
SARS-CoV CH.1.1 Variant: Genomic and Structural Insight

 ,
,  , , , , , , ,
, , , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
3. Modeling
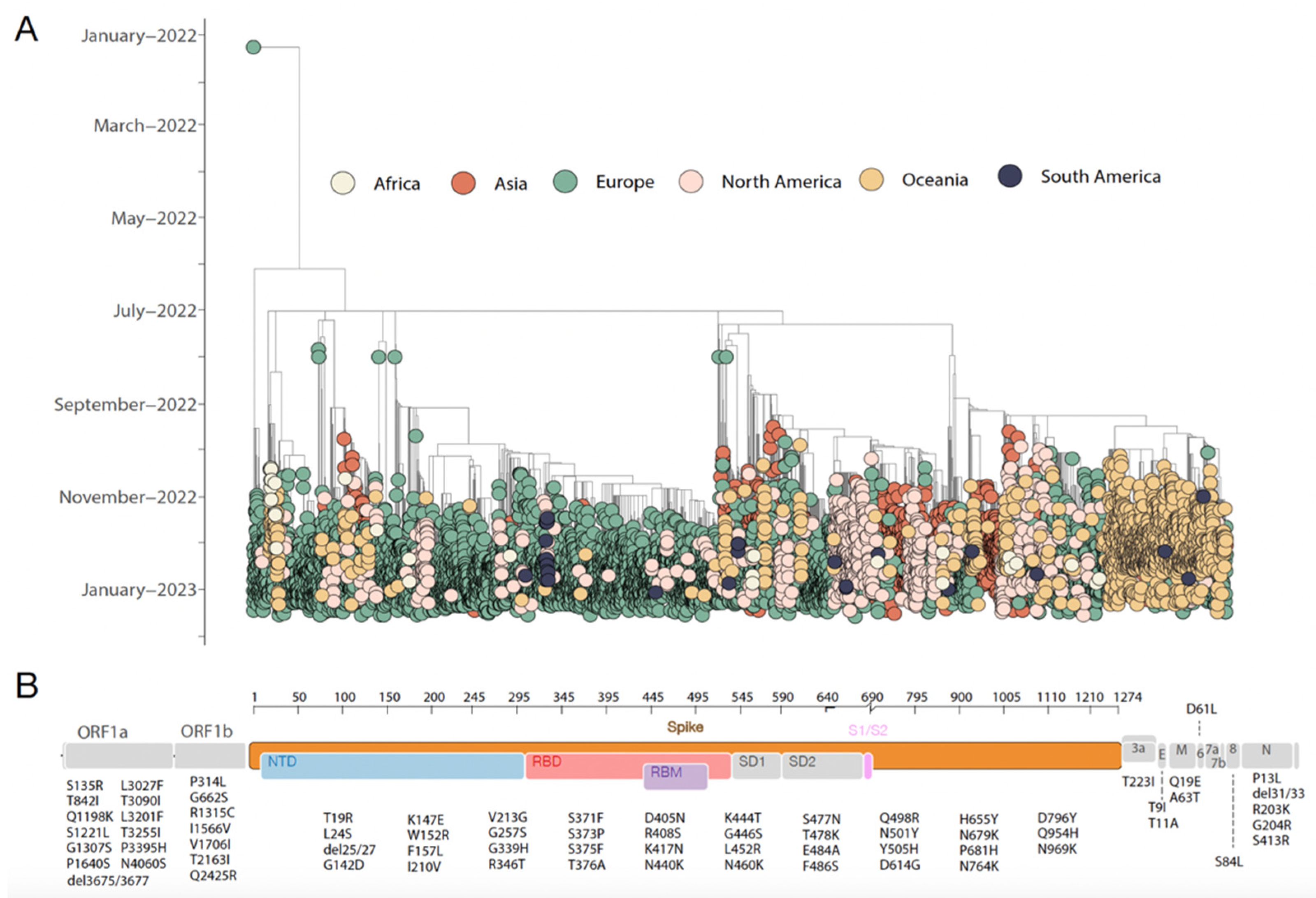
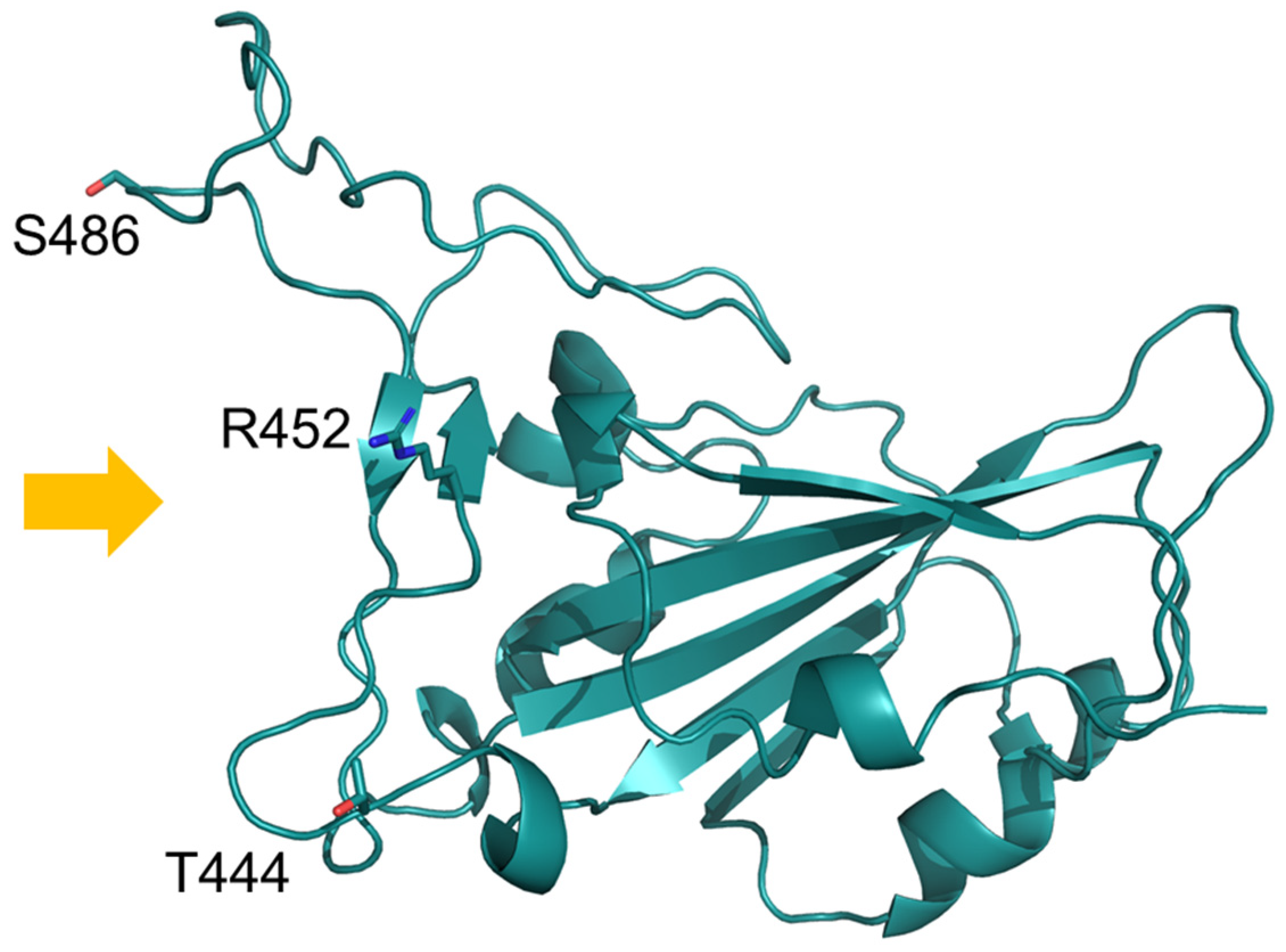
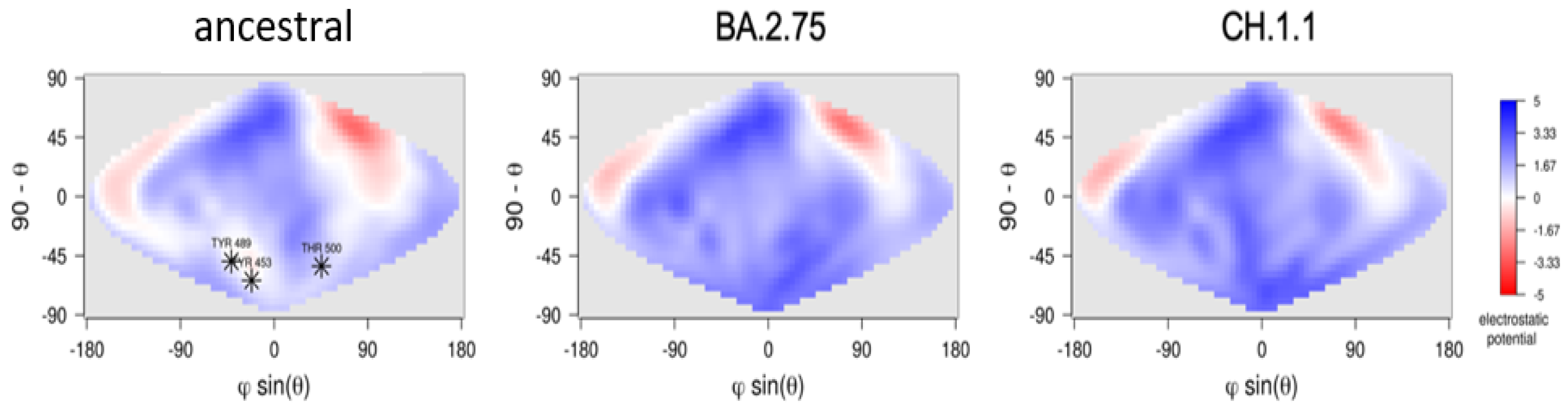
4. Results
5. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. 2023. Available online: https://covid19.who.int/ (accessed on 3 May 2023).
- Galloway, S.E.; Paul, P.; MacCannell, D.R.; Johansson, M.A.; Brooks, J.T.; MacNeil, A.; Slayton, R.B.; Tong, S.; Silk, B.J.; Armstrong, G.L.; et al. Emergence of SARS-CoV-2 B.1.1.7 Lineage—United States, December 29, 2020–January 12, 2021. Morb. Mortal. Wkly. Rep. 2021, 70, 95–99. [Google Scholar] [CrossRef]
- Giovanetti, M.; Slavov, S.N.; Fonseca, V.; Wilkinson, E.; Tegally, H.; Patané, J.S.L.; Viala, V.L.; San, E.J.; Rodrigues, E.S.; Santos, E.V.; et al. Genomic epidemiology of the SARS-CoV-2 epidemic in Brazil. Nat. Microbiol. 2022, 7, 1490–1500. [Google Scholar] [CrossRef] [PubMed]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Tegally, H.; Moir, M.; Everatt, J.; Giovanetti, M.; Scheepers, C.; Wilkinson, E.; Subramoney, K.; Makatini, Z.; Moyo, S.; Amoako, D.G.; et al. Emergence of SARS-CoV-2 Omicron lineages BA.4 and BA.5 in South Africa. Nat. Med. 2022, 28, 1785–1790. [Google Scholar] [CrossRef] [PubMed]
- Tosta, S.; Moreno, K.; Schuab, G.; Fonseca, V.; Segovia, F.M.C.; Kashima, S.; Elias, M.C.; Sampaio, S.C.; Ciccozzi, M.; Alcantara, L.C.J.; et al. Global SARS-CoV-2 genomic surveillance: What we have learned (so far). Infect. Genet. Evol. 2023, 18, 105405. [Google Scholar] [CrossRef] [PubMed]
- Qu, P.; Faraone, J.N.; Evans, J.P.; Zheng, Y.-M.; Carlin, C.; Anghelina, M.; Stevens, P.; Fernandez, S.; Jones, D.; Panchal, A.; et al. Extraordinary Evasion of Neutralizing Antibody Response by Omicron XBB.1.5, CH.1.1 and CA.3.1 Variants. bioRxiv 2023. [Google Scholar] [CrossRef]
- Khare, S.; Gurry, C.; Freitas, L.; Schultz, M.B.; Bach, G.; Diallo, A.; Akite, N.; Ho, J.; Lee, R.T.; Yeo, W.; et al. GISAID’s Role in Pandemic Response. China CDC Wkly. 2021, 3, 1049–1051. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 11. [Google Scholar] [CrossRef]
- Moshiri, N. ViralMSA: Massively scalable reference-guided multiple sequence alignment of viral genomes. Bioinformatics 2021, 37, 714–716. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Sagulenko, P.; Puller, V.; Neher, R.A. TreeTime: Maximum-likelihood phylodynamic analysis. Virus Evol. 2018, 4, vex042. [Google Scholar] [CrossRef] [PubMed]
- Aksamentov, I.; Roemer, C.; Hodcroft, E.B.; Neher, R.A. Nextclade: Clade assignment, mutation calling and quality control for viral genomes. J. Open Source Softw. 2021, 6, 3773. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Protein structure modeling with MODELLER. Methods Mol. Biol. 2017, 1654, 39–54. [Google Scholar] [CrossRef]
- Schiffrin, B.; Radford, S.E.; Brockwell, D.J.; Calabrese, A.N. PyXlinkViewer: A flexible tool for visualization of protein chemical crosslinking data within the PyMOL molecular graphics system. Protein Sci. 2020, 29, 1851–1857. [Google Scholar] [CrossRef]
- Delgado, J.; Radusky, L.G.; Cianferoni, D.; Serrano, L. FoldX 5.0: Working with RNA, small molecules and a new graphical interface. Bioinformatics 2019, 35, 4168–4169. [Google Scholar] [CrossRef]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent treatment of internal and surface residues in empirical p K a predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. Publ. Protein Sci. 2018, 27, 112–128. [Google Scholar] [CrossRef]
- Schweke, H.; Mucchielli, M.-H.; Chevrollier, N.; Gosset, S.; Lopes, A. SURFMAP: A Software for Mapping in Two Dimensions Protein Surface Features. J. Chem. Inf. Model. 2022, 62, 1595–1601. [Google Scholar] [CrossRef]
- Weng, G.; Wang, E.; Wang, Z.; Liu, H.; Zhu, F.; Li, D.; Hou, T. HawkDock: A web server to predict and analyze the protein–protein complex based on computational docking and MM/GBSA. Nucleic Acids Res. 2019, 47, W322–W330. [Google Scholar] [CrossRef]
- COVID-19 and SARS-CoV-2 Data with Variant Surveillance Reports, Data on Cases and Deaths, and a Standardized, Searchable Research Library. Outbreak.info. 2023. Available online: https://outbreak.info/ (accessed on 3 May 2023).
- Outbreak Info. Global Initiative on Sharing all Influenza Data, GISAID. 2023. Available online: https://www.gisaid.org/ (accessed on 3 May 2023).
- Ao, D.; He, X.; Hong, W.; Wei, X. The rapid rise of SARS-CoV-2 Omicron subvariants with immune evasion properties: XBB.1.5 and BQ.1.1 subvariants. MedComm. 2023, 4, e239. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Jian, F.; Wang, J.; Yu, Y.; Song, W.; Yisimayi, A.; An, R.; Chen, X.; Na Zhang, N.; Wang, Y.; et al. Imprinted SARS-CoV-2 humoral immunity induces convergent Omicron RBD evolution. Nature 2023, 614, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Ciccozzi, M.; Pascarella, S. Two sides of the same coin: The N-terminal and the receptor binding domains of SARS-CoV-2 Spike. Future Virol. 2023, 18, 25–289. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Method | BA.2.75 | CH.1.1 |
|---|---|---|
| FoldX 5.0 | −6.19 ± 0.32 | −3.51 ± 0.30 |
| MM/GBSA | −62.7 ± 0.4 | −61.8 ± 0.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bazzani, L.; Imperia, E.; Scarpa, F.; Sanna, D.; Casu, M.; Borsetti, A.; Pascarella, S.; Petrosillo, N.; Cella, E.; Giovanetti, M.; et al. SARS-CoV CH.1.1 Variant: Genomic and Structural Insight. Infect. Dis. Rep. 2023, 15, 292-298. https://doi.org/10.3390/idr15030029
Bazzani L, Imperia E, Scarpa F, Sanna D, Casu M, Borsetti A, Pascarella S, Petrosillo N, Cella E, Giovanetti M, et al. SARS-CoV CH.1.1 Variant: Genomic and Structural Insight. Infectious Disease Reports. 2023; 15(3):292-298. https://doi.org/10.3390/idr15030029
Chicago/Turabian StyleBazzani, Liliana, Elena Imperia, Fabio Scarpa, Daria Sanna, Marco Casu, Alessandra Borsetti, Stefano Pascarella, Nicola Petrosillo, Eleonora Cella, Marta Giovanetti, and et al. 2023. "SARS-CoV CH.1.1 Variant: Genomic and Structural Insight" Infectious Disease Reports 15, no. 3: 292-298. https://doi.org/10.3390/idr15030029