
Drug Resistance: An Incessant Fight against Evolutionary Strategies of Survival

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Bacterial Resistance
2.1. Bacterial Targets and Main Class of Antibiotics
2.2. Resistant Health-Threatening Bacteria
2.2.1. Klebsiella pneumoniae
2.2.2. Pseudomonas aeruginosa
2.2.3. Acinetobacter baumannii
2.2.4. Staphylococcus aureus
2.2.5. Neisseria gonorrhoeae
2.2.6. Mycobacterium tuberculosis
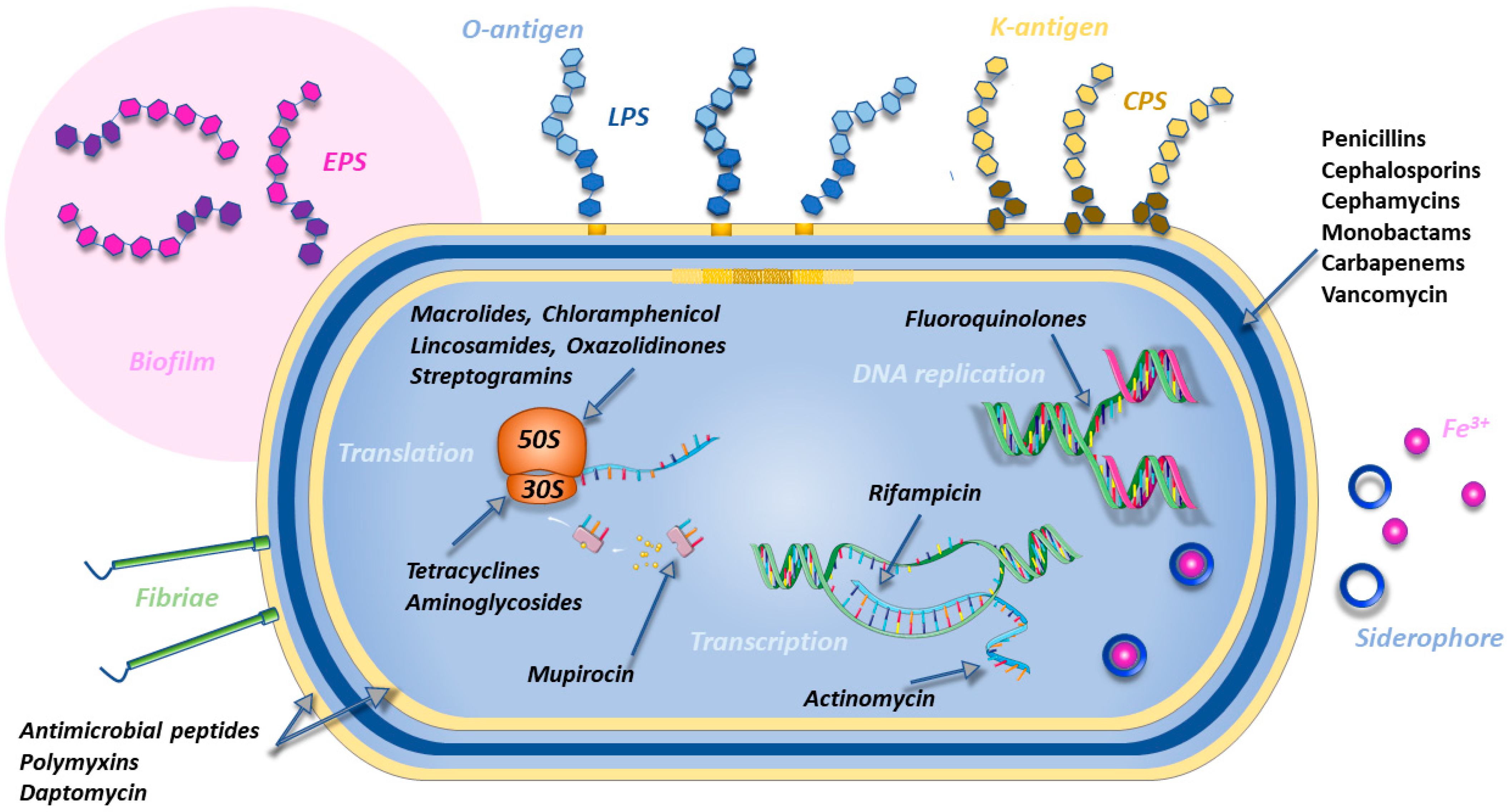
2.3. Bacterial Resistance Mechanisms
2.3.1. Target Modification
2.3.2. Antibiotic Inactivation: The Case of β-Lactamases
2.3.3. Membrane Permeability Reduction
2.3.4. Phospholipid Shedding
2.3.5. Drug Extrusion by Efflux Pumps
2.3.6. Ribosomal Protection
2.3.7. Evasion from Immune Surveillance
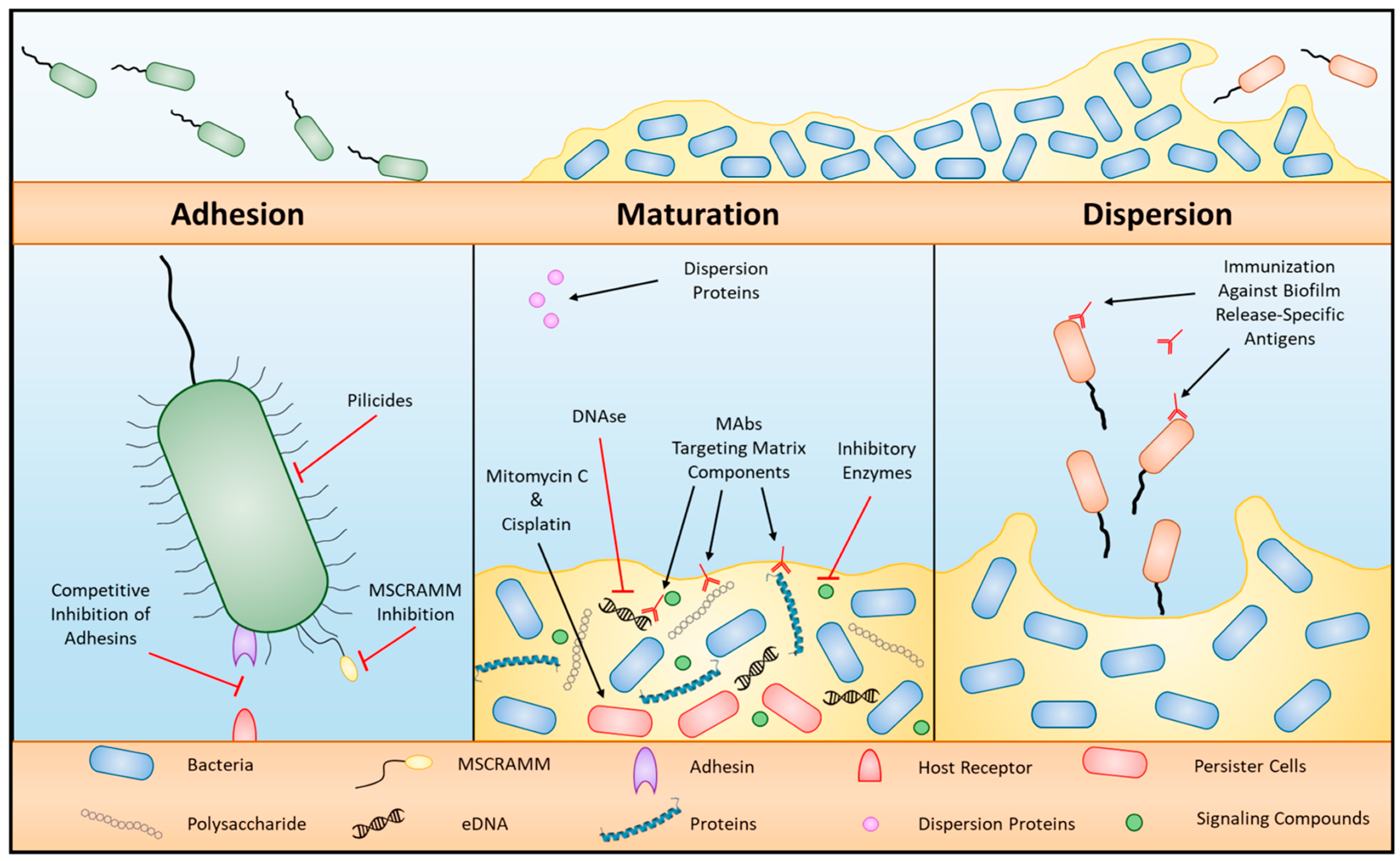
2.3.8. Biofilm Formation

2.3.9. Other Phenomena Correlated with Resistance
2.4. Strategies to Overcome Drug Resistance in Bacteria
2.4.1. Combinations of Antibiotics
2.4.2. Bacteriophage Therapy
2.4.3. Antimicrobial Peptide Therapy
2.4.4. CRISPR

2.4.5. Light-Mediated Bactericidal Techniques
2.4.6. Silver Nanoparticles
2.4.7. Microbiota Therapy
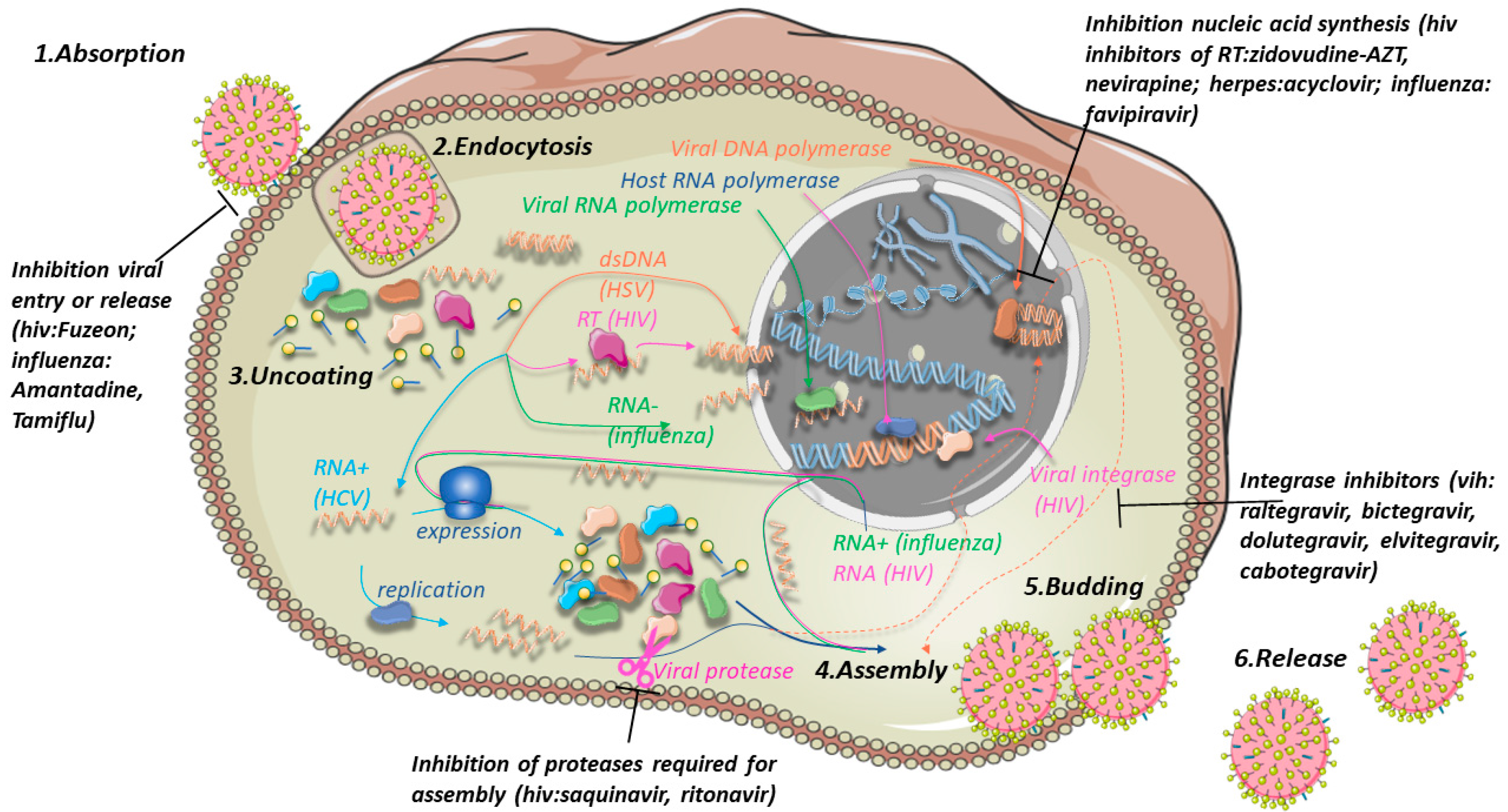
3. Viral Resistance
3.1. Examples of Resistant Viruses
3.1.1. Influenza A
3.1.2. Hepatitis C
3.1.3. Hepatitis B
3.1.4. Herpes Simplex
3.1.5. Human Cytomegalovirus
3.1.6. Human Immunodeficiency Virus
3.2. Antiviral Drugs
3.3. Strategies to Overcome Drug Resistance in Viruses
4. Resistance in Eukaryotic Microorganisms
4.1. Fungal Resistance Mechanisms
4.2. Drug Resistance in Parasites
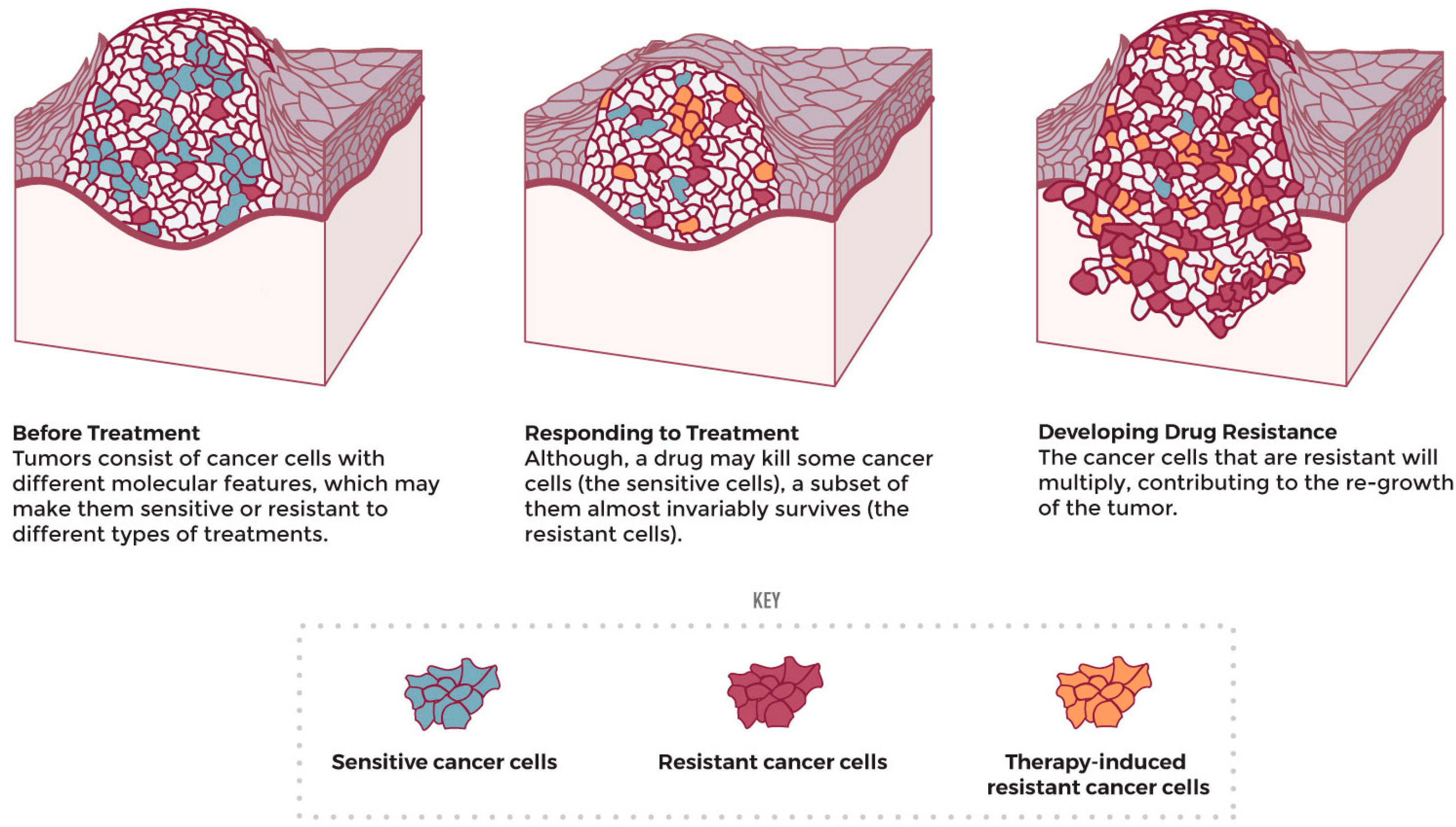
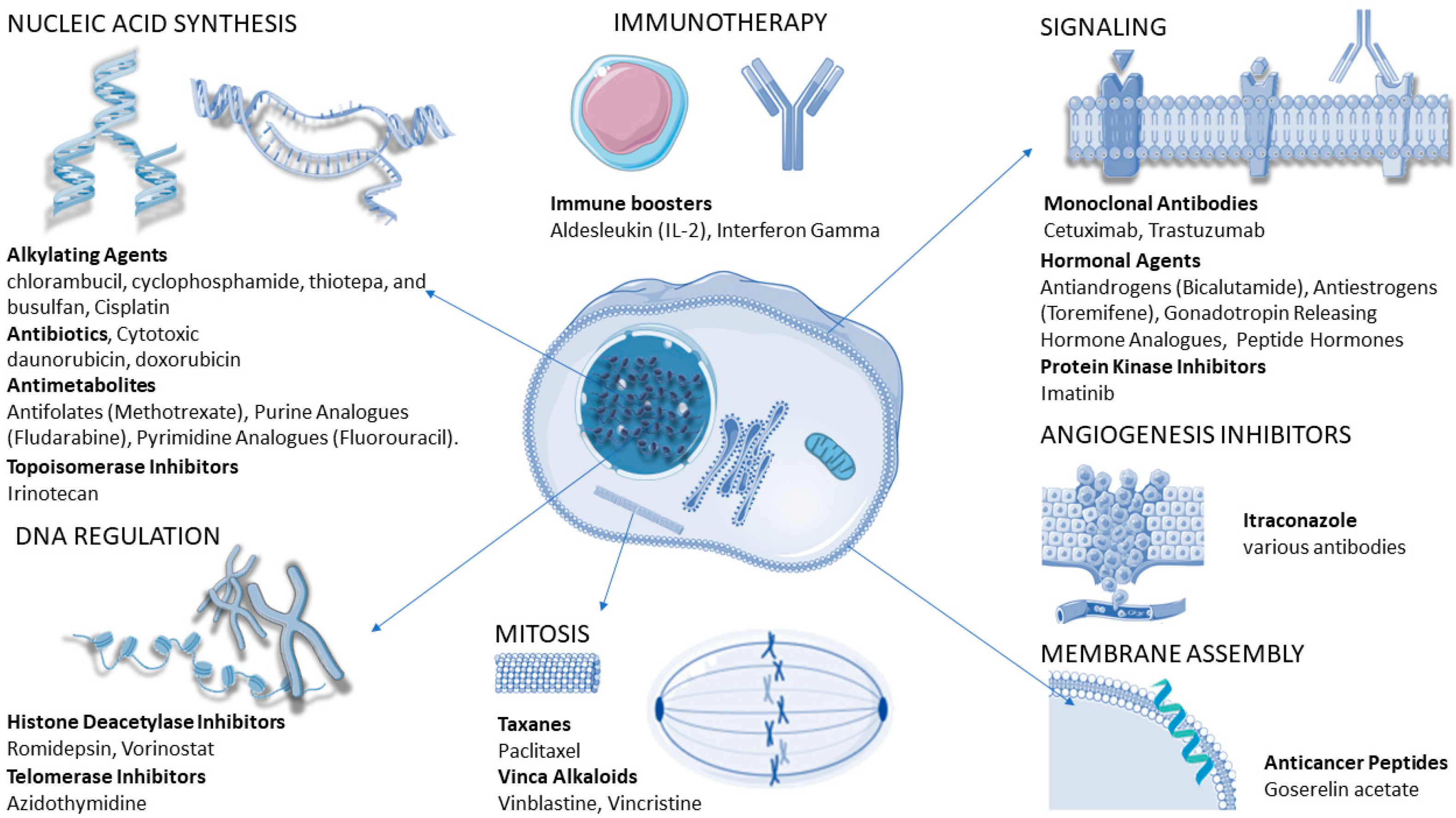
4.3. Cancer-Cell Resistance
Mitochondria in Cancer
5. Novel Drug Delivery Systems
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vasan, N.; Baselga, J.; Hyman, D.M. A View on Drug Resistance in Cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Dadgostar, P. Antimicrobial Resistance: Implications and Costs. Infect. Drug Resist. 2019, 12, 3903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emran, T.B.; Shahriar, A.; Mahmud, A.R.; Rahman, T.; Abir, M.H.; Siddiquee, M.F.-R.; Ahmed, H.; Rahman, N.; Nainu, F.; Wahyudin, E.; et al. Multidrug Resistance in Cancer: Understanding Molecular Mechanisms, Immunoprevention and Therapeutic Approaches. Front. Oncol. 2022, 12, 891652. [Google Scholar] [CrossRef] [PubMed]
- Irwin, K.K.; Renzette, N.; Kowalik, T.F.; Jensen, J.D. Antiviral Drug Resistance as an Adaptive Process. Virus Evol. 2016, 2, vew014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fairlamb, A.H.; Gow, N.A.R.; Matthews, K.R.; Waters, A.P. Drug Resistance in Eukaryotic Microorganisms. Nat. Microbiol. 2016, 1, 16092. [Google Scholar] [CrossRef] [Green Version]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef]
- Allegranzi, B.; Bagheri Nejad, S.; Combescure, C.; Graafmans, W.; Attar, H.; Donaldson, L.; Pittet, D. Burden of Endemic Health-Care-Associated Infection in Developing Countries: Systematic Review and Meta-Analysis. Lancet 2011, 377, 228–241. [Google Scholar] [CrossRef]
- Ibrahim, M.E.; Bilal, N.E.; Hamid, M.E. Increased Multi-Drug Resistant Escherichia Coli from Hospitals in Khartoum State, Sudan. Afr. Health Sci. 2012, 12, 368–375. [Google Scholar] [CrossRef] [Green Version]
- Pendleton, J.N.; Gorman, S.P.; Gilmore, B.F. Clinical Relevance of the ESKAPE Pathogens. Expert Rev. Anti. Infect. Ther. 2013, 11, 297–308. [Google Scholar] [CrossRef]
- Mulani, M.S.; Kamble, E.E.; Kumkar, S.N.; Tawre, M.S.; Pardesi, K.R. Emerging Strategies to Combat ESKAPE Pathogens in the Era of Antimicrobial Resistance: A Review. Front. Microbiol. 2019, 10, 539. [Google Scholar] [CrossRef]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, Research, and Development of New Antibiotics: The WHO Priority List of Antibiotic-Resistant Bacteria and Tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Santajit, S.; Indrawattana, N. Mechanisms of Antimicrobial Resistance in ESKAPE Pathogens. BioMed Res. Int. 2016, 2016, 2475067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trejo-Becerril, C.; Pérez-Cárdenas, E.; Taja-Chayeb, L.; Anker, P.; Herrera-Goepfert, R.; Medina-Velázquez, L.A.; Hidalgo-Miranda, A.; Pérez-Montiel, D.; Chávez-Blanco, A.; Cruz-Velázquez, J.; et al. Cancer Progression Mediated by Horizontal Gene Transfer in an In Vivo Model. PLoS ONE 2012, 7, e52754. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.-F.; Kovarova, J.; Bajzikova, M.; Bezawork-Geleta, A.; Svec, D.; Endaya, B.; Sachaphibulkij, K.; Coelho, A.R.; Sebkova, N.; Ruzickova, A.; et al. Horizontal Transfer of Whole Mitochondria Restores Tumorigenic Potential in Mitochondrial DNA-Deficient Cancer Cells. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nimmakayala, R.K.; Batra, S.K.; Ponnusamy, M.P. Unraveling the Journey of Cancer Stem Cells from Origin to Metastasis. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Raji, G.R.; Sruthi, T.V.; Edatt, L.; Haritha, K.; Sharath Shankar, S.; Sameer Kumar, V.B. Horizontal Transfer of miR-106a/b from Cisplatin Resistant Hepatocarcinoma Cells Can Alter the Sensitivity of Cervical Cancer Cells to Cisplatin. Cell. Signal. 2017, 38, 146–158. [Google Scholar] [CrossRef]
- Bush, K.; Bradford, P.A. β-Lactams and β-Lactamase Inhibitors: An Overview. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef]
- Bush, K.; Page, M.G.P. What We May Expect from Novel Antibacterial Agents in the Pipeline with Respect to Resistance and Pharmacodynamic Principles. J. Pharmacokinet. Pharmacodyn. 2017, 44, 113–132. [Google Scholar] [CrossRef]
- Brandt, C.; Braun, S.D.; Stein, C.; Slickers, P.; Ehricht, R.; Pletz, M.W.; Makarewicz, O. In Silico Serine β-Lactamases Analysis Reveals a Huge Potential Resistome in Environmental and Pathogenic Species. Sci. Rep. 2017, 7, 43232. [Google Scholar] [CrossRef] [Green Version]
- Pfennigwerth, N. Bericht Des Nationalen Referenzzentrums (NRZ) Für Gramnegative Krankenhauserreger—Zeitraum 1. Januar 2017—31. Dezember 2017. Epidemiol. Bull. 2018, 28, 263–267. [Google Scholar]
- Ramos-Martín, F.; D’Amelio, N. Biomembrane Lipids: When Physics and Chemistry Join to Shape Biological Activity. Biochimie 2022, 203, 118–138. [Google Scholar] [CrossRef]
- Liu, Y.-Y.; Wang, Y.; Walsh, T.R.; Yi, L.-X.; Zhang, R.; Spencer, J.; Doi, Y.; Tian, G.; Dong, B.; Huang, X.; et al. Emergence of Plasmid-Mediated Colistin Resistance Mechanism MCR-1 in Animals and Human Beings in China: A Microbiological and Molecular Biological Study. Lancet Infect. Dis. 2016, 16, 161–168. [Google Scholar] [CrossRef]
- Rabanal, F.; Cajal, Y. Recent Advances and Perspectives in the Design and Development of Polymyxins. Nat. Prod. Rep. 2017, 34, 886–908. [Google Scholar] [CrossRef] [PubMed]
- Corbett, D.; Wise, A.; Langley, T.; Skinner, K.; Trimby, E.; Birchall, S.; Dorali, A.; Sandiford, S.; Williams, J.; Warn, P.; et al. Potentiation of Antibiotic Activity by a Novel Cationic Peptide: Potency and Spectrum of Activity of SPR741. Antimicrob. Agents Chemother. 2017, 61, e00200-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Breij, A.; Riool, M.; Cordfunke, R.A.; Malanovic, N.; de Boer, L.; Koning, R.I.; Ravensbergen, E.; Franken, M.; van der Heijde, T.; Boekema, B.K.; et al. The Antimicrobial Peptide SAAP-148 Combats Drug-Resistant Bacteria and Biofilms. Sci. Transl. Med. 2018, 10, eaan4044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Büttner, D.; Kramer, J.S.; Klingler, F.-M.; Wittmann, S.K.; Hartmann, M.R.; Kurz, C.G.; Kohnhäuser, D.; Weizel, L.; Brüggerhoff, A.; Frank, D.; et al. Challenges in the Development of a Thiol-Based Broad-Spectrum Inhibitor for Metallo-β-Lactamases. ACS Infect. Dis. 2018, 4, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Strahilevitz, J.; Jacoby, G.A.; Hooper, D.C.; Robicsek, A. Plasmid-Mediated Quinolone Resistance: A Multifaceted Threat. Clin. Microbiol. Rev. 2009, 22, 664–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotler, M.; Becker, Y. Rifampicin and Distamycin A as Inhibitors of Rous Sarcoma Virus Reverse Transcriptase. Nat. New Biol. 1971, 234, 212–214. [Google Scholar] [CrossRef] [PubMed]
- Grado, C.; Ohlbaum, A. The Effect of Rifampicin, Actinomycin D and Mitomycin C on Poliovirus and Foot-and-Mouth Disease Virus Replication. J. Gen. Virol. 1973, 21, 297–303. [Google Scholar] [CrossRef]
- Wilson, D.N. Ribosome-Targeting Antibiotics and Mechanisms of Bacterial Resistance. Nat. Rev. Microbiol. 2014, 12, 35–48. [Google Scholar] [CrossRef]
- Bulkley, D.; Axel Innis, C.; Blaha, G.; Steitz, T.A. Revisiting the Structures of Several Antibiotics Bound to the Bacterial Ribosome. Proc. Natl. Acad. Sci. USA 2020, 108, 17158–17163. [Google Scholar]
- Polikanov, Y.S.; Aleksashin, N.A.; Beckert, B.; Wilson, D.N. The Mechanisms of Action of Ribosome-Targeting Peptide Antibiotics. Front. Mol. Biosci. 2018, 5, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chukwudi, C.U. rRNA Binding Sites and the Molecular Mechanism of Action of the Tetracyclines. Antimicrob. Agents Chemother. 2016, 60, 4433–4441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Y.; Li, B.; Zhang, T. Bacteria That Make a Meal of Sulfonamide Antibiotics: Blind Spots and Emerging Opportunities. Environ. Sci. Technol. 2018, 52, 3854–3868. [Google Scholar] [CrossRef] [PubMed]
- Fariñas, M.C.; González-Rico, C.; Fernández-Martínez, M.; Fortún, J.; Escudero-Sanchez, R.; Moreno, A.; Bodro, M.; Muñoz, P.; Valerio, M.; Montejo, M.; et al. Oral Decontamination with Colistin plus Neomycin in Solid Organ Transplant Recipients Colonized by Multidrug-Resistant Enterobacterales: A Multicentre, Randomized, Controlled, Open-Label, Parallel-Group Clinical Trial. Clin. Microbiol. Infect. 2021, 27, 856–863. [Google Scholar] [CrossRef]
- Ramos-Vivas, J.; The ENTHERE Study Group; the Group for Study of Infection in Transplantation of the Spanish Society of Infectious Diseases and Clinical Microbiology (GESITRA-SEIMC) and the Spanish Network for Research in Infectious Diseases (REIPI); Chapartegui-González, I.; Fernández-Martínez, M.; González-Rico, C.; Fortún, J.; Escudero, R.; Marco, F.; Linares, L.; et al. Biofilm Formation by Multidrug Resistant Enterobacteriaceae Strains Isolated from Solid Organ Transplant Recipients. Sci. Rep. 2019, 9, 8928. [Google Scholar]
- Bartoletti, M.; Giannella, M.; Tedeschi, S.; Viale, P. Multidrug-Resistant Bacterial Infections in Solid Organ Transplant Candidates and Recipients. Infect. Dis. Clin. N. Am. 2018, 32, 551–580. [Google Scholar] [CrossRef]
- Hobby, C.R.; Herndon, J.L.; Morrow, C.A.; Peters, R.E.; Symes, S.J.K.; Giles, D.K. Exogenous Fatty Acids Alter Phospholipid Composition, Membrane Permeability, Capacity for Biofilm Formation, and Antimicrobial Peptide Susceptibility in Klebsiella Pneumoniae. Microbiologyopen 2019, 8, e00635. [Google Scholar] [CrossRef]
- Eder, A.E.; Munir, S.A.; Hobby, C.R.; Anderson, D.M.; Herndon, J.L.; Siv, A.W.; Symes, S.J.K.; Giles, D.K. Exogenous Polyunsaturated Fatty Acids (PUFAs) Alter Phospholipid Composition, Membrane Permeability, Biofilm Formation and Motility in Acinetobacter baumannii. Microbiology 2017, 163, 1626–1636. [Google Scholar] [CrossRef]
- Dell’Annunziata, F.; Dell’Aversana, C.; Doti, N.; Donadio, G.; Dal Piaz, F.; Izzo, V.; De Filippis, A.; Galdiero, M.; Altucci, L.; Boccia, G.; et al. Outer Membrane Vesicles Derived from Are a Driving Force for Horizontal Gene Transfer. Int. J. Mol. Sci. 2021, 22. [Google Scholar] [CrossRef]
- Wang, G.; Zhao, G.; Chao, X.; Xie, L.; Wang, H. The Characteristic of Virulence, Biofilm and Antibiotic Resistance of Klebsiella pneumoniae. Int. J. Environ. Res. Public Health 2020, 17, 6278. [Google Scholar] [CrossRef] [PubMed]
- Russo, T.A.; Shon, A.S.; Beanan, J.M.; Olson, R.; MacDonald, U.; Pomakov, A.O.; Visitacion, M.P.; Hypervirulent, K. Pneumoniae Secretes More and More Active Iron-Acquisition Molecules than “Classical” K. Pneumoniae Thereby Enhancing Its Virulence. PLoS ONE 2011, 6, e26734. [Google Scholar] [CrossRef] [PubMed]
- Galani, I.; Karaiskos, I.; Giamarellou, H. Multidrug-Resistant: Mechanisms of Resistance Including Updated Data for Novel β-Lactam-β-Lactamase Inhibitor Combinations. Expert Rev. Anti. Infect. Ther. 2021, 19, 1457–1468. [Google Scholar] [CrossRef] [PubMed]
- Benamara, H.; Rihouey, C.; Jouenne, T.; Alexandre, S. Impact of the Biofilm Mode of Growth on the Inner Membrane Phospholipid Composition and Lipid Domains in Pseudomonas Aeruginosa. Biochim. Biophys. Acta 2011, 1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, Z.; Raudonis, R.; Glick, B.R.; Lin, T.-J.; Cheng, Z. Antibiotic Resistance in Pseudomonas Aeruginosa: Mechanisms and Alternative Therapeutic Strategies. Biotechnol. Adv. 2019, 37, 177–192. [Google Scholar] [CrossRef]
- Langendonk, R.F.; Neill, D.R.; Fothergill, J.L. The Building Blocks of Antimicrobial Resistance in: Implications for Current Resistance-Breaking Therapies. Front. Cell. Infect. Microbiol. 2021, 11, 665759. [Google Scholar] [CrossRef]
- Van den Bergh, B.; Fauvart, M.; Michiels, J. Formation, Physiology, Ecology, Evolution and Clinical Importance of Bacterial Persisters. FEMS Microbiol. Rev. 2017, 41, 219–251. [Google Scholar] [CrossRef]
- Behzadi, P.; Baráth, Z.; Gajdács, M. It’s Not Easy Being Green: A Narrative Review on the Microbiology, Virulence and Therapeutic Prospects of Multidrug-Resistant. Antibiotics 2021, 10, 42. [Google Scholar] [CrossRef]
- Fito-Boncompte, L.; Chapalain, A.; Bouffartigues, E.; Chaker, H.; Lesouhaitier, O.; Gicquel, G.; Bazire, A.; Madi, A.; Connil, N.; Véron, W.; et al. Full Virulence of Pseudomonas Aeruginosa Requires OprF. Infect. Immun. 2011, 79, 1176–1186. [Google Scholar] [CrossRef] [Green Version]
- Skiada, A.; Markogiannakis, A.; Plachouras, D.; Daikos, G.L. Adaptive Resistance to Cationic Compounds in Pseudomonas Aeruginosa. Int. J. Antimicrob. Agents 2011, 37, 187–193. [Google Scholar] [CrossRef]
- Peleg, A.Y.; Seifert, H.; Paterson, D.L. Acinetobacter baumannii: Emergence of a Successful Pathogen. Clin. Microbiol. Rev. 2008, 21, 538–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vrancianu, C.O.; Gheorghe, I.; Czobor, I.B.; Chifiriuc, M.C. Antibiotic Resistance Profiles, Molecular Mechanisms and Innovative Treatment Strategies of Acinetobacter baumannii. Microorganisms 2020, 8, 935. [Google Scholar] [CrossRef] [PubMed]
- Howard, A.; O’Donoghue, M.; Feeney, A.; Sleator, R.D. Acinetobacter baumannii: An Emerging Opportunistic Pathogen. Virulence 2012, 3, 243–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, C.M.; Hennon, S.W.; Feldman, M.F. Uncovering the Mechanisms of Acinetobacter baumannii Virulence. Nat. Rev. Microbiol. 2018, 16, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.G.; Yan, H.; Hancock, R.E.W. Biological Properties of Structurally Related α-Helical Cationic Antimicrobial Peptides. Infect. Immun. 1999, 67, 2005–2009. [Google Scholar] [CrossRef] [PubMed]
- Lopalco, P.; Stahl, J.; Annese, C.; Averhoff, B.; Corcelli, A. Identification of Unique Cardiolipin and Monolysocardiolipin Species in Acinetobacter baumannii. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Zhu, C.; Ren, B.; Yin, X.; Shim, S.H.; Gao, Y.; Zhu, J.; Zhao, P.; Liu, C.; Yu, R.; et al. Two Optimized Antimicrobial Peptides with Therapeutic Potential for Clinical Antibiotic-Resistant Staphylococcus aureus. Eur. J. Med. Chem. 2019, 183, 111686. [Google Scholar] [CrossRef]
- Röhrig, C.; Huemer, M.; Lorgé, D.; Luterbacher, S.; Phothaworn, P.; Schefer, C.; Sobieraj, A.M.; Zinsli, L.V.; Mairpady Shambat, S.; Leimer, N.; et al. Targeting Hidden Pathogens: Cell-Penetrating Enzybiotics Eradicate Intracellular Drug-Resistant Staphylococcus aureus. mBio 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, M.F.; Abdelkhalek, A.; Seleem, M.N. Evaluation of Short Synthetic Antimicrobial Peptides for Treatment of Drug-Resistant and Intracellular Staphylococcus aureus. Sci. Rep. 2016, 6, 29707. [Google Scholar] [CrossRef]
- Bongers, S.; Hellebrekers, P.; Leenen, L.P.H.; Koenderman, L.; Hietbrink, F. Intracellular Penetration and Effects of Antibiotics on Staphylococcus aureus Inside Human Neutrophils: A Comprehensive Review. Antibiotic 2019, 8, 54. [Google Scholar] [CrossRef] [Green Version]
- Alder, K.D.; Lee, I.; Munger, A.M.; Kwon, H.-K.; Morris, M.T.; Cahill, S.V.; Back, J.; Yu, K.E.; Lee, F.Y. Intracellular Staphylococcus aureus in Bone and Joint Infections: A Mechanism of Disease Recurrence, Inflammation, and Bone and Cartilage Destruction. Bone 2020, 141, 115568. [Google Scholar] [CrossRef]
- Salinas, N.; Colletier, J.-P.; Moshe, A.; Landau, M. Extreme Amyloid Polymorphism in Staphylococcus aureus Virulent PSMα Peptides. Nat. Commun. 2018, 9, 3512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joo, H.-S.; Fu, C.-I.; Otto, M. Bacterial Strategies of Resistance to Antimicrobial Peptides. Philos. Trans. R. Soc. B Biol. Sci. 2016, 371, 20150292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boudjemaa, R.; Cabriel, C.; Dubois-Brissonnet, F.; Bourg, N.; Dupuis, G.; Gruss, A.; Lévêque-Fort, S.; Briandet, R.; Fontaine-Aupart, M.-P.; Steenkeste, K. Impact of Bacterial Membrane Fatty Acid Composition on the Failure of Daptomycin to Kill Staphylococcus aureus. Antimicrob. Agents Chemother. 2018, 62, e00023-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koppen, B.C.; Mulder, P.P.G.; de Boer, L.; Riool, M.; Drijfhout, J.W.; Zaat, S.A.J. Synergistic Microbicidal Effect of Cationic Antimicrobial Peptides and Teicoplanin against Planktonic and Biofilm-Encased Staphylococcus aureus. Int. J. Antimicrob. Agents 2019, 53, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Jean, S.R.; Ahmed, M.; Lei, E.K.; Wisnovsky, S.P.; Kelley, S.O. Peptide-Mediated Delivery of Chemical Probes and Therapeutics to Mitochondria. Acc. Chem. Res. 2016, 49, 1893–1902. [Google Scholar] [CrossRef]
- Lei, E.K.; Pereira, M.P.; Kelley, S.O. Tuning the Intracellular Bacterial Targeting of Peptidic Vectors. Angew. Chem. Int. Ed Engl. 2013, 52, 9660–9663. [Google Scholar] [CrossRef]
- Pereira, M.P.; Shi, J.; Kelley, S.O. Peptide Targeting of an Antibiotic Prodrug toward Phagosome-Entrapped Mycobacteria. ACS Infect. Dis. 2015, 1, 586–592. [Google Scholar] [CrossRef]
- Nepal, M.; Thangamani, S.; Seleem, M.N.; Chmielewski, J. Targeting Intracellular Bacteria with an Extended Cationic Amphiphilic Polyproline Helix. Org. Biomol. Chem. 2015, 13, 5930–5936. [Google Scholar] [CrossRef]
- Seleem, M.; Thangamani, S.; Nepal, M.; Chmielewski, J. Antibacterial Activity and Therapeutic Efficacy of Fl-PRPRPL-5, a Cationic Amphiphilic Polyproline Helix, in a Mouse Model of Staphylococcal Skin Infection. Drug Des. Dev. Ther. 2015, 9, 5749. [Google Scholar] [CrossRef] [Green Version]
- Kuriakose, J.; Hernandez-Gordillo, V.; Nepal, M.; Brezden, A.; Pozzi, V.; Seleem, M.N.; Chmielewski, J. Targeting Intracellular Pathogenic Bacteria with Unnatural Proline-Rich Peptides: Coupling Antibacterial Activity with Macrophage Penetration. Angew. Chem. Int. Ed. Engl. 2013, 52, 9664–9667. [Google Scholar] [CrossRef] [PubMed]
- Rubin, D.H.F.; Ross, J.D.C.; Grad, Y.H. The Frontiers of Addressing Antibiotic Resistance in Neisseria Gonorrhoeae. Transl. Res. 2020, 220, 122–137. [Google Scholar] [CrossRef]
- Ma, K.C.; Mortimer, T.D.; Hicks, A.L.; Wheeler, N.E.; Sánchez-Busó, L.; Golparian, D.; Taiaroa, G.; Rubin, D.H.F.; Wang, Y.; Williamson, D.A.; et al. Adaptation to the Cervical Environment Is Associated with Increased Antibiotic Susceptibility in Neisseria Gonorrhoeae. Nat. Commun. 2020, 11, 4126. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Busó, L.; Golparian, D.; Corander, J.; Grad, Y.H.; Ohnishi, M.; Flemming, R.; Parkhill, J.; Bentley, S.D.; Unemo, M.; Harris, S.R. The Impact of Antimicrobials on Gonococcal Evolution. Nat. Microbiol. 2019, 4, 1941–1950. [Google Scholar] [CrossRef]
- Dobrindt, U.; Hacker, J. Whole Genome Plasticity in Pathogenic Bacteria. Curr. Opin. Microbiol. 2001, 4, 550–557. [Google Scholar] [CrossRef] [PubMed]
- de Korne-Elenbaas, J.; Bruisten, S.M.; van Dam, A.P.; Maiden, M.C.J.; Harrison, O.B. The Neisseria Gonorrhoeae Accessory Genome and Its Association with the Core Genome and Antimicrobial Resistance. Microbiol. Spectr. 2022, 10, e0265421. [Google Scholar] [CrossRef]
- Mlynarczyk-Bonikowska, B.; Kowalewski, C.; Krolak-Ulinska, A.; Marusza, W. Molecular Mechanisms of Drug Resistance and Epidemiology of Multidrug-Resistant Variants of. Int. J. Mol. Sci. 2022, 23, 10499. [Google Scholar] [CrossRef]
- Młynarczyk-Bonikowska, B.; Majewska, A.; Malejczyk, M.; Młynarczyk, G.; Majewski, S. Multiresistant Neisseria Gonorrhoeae: A New Threat in Second Decade of the XXI Century. Med. Microbiol. Immunol. 2020, 209, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Bourguignon, T.; Godinez-Leon, J.A.; Gref, R. Nanosized Drug Delivery Systems to Fight Tuberculosis. Pharmaceutics 2023, 15, 393. [Google Scholar] [CrossRef]
- Singh, R.; Dwivedi, S.P.; Gaharwar, U.S.; Meena, R.; Rajamani, P.; Prasad, T. Recent Updates on Drug Resistance in Mycobacterium tuberculosis. J. Appl. Microbiol. 2020, 128, 1547–1567. [Google Scholar] [CrossRef] [Green Version]
- Gould, K. Antibiotics: From Prehistory to the Present Day. J. Antimicrob. Chemother. 2016, 71, 572–575. [Google Scholar] [CrossRef] [PubMed]
- Goossens, S.N.; Sampson, S.L.; Van Rie, A. Mechanisms of Drug-Induced Tolerance in Mycobacterium tuberculosis. Clin. Microbiol. Rev. 2020, 34, e00141-20. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.L.; Flynn, J.L. Understanding Latent Tuberculosis: A Moving Target. J. Immunol. 2010, 185, 15–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palomino, J.C.; Martin, A. Drug Resistance Mechanisms in Mycobacterium tuberculosis. Antibiotics 2014, 3, 317–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramaswamy, S.; Musser, J.M. Molecular Genetic Basis of Antimicrobial Agent Resistance in Mycobacterium tuberculosis: 1998 Update. Tuber. Lung Dis. 1998, 79, 3–29. [Google Scholar] [CrossRef] [Green Version]
- Kadura, S.; King, N.; Nakhoul, M.; Zhu, H.; Theron, G.; Köser, C.U.; Farhat, M. Systematic Review of Mutations Associated with Resistance to the New and Repurposed Mycobacterium tuberculosis Drugs Bedaquiline, Clofazimine, Linezolid, Delamanid and Pretomanid. J. Antimicrob. Chemother. 2020, 75, 2031–2043. [Google Scholar] [CrossRef]
- Chellat, M.F.; Raguž, L.; Riedl, R. Targeting Antibiotic Resistance. Angew. Chem. Int. Ed Engl. 2016, 55. [Google Scholar] [CrossRef]
- Barlow, M.; Hall, B.G. Phylogenetic Analysis Shows That the OXA B-Lactamase Genes Have Been on Plasmids for Millions of Years. J. Mol. Evol. 2002, 55, 314–321. [Google Scholar] [CrossRef]
- Pader, V.; Hakim, S.; Painter, K.L.; Wigneshweraraj, S.; Clarke, T.B.; Edwards, A.M. Staphylococcus aureus Inactivates Daptomycin by Releasing Membrane Phospholipids. Nat. Microbiol. 2016, 2, 16194. [Google Scholar] [CrossRef] [Green Version]
- Bevers, E.M.; Williamson, P.L. Getting to the Outer Leaflet: Physiology of Phosphatidylserine Exposure at the Plasma Membrane. Physiol. Rev. 2016, 96, 605–645. [Google Scholar] [CrossRef]
- Whitfield, C. Biosynthesis of Lipopolysaccharide O Antigens. Trends Microbiol. 1995, 3, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Papo, N.; Shai, Y. A Molecular Mechanism for Lipopolysaccharide Protection of Gram-Negative Bacteria from Antimicrobial Peptides. J. Biol. Chem. 2005, 280, 10378–10387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opoku-Temeng, C.; Kobayashi, S.D.; DeLeo, F.R. Klebsiella Pneumoniae Capsule Polysaccharide as a Target for Therapeutics and Vaccines. Comput. Struct. Biotechnol. J. 2019, 17, 1360–1366. [Google Scholar] [CrossRef] [PubMed]
- Domenico, P.; Salo, R.J.; Cross, A.S.; Cunha, B.A. Polysaccharide Capsule-Mediated Resistance to Opsonophagocytosis in Klebsiella Pneumoniae. Infect. Immun. 1994, 62, 4495–4499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bales, P.M.; Renke, E.M.; May, S.L.; Shen, Y.; Nelson, D.C. Purification and Characterization of Biofilm-Associated EPS Exopolysaccharides from ESKAPE Organisms and Other Pathogens. PLoS ONE 2013, 8, e67950. [Google Scholar] [CrossRef] [Green Version]
- Lewis, K. Persister Cells, Dormancy and Infectious Disease. Nat. Rev. Microbiol. 2007, 5, 48–56. [Google Scholar] [CrossRef]
- Saxena, P.; Joshi, Y.; Rawat, K.; Bisht, R. Biofilms: Architecture, Resistance, Quorum Sensing and Control Mechanisms. Indian J. Microbiol. 2019, 59, 3–12. [Google Scholar] [CrossRef]
- Machado, I.; Silva, L.R.; Giaouris, E.D.; Melo, L.F.; Simões, M. Quorum Sensing in Food Spoilage and Natural-Based Strategies for Its Inhibition. Food Res. Int. 2020, 127, 108754. [Google Scholar] [CrossRef]
- Zhao, X.; Yu, Z.; Ding, T. Quorum-Sensing Regulation of Antimicrobial Resistance in Bacteria. Microorganisms 2020, 8, 425. [Google Scholar] [CrossRef] [Green Version]
- Mah, T.F.; O’Toole, G.A. Mechanisms of Biofilm Resistance to Antimicrobial Agents. Trends Microbiol. 2001, 9, 34–39. [Google Scholar] [CrossRef]
- Hussain, A.; Ansari, A.; Ahmad, R. Microbial Biofilms: Human Mucosa and Intestinal Microbiota. In New and Future Developments in Microbial Biotechnology and Bioengineering: Microbial Biofilms; Elsevier: Amsterdam, The Netherlands, 2020; pp. 47–60. ISBN 9780444642790. [Google Scholar]
- Wang, Z.-W.; Lee, S.-H.; Elkins, J.G.; Morrell-Falvey, J.L. Spatial and Temporal Dynamics of Cellulose Degradation and Biofilm Formation by Caldicellulosiruptor Obsidiansis and Clostridium Thermocellum. AMB Express 2011, 1, 30. [Google Scholar] [CrossRef] [Green Version]
- Beitelshees, M.; Hill, A.; Jones, C.H.; Pfeifer, B.A. Phenotypic Variation during Biofilm Formation: Implications for Anti-Biofilm Therapeutic Design. Materials 2018, 11, 1086. [Google Scholar] [CrossRef] [Green Version]
- Di Martino, P.; Sirot, D.; Joly, B.; Rich, C.; Darfeuille-Michaud, A. Relationship between Adhesion to Intestinal Caco-2 Cells and Multidrug Resistance in Klebsiella Pneumoniae Clinical Isolates. J. Clin. Microbiol. 1997, 35, 1499–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, T.A.; Olson, R.; Fang, C.-T.; Stoesser, N.; Miller, M.; MacDonald, U.; Hutson, A.; Barker, J.H.; La Hoz, R.M.; Johnson, J.R. Identification of Biomarkers for Differentiation of Hypervirulent Klebsiella Pneumoniae from Classical K. Pneumoniae. J. Clin. Microbiol. 2018, 56, e00776-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Fives-Taylor, P.M. Molecular Strategies for Fimbrial Expression and Assembly. Crit. Rev. Oral Biol. Med. 2001, 12, 101–115. [Google Scholar] [CrossRef] [Green Version]
- Althouse, C.; Patterson, S.; Fedorka-Cray, P.; Isaacson, R.E. Type 1 Fimbriae of Salmonella Enterica Serovar Typhimurium Bind to Enterocytes and Contribute to Colonization of Swine in Vivo. Infect. Immun. 2003, 71, 6446–6452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arato, V.; Raso, M.M.; Gasperini, G.; Berlanda Scorza, F.; Micoli, F. Prophylaxis and Treatment against: Current Insights on This Emerging Anti-Microbial Resistant Global Threat. Int. J. Mol. Sci. 2021, 22, 4042. [Google Scholar] [CrossRef]
- Mandal, S.M.; Roy, A.; Ghosh, A.K.; Hazra, T.K.; Basak, A.; Franco, O.L. Challenges and Future Prospects of Antibiotic Therapy: From Peptides to Phages Utilization. Front. Pharmacol. 2014, 5, 105. [Google Scholar] [CrossRef]
- Vazquez-Grande, G.; Kumar, A. Optimizing Antimicrobial Therapy of Sepsis and Septic Shock: Focus on Antibiotic Combination Therapy. Semin. Respir. Crit. Care Med. 2015, 36, 154–166. [Google Scholar] [CrossRef] [Green Version]
- Kalan, L.; Wright, G.D. Antibiotic Adjuvants: Multicomponent Anti-Infective Strategies. Expert Rev. Mol. Med. 2011, 13, e5. [Google Scholar] [CrossRef]
- Bernal, P.; Molina-Santiago, C.; Daddaoua, A.; Llamas, M.A. Antibiotic Adjuvants: Identification and Clinical Use. Microb. Biotechnol. 2013, 6, 445–449. [Google Scholar] [CrossRef] [Green Version]
- Domalaon, R.; Idowu, T.; Zhanel, G.G.; Schweizer, F. Antibiotic Hybrids: The Next Generation of Agents and Adjuvants against Gram-Negative Pathogens? Clin. Microbiol. Rev. 2018, 31, e00077-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamma, P.D.; Cosgrove, S.E.; Maragakis, L.L. Combination Therapy for Treatment of Infections with Gram-Negative Bacteria. Clin. Microbiol. Rev. 2012, 25, 450–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latz, S.; Wahida, A.; Arif, A.; Häfner, H.; Hoß, M.; Ritter, K.; Horz, H.-P. Preliminary Survey of Local Bacteriophages with Lytic Activity against Multi-Drug Resistant Bacteria. J. Basic Microbiol. 2016, 56, 1117–1123. [Google Scholar] [CrossRef] [PubMed]
- Domingo-Calap, P.; Delgado-Martínez, J. Bacteriophages: Protagonists of a Post-Antibiotic Era. Antibiotic 2018, 7, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirnay, J.-P.; Verbeken, G.; Ceyssens, P.-J.; Huys, I.; De Vos, D.; Ameloot, C.; Fauconnier, A. The Magistral Phage. Viruses 2018, 10, 64. [Google Scholar] [CrossRef] [Green Version]
- Loc-Carrillo, C.; Abedon, S.T. Pros and Cons of Phage Therapy. Bacteriophage 2011, 1, 111–114. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Novick, R.P. Phage-Mediated Intergeneric Transfer of Toxin Genes. Science 2009, 323, 139–141. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, H.; São-José, C.; Azeredo, J. Phage-Derived Peptidoglycan Degrading Enzymes: Challenges and Future Prospects for In Vivo Therapy. Viruses 2018, 10, 292. [Google Scholar] [CrossRef] [Green Version]
- Gordillo Altamirano, F.L.; Barr, J.J. Phage Therapy in the Postantibiotic Era. Clin. Microbiol. Rev. 2019, 32. [Google Scholar] [CrossRef] [Green Version]
- Maciejewska, B.; Olszak, T.; Drulis-Kawa, Z. Applications of Bacteriophages versus Phage Enzymes to Combat and Cure Bacterial Infections: An Ambitious and Also a Realistic Application? Appl. Microbiol. Biotechnol. 2018, 102, 2563–2581. [Google Scholar] [CrossRef] [Green Version]
- Abdelrahman, F.; Easwaran, M.; Daramola, O.I.; Ragab, S.; Lynch, S.; Oduselu, T.J.; Khan, F.M.; Ayobami, A.; Adnan, F.; Torrents, E.; et al. Phage-Encoded Endolysins. Antibiotics 2021, 10, 124. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, P.; Chen, L.; Guo, G.; Xiao, Y.; Chen, L.; Du, H.; Zhang, W. Identification of a Phage-Derived Depolymerase Specific for KL64 Capsule of Klebsiella Pneumoniae and Its Anti-Biofilm Effect. Virus Genes 2021, 57, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Amankwah, S.; Abdella, K.; Kassa, T. Bacterial Biofilm Destruction: A Focused Review On The Recent Use of Phage-Based Strategies with Other Antibiofilm Agents. Nanotechnol. Sci. Appl. 2021, 14, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Reuter, M.; Kruger, D.H. Approaches to Optimize Therapeutic Bacteriophage and Bacteriophage-Derived Products to Combat Bacterial Infections. Virus Genes 2020, 56, 136–149. [Google Scholar] [CrossRef]
- Chen, X.; Liu, M.; Zhang, P.; Xu, M.; Yuan, W.; Bian, L.; Liu, Y.; Xia, J.; Leung, S.S.Y. Phage-Derived Depolymerase as an Antibiotic Adjuvant Against Multidrug-Resistant. Front. Microbiol. 2022, 13, 845500. [Google Scholar] [CrossRef]
- De Smet, J.; Hendrix, H.; Blasdel, B.G.; Danis-Wlodarczyk, K.; Lavigne, R. Pseudomonas Predators: Understanding and Exploiting Phage-Host Interactions. Nat. Rev. Microbiol. 2017, 15, 517–530. [Google Scholar] [CrossRef]
- Rai, S.; Kumar, A. Bacteriophage Therapeutics to Confront Multidrug-Resistant Acinetobacter baumannii—A Global Health Menace. Environ. Microbiol. Rep. 2022, 14, 347–364. [Google Scholar] [CrossRef]
- Fischetti, V.A. Bacteriophage Endolysins: A Novel Anti-Infective to Control Gram-Positive Pathogens. Int. J. Med. Microbiol. 2010, 300, 357–362. [Google Scholar] [CrossRef] [Green Version]
- Hansen, A.M.; Bonke, G.; Larsen, C.J.; Yavari, N.; Nielsen, P.E.; Franzyk, H. Antibacterial Peptide Nucleic Acid–Antimicrobial Peptide (PNA–AMP) Conjugates: Antisense Targeting of Fatty Acid Biosynthesis. Bioconjugate Chem. 2016, 27, 863–867. [Google Scholar] [CrossRef]
- Schmidt, N.W.; Deshayes, S.; Hawker, S.; Blacker, A.; Kasko, A.M.; Wong, G.C.L. Engineering Persister-Specific Antibiotics with Synergistic Antimicrobial Functions. ACS Nano 2014, 8, 8786–8793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Baeder, D.Y.; Regoes, R.R.; Rolff, J. Predicting Drug Resistance Evolution: Insights from Antimicrobial Peptides and Antibiotics. Proc. Biol. Sci. 2018, 285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, J.; Sun, L.; Huang, S.; Zhu, C.; Li, P.; He, J.; Mackey, V.; Coy, D.H.; He, Q. The Antimicrobial Peptides and Their Potential Clinical Applications. Am. J. Transl. Res. 2019, 11, 3919–3931. [Google Scholar]
- Reuter, A.; Hilpert, C.; Dedieu-Berne, A.; Lematre, S.; Gueguen, E.; Launay, G.; Bigot, S.; Lesterlin, C. Targeted-Antibacterial-Plasmids (TAPs) Combining Conjugation and CRISPR/Cas Systems Achieve Strain-Specific Antibacterial Activity. Nucleic Acids Res. 2021, 49, 3584–3598. [Google Scholar] [CrossRef] [PubMed]
- Gholizadeh, P.; Köse, Ş.; Dao, S.; Ganbarov, K.; Tanomand, A.; Dal, T.; Aghazadeh, M.; Ghotaslou, R.; Rezaee, M.A.; Yousefi, B.; et al. How CRISPR-Cas System Could Be Used to Combat Antimicrobial Resistance. Infect. Drug Resist. 2020, 13, 1111. [Google Scholar] [CrossRef] [Green Version]
- Wan, F.; Draz, M.S.; Gu, M.; Yu, W.; Ruan, Z.; Luo, Q. Novel Strategy to Combat Antibiotic Resistance: A Sight into the Combination of CRISPR/Cas9 and Nanoparticles. Pharmaceutics 2021, 13, 352. [Google Scholar] [CrossRef]
- Fage, C.; Lemire, N.; Moineau, S. Delivery of CRISPR-Cas Systems Using Phage-Based Vectors. Curr. Opin. Biotechnol. 2021, 68, 174–180. [Google Scholar] [CrossRef]
- Wu, H.; Zhang, Z.; Ke, F.; Jia, P.; Zhu, K.; Zou, J. Cell-Penetrating Peptides and DNA-Free Genome Editing in Plants. Int. J. Plant Anim. Environ. Sci. 2021, 11, 474–484. [Google Scholar] [CrossRef]
- Wang, H.-X.; Song, Z.; Lao, Y.-H.; Xu, X.; Gong, J.; Cheng, D.; Chakraborty, S.; Park, J.S.; Li, M.; Huang, D.; et al. Nonviral Gene Editing via CRISPR/Cas9 Delivery by Membrane-Disruptive and Endosomolytic Helical Polypeptide. Proc. Natl. Acad. Sci. USA 2018, 115, 4903–4908. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Wen, Y.; Guo, X. CRISPR/Cas9 for Genome Editing: Progress, Implications and Challenges. Hum. Mol. Genet. 2014, 23, R40–R46. [Google Scholar] [CrossRef] [Green Version]
- Shabbir, M.A.; Wu, Q.; Shabbir, M.Z.; Sajid, A.; Ahmed, S.; Sattar, A.; Tang, Y.; Li, J.; Maan, M.K.; Hao, H.; et al. The CRISPR-Cas System Promotes Antimicrobial Resistance in Campylobacter Jejuni. Future Microbiol. 2018, 13, 1757–1774. [Google Scholar] [CrossRef]
- Arora, L.; Narula, A. Gene Editing and Crop Improvement Using CRISPR-Cas9 System. Front. Plant Sci. 2017, 8, 1932. [Google Scholar] [CrossRef] [Green Version]
- Peter, J.; Gwynne, M.P.G. Light as a Broad-Spectrum Antimicrobial. Front. Microbiol. 2018, 9, 119. [Google Scholar] [CrossRef]
- Mai, B.; Gao, Y.; Li, M.; Wang, X.; Zhang, K.; Liu, Q.; Xu, C.; Wang, P. Photodynamic Antimicrobial Chemotherapy for Staphylococcus aureus and Multidrug-Resistant Bacterial Burn Infection in Vitro and in Vivo. Int. J. Nanomed. 2017, 12, 5915–5931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.-Y.; Chang, K.-C.; Chen, L.-Y.; Wang, P.-C.; Chou, C.-C.; Wu, Z.-B.; Hu, A. Blue Light Irradiation Triggers the Antimicrobial Potential of ZnO Nanoparticles on Drug-Resistant Acinetobacter baumannii. J. Photochem. Photobiol. B 2018, 180, 235–242. [Google Scholar] [CrossRef] [PubMed]
- de Freitas, L.M.; Lorenzón, E.N.; Santos-Filho, N.A.; Zago, L.H.d.P.; Uliana, M.P.; de Oliveira, K.T.; Cilli, E.M.; Fontana, C.R. Antimicrobial Photodynamic Therapy Enhanced by the Peptide Aurein 1.2. Sci. Rep. 2018, 8, 4212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakonieczna, J.; Wolnikowska, K.; Ogonowska, P.; Neubauer, D.; Bernat, A.; Kamysz, W. Rose Bengal-Mediated Photoinactivation of Multidrug Resistant Pseudomonas Aeruginosa Is Enhanced in the Presence of Antimicrobial Peptides. Front. Microbiol. 2018, 9, 1949. [Google Scholar] [CrossRef]
- Dakal, T.C.; Kumar, A.; Majumdar, R.S.; Yadav, V. Mechanistic Basis of Antimicrobial Actions of Silver Nanoparticles. Front. Microbiol. 2016, 7, 1831. [Google Scholar] [CrossRef] [Green Version]
- Qayyum, S.; Oves, M.; Khan, A.U. Obliteration of Bacterial Growth and Biofilm through ROS Generation by Facilely Synthesized Green Silver Nanoparticles. PLoS ONE 2017, 12, e0181363. [Google Scholar] [CrossRef] [Green Version]
- Bavya, M.C.; Vimal Rohan, K.; Gaurav, G.B.; Srivasatava, R. Synergistic Treatment Strategies to Combat Resistant Bacterial Infections Using Schiff Base Modified Nanoparticulate—Hydrogel System. Mater. Sci. Eng. C Mater. Biol. Appl. 2019, 95, 226–235. [Google Scholar] [CrossRef]
- Wuethrich, I.; W Pelzer, B.; Khodamoradi, Y.; Vehreschild, M.J.G.T. The Role of the Human Gut Microbiota in Colonization and Infection with Multidrug-Resistant Bacteria. Gut Microbes 2021, 13, 1911279. [Google Scholar] [CrossRef] [PubMed]
- Bakkeren, E.; Huisman, J.S.; Fattinger, S.A.; Hausmann, A.; Furter, M.; Egli, A.; Slack, E.; Sellin, M.E.; Bonhoeffer, S.; Regoes, R.R.; et al. Salmonella Persisters Promote the Spread of Antibiotic Resistance Plasmids in the Gut. Nature 2019, 573, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Langdon, A.; Schwartz, D.J.; Bulow, C.; Sun, X.; Hink, T.; Reske, K.A.; Jones, C.; Burnham, C.-A.D.; Dubberke, E.R.; Dantas, G.; et al. Microbiota Restoration Reduces Antibiotic-Resistant Bacteria Gut Colonization in Patients with Recurrent Clostridioides Difficile Infection from the Open-Label PUNCH CD Study. Genome Med. 2021, 13, 28. [Google Scholar] [CrossRef] [PubMed]
- León-Sampedro, R.; DelaFuente, J.; Díaz-Agero, C.; Crellen, T.; Musicha, P.; Rodríguez-Beltrán, J.; de la Vega, C.; Hernández-García, M.; R-GNOSIS WP5 Study Group; López-Fresneña, N.; et al. Pervasive Transmission of a Carbapenem Resistance Plasmid in the Gut Microbiota of Hospitalized Patients. Nat. Microbiol. 2021, 6, 606–616. [Google Scholar] [CrossRef]
- Food, U.S. Drug Administration. Important Safety Alert Regarding Use of Fecal Microbiota for Transplantation and Risk of Serious Adverse Reactions due to Transmission of Multi-Drug Resistant Organisms; US Food & Drug Administration: Washington, DC, USA, 2019. [Google Scholar]
- Drider, D. Gut Microbiota Is an Important Source of Bacteriocins and Their In Situ Expression Can Be Explored for Treatment of Bacterial Infections. Probiotics Antimicrob. Proteins 2021, 13, 1759–1765. [Google Scholar] [CrossRef] [PubMed]
- Miani, M.; Le Naour, J.; Waeckel-Enée, E.; Verma, S.C.; Straube, M.; Emond, P.; Ryffel, B.; van Endert, P.; Sokol, H.; Diana, J. Gut Microbiota-Stimulated Innate Lymphoid Cells Support β-Defensin 14 Expression in Pancreatic Endocrine Cells, Preventing Autoimmune Diabetes. Cell Metab. 2018, 28, 557–572.e6. [Google Scholar] [CrossRef] [Green Version]
- Nuding, S.; Frasch, T.; Schaller, M.; Stange, E.F.; Zabel, L.T. Synergistic Effects of Antimicrobial Peptides and Antibiotics against Clostridium Difficile. Antimicrob. Agents Chemother. 2014, 58, 5719–5725. [Google Scholar] [CrossRef] [Green Version]
- Iketani, S.; Mohri, H.; Culbertson, B.; Hong, S.J.; Duan, Y.; Luck, M.I.; Annavajhala, M.K.; Guo, Y.; Sheng, Z.; Uhlemann, A.-C.; et al. Multiple Pathways for SARS-CoV-2 Resistance to Nirmatrelvir. Nature 2022, 613, 558–564. [Google Scholar] [CrossRef]
- Guo, K.; Barrett, B.S.; Morrison, J.H.; Mickens, K.L.; Vladar, E.K.; Hasenkrug, K.J.; Poeschla, E.M.; Santiago, M.L. Interferon Resistance of Emerging SARS-CoV-2 Variants. Proc. Natl. Acad. Sci. USA 2022, 119, e2203760119. [Google Scholar] [CrossRef]
- Grellet, E.; L’Hôte, I.; Goulet, A.; Imbert, I. Replication of the Coronavirus Genome: A Paradox among Positive-Strand RNA Viruses. J. Biol. Chem. 2022, 298, 101923. [Google Scholar] [CrossRef]
- Morin, B.; Kranzusch, P.J.; Rahmeh, A.A.; Whelan, S.P.J. The Polymerase of Negative-Stranded RNA Viruses. Curr. Opin. Virol. 2013, 3, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Fodor, E.; Keown, J.R. A Structural Understanding of Influenza Virus Genome Replication. Trends Microbiol. 2022, 31, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Galvin, H.D.; Haw, T.Y.; Nutsford, A.N.; Husain, M. Drug Resistance in Influenza A Virus: The Epidemiology and Management. Infect. Drug Resist. 2017, 10, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Vries, E.; Ison, M.G. Antiviral Resistance in Influenza Viruses: Clinical and Epidemiological Aspects. In Antimicrobial Drug Resistance; Springer International Publishing: Cham, Switzerland, 2017; pp. 1165–1183. ISBN 9783319472645. [Google Scholar]
- Takashita, E. Influenza Polymerase Inhibitors: Mechanisms of Action and Resistance. Cold Spring Harb. Perspect. Med. 2021, 11, a038687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Q.; Liu, Y.; Peng, R.; Wang, M.; Yang, W.; Song, H.; Chen, Y.; Liu, S.; Han, M.; Zhang, X.; et al. Structural Insight into RNA Synthesis by Influenza D Polymerase. Nat. Microbiol. 2019, 4, 1750–1759. [Google Scholar] [CrossRef]
- Omoto, S.; Speranzini, V.; Hashimoto, T.; Noshi, T.; Yamaguchi, H.; Kawai, M.; Kawaguchi, K.; Uehara, T.; Shishido, T.; Naito, A.; et al. Characterization of Influenza Virus Variants Induced by Treatment with the Endonuclease Inhibitor Baloxavir Marboxil. Sci. Rep. 2018, 8, 9633. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, R.; Yasuo, N.; Sekijima, M. Molecular Dynamics Simulation Reveals the Mechanism by Which the Influenza Cap-Dependent Endonuclease Acquires Resistance against Baloxavir Marboxil. Sci. Rep. 2019, 9, 17464. [Google Scholar] [CrossRef] [Green Version]
- Clark, M.P.; Ledeboer, M.W.; Davies, I.; Byrn, R.A.; Jones, S.M.; Perola, E.; Tsai, A.; Jacobs, M.; Nti-Addae, K.; Bandarage, U.K.; et al. Discovery of a Novel, First-in-Class, Orally Bioavailable Azaindole Inhibitor (VX-787) of Influenza PB2. J. Med. Chem. 2014, 57, 6668–6678. [Google Scholar] [CrossRef]
- Byrn, R.A.; Jones, S.M.; Bennett, H.B.; Bral, C.; Clark, M.P.; Jacobs, M.D.; Kwong, A.D.; Ledeboer, M.W.; Leeman, J.R.; McNeil, C.F.; et al. Preclinical Activity of VX-787, a First-in-Class, Orally Bioavailable Inhibitor of the Influenza Virus Polymerase PB2 Subunit. Antimicrob. Agents Chemother. 2015, 59, 1569–1582. [Google Scholar] [CrossRef] [Green Version]
- Sorbo, M.C.; Cento, V.; Di Maio, V.C.; Howe, A.Y.M.; Garcia, F.; Perno, C.F.; Ceccherini-Silberstein, F. Hepatitis C Virus Drug Resistance Associated Substitutions and Their Clinical Relevance: Update 2018. Drug Resist. Updat. 2018, 37, 17–39. [Google Scholar] [CrossRef]
- Li, D.K.; Chung, R.T. Overview of Direct-Acting Antiviral Drugs and Drug Resistance of Hepatitis C Virus. Methods Mol. Biol. 2019, 1911, 3–32. [Google Scholar] [PubMed]
- Zając, M.; Muszalska, I.; Sobczak, A.; Dadej, A.; Tomczak, S.; Jelińska, A. Hepatitis C—New Drugs and Treatment Prospects. Eur. J. Med. Chem. 2019, 165, 225–249. [Google Scholar] [CrossRef]
- Boson, B.; Denolly, S.; Turlure, F.; Chamot, C.; Dreux, M.; Cosset, F.-L. Daclatasvir Prevents Hepatitis C Virus Infectivity by Blocking Transfer of the Viral Genome to Assembly Sites. Gastroenterology 2017, 152, 895–907.e14. [Google Scholar] [CrossRef] [Green Version]
- Nettles, J.H.; Stanton, R.A.; Broyde, J.; Amblard, F.; Zhang, H.; Zhou, L.; Shi, J.; McBrayer, T.R.; Whitaker, T.; Coats, S.J.; et al. Asymmetric Binding to NS5A by Daclatasvir (BMS-790052) and Analogs Suggests Two Novel Modes of HCV Inhibition. J. Med. Chem. 2014, 57, 10031–10043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beloukas, A.; Geretti, A.M. Hepatitis B Virus Drug Resistance. In Antimicrobial Drug Resistance; Springer International Publishing: Cham, Switzerland, 2017; pp. 1227–1242. ISBN 9783319472645. [Google Scholar]
- Revill, P.A.; Tu, T.; Netter, H.J.; Yuen, L.K.W.; Locarnini, S.A.; Littlejohn, M. The Evolution and Clinical Impact of Hepatitis B Virus Genome Diversity. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 618–634. [Google Scholar] [CrossRef]
- Suk-Fong Lok, A. Hepatitis B Treatment: What We Know Now and What Remains to Be Researched. Hepatol. Commun. 2019, 3, 8–19. [Google Scholar] [CrossRef] [Green Version]
- Burdette, D.; Hyrina, A.; Song, Z.; Beran, R.K.; Cheung, T.; Gilmore, S.; Kobayashi, T.; Li, L.; Liu, Y.; Niedziela-Majka, A.; et al. Characterization of a Novel Capsid Assembly Modulator for the Treatment of Chronic Hepatitis B Virus Infection. Antimicrob. Agents Chemother. 2023, 67, e0134822. [Google Scholar] [CrossRef] [PubMed]
- Taverniti, V.; Ligat, G.; Debing, Y.; Kum, D.B.; Baumert, T.F.; Verrier, E.R. Capsid Assembly Modulators as Antiviral Agents against HBV: Molecular Mechanisms and Clinical Perspectives. J. Clin. Med. Res. 2022, 11, 1349. [Google Scholar] [CrossRef]
- Luo, Y.; Cheng, J.; Hu, Z.; Ban, H.; Wu, S.; Hwang, N.; Kulp, J.; Li, Y.; Du, Y.; Chang, J.; et al. Identification of Hepatitis B Virus Core Protein Residues Critical for Capsid Assembly, pgRNA Encapsidation and Resistance to Capsid Assembly Modulators. Antivir. Res. 2021, 191, 105080. [Google Scholar] [CrossRef]
- Piret, J.; Boivin, G. Herpesvirus Resistance to Antiviral Drugs. In Antimicrobial Drug Resistance; Springer International Publishing: Cham, Switzerland, 2017; pp. 1185–1211. ISBN 9783319472645. [Google Scholar]
- Schalkwijk, H.H.; Snoeck, R.; Andrei, G. Acyclovir Resistance in Herpes Simplex Viruses: Prevalence and Therapeutic Alternatives. Biochem. Pharmacol. 2022, 206, 115322. [Google Scholar] [CrossRef]
- Rabelo, V.W.-H.; Romeiro, N.C.; Paixão, I.C.N.d.P.; Abreu, P.A. Mechanism of Resistance to Acyclovir in Thymidine Kinase Mutants from Herpes Simplex Virus Type 1: A Computational Approach. J. Biomol. Struct. Dyn. 2020, 38, 2116–2127. [Google Scholar] [CrossRef] [PubMed]
- Sadowski, L.A.; Upadhyay, R.; Greeley, Z.W.; Margulies, B.J. Current Drugs to Treat Infections with Herpes Simplex Viruses-1 and -2. Viruses 2021, 13, 1228. [Google Scholar] [CrossRef]
- Tayyar, R.; Ho, D. Herpes Simplex Virus and Varicella Zoster Virus Infections in Cancer Patients. Viruses 2023, 15, 439. [Google Scholar] [CrossRef] [PubMed]
- Patro, A.R.K. Subversion of Immune Response by Human Cytomegalovirus. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piret, J.; Boivin, G. Clinical Development of Letermovir and Maribavir: Overview of Human Cytomegalovirus Drug Resistance. Antivir. Res. 2019, 163, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, C.; Boivin, G. Human Cytomegalovirus Resistance to Antiviral Drugs. Antimicrob. Agents Chemother. 2005, 49, 873–883. [Google Scholar] [CrossRef] [Green Version]
- Park, K.R.; Kim, Y.-E.; Shamim, A.; Gong, S.; Choi, S.-H.; Kim, K.K.; Kim, Y.-J.; Ahn, J.-H. Analysis of Novel Drug-Resistant Human Cytomegalovirus DNA Polymerase Mutations Reveals the Role of a DNA-Binding Loop in Phosphonoformic Acid Resistance. Front. Microbiol. 2022, 13, 771978. [Google Scholar] [CrossRef]
- Kapoor, A.; Ghosh, A.K.; Forman, M.; Hu, X.; Ye, W.; Southall, N.; Marugan, J.; Keyes, R.F.; Smith, B.C.; Meyers, D.J.; et al. Validation and Characterization of Five Distinct Novel Inhibitors of Human Cytomegalovirus. J. Med. Chem. 2020, 63, 3896–3907. [Google Scholar] [CrossRef]
- Shafer, R.W. Human Immunodeficiency Virus Type 1 Drug Resistance Mutations Update. J. Infect. Dis. 2017, 216, S843–S846. [Google Scholar] [CrossRef] [Green Version]
- Mayers, D.L.; Baxter, J.D. Clinical Implications of HIV-1 Drug Resistance. In Antimicrobial Drug Resistance; Springer International Publishing: Cham, Switzerland, 2017; pp. 1213–1225. ISBN 9783319472645. [Google Scholar]
- Cilento, M.E.; Kirby, K.A.; Sarafianos, S.G. Avoiding Drug Resistance in HIV Reverse Transcriptase. Chem. Rev. 2021, 121, 3271–3296. [Google Scholar] [CrossRef]
- Nastri, B.M.; Pagliano, P.; Zannella, C.; Folliero, V.; Masullo, A.; Rinaldi, L.; Galdiero, M.; Franci, G. HIV and Drug-Resistant Subtypes. Microorganisms 2023, 11, 221. [Google Scholar] [CrossRef]
- Catalano, A.; Iacopetta, D.; Ceramella, J.; Scumaci, D.; Giuzio, F.; Saturnino, C.; Aquaro, S.; Rosano, C.; Sinicropi, M.S. Multidrug Resistance (MDR): A Widespread Phenomenon in Pharmacological Therapies. Molecules 2022, 27, 616. [Google Scholar] [CrossRef] [PubMed]
- van Heuvel, Y.; Schatz, S.; Rosengarten, J.F.; Stitz, J. Infectious RNA: Human Immunodeficiency Virus (HIV) Biology, Therapeutic Intervention, and the Quest for a Vaccine. Toxins 2022, 14, 138. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Bandaranayake, R.M.; Cai, Y.; King, N.M.; Kolli, M.; Mittal, S.; Murzycki, J.F.; Nalam, M.N.L.; Nalivaika, E.A.; Özen, A.; et al. Molecular Basis for Drug Resistance in HIV-1 Protease. Viruses 2010, 2, 2509–2535. [Google Scholar] [CrossRef] [PubMed]
- Blanco, J.-L.; Varghese, V.; Rhee, S.-Y.; Gatell, J.M.; Shafer, R.W. HIV-1 Integrase Inhibitor Resistance and Its Clinical Implications. J. Infect. Dis. 2011, 203, 1204–1214. [Google Scholar] [CrossRef]
- Geretti, A.M.; Armenia, D.; Ceccherini-Silberstein, F. Emerging Patterns and Implications of HIV-1 Integrase Inhibitor Resistance. Curr. Opin. Infect. Dis. 2012, 25, 677–686. [Google Scholar] [CrossRef]
- Anang, S.; Richard, J.; Bourassa, C.; Goyette, G.; Chiu, T.-J.; Chen, H.-C.; Smith, A.B., 3rd; Madani, N.; Finzi, A.; Sodroski, J. Characterization of Human Immunodeficiency Virus (HIV-1) Envelope Glycoprotein Variants Selected for Resistance to a CD4-Mimetic Compound. J. Virol. 2022, 96, e0063622. [Google Scholar] [CrossRef]
- Heaton, N.S.; Randall, G. Multifaceted Roles for Lipids in Viral Infection. Trends Microbiol. 2011, 19, 368–375. [Google Scholar] [CrossRef]
- Ryu, W.-S. Virus Life Cycle. In Molecular Virology of Human Pathogenic Viruses; Elsevier: London, UK, 2017; pp. 31–45. ISBN 9780128008386. [Google Scholar]
- Huang, Y.; Yang, C.; Xu, X.-F.; Xu, W.; Liu, S.-W. Structural and Functional Properties of SARS-CoV-2 Spike Protein: Potential Antivirus Drug Development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef]
- Yamauchi, Y.; Greber, U.F. Principles of Virus Uncoating: Cues and the Snooker Ball. Traffic 2016, 17, 569–592. [Google Scholar] [CrossRef] [Green Version]
- Tabler, C.O.; Wegman, S.J.; Chen, J.; Shroff, H.; Alhusaini, N.; Tilton, J.C. The HIV-1 Viral Protease Is Activated during Assembly and Budding Prior to Particle Release. J. Virol. 2022, 96, e0219821. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.J.; Cooper, K.L.; Tappenden, P.; Rees, A.; Simpson, E.L.; Read, R.C.; Nicholson, K.G. Oseltamivir, Zanamivir and Amantadine in the Prevention of Influenza: A Systematic Review. J. Infect. 2011, 62, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Wexler-Cohen, Y.; Shai, Y. Multifaceted Action of Fuzeon as Virus–cell Membrane Fusion Inhibitor. Biochim. Et Biophys. Acta (BBA)—Biomembr. 2011, 1808, 2352–2358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Safadi, Y.; Vivet-Boudou, V.; Marquet, R. HIV-1 Reverse Transcriptase Inhibitors. Appl. Microbiol. Biotechnol. 2007, 75, 723–737. [Google Scholar] [CrossRef]
- Kłysik, K.; Pietraszek, A.; Karewicz, A.; Nowakowska, M. Acyclovir in the Treatment of Herpes Viruses—A Review. Curr. Med. Chem. 2020, 27, 4118–4137. [Google Scholar] [CrossRef]
- Shiraki, K.; Daikoku, T. Favipiravir, an Anti-Influenza Drug against Life-Threatening RNA Virus Infections. Pharmacol. Ther. 2020, 209, 107512. [Google Scholar] [CrossRef]
- Scarsi, K.K.; Havens, J.P.; Podany, A.T.; Avedissian, S.N.; Fletcher, C.V. HIV-1 Integrase Inhibitors: A Comparative Review of Efficacy and Safety. Drugs 2020, 80, 1649–1676. [Google Scholar] [CrossRef]
- Lea, A.P.; Faulds, D. Ritonavir. Drugs 2012, 52, 541–546. [Google Scholar] [CrossRef]
- Geraghty, R.J.; Aliota, M.T.; Bonnac, L.F. Broad-Spectrum Antiviral Strategies and Nucleoside Analogues. Viruses 2021, 13, 667. [Google Scholar] [CrossRef]
- Crotty, S.; Maag, D.; Arnold, J.J.; Zhong, W.; Lau, J.Y.; Hong, Z.; Andino, R.; Cameron, C.E. The Broad-Spectrum Antiviral Ribonucleoside Ribavirin Is an RNA Virus Mutagen. Nat. Med. 2000, 6, 1375–1379. [Google Scholar] [CrossRef]
- Tian, L.; Pang, Z.; Li, M.; Lou, F.; An, X.; Zhu, S.; Song, L.; Tong, Y.; Fan, H.; Fan, J. Molnupiravir and Its Antiviral Activity Against COVID-19. Front. Immunol. 2022, 13, 855496. [Google Scholar] [CrossRef]
- Domingo, E.; García-Crespo, C.; Lobo-Vega, R.; Perales, C. Mutation Rates, Mutation Frequencies, and Proofreading-Repair Activities in RNA Virus Genetics. Viruses 2021, 13, 1882. [Google Scholar] [CrossRef] [PubMed]
- Bruzzoni-Giovanelli, H.; Alezra, V.; Wolff, N.; Dong, C.-Z.; Tuffery, P.; Rebollo, A. Interfering Peptides Targeting Protein–protein Interactions: The next Generation of Drugs? Drug Discov. Today 2018, 23, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Diamond, G.; Molchanova, N.; Herlan, C.; Fortkort, J.A.; Lin, J.S.; Figgins, E.; Bopp, N.; Ryan, L.K.; Chung, D.; Adcock, R.S.; et al. Potent Antiviral Activity against HSV-1 and SARS-CoV-2 by Antimicrobial Peptoids. Pharmaceuticals 2021, 14, 304. [Google Scholar] [CrossRef] [PubMed]
- Mukai, K.; Tsai, M.; Starkl, P.; Marichal, T.; Galli, S.J. IgE and Mast Cells in Host Defense against Parasites and Venoms. Semin. Immunopathol. 2016, 38, 581–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacks, D.L.; Peters, N.C.; Bethony, J.M. Vaccines Against Parasites. In The Vaccine Book; Elsevier: Amsterdam, The Netherlands, 2016; pp. 331–360. [Google Scholar]
- Kim, S.; Hwang, J.S.; Lee, D.G. Lactoferricin B like Peptide Triggers Mitochondrial Disruption-Mediated Apoptosis by Inhibiting Respiration under Nitric Oxide Accumulation in Candida Albicans. IUBMB Life 2020, 72, 1515–1527. [Google Scholar] [CrossRef]
- Gonçalves, S.S.; Souza, A.C.R.; Chowdhary, A.; Meis, J.F.; Colombo, A.L. Epidemiology and Molecular Mechanisms of Antifungal Resistance in Candida and Aspergillus. Mycoses 2016, 59, 198–219. [Google Scholar] [CrossRef]
- Borghi, E.; Borgo, F.; Morace, G. Fungal Biofilms: Update on Resistance. In Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2016; pp. 37–47. [Google Scholar]
- Fuentefria, A.M.; Pippi, B.; Dalla Lana, D.F.; Donato, K.K.; de Andrade, S.F. Antifungals Discovery: An Insight into New Strategies to Combat Antifungal Resistance. Lett. Appl. Microbiol. 2018, 66, 2–13. [Google Scholar] [CrossRef] [Green Version]
- Kowalski, C.H.; Morelli, K.A.; Schultz, D.; Nadell, C.D.; Cramer, R.A. Fungal Biofilm Architecture Produces Hypoxic Microenvironments That Drive Antifungal Resistance. Proc. Natl. Acad. Sci. USA 2020, 117, 22473–22483. [Google Scholar] [CrossRef]
- Ramos-Martín, F.; Herrera-León, C.; Antonietti, V.; Sonnet, P.; Sarazin, C.; D’Amelio, N. The Potential of Antifungal Peptide Sesquin as Natural Food Preservative. Biochimie 2022, 203, 51–64. [Google Scholar] [CrossRef]
- Ramos-Martín, F.; Herrera-León, C.; Antonietti, V.; Sonnet, P.; Sarazin, C.; D’Amelio, N. Antimicrobial Peptide K11 Selectively Recognizes Bacterial Biomimetic Membranes and Acts by Twisting Their Bilayers. Pharmaceuticals 2020, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Capela, R.; Moreira, R.; Lopes, F. An Overview of Drug Resistance in Protozoal Diseases. Int. J. Mol. Sci. 2019, 20, 5748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Useche, Y.; Pérez, A.R.; de Meis, J.; Bonomo, A.; Savino, W. Central Nervous System Commitment in Chagas Disease. Front. Immunol. 2022, 13, 975106. [Google Scholar] [CrossRef]
- Bansal, R.; Sen, S.S.; Muthuswami, R.; Madhubala, R. Stigmasterol as a Potential Biomarker for Amphotericin B Resistance in Leishmania Donovani. J. Antimicrob. Chemother. 2020, 75, 942–950. [Google Scholar] [CrossRef]
- Argüello-García, R.; Leitsch, D.; Skinner-Adams, T.; Ortega-Pierres, M.G. Drug Resistance in Giardia: Mechanisms and Alternative Treatments for Giardiasis. Adv. Parasitol. 2020, 107, 201–282. [Google Scholar] [PubMed]
- Aley, S.B.; Zimmerman, M.; Hetsko, M.; Selsted, M.E.; Gillin, F.D. Killing of Giardia Lamblia by Cryptdins and Cationic Neutrophil Peptides. Infect. Immun. 1994, 62, 5397–5403. [Google Scholar] [CrossRef] [Green Version]
- Seshacharyulu, P.; Baine, M.J.; Souchek, J.J.; Menning, M.; Kaur, S.; Yan, Y.; Ouellette, M.M.; Jain, M.; Lin, C.; Batra, S.K. Biological Determinants of Radioresistance and Their Remediation in Pancreatic Cancer. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 69–92. [Google Scholar] [CrossRef]
- What Is Drug Resistance? Available online: https://www.cancer.gov/research/annual-plan/scientific-topics/what-is-drug-resistance-infographic (accessed on 17 March 2023).
- Schirrmacher, V. From Chemotherapy to Biological Therapy: A Review of Novel Concepts to Reduce the Side Effects of Systemic Cancer Treatment (Review). Int. J. Oncol. 2019, 54, 407–419. [Google Scholar]
- Lam, K.-O.; Kwong, D.L.W. Chemotherapy for Esophageal Adenocarcinoma. Methods Mol. Biol. 2018, 1756, 19–34. [Google Scholar]
- National Institute of Diabetes and Digestive and Kidney Diseases (U.S.). LiverTox: Clinical and Research Information on Drug-Induced Liver Injury; National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2012. [Google Scholar]
- Horwitz, S.B. Taxol (paclitaxel): Mechanisms of Action. Ann. Oncol. 1994, 5 (Suppl. 6), S3–S6. [Google Scholar]
- Huang, C.-Y.; Ju, D.-T.; Chang, C.-F.; Muralidhar Reddy, P.; Velmurugan, B.K. A Review on the Effects of Current Chemotherapy Drugs and Natural Agents in Treating Non–small Cell Lung Cancer. BioMedicine 2017, 7, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Shang, Q.; Li, W.; Guo, W.; Stojadinovic, A.; Mannion, C.; Man, Y.-G.; Chen, T. Antibiotics for Cancer Treatment: A Double-Edged Sword. J. Cancer 2020, 11, 5135–5149. [Google Scholar] [CrossRef] [PubMed]
- Teng, Q.-X.; Luo, X.; Lei, Z.-N.; Wang, J.-Q.; Wurpel, J.; Qin, Z.; Yang, D.-H. The Multidrug Resistance-Reversing Activity of a Novel Antimicrobial Peptide. Cancers 2020, 12, 1963. [Google Scholar] [CrossRef] [PubMed]
- Lohan-Codeço, M.; Barambo-Wagner, M.L.; Nasciutti, L.E.; Ribeiro Pinto, L.F.; Meireles Da Costa, N.; Palumbo, A. Molecular Mechanisms Associated with Chemoresistance in Esophageal Cancer. Cell. Mol. Life Sci. 2022, 79, 1–26. [Google Scholar] [CrossRef]
- Yang, F.; Zhao, N.; Ge, D.; Chen, Y. Next-Generation of Selective Histone Deacetylase Inhibitors. RSC Adv. 2019, 9, 19571–19583. [Google Scholar] [CrossRef] [Green Version]
- Wawruszak, A.; Kalafut, J.; Okon, E.; Czapinski, J.; Halasa, M.; Przybyszewska, A.; Miziak, P.; Okla, K.; Rivero-Muller, A.; Stepulak, A. Histone Deacetylase Inhibitors and Phenotypical Transformation of Cancer Cells. Cancers 2019, 11, 148. [Google Scholar] [CrossRef] [Green Version]
- Saraswati, A.P.; Relitti, N.; Brindisi, M.; Gemma, S.; Zisterer, D.; Butini, S.; Campiani, G. Raising the Bar in Anticancer Therapy: Recent Advances In, and Perspectives On, Telomerase Inhibitors. Drug Discov. Today 2019, 24, 1370–1388. [Google Scholar] [CrossRef]
- Guterres, A.N.; Villanueva, J. Targeting Telomerase for Cancer Therapy. Oncogene 2020, 39, 5811–5824. [Google Scholar] [CrossRef]
- Okamoto, K.; Seimiya, H. Revisiting Telomere Shortening in Cancer. Cells 2019, 8, 107. [Google Scholar] [CrossRef] [Green Version]
- Ellingsen, E.B.; Mangsbo, S.M.; Hovig, E.; Gaudernack, G. Telomerase as a Target for Therapeutic Cancer Vaccines and Considerations for Optimizing Their Clinical Potential. Front. Immunol. 2021, 12, 682492. [Google Scholar] [CrossRef]
- Mahadik, N.; Bhattacharya, D.; Padmanabhan, A.; Sakhare, K.; Narayan, K.P.; Banerjee, R. Targeting Steroid Hormone Receptors for Anti-Cancer Therapy-A Review on Small Molecules and Nanotherapeutic Approaches. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2022, 14, e1755. [Google Scholar] [CrossRef] [PubMed]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-Targeted Cancer Therapies: Progress, Challenges and Future Directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunda, N.K. Antimicrobial Peptides as Novel Therapeutics for Non-Small Cell Lung Cancer. Drug Discov. Today 2020, 25, 238–247. [Google Scholar] [CrossRef]
- Wu, D.; Gao, Y.; Qi, Y.; Chen, L.; Ma, Y.; Li, Y. Peptide-Based Cancer Therapy: Opportunity and Challenge. Cancer Lett. 2014, 351, 13–22. [Google Scholar] [CrossRef]
- Merchant, T.E.; de Graaf, P.W.; Minsky, B.D.; Obertop, H.; Glonek, T. Esophageal Cancer Phospholipid Characterization by 31P NMR. NMR Biomed. 1993, 6, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Abbassi-Ghadi, N.; Antonowicz, S.S.; McKenzie, J.S.; Kumar, S.; Huang, J.; Jones, E.A.; Strittmatter, N.; Petts, G.; Kudo, H.; Court, S.; et al. De Novo Lipogenesis Alters the Phospholipidome of Esophageal Adenocarcinoma. Cancer Res. 2020, 80, 2764–2774. [Google Scholar] [CrossRef]
- Liu, R.; Peng, Y.; Li, X.; Wang, Y.; Pan, E.; Guo, W.; Pu, Y.; Yin, L. Identification of Plasma Metabolomic Profiling for Diagnosis of Esophageal Squamous-Cell Carcinoma Using an UPLC/TOF/MS Platform. Int. J. Mol. Sci. 2013, 14, 8899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos-Martín, F.; Herrera-León, C.; D’Amelio, N. Bombyx Mori Cecropin D Could Trigger Cancer Cell Apoptosis by Interacting with Mitochondrial Cardiolipin. Biochim. Biophys. Acta Biomembr. 2022, 1864, 184003. [Google Scholar] [CrossRef]
- Ramos-Martín, F.; Herrera-León, C.; D’Amelio, N. Molecular Basis of the Anticancer, Apoptotic and Antibacterial Activities of Bombyx Mori Cecropin A. Arch. Biochem. Biophys. 2022, 715, 109095. [Google Scholar] [CrossRef]
- Ramos-Martín, F.; D’Amelio, N. Molecular Basis of the Anticancer and Antibacterial Properties of CecropinXJ Peptide: An In Silico Study. Int. J. Mol. Sci. 2021, 22, 691. [Google Scholar] [CrossRef]
- Herrera-León, C.; Ramos-Martín, F.; Antonietti, V.; Sonnet, P.; D’Amelio, N. The Impact of Phosphatidylserine Exposure on Cancer Cell Membranes on the Activity of the Anticancer Peptide HB43. FEBS J. 2022, 289, 1984–2003. [Google Scholar] [CrossRef]
- Findlay, E.G.; Currie, A.J.; Zhang, A.; Ovciarikova, J.; Young, L.; Stevens, H.; McHugh, B.J.; Canel, M.; Gray, M.; Milling, S.W.F.; et al. Exposure to the Antimicrobial Peptide LL-37 Produces Dendritic Cells Optimized for Immunotherapy. Oncoimmunology 2019, 8, 1608106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Tang, L.; Tian, Y.; Ji, X.; Hu, Q.; Zhou, B.; Zhenyu, D.; Heng, X.; Yang, L. Cholesterol-Modified DP7 Enhances the Effect of Individualized Cancer Immunotherapy Based on Neoantigens. Biomaterials 2020, 241, 119852. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.-W.; Garris, C.; Pfirschke, C.; Rickelt, S.; Arlauckas, S.; Siwicki, M.; Kohler, R.H.; Weissleder, R.; Sundvold-Gjerstad, V.; Sveinbjørnsson, B.; et al. LTX-315 Sequentially Promotes Lymphocyte-Independent and Lymphocyte-Dependent Antitumor Effects. Cell Stress Chaperones 2019, 3, 348–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer Immunoediting and Resistance to T Cell-Based Immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef]
- Islam, F.; Gopalan, V.; Lam, A.K. RNA Interference-Mediated Gene Silencing in Esophageal Adenocarcinoma. Methods Mol. Biol. 2018, 1756, 269–279. [Google Scholar]
- Kara, G.; Calin, G.A.; Ozpolat, B. RNAi-Based Therapeutics and Tumor Targeted Delivery in Cancer. Adv. Drug Deliv. Rev. 2022, 182, 114113. [Google Scholar] [CrossRef]
- Amin, M.; Islam, F.; Gopalan, V.; Lam, A.K. Detection and Quantification of MicroRNAs in Esophageal Adenocarcinoma. Methods Mol. Biol. 2018, 1756, 257–268. [Google Scholar]
- Tian, W.; Li, B.; Zhang, X.; Dang, W.; Wang, X.; Tang, H.; Wang, L.; Cao, H.; Chen, T. Suppression of Tumor Invasion and Migration in Breast Cancer Cells Following Delivery of siRNA against Stat3 with the Antimicrobial Peptide PR39. Oncol. Rep. 2012, 28, 1362–1368. [Google Scholar] [CrossRef] [Green Version]
- Herrera-León, C.; Ramos-Martín, F.; El Btaouri, H.; Antonietti, V.; Sonnet, P.; Martiny, L.; Zevolini, F.; Falciani, C.; Sarazin, C.; D’Amelio, N. The Influence of Short Motifs on the Anticancer Activity of HB43 Peptide. Pharmaceutics 2022, 14, 1089. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, D.; Mussnich, P.; Arra, C.; Battista, S.; Fusco, A. Critical Role of HMGA Proteins in Cancer Cell Chemoresistance. J. Mol. Med. 2017, 95, 353–360. [Google Scholar] [CrossRef]
- Pegoraro, S.; Ros, G.; Sgubin, M.; Petrosino, S.; Zambelli, A.; Sgarra, R.; Manfioletti, G. Targeting the Intrinsically Disordered Architectural High Mobility Group A (HMGA) Oncoproteins in Breast Cancer: Learning from the Past to Design Future Strategies. Expert Opin. Ther. Targets 2020, 24, 953–969. [Google Scholar] [CrossRef] [PubMed]
- Jonathan, E.; Constance, C.S.L. Targeting Malignant Mitochondria with Therapeutic Peptides. Ther. Deliv. 2012, 3, 961. [Google Scholar]
- Badrinath, N.; Yoo, S.Y. Mitochondria in Cancer: In the Aspects of Tumorigenesis and Targeted Therapy. Carcinogenesis 2018, 39, 1419–1430. [Google Scholar] [CrossRef]
- Krishnan, K.J.; Greaves, L.C.; Reeve, A.K.; Turnbull, D. The Ageing Mitochondrial Genome. Nucleic Acids Res. 2007, 35, 7399. [Google Scholar] [CrossRef] [Green Version]
- Lamb, R.; Harrison, H.; Hulit, J.; Smith, D.L.; Lisanti, M.P.; Sotgia, F. Mitochondria as New Therapeutic Targets for Eradicating Cancer Stem Cells: Quantitative Proteomics and Functional Validation via MCT1/2 Inhibition. Oncotarget 2014, 5, 11029–11037. [Google Scholar] [CrossRef] [Green Version]
- Hickey, J.L.; Ruhayel, R.A.; Barnard, P.J.; Baker, M.V.; Berners-Price, S.J.; Filipovska, A. Mitochondria-Targeted Chemotherapeutics: The Rational Design of gold(I) N-Heterocyclic Carbene Complexes That Are Selectively Toxic to Cancer Cells and Target Protein Selenols in Preference to Thiols. J. Am. Chem. Soc. 2008, 130, 12570–12571. [Google Scholar] [CrossRef] [PubMed]
- Xiang, S.; Zhang, K.; Yang, G.; Gao, D.; Zeng, C.; He, M. Mitochondria-Targeted and Resveratrol-Loaded Dual-Function Titanium Disulfide Nanosheets for Photothermal-Triggered Tumor Chemotherapy. Nanoscale Res. Lett. 2019, 14, 1–10. [Google Scholar] [CrossRef]
- Zhang, C.; Guan, R.; Liao, X.; Ouyang, C.; Rees, T.W.; Liu, J.; Chen, Y.; Ji, L.; Chao, H. A Mitochondria-Targeting Dinuclear Ir-Ru Complex as a Synergistic Photoactivated Chemotherapy and Photodynamic Therapy Agent against Cisplatin-Resistant Tumour Cells. Chem. Commun. 2019, 55, 12547–12550. [Google Scholar] [CrossRef]
- Yu, C.Y.Y.; Xu, H.; Ji, S.; Kwok, R.T.K.; Lam, J.W.Y.; Li, X.; Krishnan, S.; Ding, D.; Tang, B.Z. Mitochondrion-Anchoring Photosensitizer with Aggregation-Induced Emission Characteristics Synergistically Boosts the Radiosensitivity of Cancer Cells to Ionizing Radiation. Adv. Mater. 2017, 29, 1606167. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.P.; Kelley, S.O. Maximizing the Therapeutic Window of an Antimicrobial Drug by Imparting Mitochondrial Sequestration in Human Cells. J. Am. Chem. Soc. 2011, 133, 3260–3263. [Google Scholar] [CrossRef] [PubMed]
- Mehta, M.; Deeksha; Sharma, N.; Vyas, M.; Khurana, N.; Maurya, P.K.; Singh, H.; Andreoli de Jesus, T.P.; Dureja, H.; Chellappan, D.K.; et al. Interactions with the Macrophages: An Emerging Targeted Approach Using Novel Drug Delivery Systems in Respiratory Diseases. Chem. Biol. Interact. 2019, 304, 10–19. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, G.; Tiwari, R.; Sriwastawa, B.; Bhati, L.; Pandey, S.; Pandey, P.; Bannerjee, S.K. Drug Delivery Systems: An Updated Review. Int. J. Pharm. Investig. 2012, 2, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calixto, G.M.F.; de Annunzio, S.R.; Victorelli, F.D.; Frade, M.L.; Ferreira, P.S.; Chorilli, M.; Fontana, C.R. Chitosan-Based Drug Delivery Systems for Optimization of Photodynamic Therapy: A Review. AAPS PharmSciTech 2019, 20, 253. [Google Scholar] [CrossRef]
- Dubashynskaya, N.V.; Bokatyi, A.N.; Dobrodumov, A.V.; Kudryavtsev, I.V.; Trulioff, A.S.; Rubinstein, A.A.; Aquino, A.D.; Dubrovskii, Y.A.; Knyazeva, E.S.; Demyanova, E.V.; et al. Succinyl Chitosan-Colistin Conjugates as Promising Drug Delivery Systems. Int. J. Mol. Sci. 2022, 24, 166. [Google Scholar] [CrossRef]
- Canaparo, R.; Foglietta, F.; Giuntini, F.; Della Pepa, C.; Dosio, F.; Serpe, L. Recent Developments in Antibacterial Therapy: Focus on Stimuli-Responsive Drug-Delivery Systems and Therapeutic Nanoparticles. Molecules 2019, 24, 1991. [Google Scholar] [CrossRef] [Green Version]
- Tenchov, R.; Bird, R.; Curtze, A.E.; Zhou, Q. Lipid Nanoparticles—From Liposomes to mRNA Vaccine Delivery, a Landscape of Research Diversity and Advancement. ACS Nano 2021, 15, 16982–17015. [Google Scholar] [CrossRef]
- Gregoriadis, G. Liposomes and mRNA: Two Technologies Together Create a COVID-19 Vaccine. Med. Drug Discov. 2021, 12, 100104. [Google Scholar] [CrossRef]
- Gbian, D.L.; Omri, A. Lipid-Based Drug Delivery Systems for Diseases Managements. Biomedicines 2022, 10, 2137. [Google Scholar] [CrossRef]
- Ferreira, M.; Ogren, M.; Dias, J.N.R.; Silva, M.; Gil, S.; Tavares, L.; Aires-da-Silva, F.; Gaspar, M.M.; Aguiar, S.I. Liposomes as Antibiotic Delivery Systems: A Promising Nanotechnological Strategy against Antimicrobial Resistance. Molecules 2021, 26, 2047. [Google Scholar] [CrossRef]
- Nwabuife, J.C.; Pant, A.M.; Govender, T. Liposomal Delivery Systems and Their Applications against Staphylococcus aureus and Methicillin-Resistant Staphylococcus aureus. Adv. Drug Deliv. Rev. 2021, 178, 113861. [Google Scholar] [CrossRef] [PubMed]
- Smola, M.; Vandamme, T.; Sokolowski, A. Nanocarriers as Pulmonary Drug Delivery Systems to Treat and to Diagnose Respiratory and Non Respiratory Diseases. Int. J. Nanomed. 2008, 3, 1–19. [Google Scholar]
- Li, Y.-J.; Wu, J.-Y.; Liu, J.; Xu, W.; Qiu, X.; Huang, S.; Hu, X.-B.; Xiang, D.-X. Artificial Exosomes for Translational Nanomedicine. J. Nanobiotechnology 2021, 19, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Millan, C.; Calvo Díaz, C.; Lanao, J.M.; Colino, C.I. Advances in Exosomes-Based Drug Delivery Systems. Macromol. Biosci. 2021, 21, e2000269. [Google Scholar] [CrossRef]
- Jacob, S.; Nair, A.B.; Shah, J. Emerging Role of Nanosuspensions in Drug Delivery Systems. Biomater Res 2020, 24, 3. [Google Scholar] [CrossRef] [Green Version]
- Xin, J.; Qin, M.; Ye, G.; Gong, H.; Li, M.; Sui, X.; Liu, B.; Fu, Q.; He, Z. Hydrophobic Ion Pairing-Based Self-Emulsifying Drug Delivery Systems: A New Strategy for Improving the Therapeutic Efficacy of Water-Soluble Drugs. Expert Opin. Drug Deliv. 2023, 20, 1–11. [Google Scholar] [CrossRef]
- Herman, L.; De Smedt, S.C.; Raemdonck, K. Pulmonary Surfactant as a Versatile Biomaterial to Fight COVID-19. J. Control. Release 2022, 342, 170–188. [Google Scholar] [CrossRef]
- García-Mouton, C.; Parra-Ortiz, E.; Malmsten, M.; Cruz, A.; Pérez-Gil, J. Pulmonary Surfactant and Drug Delivery: Vehiculization of a Tryptophan-Tagged Antimicrobial Peptide over the Air-Liquid Interfacial Highway. Eur. J. Pharm. Biopharm. 2022, 180, 33–47. [Google Scholar] [CrossRef]
- Hidalgo, A.; Garcia-Mouton, C.; Autilio, C.; Carravilla, P.; Orellana, G.; Islam, M.N.; Bhattacharya, J.; Bhattacharya, S.; Cruz, A.; Pérez-Gil, J. Pulmonary Surfactant and Drug Delivery: Vehiculization, Release and Targeting of Surfactant/tacrolimus Formulations. J. Control. Release 2021, 329, 205–222. [Google Scholar] [CrossRef]
- Oh, C.; Jeon, O.H.; Han, J.; Kim, K.; Kim, H.K.; Park, J.-H. Pulmonary Surfactant-Based Paclitaxel-Loaded Nanovesicles for Inhalation Therapy of Lung Adenocarcinoma. ACS Appl. Nano Mater. 2023, 6, 3693–3703. [Google Scholar] [CrossRef]
- Sousa, M.G.C.; Rezende, T.M.B.; Franco, O.L. Nanofibers as Drug-Delivery Systems for Antimicrobial Peptides. Drug Discov. Today 2021, 26, 2064–2074. [Google Scholar] [CrossRef] [PubMed]
- Stocco, T.D.; Bassous, N.J.; Zhao, S.; Granato, A.E.C.; Webster, T.J.; Lobo, A.O. Nanofibrous Scaffolds for Biomedical Applications. Nanoscale 2018, 10, 12228–12255. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Ji, H.; Kong, X.; Lei, P.; Yang, Q.; Wu, W.; Jin, L.; Sun, D. Bacterial Ghosts-Based Vaccine and Drug Delivery Systems. Pharmaceutics 2021, 13, 1892. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Xuan, M.; Zhao, P.; Loznik, M.; Chen, J.; Kiessling, F.; Zheng, L.; Herrmann, A. Ultrasound Responsive Microcapsules for Antibacterial Nanodrug Delivery. Nano Res. 2022, 16, 2738–2748. [Google Scholar] [CrossRef]
- Field, R.D.; Jakus, M.A.; Chen, X.; Human, K.; Zhao, X.; Chitnis, P.V.; Sia, S.K. Ultrasound-Responsive Aqueous Two-Phase Microcapsules for On-Demand Drug Release. Angew. Chem. Int. Ed. Engl. 2022, 61, e202116515. [Google Scholar] [CrossRef]
- Haupt, K.; Medina Rangel, P.X.; Bui, B.T.S. Molecularly Imprinted Polymers: Antibody Mimics for Bioimaging and Therapy. Chem. Rev. 2020, 120, 9554–9582. [Google Scholar] [CrossRef]
- Mier, A.; Maffucci, I.; Merlier, F.; Prost, E.; Montagna, V.; Ruiz-Esparza, G.U.; Bonventre, J.V.; Dhal, P.K.; Tse Sum Bui, B.; Sakhaii, P.; et al. Molecularly Imprinted Polymer Nanogels for Protein Recognition: Direct Proof of Specific Binding Sites by Solution STD and WaterLOGSY NMR Spectroscopies. Angew. Chem. Int. Ed. Engl. 2021, 60, 20849–20857. [Google Scholar] [CrossRef]
- Medina Rangel, P.X.; Mier, A.; Moroni, E.; Merlier, F.; Gheber, L.A.; Vago, R.; Maffucci, I.; Tse Sum Bui, B.; Haupt, K. Molecularly Imprinted Polymer Nanogels Targeting the HAV Motif in Cadherins Inhibit Cell-Cell Adhesion and Migration. J. Mater. Chem. B Mater. Biol. Med. 2022, 10, 6688–6697. [Google Scholar] [CrossRef]
- Tse Sum Bui, B.; Haupt, K. Molecularly Imprinted Polymer Hydrogel Nanoparticles: Synthetic Antibodies for Cancer Diagnosis and Therapy. Chembiochem 2022, 23, e202100598. [Google Scholar] [CrossRef]
- Tse Sum Bui, B.; Auroy, T.; Haupt, K. Fighting Antibiotic-resistant Bacteria: Promising Strategies Orchestrated by Molecularly Imprinted Polymers. Angew. Chem. Weinh. Bergstr. Ger. 2022, 134, e202106493. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramos-Martín, F.; D’Amelio, N. Drug Resistance: An Incessant Fight against Evolutionary Strategies of Survival. Microbiol. Res. 2023, 14, 507-542. https://doi.org/10.3390/microbiolres14020037
Ramos-Martín F, D’Amelio N. Drug Resistance: An Incessant Fight against Evolutionary Strategies of Survival. Microbiology Research. 2023; 14(2):507-542. https://doi.org/10.3390/microbiolres14020037
Chicago/Turabian StyleRamos-Martín, Francisco, and Nicola D’Amelio. 2023. "Drug Resistance: An Incessant Fight against Evolutionary Strategies of Survival" Microbiology Research 14, no. 2: 507-542. https://doi.org/10.3390/microbiolres14020037