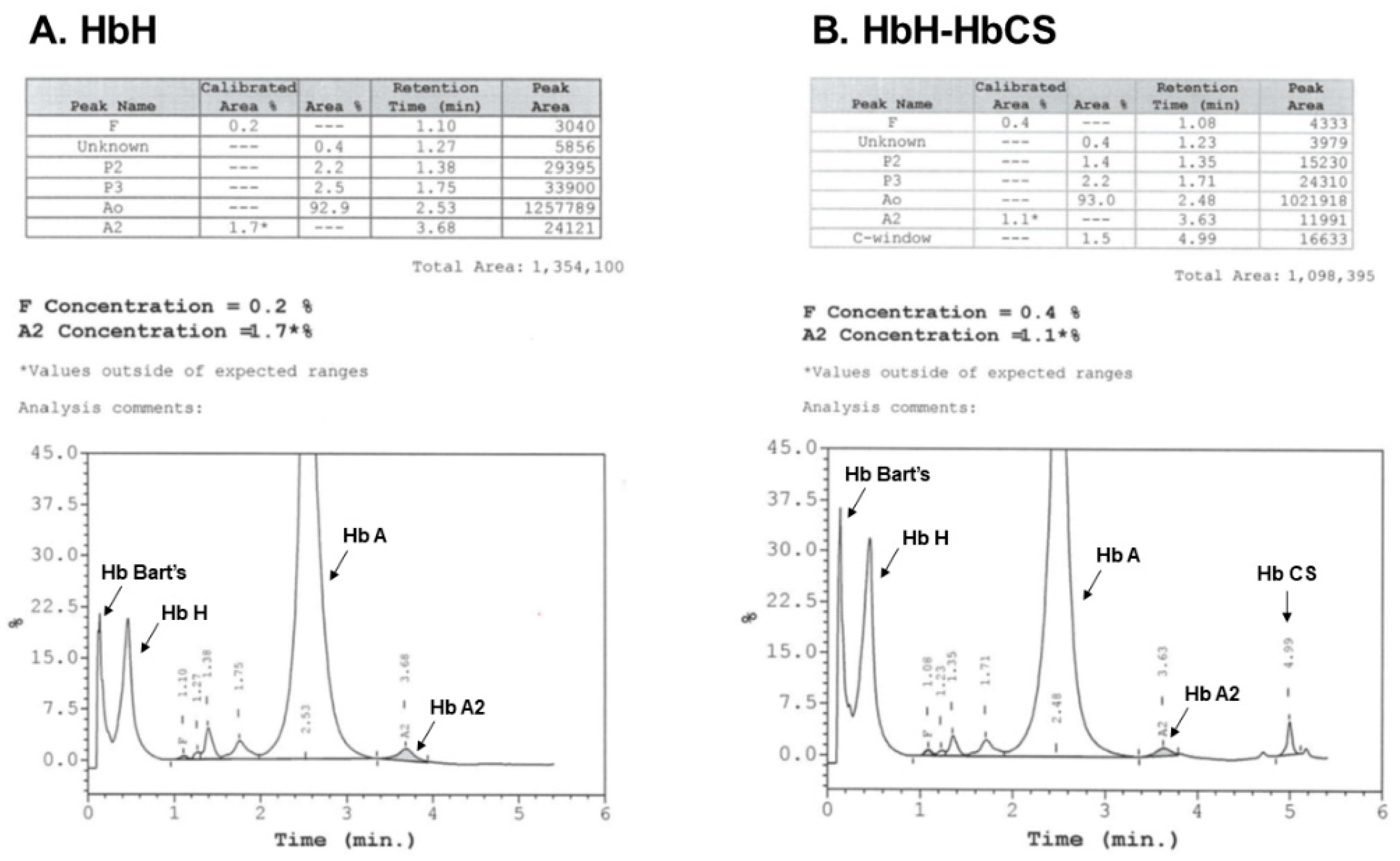
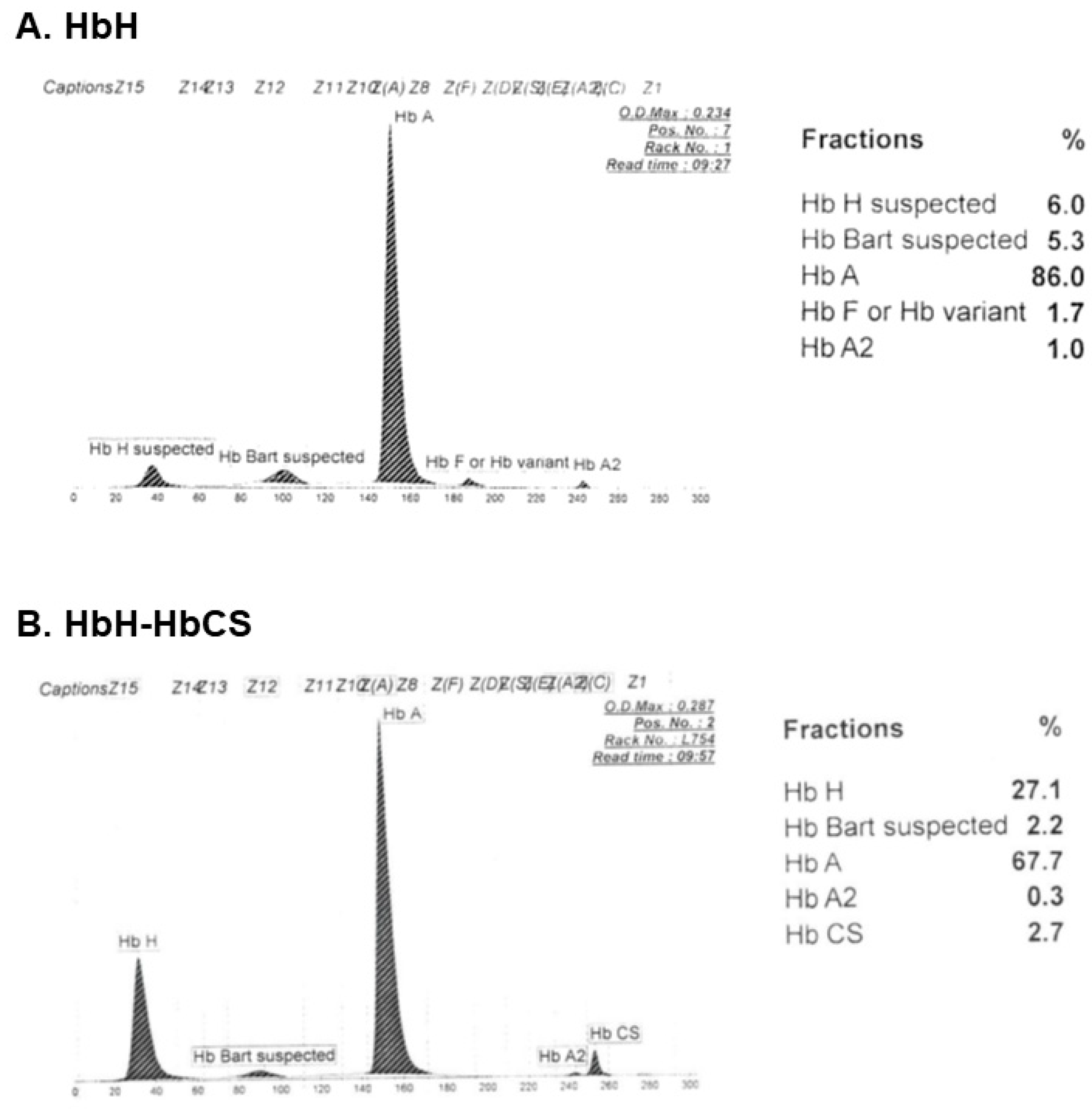
3.3. Hb Analysis
3.4. Molecular Diagnosis
4. Current General Management of HbH Disease
5. Challenges in Transfusion Management of HbH Disease
6. Impact of HbH on Older Patients and Challenges in Management of Iron Overload
7. Current and Ongoing Natural History-Modifying Treatments of HbH Disease
8. The Paradigm Changes for BHFS
- Mothers carrying BHFS fetuses often experience obstetric complications, such as preterm delivery, polyhydramnios and preeclampsia. This occurred irrespective of whether the fetuses received IUTs (intrauterine transfusion, exchange transfusion and/or in utero stem cell transplantation).
- Most survivors experienced stormy neonatal periods. However, IUTs significantly resulted in decreased fetal growth restriction, better Apgar scores and decreased length of mechanical ventilator required.
- IUTs significantly decreased the chance of the survivors being born with hydropic features.
- Up to 64% of the survivors had associated congenital abnormalities, urogenital abnormalities, including hypospadias and ambiguous genitalia, being the most common, followed by limb abnormalities.
- Approximately one half of the survivors had growth impairment, regardless of IUTs received.
- Up to 80% of the survivors had normal or only mildly delayed neurodevelopmental outcomes.
- All of the survivors became transfusion-dependent shortly after birth and this continued lifelong, unless receiving successful HSCT.
9. Treatment Strategies for Survivors of BHFS
9.1. Blood Transfusion
9.2. Iron Overload Monitoring and Chelation Therapy
9.3. HSCT
10. Recently Proposed Perinatal Management and Intrauterine HSCT for Affected Fetuses with BHFS
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Harteveld, C.L.; Higgs, D.R. α-thalassaemia. Orphanet J. Rare Dis. 2010, 5, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enevold, A.; Alifrangis, M.; Sanchez, J.J.; Carneiro, I.; Roper, C.; Børsting, C.; Lusingu, J.; Vestergaard, L.S.; Lemnge, M.M.; Morling, N.; et al. Associations between alpha+-thalassemia and Plasmodium falciparum malarial infection in northeastern Tanzania. J. Infect. Dis. 2007, 196, 451–459. [Google Scholar] [CrossRef] [Green Version]
- Kariuki, S.N.; Williams, T.N. Human genetics and malaria resistance. Hum. Genet. 2020, 139, 801–811. [Google Scholar] [CrossRef] [Green Version]
- Weatherall, D.J. Phenotype—Genotype relationships in monogenic disease: Lessons from the thalassaemias. Nat. Rev. Genet. 2001, 2, 245–255. [Google Scholar] [CrossRef]
- Weatherall, D.J. The Evolving Spectrum of the Epidemiology of Thalassemia. Hematol. Oncol. Clin. N. Am. 2018, 32, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Fucharoen, S.; Winichagoon, P. Thalassemia in SouthEast Asia: Problems and strategy for prevention and control. Southeast Asian J. Trop. Med. Public Health 1992, 23, 647–655. [Google Scholar]
- Kulaphisit, M.; Kampuansai, J.; Leecharoenkiat, K.; Wathikthinnakon, M.; Kangwanpong, D.; Munkongdee, T.; Svasti, S.; Fucharoen, S.; Smith, D.R.; Lithanatudom, P. A comprehensive ethnic-based analysis of alpha thalassaemia allelle frequency in northern Thailand. Sci. Rep. 2017, 7, 4690. [Google Scholar] [CrossRef] [PubMed]
- Fucharoen, S.; Winichagoon, P. Problems of thalassemia in Thailand. ICMR Ann. 1988, 8, 29–33. [Google Scholar]
- Hockham, C.; Ekwattanakit, S.; Bhatt, S.; Penman, B.S.; Gupta, S.; Viprakasit, V.; Piel, F.B. Estimating the burden of α-thalassaemia in Thailand using a comprehensive prevalence database for Southeast Asia. eLife 2019, 8, e40580. [Google Scholar] [CrossRef] [PubMed]
- Fucharoen, S.; Weatherall, D.J. Progress toward the Control and Management of the Thalassemias. Hematol. Oncol. Clin. N. Am. 2016, 30, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Michlitsch, J.; Azimi, M.; Hoppe, C.; Walters, M.C.; Lubin, B.; Lorey, F.; Vichinsky, E. Newborn screening for hemoglobinopathies in California. Pediatr. Blood Cancer 2008, 52, 486–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vichinsky, E.P. Changing Patterns of Thalassemia Worldwide. Ann. N. Y. Acad. Sci. 2005, 1054, 18–24. [Google Scholar] [CrossRef]
- Vichinsky, E.P.; MacKlin, E.A.; Waye, J.S.; Lorey, F.; Olivieri, N.F. Changes in the Epidemiology of Thalassemia in North America: A New Minority Disease. Pediatrics 2005, 116, e818–e825. [Google Scholar] [CrossRef] [Green Version]
- Lorey, F.; Cunningham, G.; Vichinsky, E.P.; Lubin, B.H.; Witkowska, H.E.; Matsunaga, A.; Azimi, M.; Sherwin, J.; Eastman, J.; Farina, F.; et al. Universal Newborn Screening for Hb H Disease in California. Genet. Test. 2001, 5, 93–100. [Google Scholar] [CrossRef]
- Amato, A.; Giordano, P.C. Screening and genetic diagnosis of hemoglobinopathies in southern and northern europe: Two examples. Mediterr. J. Hematol. Infect. Dis. 2009, 1, e2009007. [Google Scholar] [CrossRef]
- Fucharoen, S.; Viprakasit, V. Hb H disease: Clinical course and disease modifiers. Hematol. Am. Soc. Hematol. Educ. Program 2009, 2009, 26–34. [Google Scholar] [CrossRef] [Green Version]
- Traivaree, C.; Boonyawat, B.; Monsereenusorn, C.; Rujkijyanont, P.; Photia, A. Clinical and molecular genetic features of Hb H and AE Bart’s diseases in central Thai children. Appl. Clin. Genet. 2018, ume11, 23–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viprakasit, V.; Tanphaichitr, V.S.; Pung-Amritt, P.; Petrarat, S.; Suwantol, L.; Fisher, C.; Higgs, D.R. Clinical phenotypes and molecular characterization of Hb H-Paksé disease. Haematologica 2002, 87, 117–125. [Google Scholar]
- Liao, C.; Li, J.; Xie, X.-M.; Zhou, J.-Y.; Li, D.-Z. Diversity in clinical presentation of hemoglobin H disease induced by the SEA deletion and the hemoglobin Quong Sze. Ann. Hematol. 2009, 88, 1145–1147. [Google Scholar] [CrossRef] [PubMed]
- Sanpakit, K.; Viprakasit, V. Variable genotype-phenotype correlations in patients with a rare nondeletional α-thalassemia; Hb Pak Num Po (HBA1: c.396_397insT). J. Pediatr. Hematol. Oncol. 2014, 36, e185–e189. [Google Scholar] [CrossRef]
- Viprakasit, V.; Tanphaichitr, V.S.; Veerakul, G.; Chinchang, W.; Petrarat, S.; Pung-Amritt, P.; Higgs, D.R. Co-inheritance of Hb Pak Num Po, a novel alpha1 gene mutation, and alpha0 thalassemia associated with transfusion-dependent Hb H disease. Am. J. Hematol. 2004, 75, 157–163. [Google Scholar] [CrossRef]
- Zainal, N.Z.; Alauddin, H.; Ahmad, S.; Hussin, N.H. α-Thalassemia with Haemoglobin Adana mutation: Prenatal diagnosis. Malays. J. Pathol. 2014, 36, 207–211. [Google Scholar]
- Surapolchai, P.; Sirachainan, N.; So, C.-C.; Hongeng, S.; Pakakasama, S.; Anurathapan, U.; Chuansumrit, A. Curative Stem Cell Transplantation for Severe Hb H Disease Manifesting From Early Infancy: Phenotypic and Genotypic Analyses. Hemoglobin 2015, 40, 70–73. [Google Scholar] [CrossRef]
- Sanguansermsri, T.; Matragoon, S.; Changloah, L.; Flatz, G. Hemoglobin Suan-Dok (alpha 2 109 (G16) Leu replaced by Arg beta 2): An unstable variant associated with alpha-thalassemia. Hemoglobin 1979, 3, 161–174. [Google Scholar] [CrossRef]
- Weiss, I.; Cash, F.E.; Coleman, M.B.; Pressley, A.; Adams, J.G.; Sanguansermsri, T.; Liebhaber, S.A.; Steinberg, M. Molecular basis for alpha-thalassemia associated with the structural mutant hemoglobin Suan-Dok (alpha 2 109leu----arg). Blood 1990, 76, 2630–2636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vichinsky, E.P. Clinical manifestations of α-thalassemia. Cold Spring Harb. Perspect. Med. 2013, 3, a011742. [Google Scholar] [CrossRef]
- Lal, A.; Goldrich, M.L.; Haines, D.A.; Azimi, M.; Singer, S.T.; Vichinsky, E.P. Heterogeneity of Hemoglobin H Disease in Childhood. N. Engl. J. Med. 2011, 364, 710–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chui, D.; Fucharoen, S.; Chan, V. Hemoglobin H disease: Not necessarily a benign disorder. Blood 2003, 101, 791–800. [Google Scholar] [CrossRef] [Green Version]
- Laosombat, V.; Viprakasit, V.; Chotsampancharoen, T.; Wongchanchailert, M.; Khodchawan, S.; Chinchang, W.; Sattayasevana, B. Clinical features and molecular analysis in Thai patients with HbH disease. Ann. Hematol. 2009, 88, 1185–1192. [Google Scholar] [CrossRef]
- Origa, R.; Sollaino, M.C.; Giagu, N.; Barella, S.; Campus, S.; Mandas, C.; Bina, P.; Perseu, L.; Galanello, R. Clinical and molecular analysis of haemoglobin H disease in Sardinia: Haematological, obstetric and cardiac aspects in patients with different genotypes. Br. J. Haematol. 2007, 136, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Vichinsky, E. Advances in the treatment of alpha-thalassemia. Blood Rev. 2012, 26 (Suppl. S1), S31–S34. [Google Scholar] [CrossRef] [PubMed]
- Charoenkwan, P.; Sirichotiyakul, S.; Chanprapaph, P.; Tongprasert, F.; Taweephol, R.; Sae-Tung, R.; Sanguansermsri, T. Anemia and Hydrops in a Fetus With Homozygous Hemoglobin Constant Spring. J. Pediatr. Hematol. Oncol. 2006, 28, 827–830. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Zheng, C.; Meng, D.; Chen, R.; Zhang, Q.; Tian, X.; Chen, S. Hb H Hydrops Fetalis Syndrome Caused by Association of the—(SEA) Deletion and Hb Constant Spring (HBA2: c.427T > C) Mutation in a Chinese Family. Hemoglobin 2015, 39, 216–219. [Google Scholar] [CrossRef]
- Li, D.Z.; Liao, C.; Li, J.; Xie, X.M.; Huang, Y.N.; Wu, Q.C. Hemoglobin H hydrops fetalis syndrome resulting from the association of the—SEA deletion and the alphaQuong Szealpha mutation in a Chinese woman. Eur. J. Haematol. 2005, 75, 259–261. [Google Scholar] [CrossRef]
- Henderson, S.; Pitman, M.; McCarthy, J.; Molyneux, A.; Old, J. Molecular prenatal diagnosis of Hb H Hydrops Fetalis caused by haemoglobin Adana and the implications to antenatal screening for α-thalassaemia. Prenat. Diagn. 2008, 28, 859–861. [Google Scholar] [CrossRef]
- Fucharoen, S.; Weatherall, D.J. The Hemoglobin E Thalassemias. Cold Spring Harb. Perspect. Med. 2012, 2, a011734. [Google Scholar] [CrossRef] [PubMed]
- Turbpaiboon, C.; Wilairat, P. Alpha-hemoglobin stabilizing protein: Molecular function and clinical correlation. Front. Biosci. 2010, 15, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Higgs, D.; Garrick, D.; Anguita, E.; Gobbi, M.; Hughes, J.; Muers, M.; Vernimmen, D.; Lower, K.; Law, M.; Argentaro, A.; et al. Understanding α-Globin Gene Regulation: Aiming to Improve the Management of Thalassemia. Ann. N. Y. Acad. Sci. 2005, 1054, 92–102. [Google Scholar] [CrossRef]
- Surapolchai, P.; Chuansumrit, A.; Sirachainan, N.; Kadegasem, P.; Leung, K.-C.; So, C.-C. A molecular study on the role of alpha-hemoglobin-stabilizing protein in hemoglobin H disease. Ann. Hematol. 2017, 96, 1005–1014. [Google Scholar] [CrossRef]
- Schrier, S.L.; Bunyaratvej, A.; Khuhapinant, A.; Fucharoen, S.; Aljurf, M.; Snyder, L.M.; Keifer, C.R.; Ma, L.; Mohandas, N. The unusual pathobiology of hemoglobin constant spring red blood cells. Blood 1997, 89, 1762–1769. [Google Scholar] [CrossRef] [Green Version]
- Fucharoen, S.; Winichagoon, P.; Piankijagum, A. Standardization on laboratory diagnosis of thalassemia and abnormal hemoglobin. Southeast Asian J. Trop. Med. Public Health 1999, 30 (Suppl. S3), 90–98. [Google Scholar] [PubMed]
- Winichagoon, P.; Svasti, S.; Munkongdee, T.; Chaiya, W.; Boonmongkol, P.; Chantrakul, N.; Fucharoen, S. Rapid diagnosis of thalassemias and other hemoglobinopathies by capillary electrophoresis system. Transl. Res. 2008, 152, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Munkongdee, T.; Chen, P.; Winichagoon, P.; Fucharoen, S.; Paiboonsukwong, K. Update in Laboratory Diagnosis of Thalassemia. Front. Mol. Biosci. 2020, 7, 74. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Zhou, J.-Y.; Xie, X.-M.; Tang, H.-S.; Li, R.; Li, D.-Z. Newborn Screening for Hb H Disease by Determination of Hb Bart’s Using the Sebia Capillary Electrophoresis System in Southern China. Hemoglobin 2013, 38, 73–75. [Google Scholar] [CrossRef] [PubMed]
- Ma, E.; Chan, A.Y.; Au, W.Y.; Yeung, Y.M.; Chan, L.C. Diagnosis of concurrent hemoglobin H disease and heterozygous beta-thalassemia. Haematologica 2001, 86, 432–433. [Google Scholar] [PubMed]
- Galanello, R.; Paglietti, E.; Melis, M.A.; Crobu, M.G.; Addis, M.; Moi, P.; Cao, A. Interaction of heterozygous beta zero-thalassemia with single functional alpha-globin gene. Am. J. Hematol. 1988, 29, 63–66. [Google Scholar] [CrossRef]
- de Mare, A.; Groeneger, A.H.; Schuurman, S.; van den Bergh, F.A.; Slomp, J. A rapid single-tube multiplex polymerase chain reaction assay for the seven most prevalent alpha-thalassemia deletions and alphaalphaalpha(anti 3.7) alpha-globin gene triplication. Hemoglobin 2010, 34, 184–190. [Google Scholar] [CrossRef]
- Tan, A.S.-C.; Quah, T.C.; Low, P.S.; Chong, S.S. A rapid and reliable 7-deletion multiplex polymerase chain reaction assay for α-thalassemia. Blood 2001, 98, 250–251. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.T.; Old, J.M.; Miles, K.; Fisher, C.; Weatherall, D.J.; Clegg, J.B. Rapid detection of α-thalassaemia deletions and α-globin gene triplication by multiplex polymerase chain reactions. Br. J. Haematol. 2000, 108, 295–299. [Google Scholar] [CrossRef]
- Chong, S.S.; Boehm, C.D.; Higgs, D.R.; Cutting, G.R. Single-tube multiplex-PCR screen for common deletional determinants of alpha-thalassemia. Blood 2000, 95, 360–362. [Google Scholar] [CrossRef]
- Singsanan, S.; Fucharoen, G.; Savongsy, O.; Sanchaisuriya, K.; Fucharoen, S. Molecular characterization and origins of Hb Constant Spring and Hb Paksé in Southeast Asian populations. Ann. Hematol. 2007, 86, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.L.; Sui, H.; Liu, Y.J.; Pan, J.J.; Liu, Y.H.; Lou, J.W. Molecular and Hematological Characterization of a Novel Translation Initiation Codon Mutation of the α2-Globin Gene (ATG>ATC or HBA2: c.3G>C). Hemoglobin 2019, 43, 241–244. [Google Scholar] [CrossRef]
- Schouten, J.P.; McElgunn, C.J.; Waaijer, R.; Zwijnenburg, D.; Diepvens, F.; Pals, G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002, 30, e57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakavachara, P.; Viprakasit, V. Children with hemoglobin E/β-thalassemia have a high risk of being vitamin D deficient even if they get abundant sun exposure: A study from thailand. Pediatr. Blood Cancer 2013, 60, 1683–1688. [Google Scholar] [CrossRef]
- Taher, A.; Musallam, K.; El-Beshlawy, A.; Karimi, M.; Daar, S.; Belhoul, K.; Saned, M.-S.; Graziadei, G.; Cappellini, M.D. Age-related complications in treatment-naïve patients with thalassaemia intermedia. Br. J. Haematol. 2010, 150, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Musallam, K.M.; Karimi, M.; El-Beshlawy, A.; Belhoul, K.; Daar, S.; Saned, M.-S.; El-Chafic, A.-H.; Fasulo, M.R.; Cappellini, M.D. Overview on practices in thalassemia intermedia management aiming for lowering complication rates across a region of endemicity: The OPTIMAL CARE study. Blood 2010, 115, 1886–1892. [Google Scholar] [CrossRef] [Green Version]
- Taher, A.T.; Vichinsky, E.; Musallam, K.M.; Cappellini, M.D.; Viprakasit, V. Guildlines for the Management of Non Transfusion Dependent Thalassemia (NTDT), 2nd ed.; Thalassemia International Federation: Nicosia, Cyprus, 2017. [Google Scholar]
- Cappellini, M.D.; Farmakis, D.; Porter, J.; Taher, A. 2021 Guidelines for the Management of Transfusion Dependent Thalassemia (TDT), 4th ed.; Thalassemia International Federation: Nicosia, Cyprus, 2021. [Google Scholar]
- Amid, A.; Chen, S.; Brien, W.; Kirby-Allen, M.; Odame, I. Optimizing chronic transfusion therapy for survivors of hemoglobin Barts hydrops fetalis. Blood 2016, 127, 1208–1211. [Google Scholar] [CrossRef] [Green Version]
- Bou-Fakhredin, R.; Bazarbachi, A.-H.; Chaya, B.; Sleiman, J.; Cappellini, M.D.; Taher, A.T. Iron Overload and Chelation Therapy in Non-Transfusion Dependent Thalassemia. Int. J. Mol. Sci. 2017, 18, 2778. [Google Scholar] [CrossRef] [Green Version]
- Pennell, D.J.; Porter, J.B.; Piga, A.; Lai, Y.; El-Beshlawy, A.; Belhoul, K.M.; Elalfy, M.; Yesilipek, A.; Kilinç, Y.; Lawniczek, T.; et al. A 1-year randomized controlled trial of deferasirox vs deferoxamine for myocardial iron removal in β-thalassemia major (CORDELIA). Blood 2014, 123, 1447–1454. [Google Scholar] [CrossRef] [Green Version]
- Ang, A.L.; Le, T.T.; Tan, R.S. HbH Constant Spring disease has lower serum ferritin relative to liver iron concentration (LIC): Importance of LIC measurement and potential impact on serum ferritin thresholds for iron chelation. Br. J. Haematol. 2016, 176, 986–988. [Google Scholar] [CrossRef]
- Musallam, K.M.; Cappellini, M.D.; Daar, S.; Karimi, M.; El-Beshlawy, A.; Graziadei, G.; Magestro, M.; Wulff, J.; Pietri, G.; Taher, A.T. Serum ferritin level and morbidity risk in transfusion-independent patients with β-thalassemia intermedia: The ORIENT study. Haematologica 2014, 99, e218–e221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taher, A.T.; Porter, J.B.; Viprakasit, V.; Kattamis, A.; Chuncharunee, S.; Sutcharitchan, P.; Siritanaratkul, N.; Origa, R.; Karakas, Z.; Habr, D.; et al. Defining serum ferritin thresholds to predict clinically relevant liver iron concentrations for guiding deferasirox therapy when MRI is unavailable in patients with non-transfusion-dependent thalassaemia. Br. J. Haematol. 2015, 168, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Porter, J.B.; Viprakasit, V.; Kattamis, A.; Chuncharunee, S.; Sutcharitchan, P.; Siritanaratkul, N.; Galanello, R.; Karakas, Z.; Lawniczek, T.; et al. Deferasirox effectively reduces iron overload in non-transfusion-dependent thalassemia (NTDT) patients: 1-year extension results from the THALASSA study. Ann. Hematol. 2013, 92, 1485–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amid, A.; Chen, S.; Athale, U.; Charpentier, K.; Merelles-Pulcini, M.; Odame, I.; Kirby-Allen, M. Iron overload in transfusion-dependent survivors of hemoglobin Bart’s hydrops fetalis. Haematologica 2018, 103, e184–e187. [Google Scholar] [CrossRef]
- Singer, S.T.; Kim, H.-Y.; Olivieri, N.F.; Kwiatkowski, J.L.; Coates, T.D.; Carson, S.; Neufeld, E.; Cunningham, M.J.; Giardina, P.J.; Mueller, B.U.; et al. Hemoglobin H-constant spring in North America: An alpha thalassemia with frequent complications. Am. J. Hematol. 2009, 84, 759–761. [Google Scholar] [CrossRef] [Green Version]
- Luu, S.; Spelman, D.; Woolley, I.J. Post-splenectomy sepsis: Preventative strategies, challenges, and solutions. Infect. Drug Resist. 2019, ume 12, 2839–2851. [Google Scholar] [CrossRef] [Green Version]
- Tso, S.C.; Chan, T.K.; Todd, D. Venous Thrombosis in Haemoglobin H Disease after Splenectomy. Aust. N. Z. J. Med. 1982, 12, 635–638. [Google Scholar] [CrossRef]
- Hirsh, J.; Dacie, J.V. Persistent Post-Splenectomy Thrombocytosis and Thrombo-embolism: A Consequence of Continuing Anaemia. Br. J. Haematol. 1966, 12, 44–53. [Google Scholar] [CrossRef]
- Songdej, D.; Babbs, C.; Higgs, D.R. An international registry of survivors with Hb Bart’s hydrops fetalis syndrome. Blood 2017, 129, 1251–1259. [Google Scholar] [CrossRef] [Green Version]
- MacKenzie, T.C.; Amid, A.; Angastiniotis, M.; Butler, C.; Gilbert, S.; Gonzalez, J.; Keller, R.L.; Kharbanda, S.; Kirby-Allen, M.; Koenig, B.A.; et al. Consensus statement for the perinatal management of patients with α thalassemia major. Blood Adv. 2021, 5, 5636–5639. [Google Scholar] [CrossRef]
- Amid, A.; Barrowman, N.; Odame, I.; Kirby-Allen, M. Optimizing transfusion therapy for survivors of Haemoglobin Bart’s hydrops fetalis syndrome: Defining the targets for haemoglobin-H fraction and “functional” haemoglobin level. Br. J. Haematol. 2022, 197, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Horvei, P.; MacKenzie, T.; Kharbanda, S. Advances in the management of α-thalassemia major: Reasons to be optimistic. Hematology 2021, 2021, 592–599. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, T.C.; Frascoli, M.; Sper, R.; Lianoglou, M.B.R.; Velez, J.G.; Dvorak, C.C.; Kharbanda, S.; Vichinsky, E. In Utero Stem Cell Transplantation in Patients with Alpha Thalassemia Major: Interim Results of a Phase 1 Clinical Trial. Blood 2020, 136, 1. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Clinical Characteristics | Deletional HbH | Non-Deletional HbH |
|---|---|---|
| Symptomatic patients (%) | 40 | 60 |
| Age at diagnosis (approximate range, y) | 1 to 7 | <1 to 5 |
| History of blood transfusion (%) | 10 to 29 | 50 to 60 |
| Age at first transfusion (approximate range, y) | 2 to 17 | <1 to 5 |
| TDT during childhood (%) | Rare | 20 to 30 |
| Facial bone changes (%) | 2 to 3 | 20 to 30 |
| Growth retardation (%) | Rare | 15 to 20 |
| Splenomegaly (%) | 10 to 20 | 20 to 30 |
| Gallstones (%) | ~10 | 20 to 30 |
| Acute hemolysis following infection/inflammation | Periodically | More common |
| Baseline Hb level (approximate range, g/dL) | 9 to 11 | 7 to 9 |
| Type | Specific Regions Found | Clinical Significance |
|---|---|---|
| HbH-HbCS (--/αCSα) | Southeast Asia, China, Mediterranean | Approximately 20% had more severe phenotypes (facial bone change, splenomegaly, growth impairment) and required frequent transfusions |
| HbH-HbPS (--/αPSα) | Lao PDR, Thailand | Most hematologic findings and clinical courses resemble those of HbH-HbCS, proportion of HbH was higher in HbH-HbPS |
| HbH-HbQS (--/αQSα) | Southeast Asia, China | Clinical phenotypes ranged from hydrops fetalis to TDT and NTDT |
| HbH-HbPNP (--/αPNPα) i | Thailand | Most reported cases were transfusion-dependent |
| HbH-HbAdana (--/αAdanaα) | Malaysia, Indonesia, China | Hydrops fetalis or TDT |
| HbH-PolyA (--/αPolyAα) | Greece Saudi Arabia, Iran, Türkiye | Transfusion-dependent from early infancy |
| HbH-HbSuan-Dok (--/αSuan-Dokα) | Thailand | Chance of TDT and possibly hydrops fetalis |
| Treatment | Possible Roles | Pros | Cons | Remarks |
|---|---|---|---|---|
| Splenectomy | Marked splenomegaly, hypersplenism, areas with extremely limited access to blood product and iron chelator | Evidenced to increase baseline Hb level, able to transform transfusion-dependent to non-transfusion dependent HbH in some cases | Overwhelming post-splenectomy infection, increased risk of thromboembolic events, surgical complications | Especially effective in HbH-HbCS (although not among all patients), its effectiveness in other HbH genotypes is unclear. Postoperative LMWH and life-long low-dose aspirin should be considered for prophylaxis of thromboembolic complications. |
| HSCT | Transfusion-dependent non-deletional HbH (rare genotypes) | Curative therapy | Donor availability, transplant-related morbidities and mortalities | May also be considered among patients with transfusion-dependent HbH-HbCS failing to respond to splenectomy or who prefer HSCT upon availability of HLA-matched related donors. |
| Mitapivat * (Oral red-cell-specific pyruvate kinase activator) | Non-transfusion dependent HbH | Potential oral agent that can decrease ineffective erythropoiesis marker, prolong red cell survival and possibly decrease iron overload | More data on short- and long-term side effects are needed, not yet available in the market | Phase 2 clinical trial in NTDT (NCT03692052)- 5 of patients with HbH had increased Hb level ≥1 g/dL by 3 weeks. |
| Gene therapy ** | Severe α-thalassemia | Potentially ameliorate severity | Currently unknown | Preclinical phase |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Songdej, D.; Fucharoen, S. Alpha-Thalassemia: Diversity of Clinical Phenotypes and Update on the Treatment. Thalass. Rep. 2022, 12, 157-172. https://doi.org/10.3390/thalassrep12040020
Songdej D, Fucharoen S. Alpha-Thalassemia: Diversity of Clinical Phenotypes and Update on the Treatment. Thalassemia Reports. 2022; 12(4):157-172. https://doi.org/10.3390/thalassrep12040020
Chicago/Turabian StyleSongdej, Duantida, and Suthat Fucharoen. 2022. "Alpha-Thalassemia: Diversity of Clinical Phenotypes and Update on the Treatment" Thalassemia Reports 12, no. 4: 157-172. https://doi.org/10.3390/thalassrep12040020