
Core Genome Sequencing Analysis of E. coli O157:H7 Unravelling Genetic Relatedness among Strains from Cattle, Beef, and Humans in Bishoftu, Ethiopia
 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sources and Detection of E. coli O157
2.2. Pulsed-Field Gel Electrophoresis
2.3. Whole-Genome Sequencing
2.4. Core Genome MLST Analysis
2.5. Comparison of E. coli O157:H7 in EnteroBase
2.6. In Silico Identification of Genes Linked to Serotype, Virulence, Antibiotic Resistance, and Plasmids
3. Results
3.1. Pulsed-Field Gel Electrophoresis
3.2. Core Genome MLST Analysis
3.3. Comparison of E. coli O157:H7 Genomes in EnteroBase
3.4. In Silico Identification of Genes Linked to Serotype, Virulence, Antibiotic Resistance, and Plasmids
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Croxen, M.A.; Law, R.J.; Scholz, R.; Keeney, K.M.; Wlodarska, M.; Finlay, B.B. Recent advances in understanding enteric pathogenic Escherichia coli. Clin. Microbiol. Rev. 2013, 26, 822–880. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Payne, M.; Kaur, S.; Lan, R. Improved Genomic Identification, Clustering, and Serotyping of Shiga Toxin-Producing Escherichia coli Using Cluster/Serotype-Specific Gene Markers. Front. Cell Infect. Microbiol. 2021, 11, 772574. [Google Scholar] [CrossRef] [PubMed]
- Gambushe, S.M.; Zishiri, O.T.; El Zowalaty, M.E. Review of Escherichia coli O157:H7 Prevalence, Pathogenicity, Heavy Metal and Antimicrobial Resistance, African Perspective. Infect. Drug Resist. 2022, 15, 4645–4673. [Google Scholar] [CrossRef] [PubMed]
- Valilis, E.; Ramsey, A.; Sidiq, S.; DuPont, H.L. Non-O157 Shiga toxin-producing Escherichia coli-A poorly appreciated enteric pathogen: Systematic review. Int. J. Infect. Dis. 2018, 76, 82–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umpierrez, A.; Bado, I.; Oliver, M.; Acquistapace, S.; Etcheverria, A.; Padola, N.L.; Vignoli, R.; Zunino, P. Zoonotic Potential and Antibiotic Resistance of Escherichia coli in Neonatal Calves in Uruguay. Microbes. Environ. 2017, 32, 275–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, K.E.; Thorpe, C.M.; Sears, C.L. The emerging clinical importance of non-O157 Shiga toxin-producing Escherichia coli. Clin. Infect. Dis. 2006, 43, 1587–1595. [Google Scholar] [CrossRef]
- Havelaar, A.H.; Sapp, A.C.; Amaya, M.P.; Nane, G.F.; Morgan, K.M.; Devleesschauwer, B.; Grace, D.; Knight-Jones, T.; Kowalcyk, B.B. Burden of foodborne disease due to bacterial hazards associated with beef, dairy, poultry meat, and vegetables in Ethiopia and Burkina Faso, 2017. Front. Sustain. Food Syst. 2022, 6, 521. [Google Scholar] [CrossRef]
- Gyles, C.L. Shiga toxin-producing Escherichia coli: An overview. J. Anim. Sci. 2007, 85, E45–E62. [Google Scholar] [CrossRef]
- Tourret, J.; Willing, B.P.; Croxen, M.A.; Dufour, N.; Dion, S.; Wachtel, S.; Denamur, E.; Finlay, B.B. Small Intestine Early Innate Immunity Response during Intestinal Colonization by Escherichia coli Depends on Its Extra-Intestinal Virulence Status. PLoS ONE 2016, 11, e0153034. [Google Scholar] [CrossRef] [Green Version]
- Nichols, M.C.; Gacek, P.; Phan, Q.; Gambino-Shirley, K.J.; Gollarza, L.M.; Schroeder, M.N.; Mercante, A.; Mullins, J.; Blackstock, A.; Laughlin, M.E.; et al. Agritourism and Kidding Season: A Large Outbreak of Human Shiga Toxin-Producing Escherichia coli O157 (STEC O157) Infections Linked to a Goat Dairy Farm-Connecticut, 2016. Front. Vet. Sci. 2021, 8, 744055. [Google Scholar] [CrossRef]
- Heredia, N.; Garcia, S. Animals as sources of food-borne pathogens: A review. Anim. Nutr. 2018, 4, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.F.; Tauxe, R.V.; Hedberg, C.W. The growing burden of foodborne outbreaks due to contaminated fresh produce: Risks and opportunities. Epidemiol. Infect. 2009, 137, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Rani, A.; Ravindran, V.B.; Surapaneni, A.; Mantri, N.; Ball, A.S. Review: Trends in point-of-care diagnosis for Escherichia coli O157:H7 in food and water. Int. J. Food Microbiol. 2021, 349, 109233. [Google Scholar] [CrossRef] [PubMed]
- Tack, D.M.; Kisselburgh, H.M.; Richardson, L.C.; Geissler, A.; Griffin, P.M.; Payne, D.C.; Gleason, B.L. Shiga Toxin-Producing Escherichia coli Outbreaks in the United States, 2010–2017. Microorganisms 2021, 9, 1529. [Google Scholar] [CrossRef] [PubMed]
- Pires, S.M.; Majowicz, S.; Gill, A.; Devleesschauwer, B. Global and regional source attribution of Shiga toxin-producing Escherichia coli infections using analysis of outbreak surveillance data. Epidemiol. Infect. 2019, 147, e236. [Google Scholar] [CrossRef] [Green Version]
- Panel, E.B.K.; Koutsoumanis, A.; Allende, A.; Alvarez-Ordóñez, S.; Bover-Cid, M.; Chemaly, R.; Davies, A.; De Cesare, L.; Herman, F.; Hilbert, R.; et al. Pathogenicity assessment of Shiga toxin-producing Escherichia coli (STEC) and the public health risk posed by contamination of food with STEC. EFSA J. 2020, 18, 5967. [Google Scholar] [CrossRef]
- Sapp, A.C.; Amaya, M.P.; Havelaar, A.H.; Nane, G.F. Attribution of country level foodborne disease to food group and food types in three African countries: Conclusions from a structured expert judgment study. PLoS Negl. Trop. Dis. 2022, 16, e0010663. [Google Scholar] [CrossRef] [PubMed]
- Gutema, F.D.; Rasschaert, G.; Agga, G.E.; Jufare, A.; Duguma, A.B.; Abdi, R.D.; Duchateau, L.; Crombe, F.; Gabriel, S.; De Zutter, L. Occurrence, Molecular Characteristics, and Antimicrobial Resistance of Escherichia coli O157 in Cattle, Beef, and Humans in Bishoftu Town, Central Ethiopia. Foodborne Pathog. Dis. 2021, 18, 1–7. [Google Scholar] [CrossRef]
- utema, F.D.; Abdi, R.D.; Agga, G.E.; Firew, S.; Rasschaert, G.; Mattheus, W.; Crombe, F.; Duchateau, L.; Gabriël, S.; De Zutter, L. Assessment of beef carcass contamination with Salmonella and E. coli O157 in slaughterhouses in Bishoftu, Ethiopia. Int. J. Food Contam. 2021, 8, 1–9. [Google Scholar]
- Standard operating procedure for PulseNet PFGE of Escherichia coli O157:H7, Escherichia coli non-O157 (STEC), Salmonella serotypes, Shigella sonnei and Shigella flexneri; CDC PulseNet: Atlanta, GA, USA, 2017; pp. 1–16.
- Ferrari, R.G.; Panzenhagen, P.H.N.; Conte, C.A. Phenotypic and Genotypic Eligible Methods for Salmonella Typhimurium Source Tracking. Front. Microbiol. 2017, 8, 1591. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.L.; Orsi, R.H.; Luo, H.; Ge, C.T.; Zhang, G.T.; Baker, R.C.; Stevenson, A.; Wiedmann, M. Assessment and Comparison of Molecular Subtyping and Characterization Methods for Salmonella. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Nadon, C.; Van Walle, I.; Gerner-Smidt, P.; Campos, J.; Chinen, I.; Concepcion-Acevedo, J.; Gilpin, B.; Smith, A.M.; Kam, K.M.; Perez, E.; et al. PulseNet International: Vision for the implementation of whole genome sequencing (WGS) for global food-borne disease surveillance. Eurosurveillance 2017, 22, 13–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Alikhan, N.F.; Mohamed, K.; Fan, Y.; Agama Study, G.; Achtman, M. The EnteroBase user’s guide, with case studies on Salmonella transmissions, Yersinia pestis phylogeny, and Escherichia core genomic diversity. Genome Res. 2020, 30, 138–152. [Google Scholar] [CrossRef] [Green Version]
- De Rauw, K.; Buyl, R.; Jacquinet, S.; Pierard, D. Risk determinants for the development of typical haemolytic uremic syndrome in Belgium and proposition of a new virulence typing algorithm for Shiga toxin-producing Escherichia coli. Epidemiol. Infect. 2018, 147, e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, X.N.; Zhang, J.; Hua, Y.; Jernberg, C.; Xiong, Y.W.; French, N.; Lofgren, S.; Hedenstrom, I.; Ambikan, A.; Mernelius, S.; et al. Genomic Insights Into Clinical Shiga Toxin-Producing Escherichia coli Strains: A 15-Year Period Survey in Jonkoping, Sweden. Front. Microbiol. 2021, 12, 627861. [Google Scholar] [CrossRef] [PubMed]
- Jagadeesan, B.; Gerner-Smidt, P.; Allard, M.W.; Leuillet, S.; Winkler, A.; Xiao, Y.; Chaffron, S.; Van Der Vossen, J.; Tang, S.; Katase, M.; et al. The use of next generation sequencing for improving food safety: Translation into practice. Food Microbiol. 2019, 79, 96–115. [Google Scholar] [CrossRef] [PubMed]
- Chekabab, S.M.; Paquin-Veillette, J.; Dozois, C.M.; Harel, J. The ecological habitat and transmission of Escherichia coli O157:H7. FEMS Microbiol. Lett. 2013, 341, 1–12. [Google Scholar] [CrossRef]
- Ibrahim, E.M.E.; El-Liethy, M.A.; Abia, A.L.K.; Hemdan, B.A.; Shaheen, M.N. Survival of E. coli O157:H7, Salmonella Typhimurium, HAdV2 and MNV-1 in river water under dark conditions and varying storage temperatures. Sci. Total Environ. 2019, 648, 1297–1304. [Google Scholar] [CrossRef]
- Zelalem, A.; Abegaz, K.; Kebede, A.; Terefe, Y.; Vipham, J.L. Investigation on Salmonella enterica, Escherichia coli, and coliforms in beef from Ethiopian abattoirs: A potential risk of meat safety. Food Sci. Nutr. 2022, 10, 1714–1724. [Google Scholar] [CrossRef]
- Besser, J.M.; Carleton, H.A.; Trees, E.; Stroika, S.G.; Hise, K.; Wise, M.; Gerner-Smidt, P. Interpretation of Whole-Genome Sequencing for Enteric Disease Surveillance and Outbreak Investigation. Foodborne Pathog. Dis. 2019, 16, 504–512. [Google Scholar] [CrossRef]
- Scheutz, F. Taxonomy Meets Public Health: The Case of Shiga Toxin-Producing Escherichia coli. Microbiol. Spectr. 2014, 2, 15–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pakbin, B.; Bruck, W.M.; Rossen, J.W.A. Virulence Factors of Enteric Pathogenic Escherichia coli: A Review. Int. J. Mol. Sci. 2021, 22, 9922. [Google Scholar] [CrossRef]
- Bielaszewska, M.; Kock, R.; Friedrich, A.W.; von Eiff, C.; Zimmerhackl, L.B.; Karch, H.; Mellmann, A. Shiga Toxin-Mediated Hemolytic Uremic Syndrome: Time to Change the Diagnostic Paradigm? Plos ONE 2007, 2, e1024. [Google Scholar] [CrossRef]
- Ferdous, M.; Zhou, K.; Mellmann, A.; Morabito, S.; Croughs, P.D.; de Boer, R.F.; Kooistra-Smid, A.M.D.; Rossen, J.W.A.; Friedrich, A.W. Is Shiga Toxin-Negative Escherichia coli O157:H7 Enteropathogenic or Enterohemorrhagic Escherichia coli? Comprehensive Molecular Analysis Using Whole-Genome Sequencing. J. Clin. Microbiol. 2015, 53, 3530–3538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, P.; Dey, M.; Abe, A.; Takeda, T. Isogenic strain of Escherichia coli O157: H7 that has lost both Shiga toxin 1 and 2 genes. Clin. Diagn. Lab. Immunol. 2001, 8, 711–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellmann, A.; Lu, S.; Karch, H.; Xu, J.G.; Harmsen, D.; Schmidt, M.A.; Bielaszewska, M. Recycling of Shiga toxin 2 genes in sorbitol-fermenting enterohemorrhagic Escherichia coli O157: NM. Appl. Environ. Microbiol. 2008, 74, 67–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senthakumaran, T.; Brandal, L.T.; Lindstedt, B.A.; Jorgensen, S.B.; Charnock, C.; Tunsjo, H.S. Implications of stx loss for clinical diagnostics of Shiga toxin-producing Escherichia coli. Eur. J. Clin. Microbiol. Infect. Dis. 2018, 37, 2361–2370. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Torres, A.G. Enteropathogenic Escherichia coli: Foe or innocent bystander? Clin. Microbiol. Infect. 2015, 21, 729–734. [Google Scholar] [CrossRef] [Green Version]
- Hernandes, R.T.; Elias, W.P.; Vieira, M.A.; Gomes, T.A. An overview of atypical enteropathogenic Escherichia coli. FEMS Microbiol. Lett. 2009, 297, 137–149. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.; Bibi, E. MdfA, an Escherichia coli multidrug resistance protein with an extraordinarily broad spectrum of drug recognition. J. Bacteriol. 1997, 179, 2274–2280. [Google Scholar] [CrossRef] [Green Version]
- Hickman, R.A.; Agarwal, V.; Sjostrom, K.; Emanuelson, U.; Fall, N.; Sternberg-Lewerin, S.; Jarhult, J.D. Dissemination of Resistant Escherichia coli Among Wild Birds, Rodents, Flies, and Calves on Dairy Farms. Front. Microbiol. 2022, 13, 974. [Google Scholar] [CrossRef] [PubMed]
- Vanstokstraeten, R.; Crombe, F.; Pierard, D.; Castillo Moral, A.; Wybo, I.; De Geyter, D.; Janssen, T.; Caljon, B.; Demuyser, T. Molecular characterization of extraintestinal and diarrheagenic Escherichia coli blood isolates. Virulence 2022, 13, 2032–2041. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
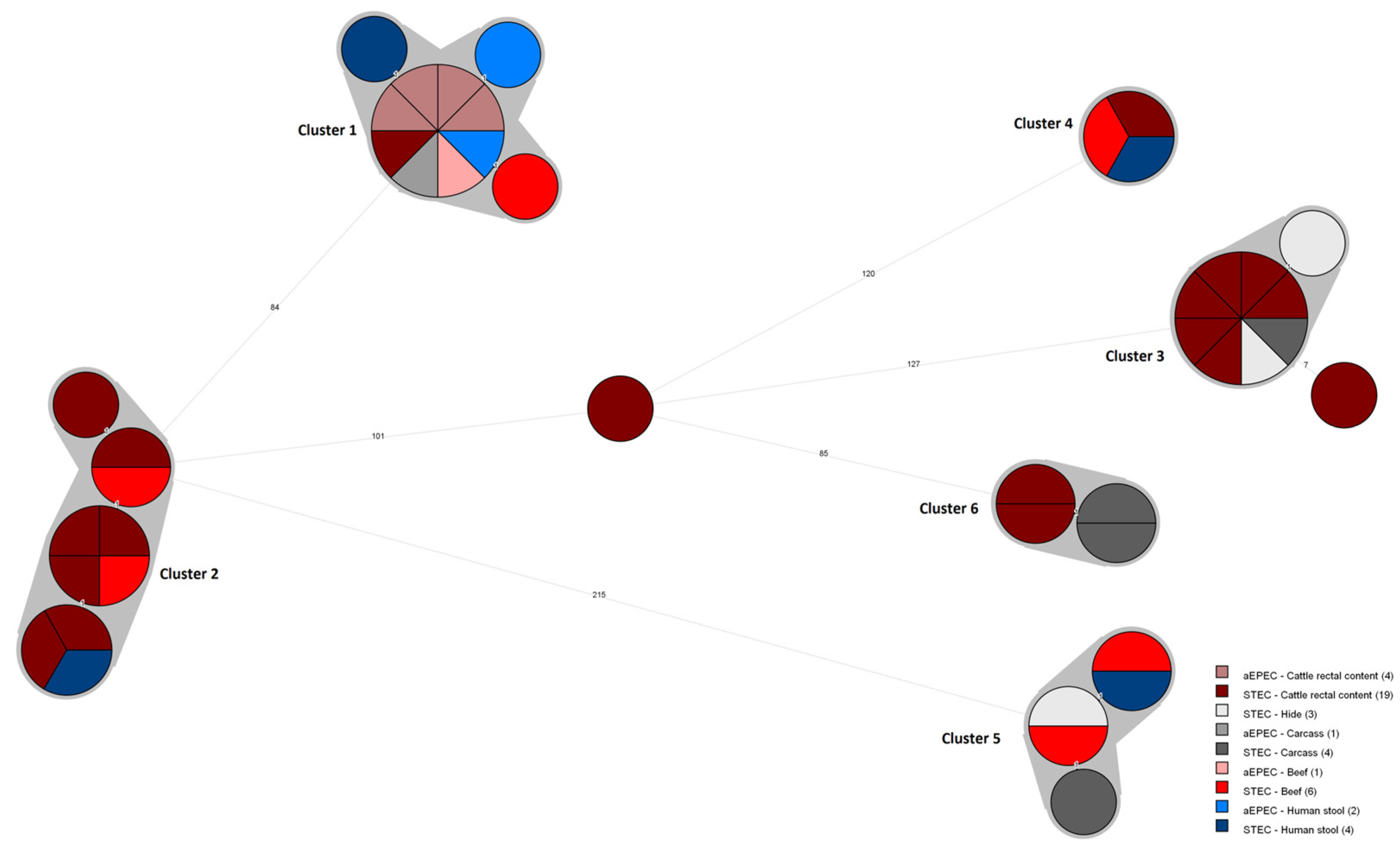{kind=link}
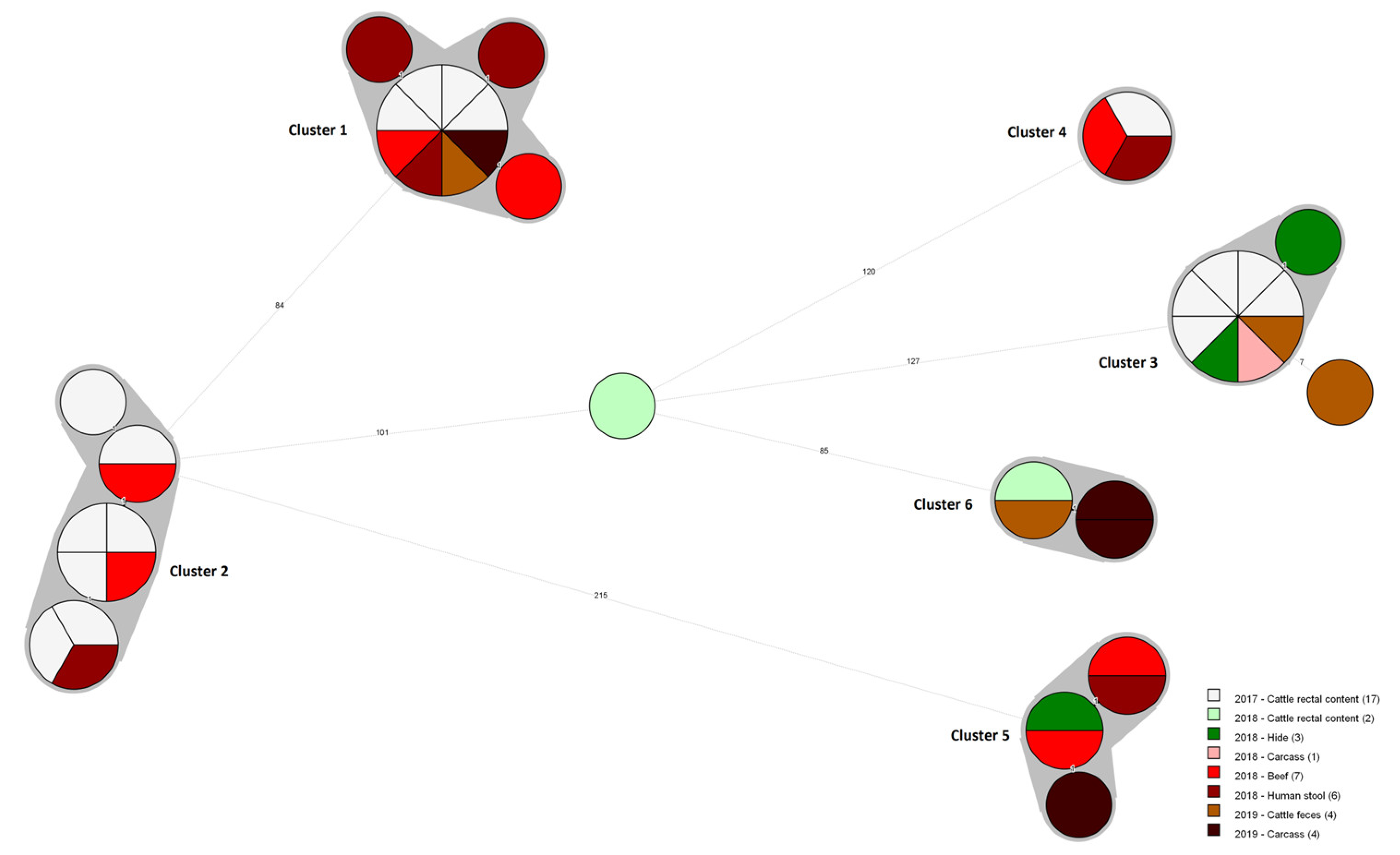{kind=link}
| HC100 1 | HC50 | HC20 | HC10 | HC5 | Origin | Sample Type | Country of Isolation | Collection Year |
|---|---|---|---|---|---|---|---|---|
| 1022 | 15671 | 147247 | 147247 | 147247 | Cattle | Beef tartare, carcass | Belgium | 2005 |
| 15671 | 147247 | 147247 | 147247 | Cattle, human | Hide, carcass, beef, stool | Ethiopia | 2018, 2019 | |
| 16335 | 205844 | 205844 | 205844 | 205844 | Cattle | Rectal content, hide, carcass | Ethiopia | 2017, 2018, 2019 |
| 205844 | 205844 | 205844 | 205846 | Cattle | Rectal content | Ethiopia | 2019 | |
| 205844 | 205844 | 205844 | 211075 | Human | NS | United Kingdom | 2022 | |
| 30437 | 30437 | 30437 | 30437 | 30437 | Human | NS | United Kingdom | 2016 |
| 205843 | 205843 | 205843 | 205843 | Cattle, human | Rectal content, beef, stool | Ethiopia | 2017, 2018 | |
| 205845 | 205845 | 205845 | 205845 | Cattle | Rectal content, carcass | Ethiopia | 2018, 2019 | |
| 205847 | 205847 | 205847 | 205847 | Cattle | Rectal content | Ethiopia | 2018 | |
| 205871 | 205871 | 205871 | 205871 | Cattle, human | Rectal content, beef, stool | Ethiopia | 2017, 2018 | |
| 37630 | 37630 | 37630 | 37630 | 37630 | Human | NS | United Kingdom | 2016 |
| 205870 | 205870 | 205870 | 205870 | Cattle, human | Rectal content, carcass, beef, stool | Ethiopia | 2017, 2018, 2019 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gutema, F.D.; De Zutter, L.; Piérard, D.; Hinckel, B.; Imamura, H.; Rasschaert, G.; Abdi, R.D.; Agga, G.E.; Crombé, F. Core Genome Sequencing Analysis of E. coli O157:H7 Unravelling Genetic Relatedness among Strains from Cattle, Beef, and Humans in Bishoftu, Ethiopia. Microbiol. Res. 2023, 14, 148-160. https://doi.org/10.3390/microbiolres14010013
Gutema FD, De Zutter L, Piérard D, Hinckel B, Imamura H, Rasschaert G, Abdi RD, Agga GE, Crombé F. Core Genome Sequencing Analysis of E. coli O157:H7 Unravelling Genetic Relatedness among Strains from Cattle, Beef, and Humans in Bishoftu, Ethiopia. Microbiology Research. 2023; 14(1):148-160. https://doi.org/10.3390/microbiolres14010013
Chicago/Turabian StyleGutema, Fanta D., Lieven De Zutter, Denis Piérard, Bruno Hinckel, Hideo Imamura, Geertrui Rasschaert, Reta D. Abdi, Getahun E. Agga, and Florence Crombé. 2023. "Core Genome Sequencing Analysis of E. coli O157:H7 Unravelling Genetic Relatedness among Strains from Cattle, Beef, and Humans in Bishoftu, Ethiopia" Microbiology Research 14, no. 1: 148-160. https://doi.org/10.3390/microbiolres14010013